

Abstract
With the current trajectory of the 2019‐nCoV outbreak unknown, public health and medicinal measures will both be needed to contain spreading of the virus and to optimize patient outcomes. Although little is known about the virus, an examination of the genome sequence shows strong homology with its better‐studied cousin, SARS‐CoV. The spike protein used for host cell infection shows key nonsynonymous mutations that might hamper the efficacy of previously developed therapeutics but remains a viable target for the development of biologics and macrocyclic peptides. Other key drug targets, including RNA‐dependent RNA polymerase and coronavirus main proteinase (3CLpro), share a strikingly high (>95 %) homology to SARS‐CoV. Herein, we suggest four potential drug candidates (an ACE2‐based peptide, remdesivir, 3CLpro‐1 and a novel vinylsulfone protease inhibitor) that could be used to treat patients suffering with the 2019‐nCoV. We also summarize previous efforts into drugging these targets and hope to help in the development of broad‐spectrum anti‐coronaviral agents for future epidemics.
Keywords: antiviral agents, coronavirus, 2019-nCoV, SARS, spike proteins, RdRp, 3CLpro
What′s sauce for the goose? Little is known about the coronavirus causing the current outbreak; however, it shares strong sequence homology with its better‐studied cousin SARS‐CoV. Based on previous studies of targeting SARS‐CoV, we suggest four potential candidates that could be used to drug the viral spike protein, RNA‐dependent RNA polymerase, and coronavirus main proteinase.
Introduction
The 2019 novel coronavirus (2019‐nCoV) is a newly emerged human‐infectious coronavirus (CoV) that originated in a Wuhan seafood market but has quickly spread in and beyond China.1 As of February 4th 2020, there have been more than 20 000 diagnosed cases and 426 confirmed deaths (Xinhua News). As the pathogenesis of this virus is yet to be understood, there are few treatment options available to healthcare professionals who are fighting this epidemic at the front line. Praise needs to be given to Chinese researchers who have acted quickly to isolate and sequence the virus. The availability of the virus genome sequence (GenBank ID: MN908947.3) makes it possible to identify treatments. Although it is essential to develop vaccines, small molecules, and biological therapeutics to specifically target the 2019‐nCoV virus, it is unlikely that any effort made at the moment will benefit patients in the current outbreak. However, 2019‐nCoV shares 82 % sequence identity with severe acute respiratory syndrome‐related coronavirus (SARS‐CoV, GenBank ID: NC_004718.3) and more than 90 % sequence identity in several essential enzymes (see figures below). Therefore, what we have learned from several medicinal chemistry studies on SARS‐CoV and the Middle East Respiratory Syndrome (MERS‐CoV) may be directly used to help us treat 2019‐nCoV. CoV relies on its spike proteins to bind a host cell‐surface receptor for entry (Figure 1).2 For 2019‐nCoV, it is evident that this receptor is angiotensin‐converting enzyme 2 (ACE2).3 After the virus′ entry into the host cell, its positive genomic RNA attaches directly to the host ribosome for the translation of two large, coterminal polyproteins that are processed by proteolysis into components for packaging new virions.4 Two proteases that participate in this proteolysis process are the coronavirus main proteinase (3CLpro) and the papain‐like protease (PLpro).5 In order to replicate the RNA genome, the CoV encodes a replicase that is an RNA‐dependent RNA polymerase (RdRp).6 These four proteins are essential for the pathogen. Therapeutics currently targeting spike, RdRp, 3CLpro, and PLpro are possible treatments for 2019‐nCoV. In this viewpoint, we shall analyze similarities in spike, RdRp, 3CLpro, and PLpro proteins between 2019‐nCoV and SARS‐CoV, and suggest possible prevention and treatment options. Most compounds discussed will be experimental compounds and drug candidates; for a review of repurposed drugs for treating coronaviruses and other viruses, see Li et al.7 As little is known so far about the virulence of this virus, we shall also discuss the interactions between spike and ACE2 that might challenge the current view that 2019‐nCoV is less virulent than SARS‐CoV owing to weaker interactions between spike and ACE2.
Figure 1.

