

Abstract
The helicase from severe acute respiratory syndrome coronavirus (SARS‐CoV) possesses NTPase, duplex RNA/DNA‐unwinding and RNA‐capping activities that are essential for viral replication and proliferation. Here, we have isolated DNA aptamers against the SARS‐CoV helicase from a combinatorial DNA library. These aptamers show two distinct classes of secondary structure, G‐quadruplex and non‐G‐quadruplex, as shown by circular dichroism and gel electrophoresis. All of the aptamers that were selected stimulated ATPase activity of the SARS‐CoV helicase with low‐nanomolar apparent K m values. Intriguingly, only the non‐G‐quadruplex aptamers showed specific inhibition of helicase activities, whereas the G‐quadruplex aptamers did not inhibit helicase activities. The non‐G‐quadruplex aptamer with the strongest inhibitory potency was modified at the 3′‐end with biotin or inverted thymidine, and the modification increased its stability in serum, particularly for the inverted thymidine modification. Structural diversity in selection coupled to post‐selection stabilisation has provided new insights into the aptamers that were selected for a helicase target. These aptamers are being further developed to inhibit SARS‐CoV replication.
Keywords: antiviral agents, aptamers, DNA structures, g‐quadruplexes, nuclease resistance
Non‐Hoogsteen or Hoogsteen? DNA aptamers evolved against the SCV helicase were split into two classes: non‐G‐quadruplex and G‐quadruplex. The non‐G‐quadruplex aptamers inhibited the helicase activities but the G‐quadruplex aptamers did not. Understanding this structure‐based inhibition will be critical for further aptamer development against helicases.
Introduction
Severe acute respiratory syndrome coronavirus (SARS‐CoV) is the major ætiologic agent of an endemic atypical pneumonia. Within a year, more than 8000 cases of SARS and at least 774 deaths were recorded worldwide due to the rapid transmission of the virus by aerosols and by the high mortality rate that is associated with infection.1–4 The SARS‐CoV genome was rapidly sequenced after initial identification,5, 6 and showed a close relationship to the group 2 coronaviruses.7 Although surveillance and infection‐control measures successfully contained the spread of SARS in humans, SARS‐CoV‐like viruses have been identified in bats as a natural reservoir of the virus.8 SARS‐CoV is also able to infect cats and Himalayan palm civets; this further warns of other possible routes for interspecies transmission.9 Currently, the mainstream treatment strategies of SARS involve broad‐spectrum antibiotics, antiviral agents and immunomodulatory therapy, but few drugs are effective against the virus.10 To improve therapeutic options, the development of a wider variety of drugs and more effective methods to combat SARS is important.
SARS‐CoV has a single positive‐stranded RNA genome of ∼29.7 kb in length that encodes two large replicative polyproteins, pp1a (486 kDa) and pp1ab (790 kDa). These precursor polyproteins are processed into a range of structural and nonstructural proteins by viral cysteine proteases (PL2pro and 3CLpro).11 The SARS‐CoV helicase contains three major domains: a putative N‐terminal metal‐binding domain (MBD), a hinge domain and a NTPase/helicase domain. Because the SARS‐CoV helicase is absolutely necessary for subsequent viral replication and proliferation, it is thought to be an attractive target for new anti‐SARS‐CoV drugs. Indeed, viral helicases are proven drug targets due to the success of helicase inhibitors in animal models of herpes simplex virus (HSV) and in the treatment of hepatitis C.12, 13
In previous work, we purified and biochemically characterised the SARS‐CoV helicase, thereby demonstrating that the enzyme belongs to superfamily 1.14 The enzyme unwinds both RNA and DNA duplexes in a 5′ to 3′ polarity by using any NTP or dNTP energy source and has RNA‐capping enzymatic activity.15 We also identified various small‐molecule inhibitors from compound libraries,16 including adamantine‐derived bananins17 and bismuth‐based compounds that target the metal‐binding domain.18, 19
Small‐molecule inhibitors can have therapeutic impact, but their action is confined to a small surface area of a target, and thus single amino acid changes can lead to significantly reduced efficacy. We have observed the evolution of drug‐resistant strains during tests on SARS‐CoV by using our small molecules. Therefore, we employed an in vitro selection strategy, namely, Systematic Evolution of Ligands by EXponential enrichment (SELEX) to isolate DNA sequences that have a high affinity for the SARS‐CoV helicase.20, 21 This approach was previously reported for inhibition of HIV and SIVcpz reverse transcriptase, and was used to overcome the resistance issue.22 Although natural phosphodiester DNA or RNA aptamers are nuclease‐sensitive (and estimated to have a lifetime of a few minutes in blood), they can be chemically modified for stabilisation post‐SELEX. In this study, we also protected the 3′‐end of the aptamer by capping with inverted thymidine or biotin after SELEX.
By using SELEX, we isolated DNA aptamers that show two distinct structures by circular dichroism and gel electrophoresis. Both stimulate ATPase activity with low apparent K m values, but only the non‐G‐quadruplex aptamers efficiently and specifically inhibit the helicase‐unwinding activity of the SARS‐CoV helicase. In comparison, G‐quadruplex‐based aptamers did not inhibit helicase activities. One non‐G‐quadruplex aptamer was then further modified by biotin or inverted‐thymidine capping and evaluated in terms of its inhibition and stability in serum.