Lifecycle of a coronavirus entering a host cell and replicating inside. The (+)‐stranded RNA is released upon viral entry; this starts the process of generating the viral coat and replicating the RNA genome.
The Spike Protein
Both 2019‐nCoV and SARS‐CoV encode a large (2019‐nCoV: 1253 aa; SARS‐CoV: 1273 aa) spike protein. The sequence identity of this protein between the two origins is 76 %. A large variation exists at the N terminus (Figure 2 A). The spike protein has two regions, S1 and S2. For the SARS‐CoV, there is a receptor binding domain (RBD) in the S1 region that interacts with ACE2 with high affinity. The current assumption is that 2019‐nCoV also engages this RBD to bind ACE2 for entry into its human host cell. The alignment of the RBD from the two origins shows 73.5 % sequence identity (Figure 2 A). However, many nonconserved mutations that interact directly with ACE2 have accumulated in the two structural regions (1 and 2 in Figure 2 B).2 Both crystal and cryo‐EM structures of the SARS‐CoV spike‐ACE2 complex (PDB IDs: 2AJF and 6ACD) have showed that only regions 1 and 2 engage in hydrogen bonding and hydrophobic interactions with ACE2. As many residues in these two regions have been replaced in 2019‐nCoV, this will lead to a loss of some of these interactions. It has also been predicted that the 2019‐nCoV RBD interacts with ACE2 more weakly than the SARS‐CoV RBD.3 However, both regions are highly looped structures. Large variations in the two regions will inevitably lead to structural rearrangements that potentiate novel and possibly even stronger interactions with ACE2. For region 2, there is almost no similarity in the sequence between the two virus origins; however, it is premature to presume that the protein will fold in the same way to interact with ACE2. Assuming that region 2 of 2019‐nCoV folds in the same way as that in SARS‐CoV, F486 will be placed right at the position that engages in strong hydrophobic interactions with both L79 and M82 in ACE2 (Figure 2 C). These interactions do not exist in the SARS RBD–ACE2 complex due to the significantly smaller L472 in that position. Another residue that potentially engages in strong hydrogen‐bonding interactions is Q474, which is at the correct distance to engage a rearranged Q24 in ACE2 (Figure 2 C). In region 1, the critical residue Y484 in SARS‐CoV is replaced by Q498 in 2019‐nCoV. However, P499, a proline that is a known secondary‐structure disruptor, is expected to lead to a structural rearrangement of region 1 in the SARS‐CoV RBD. In combination with Q498 and N501, new hydrogen‐bonding interactions that might involve K353 and other residues in ACE2 could form (Figure 2 D). Another notable difference between 2019‐nCoV and SARS‐CoV in the RBD is K417(2019‐nCoV)/V404(SARS‐CoV). This is a residue in the middle of the concave RBD binding surface that involves no interaction between SARS‐CoV and ACE2. However, the long, positively charged K417 could potentially engage in strong hydrogen bonding and salt‐bridge interactions with H34 and D30, respectively, in ACE2 (Figure 2 E). Although molecular simulation may be used to analyze all these possible interactions in detail, uncertainty about them will persist until the structure of the 2019‐nCoV‐RBD–ACE2 complex is determined by crystallography or cryo‐EM. Given the urgency of the matter, there must be multiple research groups working on this. We hope to see their results within a short time. Alternatively, the 2019‐nCoV RBD could be expressed, and its affinity toward ACE2 could be independently determined biochemically and compared with that of the SARS‐CoV RBD. Before any solid experimental results are available, any claims about the weaker binding of the 2019‐nCoV RBD toward ACE2 than that of the SARS‐CoV RBD is premature.
Figure 2.

A) Sequence alignment for the amino acids between the 2019‐nCoV and SARS‐CoV spike RBD domains. Conserved (pink arrows) and nonconserved (black arrows) mutations are highlighted. Gray: hydrophobic aliphatic, orange: neutral aromatic, yellow: thiol and sulfide, green: hydroxy, red: basic, blue: carboxylic acid, brown: primary amide, pink: proline. B) Various binding interactions between the 2019‐nCov spike protein (pink) and ACE2 (blue; spike protein homology model built by using Modeller, based upon PDB ID: 2AJF) in regions 1 and 2. C)–E) Zoomed in views of several spike protein–ACE2 interactions depicted in (B). Residues before the forward slash refer to 2019‐nCoV; the amino acid after the slash refers to its corresponding identity in SARS‐CoV.