Results
Selection of DNA aptamers binding to the SARS‐CoV helicase
Because the SARS‐CoV helicase was overexpressed with a N‐terminal hexahistidine tag, we immobilised the protein on the Ni‐NTA magnetic beads (the solid support for affinity chromatography) and screened for aptamers from a single‐stranded DNA library that contained N30 random core sequences (Figure 1). This method presented a homogeneous population to the aptamer pool and coupled the selection process to facile affinity chromatography binding and elution steps; this decreases the chance of selecting contaminants. To increase the stringency of selection, excess amounts of polydeoxynucleotide (dIdC) were added to the selection buffer to eliminate nonspecifically bound aptamers. Aptamers that bind to the magnetic beads but not the protein itself were eliminated during counter‐selection steps by using the magnetic beads alone. Enrichment of a pool that specifically bound to the SARS‐CoV helicase was checked after the seventh and 15th rounds of selection, but little sequence enrichment was observed. After 20 rounds of selection, the selected oligonucleotides were subsequently amplified by PCR by using unmodified primers to allow for cloning of the aptamer pool. As a result, 26 individual aptamer clones were sequenced, and it was shown that most of the sequences were guanosine rich (Figure 2). Because G‐rich‐characteristic sequences are likely to fold into a G‐quadruplex structure (Figure 3 B), formation of this structure by the aptamers was predicted by bioinformatic analysis and classified into two main groups. In Figure 2 A, 17 sequences were identified that were unlikely to form a G‐quadruplex structure and then further grouped by multiple‐sequence alignment. In group A, aptamers NG1 and NG2 were similar (with only a few base variations), and NG3 had a few further differences. Among group B, high sequence homology was observed at the 3′‐end with a highly conserved sequence (5′‐GTTAGTGTGTT‐3′). Aptamers in group C were apparently orphan sequences that were not closely related to the other groups. In contrast to non‐G‐quadruplex‐forming aptamers, the G‐quadruplex‐forming aptamers had less sequence homology; this suggests that they might use a different mechanism for binding (Figure 2 B). However, a repeat pattern of GG(N) x GG, in which N is a deoxynucleotide and x is the number of repeats, appeared; this pattern is likely to fold into a G‐quadruplex structure. Five aptamers (NG1, NG3, NG8, G5 and G8) including representatives of each group were further characterised.
Figure 1.
DNA aptamer selection strategy. His‐tagged SARS‐CoV helicase was immobilised on Ni‐NTA magnetic beads. The library was incubated with the target beads for binding. Unbound oligonucleotides were washed away, and the bound ones were eluted with the target by imidazole. The selected binders were amplified by PCR by using biotinylated primers. ssDNA was subsequently purified from the PCR product, resulting in an enriched DNA pool, which was used in the next SELEX round. After the last round, the selected aptamers were cloned, sequenced and characterised.
Figure 2.

Sequences of the aptamers that were isolated from the ssDNA pool after twenty rounds of selection against SARS‐CoV helicase. Two groups of sequences were classified by the presence of G‐quadruplex structure that was predicted by QGRS mapper. A) Multiple sequence alignment of non‐G‐quadruplex aptamers by clustalW2. B) Multiple sequence alignment of G‐quadruplex‐forming aptamers. Guanine nucleotides that participated in formation of G‐quadruplex structure were predicted by QGRS mapper and are in bold type‐face and underlined.
Figure 3.
Secondary structure analysis of selected DNA aptamers. A) CD spectrometry was performed for the putative non‐G‐quadruplex aptamers NG1, NG3 and NG8 and putative G‐quadruplex aptamers G5 and G8 in 10 mm Tris–HCl buffer pH 7.5 and 100 mm KCl buffer solution. The concentration of oligonucleotides was 10 μm. B) G‐quadruplex structure. Four guanine bases interact in a square planar configuration to form a G‐quadruplex. Each base interacts with adjacent bases through two hydrogen bonds by Hoogsteen‐like hydrogen bonding. CD spectra of C) aptamer G5 and D) aptamer G8 measured in the presence of either NaCl or KCl buffer solution. E) Mobility of the fluorescently labelled oligonucleotides in a 20 % polyacrylamide gel containing 8 m urea. F) Mobility of folded aptamers on 16 % native polyacrylamide gel supplemented with 50 mm KCl.
Secondary structural analysis of selected DNA aptamers
The secondary structures of five representative aptamers were experimentally analysed by biophysical methods. Circular dichroism spectroscopy and gel electrophoresis were employed to investigate whether the G‐quadruplex structure was formed. Putative non‐G‐quadruplex aptamers NG1, NG3 and NG8 did not have guanine repeats in their sequence, and their CD spectra showed positive peaks at 280 nm (Figure 3 A). This spectrum is characteristic of the canonical B‐form structure.23 However, putative G‐quadruplex aptamers G5 and G8 have guanine‐rich characteristics, and their CD spectra differed from typical B‐form CD spectra. Aptamer G5 formed a parallel G‐quadruplex structure that was typified by a positive maximum peak near 265 nm and a negative peak at 240 nm.24 Aptamer G8 showed a signature of antiparallel G‐quadruplex structure that was characterised by a long‐wavelength positive maximum peak near 295 nm, similar to the previously reported thrombin‐binding aptamer (Figure 3 A).25 Schematic depictions of parallel and antiparallel strand orientation of the G‐quadruplex structures are shown in Figures 3 C and D respectively. Interestingly, no specific peaks at these wavelengths were detected when the aptamers were incubated in the presence of sodium chloride buffer (Figures 3 C and D); this is consistent with previous reports that the G‐quadruplex structure is more stable in the presence of potassium ions than sodium ions.26 KCl was present during aptamer selection so that the potassium‐bound structures are those relevant to binding to the SCV helicase.