The roles of the SARS‐CoV spike protein in receptor binding and membrane fusion make it an ideal target for vaccine and antiviral development. The development of SARS vaccines based on the spike protein has been summarized in several previous reviews.8, 9, 10, 11, 12 Several strategies including live‐attenuated SARS‐CoV, killed SARS‐CoV, DNA vaccines and viral vectored vaccines have been successfully used to vaccinate against animal SARS‐CoVs.8, 13, 14 Similar ideas could be applied in developing 2019‐nCoV vaccines. Alternative approaches are to directly use the 2019‐nCoV RBD in combination with immunity‐promoting adjuvants as a vaccine to trigger the human body to develop antibodies for the 2019‐nCoV RBD, thereby neutralizing the virus.15
Although there are published results about therapeutic antibodies and peptides developed to neutralize the SARS‐CoV spike protein, they are expected to have little use in neutralizing 2019‐nCoV. As discussed above, the two engaging regions in the spike RBD for binding ACE2 are very different between SARS‐CoV and 2019‐nCoV. Antibodies and peptides targeting two regions in the SARS‐CoV RBD are expected to interact weakly with the 2019‐nCoV RBD. Novel antibodies and therapeutic peptides that interact potently with the 2019‐nCoV RBD can be used to block its interaction with ACE2. Several research groups, including ours, have developed methods for building macrocyclic peptide libraries and applying them to the quick identification of macrocyclic peptide ligands for drug targets.16, 17, 18, 19, 20, 21 Application of these libraries to search for potent ligands for the 2019‐nCoV RBD or the two ACE2‐engaging peptide regions will potentially lead to rapid discovery of anti‐2019‐nCoV macrocyclic peptides. Although our group has initiated this effort, the lengthy drug‐discovery process will not make it possible to help patients in the current epidemic in this way. Learning from the study of SARS‐CoV, a possible alternative is the direct use of peptides derived from the 2019‐nCoV RBD and ACE2. Peptides derived from both the SARS‐CoV RBD and ACE2 have been developed as novel therapeutics against SARS‐CoV infection by blocking SARS‐CoV RBD–ACE2 binding. For example, a peptide that overlaps the RBD sequence (aa 471–503) can specifically block the binding of ACE2 to the SARS‐CoV RBD and inhibit the entry of SARS‐CoV into Vero cells with an IC50 of 41.6 μm.22 One peptide comprising two ACE2 motifs (aa 22–44 and 351–357) linked by glycine exhibited potent anti‐SARS activity with an IC50 value of 0.1 μm.23 Before any potent therapeutics to neutralize the 2019‐nCoV RBD–ACE2 interaction are available, a possible quick solution to blocking this interaction is to use 2019‐nCoV RBD‐based peptides and cocktails thereof.