As a second line of evidence, the topologies of these quadruplex aptamers were investigated by comparing their mobilities by polyacrylamide gel electrophoresis. The oligonucleotides ran as single bands on denaturing polyacrylamide gels that contained 8 m urea with mobilities that depended on their denatured size (Figure 3 E). By native polyacrylamide gel (Figure 3 F), aptamers NG1, NG3 and NG8 and random sequence migrated as a single band; this is consistent with a lack of G‐quadruplex structure by CD. The thrombin‐binding aptamer had a smaller molecular size and therefore migrated faster. Aptamer G8 (putatively anti‐parallel G‐quadruplex and monomeric by CD) migrated as a single band by gel, whereas aptamer G5 (putatively parallel G‐quadruplex and multimeric by CD) migrated as two bands. The gel electrophoresis data, which shows a slow‐migrating multimeric band for G5, is consistent with the previous CD data supporting a multimeric parallel G‐quadruplex structure for G5.
Inhibition of SARS‐CoV helicase enzymatic activity
We assayed the inhibitory activities of the aptamers against the SARS‐CoV helicase activities. We conducted a helicase assay based on fluorescence resonance energy transfer (FRET) between the partially labelled duplex DNA substrate with fluorophore Cy3 and the quencher black hole quencher 2 (BHQ‐2) as previously described.17, 18 The principle behind this assay is shown in Figure 4 A. Because SARS‐CoV helicase is able to unwind both DNA and RNA duplexes that contain 5′ single‐stranded overhangs,14, 15 the DNA substrate contained a single‐stranded DNA region at their 5′‐ends. The SARS‐CoV helicase was then titrated with increasing concentrations of the selected aptamers. The data were collected and fitted to a logistic equation (Figure 4 B). All the selected aptamers showed inhibition of the SARS‐CoV helicase but differed in their IC50 values. Non‐G‐quadruplex‐forming aptamers NG1, NG3 and NG8 were observed to inhibit duplex substrate unwinding with IC50 values of 87.7, 120.8 and 91.0 nm, respectively (Table 1). However, G‐quadruplex‐forming aptamers G5 and G8 appeared to have little inhibitory activity.
Figure 4.
Inhibition of SARS‐CoV helicase partial duplex DNA unwinding activity. A) Schematic showing the principles behind the FRET‐based fluorimetric assay of helicase activity. B) Unwinding was performed in the presence of various concentrations of the five different aptamers and one random sequence. C) Observation of helicase activity of E. coli DnaB in the absence and presence of 1 μm or 5 μm aptamer NG1, NG3 and NG8. BSA (35 μg) was added as a negative control. The FRET assay was used to observe helicase activity by fluorescence over time. Points shown are an average of triplicate experiments. Error bars represents the standard deviation of triplicate measurements.
Table 1.
The publisher did not receive permission from the copyright owner to include this object in this version of this product. Please refer either to the publisher's own online version of this product or the printed product where one exists.
Specificity of inhibitory SARS‐CoV helicase binding aptamers
To investigate whether the aptamers are general helicase inhibitors, aptamers NG1, NG3 and NG8 were subjected to helicase assays by using Escherichia coli DnaB helicase which also has 5′ to 3′ unwinding directionality. The cloning, expression and purification of this protein were reported previously.17 We found that 1 μm of the aptamers did not inhibit E. coli DnaB in our FRET‐based assay (Figure 4 C), whereas this concentration of aptamers inhibited the SARS‐CoV helicase by more than 90 % (Figure 4 B). A 5 μm concentration of aptamer NG8 also did not show a significant effect on DnaB nucleic acid unwinding activity. Using bovine serum albumin (BSA) instead of helicase did not show the partial duplex DNA unwinding in this experiment. These results suggest that the selected aptamers do not act as general helicase inhibitors.
Effect of aptamers on SARS‐CoV helicase ATPase activity
All helicases bind NTP and exhibit nucleic acid dependent, intrinsic NTPase activity that is necessary for energetically stable duplex unwinding. We investigated the effect of the aptamers on NTP hydrolysis by using a previously developed colorimetric assay that was based on complexation of triphenylmethane (TPM) dye malachite green and ammonium molybdate.14, 17, 27 Interestingly, none of the five selected aptamers inhibited ATPase activity, but instead stimulated ATPase activity at an even lower concentration than a random nucleic acid sequence (Figure 5). This might suggest that aptamers bind into the normal nucleic acid binding site, and then “lock” the enzyme in the conformation for high ATPase turnover. As is a general feature for helicases, NTPase activity of the SARS‐CoV helicase is stimulated in the presence of nucleic acid.14
Figure 5.
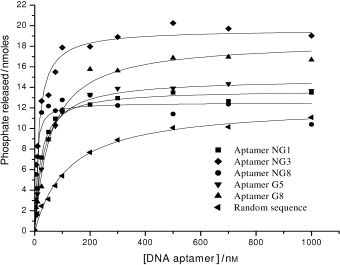
Determination of the apparent strength of binding of aptamers to the SARS‐CoV helicase by hydrolysis of ATP in the presence of varying concentrations of different aptamers. A colorimetric assay was used to measure the amount of phosphate release due to ATP to ADP hydrolysis. Data were shown after subtraction of basal ATPase phosphate release and the ATPase activities in the presence of each aptamer were fitted to a simple Michaelis–Menten model by using average values from three independent determinations.
A set of apparent K m values that were obtained from the half‐maximal stimulation of the ATPase assay was measured for the strength of aptamer binding to the SARS‐CoV helicase. The ATPase activity of the enzyme displayed simple single‐site behaviour in the presence of different aptamers (Figure 5). The apparent half‐stimulation values for the binding of aptamers NG1, NG3, NG8, G5 and G8 were 20.8, 13.3, 5.4, 30.9 and 56.5 nm, respectively, and were obtained from a simple Michaelis–Menten fit to the ATPase data (Figure 5 and Table 1). The non‐G‐quadruplex aptamers (NG1, NG3 and NG8) bound more tightly than the G‐quadruplex‐forming aptamers (G5 and G8). As the SARS‐CoV helicase is a nucleic acid binding protein, a random sequence was also shown to bind to the protein, but less tightly (with an apparent K m value 122 nm, which is 22‐fold weaker binding affinity than observed for aptamer NG8).
Stability of modified aptamers in Foetal Bovine Serum (FBS)
Natural phosphodiester DNA or RNA ligands are susceptible to degradation by nucleases, which makes them unsuitable for therapeutic use. Capping the 3′‐end is one of the commonly used approaches to block 3′ to 5′ exonuclease attack. Therefore, the stability of aptamer NG8 that was modified with 3′‐biotin or 3′‐inverted thymidine was evaluated in DMEM that was supplemented with 5 and 10 % heat‐inactivated FBS (Figure 6). Under both 5 and 10 % serum conditions, unmodified aptamer NG8 was quickly digested by nuclease in serum (Figures 6 C and F). In contrast, the 3′‐biotin and 3′‐inverted thymidine aptamer NG8 was strongly resistant to nuclease attack in serum. In the case of the 3′‐inverted thymidine aptamer, the oligo remained intact for up to 72 and 31 h in 5 and 10 % FBS respectively (Figures 6 A and D). The 3′‐inverted thymidine modification had a higher stability than the 3′‐biotin modification, but both modifications were significantly more stable than the unmodified aptamer.
Figure 6.
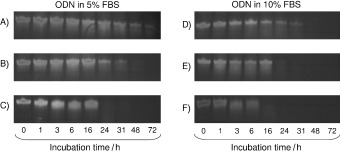
Stability of 3′‐inverted thymidine‐, 3′‐biotin‐modified and unmodified NG8 in 5 and 10 % FBS. Aptamer NG8 with various modifications was mixed with 5 or 10 % FBS and incubated at 37 °C. Aliquots of the reaction mixture were taken at different time points and loaded into 20 % denaturing urea PAGE; A) 3′‐inverted thymidine aptamer NG8; B) 3′‐biotin aptamer NG8 and C) unmodified aptamer NG8 in 5 % FBS; D) 3′‐inverted thymidine aptamer NG8; E) 3′‐biotin aptamer NG8 and F) unmodified aptamer NG8 in 10 % FBS.
Biochemical assays of modified aptamers
We examined whether modification of aptamer NG8 with either 3′‐inverted thymidine or 3′‐biotin might have any effect on its inhibitory activities against the SCV helicase. In Figure 7 A, both modified aptamers inhibited the nucleic acid unwinding activity of SCV helicase with slightly lower IC50 values than unmodified ones (Table 1), whereas a modified random sequence control showed no inhibition. Specificity analysis by using E. coli DnaB revealed that the modified aptamers did not show general helicase inhibition; this suggests that the modified aptamers retained their specificity (Figure 7 B). In the ATPase assay, the modified aptamers stimulated ATPase activity in a similar fashion to the unmodified aptamers (Figure 7 C and Table 1). Our results suggest that the 3′‐modification has little impact on the inhibitory activity of aptamer NG8.
Figure 7.
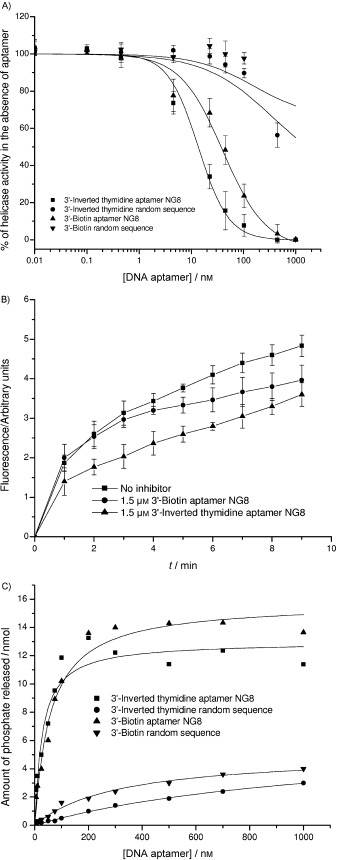
Biochemical assays of modified aptamers NG8. A) Inhibition of helicase activity of the SCV helicase in the presence of various concentrations of the modified aptamers. B) Specificity of modified aptamers by using E. coli DnaB. C) Effect of modified aptamers on the ATPase activity of SCV helicase.
Discussion and Conclusions
We have shown the selection of DNA aptamers that bind to SARS‐CoV helicase by using Ni‐NTA magnetic beads as an immobilisation matrix by systematic evolution of ligands by exponential enrichment (SELEX) method. DNA aptamers were structurally characterised and found to be either G‐quadruplex or non‐G‐quadruplex by circular dichroism and gel electrophoresis. Five aptamers were further characterised, and all showed high apparent binding affinity to the SARS‐CoV helicase and stimulated ATPase activity. This would suggest that the aptamers bind to the nucleic acid binding sites, and lock them into a high ATPase turnover conformation. However, the G‐quadruplex aptamers were unable to inhibit helicase‐unwinding activity; this suggests that the G‐quadruplex structure is incapable of locking the conformation.