RNA‐dependent RNA polymerase
Although 2019‐nCoV and SARS‐CoV share 82 % sequence identity at their genomic RNA level, their RdRp proteins share a remarkable 96 % sequence identity (Figure 3 A). RdRp involves a very large and deep groove as an active site for the polymerization of RNA. Residues that show variations between the 2019‐nCoV and SARS‐CoV RdRps are mostly distal to this active site (Figure 3 B).24 This high sequence conservation between the two enzymes makes it very likely that any potent agents developed for the SARS‐CoV RdRp will exhibit equal potency and efficacy on the 2019‐nCoV RdRp. Although not extensively explored, several agents exist that target the SARS‐CoV RdRp or its catalyzed polymerization process. One such compound found to show antiviral activity was aurintricarboxylic acid (ATA; Figure 4). ATA is an anionic polymer shown to bind to a variety of protein targets, including gp120 of HIV‐1 and HIV‐2, that has been demonstrated to prevent SARS‐CoV replication (IC50=0.2 mg mL−1).25, 26, 27 Despite computational models validated against known ATA targets predicting RdRp as the bound target, no experimental evidence has demonstrated this relationship.28 Beyond this exception, the remaining RdRp inhibitors have been nucleoside analogues, and these provide the most promising avenue towards disrupting viral RNA replication. The nucleoside analogue ribavirin (RBV, Figure 4) has been tested against SARS‐CoV, and in SARS‐ and MERS‐infected patients.29, 30, 31, 32 At best, efficacy with RBV was inconclusive, with some studies showing a worsening of patient outcomes (as reviewed by Stockman, et al.).33 Exonuclease activity by the enzyme nsp14 has been shown to be able to remove mismatches as well as incorporated nucleoside analogues, and inactivation of nsp14′s exonuclease activity has been shown to increase the efficacy of nucleosides like RBV.32, 34 In order to develop nucleoside analogues to effectively inhibit viral RNA replication, the nucleoside must either evade detection by the exonuclease or must outcompete exonuclease activity. Remdesivir (GS‐5734) is an excellent example of the latter. An adenosine analogue prodrug with a 1′‐nitrile, it displayed potent efficacy against SARS and MERS in human airway epithelial (HAE) cell models and in mice (IC50=0.069 and 0.074 μm for SARS‐CoV and MERS‐CoV, respectively, in HAE).35 Broad‐spectrum activity against various bat coronaviruses was also demonstrated.35 The susceptibility of CoV to remdesivir was shown to be increased in strains with inactivated exonuclease activity.36 CoV resistance to remdesivir was studied in the model β‐coronavirus, murine hepatitis virus (MHV). MHV passaged in the presence of the parent nucleoside, GS‐441524 (which contains a 5′‐hydroxy group instead of a phosphoramidate), developed two mutations in the RdRp, F476L and V553L. These mutations conferred a 5.6‐fold increase in resistance to remdesivir in MHV and a sixfold increase in resistance when the homologous mutations were introduced to the RdRp of SARS‐CoV (0.01 vs. 0.06 μm). Mice infected with this resistant SARS‐CoV had significantly lower lung viral titers 4 days post‐infection.36 Altogether, remdesivir has been shown to outcompete the proofreading ability of nsp12, and mutations that confer resistance attenuate virulence. Efforts towards drugging coronavirus in a RdRp manner should provide a basis not only to develop therapeutics for 2019‐nCoV, but could provide broad‐spectrum antivirals useful for future CoV outbreaks.
Figure 3.

A) Sequence alignment for the amino acids between the 2019‐nCoV RdRp and the SARS‐CoV RdRp. Conserved (pink arrows) and nonconserved (black arrows) mutations are highlighted. Gray: hydrophobic aliphatic, orange: neutral aromatic, yellow: thiol and sulfide, green: hydroxy, red: basic, blue: carboxylic acid, brown: primary amide, pink: proline. B) Crystal structure of the SARS‐CoV RdRp active site (PDB ID: 6NUS).
Figure 4.
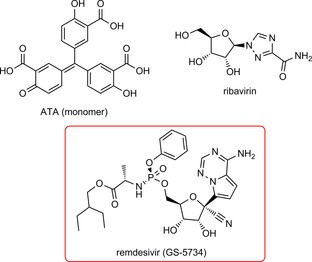
Structure of compounds inhibiting SARS‐CoV viral replication through the mechanistic action of RdRp. The most promising candidate, remdesivir, is highlighted in the red box.