Previously, RNA aptamers that were selected against the HCV NS3 helicase domain that belongs to superfamily 2 with 3′ to 5′ polarity, showed consensus stem–loop structures.28 A hypothetical schematic model was suggested in which it was proposed that the aptamers acted as a decoy by competitively binding to the nucleic acid binding domain and inhibiting the translocation step due to the conserved stem–loop structure.28 Although the exact model mechanism of our SCV helicase aptamer remains to be extensively studied, the aptamers NG1, NG3 and NG8 might perform in a similar way in that the helicase first binds to the 5′‐overhangs of the DNA aptamers and translocates in a 5′ to 3′ direction by using the ATP energy source. When the enzyme meets the aptamer, the enzyme is locked in a conformation; this explains the high ATPase turnover. ATPase activities of both SARS‐CoV and HCV helicases have been previously reported to be highly stimulated by oligomeric nucleic acids; this illustrates why the aptamer actually stimulates rather than inhibits ATP hydrolysis.14, 29
Aptamers NG1, NG3 and NG8 were shown to be potent aptamers with relatively high binding strength and inhibition. Interestingly, some aptamers (G5 and G8) were isolated from the same library but revealed sequences that were typical of G‐quadruplex structure with guanine duplets that were separated by two or three deoxynucleotides (Figure 2), and the G‐quadruplex structure was confirmed by circular dichroism and gel electrophoresis. Such sequences have been selected in previous aptamer studies, for example DNA aptamers against thrombin, and RNA aptamers against prions,30, 31 and are stabilised by the potassium ions that were also present in our selection buffer.32–34 However, this is the first demonstration of structural specificity in enzymatic inhibition by aptamers that were derived from the same pool under similar conditions. Furthermore, we stabilised one of the aptamers with 3′‐inverted thymidine and 3′‐biotin modifications and showed that the aptamer was still potent enough to inhibit the SCV helicase, and in particular that the 3′‐inverted thymidine modification resulted in a significantly improved half life in serum.
RNA aptamers against the SARS‐CoV helicase were recently reported.35 Stem–loop RNA aptamers were identified with an IC50 for helicase inhibition of 1.2 nm, but no G‐quadruplex families were reported. Similarly to our aptamers, these RNA aptamers also only inhibited helicase activities and did not inhibit ATPase activities; this suggests that this pattern of behaviour is common to aptamers that were selected against helicase targets. The RNA aptamers do provide higher structural diversities that help inhibit with a slightly stronger IC50 than DNA aptamers against the same target. However, natural RNA is very susceptible to nuclease attack and is digested in less than 5 minutes in vivo; this makes RNA incompatible for many applications without stabilisation.
This work revealed differential activities against the SARS‐CoV helicase by G‐quadruplex and non‐G‐quadruplex aptamers that were isolated from the same library pool. The non‐G‐quadruplex aptamers strongly and specifically inhibited helicase activities, but the G‐quadruplex aptamers did not. However, all of the selected aptamers stimulated ATPase activity strongly. The strength of binding and IC50 values were in the low‐nanomolar range, which is comparable to other aptamers that target nucleic acid binding proteins.28, 36 The aptamer that shows the best potency in the characterisation was modified by 3′‐end capping with inverted thymidine and biotin. The modifications made the aptamer resistant to nuclease attack and retained the aptamer's potency. This study provides new mechanistic insight into the structure and function relationships of aptamers on a helicase target, and these aptamers will be further investigated as a new approach for the inhibition of SARS‐CoV replication.
Experimental Section
Oligonucleotides: Aptamers that were modified with biotin and inverted thymidine at 3′‐end were synthesised by Molecular informatrix Laboratory Ltd (MiL, Hong Kong) and purified by HPLC, unless otherwise specified. The same manufacturer was used to synthesise all of the primers and aptamers that were applied in this study. Thrombin‐binding aptamer (TBA) and random sequence were designed as controls respectively as follows: TBA, 5′‐GGT TGG TGT GGT TGG‐3′; Random sequence, 5′‐GAA GGC CGT TCT CAG TGA ACA ACA AAA ACT‐3′
Cloning, expression and purification of the SARS‐CoV helicase and magnetic bead preparation: The SARS‐CoV helicase domain (nsp13‐pp1ab, accession number NP_828870, originally denoted as nsp10) was cloned, expressed and purified as previously described.14 Protein‐bound Ni‐NTA magnetic beads were prepared by first equilibrating a 5 % slurry (45 μL; ∼13 μg protein binding capacity) of Ni‐NTA magnetic beads (Qiagen, Hilden, Germany) into buffer A (50 mm Tris‐HCl pH 6.8, 150 mm KCl, 20 mm imidazole, 0.05 % Tween‐20, 0.1 % Triton X‐100). The equilibrated beads were resuspended in buffer A (100 μL) and purified SARS‐CoV helicase (0.25 mg mL−1, 200 μL) was added and mixed by rotation for 20 min at 4 °C. The excess SARS‐CoV helicase in the supernatant was washed away by buffer A, and the magnetic beads were then further washed buffer A, (3×1 mL), concentrated to 0.25 μg μL−1 SARS‐CoV helicase in buffer A and stored at 4 °C.