3CLpro and PLpro
3CLpro and PLpro are two proteases that process the polypeptide translation product from the genomic RNA into the structural and nonstructural protein components vital for the replication and packaging of a new generation of viruses. PLpro also serves as a deubiquitinase that functions to deubiquitylate host cell proteins such as interferon regulatory factor 3 (IRF3) as well as to inactivate the pathway for nuclear factor κ‐light‐chain‐enhancer of activated B cells (NF‐κB).37 This leads to immune suppression in the cells of the host being infected by the virus. Because both proteases are vital to the virus for replication and controlling the host cell, they are viable targets for antiviral agents. Similar to the RdRp protein, 2019‐nCoV and SARS‐CoV share a remarkable 96 % sequence identity in their decoded 3CLpro (Figure 5 A). 3CLpro naturally forms a dimer, and each monomer contains two regions: the N‐terminal catalytic region and the C‐terminal region (Figure 5 B).37, 38 Most residues in the catalytic region that display variations between two origins are on the protein surface. Although S46(2019‐nCoV)/A(SARS‐CoV) might possibly interact with either substrates or inhibitors that bind to the active site, the small structural change from A to S is expected not to interfere significantly with the binding of small‐molecule inhibitors to the active site of 2019‐nCoV 3CLpro (Figure 5 C). Small‐molecule agents that potently inhibit the SARS‐CoV 3CLpro are expected to function similarly toward the 2019‐nCoV 3CLpro. Unlike 3CLpro, PLpro from the two origins shares only 83 % sequence identity (Figure 6 A). Residue variations between the two origins cover almost all the surface of PLpro. These substantial variations in amino acid composition are expected to influence how the two PLpro enzymes interact with their ligands. However, the three secondary‐structure components that form the active site do not vary in the two PLpro proteins (Figure 6 B).39 It is possible that an inhibitor developed for the SARS‐CoV PLpro would also work for the 2019‐nCoV PLpro.
Figure 5.
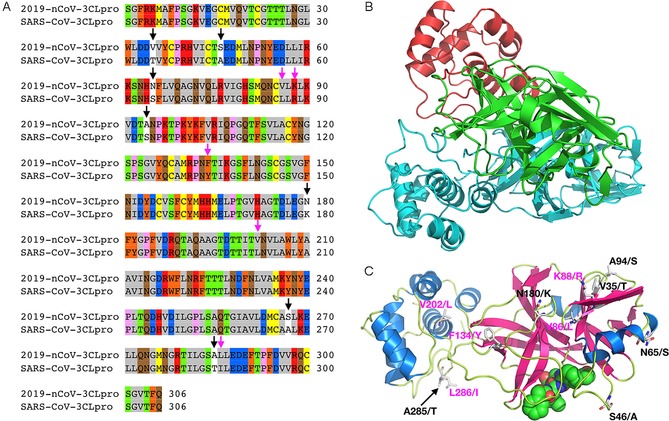
A) Sequence alignment for the amino acids between the 2019‐nCoV 3CLpro and the SARS‐CoV 3CLpro. Conserved (pink arrows) and nonconserved (black arrows) mutations are highlighted. Gray: hydrophobic aliphatic, orange: neutral aromatic, yellow: thiol and sulfide, green: hydroxy, red: basic, blue: carboxylic acid, brown: primary amide, pink: proline. B) A 2019‐nCoV 3CLpro structure modeled by using Modeller based on the SARS‐CoV 3CLpro structure (PDB ID: 2A5I); green: catalytic domain of the first monomeric unit, red: C‐terminal domain of the first monomeric unit, cyan: second monomeric unit. C) An alternative view of the predicted 2019‐nCoV 3CLpro monomeric structure (color‐coded by secondary structure) bound to an aza‐peptide inhibitor, showing conserved (pink) and nonconserved (black) mutations (not shown: S267/A). Residues before the forward slash refer to 2019‐nCoV; the amino acid after the slash refers to its corresponding identity in SARS‐CoV.
Figure 6.

A) Sequence alignment for the amino acids between the 2019‐nCoV PLpro and the SARS‐CoV PLpro. Conserved (pink arrows) and nonconserved (black arrows) mutations are highlighted. Gray: hydrophobic aliphatic, orange: neutral aromatic, yellow: thiol and sulfide, green: hydroxy, red: basic, blue: carboxylic acid, brown: primary amide, pink: proline. B) Crystal structure of the SARS‐CoV PLpro in complex with ubiquitin aldehyde (PDB ID: 4MM3).