In vitro selection: In vitro selection was performed immediately after immobilisation of the SARS‐CoV helicase to prevent protein oxidation. The selection of SARS‐CoV‐helicase‐binding aptamer relied on magnetic separation with the helicase that was immobilised on the Ni‐NTA magnetic beads (Qiagen). The starting point of the selection process was a random degenerate ssDNA oligonucleotide library that was chemically synthesised and purified by HPLC. This library, referred to as “SelexApt”, was composed of 30 random nucleotides that were flanked by sequences that were suitable for amplification: 5′‐CCG TAA TAC GAC TCA CTA TAG GGG AGC TCG GTA CCG AAT TC‐(N30)‐AAG CTT TGC AGA GAG GAT CCT T‐3′. Primers that anneal to the 5′‐ and 3′‐sequences flanking the degenerate region of SelexApt used during the selection and cloning were: “SelexF”, 5′‐CCG TAA TAC GAC TCA CTA TAG GGG AGC TCG GTA CCG AAT TC‐3′; “SelexR”, 5′‐AAG GAT CCT CTC TGC AAA GCT T‐3′; in non‐biotinylated and 5′‐biotinylated forms, respectively (HPLC purified). Iterative rounds of aptamer selection and amplification during the SELEX process were modified from the previous protocol.37 In the first round of selection, one nanomole of “SelexApt” was diluted in buffer A (100 μL) in a PCR tube, heated to 90 °C for 5 min and cooled at 4 °C. The library was added to buffer A (2 mL) that contained bovine serum albumin (BSA, 1 μg mL−1), dIdC (0.1 μg mL−1) and bead‐bound SARS‐CoV Helicase (100 pmol). The material was incubated with rotation for 30 min at 25 °C. The tubes were then applied to a magnet separator (Dynal, Oslo, Norway) to remove unbound nucleic acids in the supernatant. The protein and the beads were washed buffer A (10× 1 mL) and mixed by gentle inversion for each wash step. The helicase and bound aptamers were eluted from the Ni‐NTA magnetic beads with buffer B (30 μL; 50 mm Tris‐HCl pH 6.8, 150 mm KCl, 500 mm imidazole, 0.02 % Tween‐20, 0.1 % Triton) and transferred to PCR tubes for amplification. PCR reactions in a 100 μL volume contained Pfx polymerase (1.25 U; Invitrogen), primers “SelexF” and biotinylated “SelexR” (1 μm), dNTPs (0.1 mm), MgSO4 (0.5 mm) and enhancer solution (0.1×). Amplification conditions were 2 min at 95 °C; 15 cycles of 30 s at 95 °C; 30 s at 54.8 °C; 30 s at 68 °C; 2 min at 68 °C. After each amplification step, the PCR product (90 μL) and NaCl (5 m, 23 μL) were mixed with M‐280 streptavidin magnetic beads (1 mg; Dynal) for 10 min at room temperature, then washed with buffer A (3×1 mL). Single‐stranded aptamers (non‐biotinylated strand) were separated from the immobilised complementary strand by using a 5 min incubation of fresh NaOH (100 mm, 50 μL). The tubes were applied to a magnet separator and the ssDNA was removed and diluted into buffer A (50 μL). The next round of selection was performed only when the concentration of the eluent was higher than that of the last wash. We also checked an enrichment of ssDNA that bound to SARS‐CoV helicase that had been immobilised on magnetic beads at rounds 7, 15 and 20.
Cloning, sequencing and classification: After round 20, the material was amplified by PCR with SelexF and non‐biotinylated SelexR primers, and the products were purified with a PCR clean‐up system (Qiagen), cloned into pCR®‐Blunt II‐TOPO® vector (Invitrogen) and heat‐shock transformed into E. coli DH10α competent cells. Colonies were picked for each sample, and the plasmids were purified by Qiaprep spin mini‐prep (Qiagen). The plasmids were sequenced by using a M13 reverse primer in the Big‐dye Terminator kit. The presence of G‐quadruplex structure in aptamers was predicted by quadruplex‐forming G‐rich sequences (QGRS) mapper.38 Multiple sequence alignment was performed by ClustalW2.39
Circular dichroism (CD) spectroscopy: Oligonucleotides (10 μm), were resuspended in Tris–HCl (10 mm, pH 7.5) buffer that contained KCl (100 mm) or NaCl (100 mm). Samples were heated at 90 °C for 5 min, followed by gradual cooling to room temperature. CD spectra were collected on a JASCO J720 spectropolarimeter (JASCO, Tokyo, Japan) at 320–200 nm, by using 4 scans at 100 nm min−1, 1 s response time, 1 nm bandwidth. Quartz cells with an optical path length of 1 mm were used for the measurements.
Gel electrophoresis: Oligonucleotide samples were labelled with fluorescein isothiocyanate (FITC) at the 3′‐end and prepared in sodium phosphate buffer (50 mm) supplemented with potassium chloride (100 mm). The oligonucleotide concentration was 5 μm. Prior to performing the gel assay, the purity of the commercially synthesised oligomers was checked by running them on 20 % polyacrylamide gels that contained urea (8 m). For non‐denaturing gel assays, the samples (at 5 μm strand concentration) were heated at 95 °C for 5 min and incubated at 60 °C for 15 h. 16 % native polyacrylamide gel electrophoresis was performed in Tris/borate/EDTA (TBE) buffer (1×) that was supplemented with KCl (50 mm). Bands in the gels were visualised under UV transillumination.
Assays of aptamer activities: Concentration and quality of chemically synthesised aptamers were accurately measured by A 260 and 15 % urea polyacrylamide gel electrophoresis respectively. Apparent K m values were obtained by determining the concentration that gave half‐maximal stimulation of ATPase activity. The ATPase assay was performed by measuring the amount of phosphate release by using a colorimetric method that was based on complexation with malachite green and ammonium molybdate (AM/MG reagent) as we described previously.14 The helicase assay was based on FRET between the fluorophore Cy3 and black hole quencher 2 (BHQ‐2) as we described previously, suitable for a 5′ to 3′ helicase.17 Two oligomers were synthesised and purified by HPLC: DT20Cy3 (5′‐(DT20Cy3)TTT TTT TTT TTT TTT TTT TTC GAG CAC CGC TGC GGC TGC ACC(Cy3)‐3′), and Release BHQ‐2 (5′‐(BHQ2)GGT GCA GCC GCA GCG GTG CTC G‐3′) (Proligo, Boulder, CO, USA). The two oligomers were annealed by mixing a 1:1.2 ratio of DT20Cy3/ReleaseBHQ‐2 at a concentration of 8.2 μm (of DT20Cy3) in Tris‐HCl (10 mm, pH 8.5), heating to 90 °C, then cooling slowly to 40 °C over 1 h. The reaction was carried out in a 1 mL volume of DT20Cy3/ReleaseBHQ‐2 (5 nm), Release oligomer (10 nm, 5′‐GGT GCA GCC GCA GCG GTG CTC G‐3′), BSA (0.1 mg mL−1), SCV helicase (2 nm), MgCl2 (5 mm), KCl (100 mm) KCl and Tris‐HCl (50 mm, pH 6.8) at 25 °C for 1 min. The reaction was initiated by the addition of ATP (0.5 mm). The change in fluorescence (excitation 550 nm, emission 570 nm) after 1 min was used to monitor the extent of unwinding of the duplex. The E. coli DnaB FRET assay was carried out with DnaB (10 μg) under the same conditions. Controls to determine the specificity by using the E. coli DnaB helicase were performed as we described previously.17 Data were fitted by using Origin 6.0 (Microcal Software, Northampton, USA).