Over the last two decades, much of the research in drugging SARS‐CoV has focused on the development of small‐molecule, peptide, and peptidomimetic inhibitors of 3CLpro and PLpro. Many of the inhibitors are in the micromolar range in terms of binding to and inhibiting the two proteases.40, 41, 42, 43, 44, 45 However, a few low‐nm‐range inhibitors have been identified that can be used in combination with other protease‐inhibitor therapies to help combat the virus.46 For the purpose of this section, the inhibitors will be divided into categories based on the proteases that are inhibited to stop the virus from taking control of the host cells. Each of the compounds was tested in terms of a SARS‐CoV, a MERS‐CoV, or a deubiquitylate cell model. Several hundred small molecules have been developed to inhibit 3CLpro and PLpro; however, these are the most potent since the early 2000s. The classifications with structures and inhibitory concentrations are summarized in Figure 7. These compounds are in the low‐μm range in terms of inhibition, leaving room for further development. However, extensive SAR studies have already been performed on these final‐stage products that guide the researcher in knowing which substituents to modify when targeting 2019‐nCoV. This summary can also guide both researchers and health professionals in using combinational therapy with two or more of these compounds, as this has already been done in terms of treating people suffering with a CoV infection. One of these compounds (highlighted as 3CLpro‐1 in Figure 7) has an IC50 value against SARS‐CoV of 200 nm.42 This potency could be adequate to combat 2019‐nCoV.
Figure 7.

A representation of the top CoV protease inhibitors providing a scaffold to perform SAR studies in terms of designing novel small‐molecule protease inhibitors for 2019‐nCoV[40–45, 49]. 3CLpro‐1, the most potent inhibitor, is highlighted.
Both 3CLpro and PLpro are cysteine proteases; therefore covalent inhibitors with high potencies could potentially be developed for specific targeting. Recently, Simmons and co‐workers developed a class of potential covalent cysteine protease inhibitors that specifically target CoV entry.46 No direct relationship to 3CLpro and PLpro was drawn; however, this class of vinylsulfone small molecule was able to inhibit replication of the virus in the nanomolar range. Simmons′ group discovered that inhibition of serine proteases (by using camostat) as well as cysteine protease (by using their vinylsulfone protease inhibitors) is able to combat SARS‐CoV. The survival of mice suffering from SARS‐CoV who were treated with this combination therapy significantly increased in comparison to the control group. The group studied several variations of vinylsulfone small molecules, which are shown below with their corresponding IC50 values in Figure 8 (compounds A to C). Once again, these vinylsulfone small molecules provide an additional scaffold for SAR development. They can also be tested against the specific 3CLpro and PLpro in order to further elucidate their specific mechanism of action. Given their high potency against SARS‐CoV, it is possible that they are equally potent against 2019‐nCoV.
Figure 8.
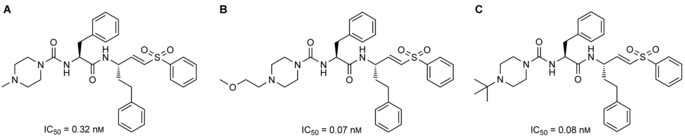
Lead vinylsulfone protease inhibitors that prevent the entry of CoV and, in combination with camostat, increase the survival rate of mice infected with SARS‐CoV.
More distant members of the orthocoronavinae subfamily can also provide inspiration for new therapeutic regimens, such as with studies done with feline coronavirus (FCoV) and its mutated form, feline infectious peritonitis virus (FIPV). The peptidyl bisulfite adducts GC376 and NPI64 (Figure 9) were both found to be potent inhibitors of FIPV replication at 0.04 μm.47 3CLpro of FIPV and SARS‐CoV share about 50 % sequence identity, but the overall structure is conserved. In a FRET‐based activity assay, the IC50 value of GC376 against recombinant SARS 3CLpro was found to be 4.9 times that against FIPV 3CLpro (4.35 vs. 0.72 μm, respectively).48 In combination with a low compound cytotoxicity (CC50>150 μm and CC50=61.91 μm in CRFK cells for GC376 and NPI64, respectively), these masked‐aldehyde warheads should be investigated for efficacy on the 3CLpro of 2019‐nCoV as soon as possible.
Figure 9.

Peptidyl bisulfide adducts that have been demonstrated to prevent viral replication in the feline coronavirus FIPV. The most promising candidate, GC376, was shown to produce similar levels of inhibition against SARS‐CoV 3CLpro in a FRET‐based activity assay.