Stability of modified oligonucleotides in foetal bovine serum (FBS): Unmodified, 3′‐biotin and 3′‐inverted thymidine‐modified aptamer NG8 were incubated in 5 % and 10 % FBS at 37 °C. Aliquots of the reaction were removed at different time intervals for electrophoresis and reactions were stopped by adding formamide gel loading buffer to each sample. All samples were then run in 20 % urea PAGE in TBE buffer (1×) and visualised by staining with SYBR Gold nucleic acid stain (Molecular Probes, Eugene, OR, USA).
Acknowledgements
This research was funded by the Hong Kong Research Grants Council (RGC) under CERG grant HKU 7589/05M. We thank Dr. J. Huang for useful discussion of the manuscript and Prof. H. Sun for advice regarding the circular dichroism experiments.
References
- 1. Peiris J. S., Lai S. T., Poon L. L., Guan Y., Yam L. Y., Lim W., Nicholls J., Yee W. K., Yan W. W., Cheung M. T., Cheng V. C., Chan K. H., Tsang D. N., Yung R. W., Ng T. K., Yuen K. Y., Lancet 2003, 361, 1319–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fouchier R. A., Kuiken T., Schutten M., van Amerongen G., van Doornum G. J., Hoogen B. G. van den, Peiris M., Lim W., Stohr K., Osterhaus A. D., Nature 2003, 423, 240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kuiken T., Fouchier R. A., Schutten M., Rimmelzwaan G. F., van Amerongen G., van Riel D., Laman J. D., de Jong T., van Doornum G., Lim W., Ling A. E., Chan P. K., Tam J. S., Zambon M. C., Gopal R., Drosten C., van der Werf S., Escriou N., Manuguerra J. C., Stohr K., Peiris J. S., Osterhaus A. D., Lancet 2003, 362, 263–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ksiazek T. G., Erdman D., Goldsmith C. S., Zaki S. R., Peret T., Emery S., Tong S., Urbani C., Comer J. A., Lim W., Rollin P. E., Dowell S. F., Ling A. E., Humphrey C. D., Shieh W. J., Guarner J., Paddock C. D., Rota P., Fields B., DeRisi J., Yang J. Y., Cox N., Hughes J. M., LeDuc J. W., Bellini W. J., Anderson L. J., N. Engl. J. Med. 2003, 348, 1953–1966. [DOI] [PubMed] [Google Scholar]
- 5. Rota P. A., Oberste M. S., Monroe S. S., Nix W. A., Campagnoli R., Icenogle J. P., Penaranda S., Bankamp B., Maher K., Chen M. H., Tong S., Tamin A., Lowe L., Frace M., DeRisi J. L., Chen Q., Wang D., Erdman D. D., Peret T. C., Burns C., Ksiazek T. G., Rollin P. E., Sanchez A., Liffick S., Holloway B., Limor J., McCaustland K., Olsen‐Rasmussen M., Fouchier R., Gunther S., Osterhaus A. D., Drosten C., Pallansch M. A., Anderson L. J., Bellini W. J., Science 2003, 300, 1394–1399. [DOI] [PubMed] [Google Scholar]
- 6. Marra M. A., Jones S. J., Astell C. R., Holt R. A., Brooks‐Wilson A., Butterfield Y. S., Khattra J., Asano J. K., Barber S. A., Chan S. Y., Cloutier A., Coughlin S. M., Freeman D., Girn N., Griffith O. L., Leach S. R., Mayo M., McDonald H., Montgomery S. B., Pandoh P. K., Petrescu A. S., Robertson A. G., Schein J. E., Siddiqui A., Smailus D. E., Stott J. M., Yang G. S., Plummer F., Andonov A., Artsob H., Bastien N., Bernard K., Booth T. F., Bowness D., Czub M., Drebot M., Fernando L., Flick R., Garbutt M., Gray M., Grolla A., Jones S., Feldmann H., Meyers A., Kabani A., Li Y., Normand S., Stroher U., Tipples G. A., Tyler S., Vogrig R., Ward D., Watson B., Brunham R. C., Krajden M., Petric M., Skowronski D. M., Upton C., Roper R. L., Science 2003, 300, 1399–1404. [DOI] [PubMed] [Google Scholar]
- 7. Mayor L. R., Fleming K. P., Muller A., Balding D. J., Sternberg M. J., J. Mol. Biol. 2004, 340, 991–1004. [DOI] [PubMed] [Google Scholar]
- 8. Hon C.‐C., Lam T.‐Y., Shi Z.‐L., Drummond A. J., Yip C.‐W., Zeng F., Lam P.‐Y., Leung F. C.‐C., J. Virol. 2008, 82, 1819–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Martina B. E., Haagmans B. L., Kuiken T., Fouchier R. A., Rimmelzwaan G. F., van Amerongen G., Peiris J. S., Lim W., Osterhaus A. D., Nature 2003, 425, 915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Stockman L. J., Bellamy R., Garner P., PLoS Med. 2006, 3, e343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ziebuhr J., Curr. Opin. Microbiol. 2004, 7, 412–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Betz U. A., Fischer R., Kleymann G., Hendrix M., Rubsamen‐Waigmann H., Antimicrob. Agents Chemother. 2002, 46, 1766–1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kwong A. D., Rao B. G., Jeang K. T., Nat. Rev. Drug. Discov. 2005, 4, 845–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tanner J. A., Watt R. M., Chai Y. B., Lu L. Y., Lin M. C., Peiris J. S., Poon L. L., Kung H. F., Huang J. D., J. Biol. Chem. 2003, 278, 39578–39582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ivanov K. A., Thiel V., Dobbe J. C., van der Meer Y., Snijder E. J., Ziebuhr J., J. Virol. 2004, 78, 5619–5632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kao R. Y., Tsui W. H., Lee T. S., Tanner J. A., Watt R. M., Huang J. D., Hu L., Chen G., Chen Z., Zhang L., He T., Chan K. H., Tse H., To A. P., Ng L. W., Wong B. C., Tsoi H. W., Yang D., Ho D. D., Yuen K. Y., Chem. Biol. 2004, 11, 1293–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tanner J. A., Zheng B. J., Zhou J., Watt R. M., Jiang J. Q., Wong K. L., Lin Y. P., Lu L. Y., He M. L., Kung H. F., Kesel A. J., Huang J. D., Chem. Biol. 2005, 12, 303–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yang N., Tanner J. A., Zheng B.‐J., Watt R. M., He M.‐L., Lu L.‐Y., Jiang J.‐Q., Shum K.‐T., Lin Y.‐P., Wong K.‐L., Lin M. C. M., Kung H.‐F., Sun H., Huang J.‐D., Angew. Chem. 2007, 119, 6584–6588; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2007, 46, 6464–6468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yang N., Tanner J. A., Wang Z., Huang J. D., Zheng B. J., Zhu N., Sun H., Chem. Commun. (Cambridge) 2007, 4413–4415. [DOI] [PubMed] [Google Scholar]
- 20. Tuerk C., Gold L., Science 1990, 249, 505–510. [DOI] [PubMed] [Google Scholar]
- 21. Ellington A. D., Szostak J. W., Nature 1990, 346, 818–822. [DOI] [PubMed] [Google Scholar]
- 22. Kissel J. D., Held D. M., Hardy R. W., Burke D. H., AIDS. Res. Hum. Retroviruses. 2007, 23, 699–708. [DOI] [PubMed] [Google Scholar]
- 23. Fukuda H., Katahira M., Tanaka E., Enokizono Y., Tsuchiya N., Higuchi K., Nagao M., Nakagama H., Genes Cells 2005, 10, 953–962. [DOI] [PubMed] [Google Scholar]
- 24. Hardin C. C., Watson T., Corregan M., Bailey C., Biochemistry 1992, 31, 833–841. [DOI] [PubMed] [Google Scholar]
- 25. Nagatoishi S., Tanaka Y., Tsumoto K., Biochem. Biophys. Res. Commun. 2007, 352, 812–817. [DOI] [PubMed] [Google Scholar]
- 26. Rachwal P. A., Findlow I. S., Werner J. M., Brown T., Fox K. R., Nucleic Acids Res. 2007, 35, 4214–4222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Baykov A. A., Evtushenko O. A., Avaeva S. M., Anal. Biochem. 1988, 171, 266–270. [DOI] [PubMed] [Google Scholar]
- 28. Nishikawa F., Funaji K., Fukuda K., Nishikawa S., Oligonucleotides 2004, 14, 114–129. [DOI] [PubMed] [Google Scholar]
- 29. Suzich J. A., Tamura J. K., Palmer‐Hill F., Warrener P., Grakoui A., Rice C. M., Feinstone S. M., Collett M. S., J. Virol. 1993, 67, 6152–6158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Vairamani M., Gross M. L., J. Am. Chem. Soc. 2003, 125, 42–43. [DOI] [PubMed] [Google Scholar]
- 31. Weiss S., Proske D., Neumann M., Groschup M. H., Kretzschmar H. A., Famulok M., Winnacker E. L., J. Virol. 1997, 71, 8790–8797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bryan T. M., Jarstfer M. B., Methods 2007, 43, 332–339. [DOI] [PubMed] [Google Scholar]
- 33. Oganesian L., Graham M. E., Robinson P. J., Bryan T. M., Biochemistry 2007, 46, 11279–11290. [DOI] [PubMed] [Google Scholar]
- 34. Mergny J. L., Helene C., Nat. Med. 1998, 4, 1366–1367. [DOI] [PubMed] [Google Scholar]
- 35. Jang K. J., Lee N.‐R., Yeo W.‐S., Jeong Y.‐J., Kim D.‐E., Biochem. Biophys. Res. Commun. 2008, 366, 738–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nimjee S. M., Rusconi C. P., Sullenger B. A., Annu. Rev. Med. 2005, 56, 555–583. [DOI] [PubMed] [Google Scholar]
- 37. Murphy M. B., Fuller S. T., Richardson P. M., Doyle S. A., Nucleic Acids Res. 2003, 31, e110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kikin O., D′Antonio L., Bagga P. S., Nucleic Acids Res. 2006, 34, W676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Larkin M. A., Blackshields G., Brown N. P., Chenna R., McGettigan P. A., McWilliam H., Valentin F., Wallace I. M., Wilm A., Lopez R., Thompson J. D., Gibson T. J., Higgins D. G., Bioinformatics 2007, 23, 2947–2948. [DOI] [PubMed] [Google Scholar]