Conclusions
2019‐nCoV and SARS‐CoV share very high sequence identity in their RdRp and 3CLpro proteins. Previous work has resulted in the discovery of some potent small‐molecule therapeutics based on these two proteins for SARS‐CoV. We envision that remdesivir and 3CLpro‐1 could be directly applied to treat 2019‐nCoV. As remdesivir is a drug undergoing a clinical trial, the authorities in China could negotiate with Gilead for the possible use of this drug for patients suffering with 2019‐nCoV. Other potential small‐molecule therapeutics for 2019‐nCoV are the molecules shown in Figures 8 and 9. The 2019‐nCoV spike RBD is significantly different from the SARS‐CoV spike RBD, especially in two regions when binding to ACE2. This difference effectively rules out the use of previously developed antibodies and therapeutic peptides for the SARS‐CoV spike RBD. However, a possible quick solution to inhibit the RBD–ACE2 interaction so as to prevent the infection is to use peptides derived from RBD and ACE2 and cocktails thereof.
Conflict of interest
The authors declare no conflict of interest.
Acknowledgements
Research support in the Texas A&M Drug Discovery Center is provided by the National Institutes of Health (grants R01GM121584 and R01GM127575), the Cancer Prevention & Research Institute of Texas (grant RP170797), and the Welch Foundation (grant A‐1715). We thank Sunshine Zea Leeuwon for providing assistance in designing all the figures.
J. S. Morse, T. Lalonde, S. Xu, W. R. Liu, ChemBioChem 2020, 21, 730.
A previous version of this manuscript has been deposited on a preprint server (https://doi.org/10.26434/chemrxiv.11728983.v1)
Contributor Information
Dr. Shiqing Xu, Email: shiqing.xu@tamu.edu.
Prof. Dr. Wenshe Ray Liu, Email: wliu@chem.tamu.edu.
References
- 1. Zhou P., Yang X. L., Wang X. G., Hu B., Zhang L., Zhang W., Si H. R., Zhu Y., Li B., Huang C. L., Chen H. D., Chen J., Luo Y., Guo H., Jiang R. D., Liu M. Q., Chen Y., Shen X. R., Wang X., Zheng X. S., Zhao K., Chen Q. J., Deng F., Liu L. L., Yan B., Zhan F. X., Wang Y. Y., Xiao G., Shi Z., bioRxiv 2020, https://www.biorxiv.org/content/10.1101/2020.01.22.914952v2 (accessed Jan. 25, 2020). [Google Scholar]
- 2. Li F., Li W., Farzan M., Harrison S. C., Science 2005, 309, 1864–1868. [DOI] [PubMed] [Google Scholar]
- 3. Dong N., Yang X., Ye L., Chen K., Chan E. W. C., Yang M., Chen S., bioRxiv 2020, https://www.biorxiv.org/content/10.1101/2020.01.20.913368v2 (accessed Jan. 25, 2020). [Google Scholar]
- 4. Baranov P. V., Henderson C. M., Anderson C. B., Gesteland R. F., Atkins J. F., Howard M. T., Virology 2005, 332, 498–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ziebuhr J., Snijder E. J., Gorbalenya A. E., J. Gen. Virol. 2000, 81, 853–879. [DOI] [PubMed] [Google Scholar]
- 6. Xu X., Liu Y., Weiss S., Arnold E., Sarafianos S. G., Ding J., Nucleic Acids Res. 2003, 31, 7117–7130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Li C. C., Wang X. J., Wang H. C. R., Drug Discovery Today 2019, 24, 726–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cavanagh D., Avian Pathol. 2003, 32, 567–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Du L., He Y., Zhou Y., Liu S., Zheng B. J., Jiang S., Nat. Rev. Microbiol. 2009, 7, 226–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jiang S., He Y., Liu S., Emerging Infect. Dis. 2005, 11, 1016–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Taylor D. R., Vaccine 2006, 24, 863–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Roper R. L., Rehm K. E., Expert Rev. Vaccines 2009, 8, 887–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Holmes K. V., J. Clin. Invest. 2003, 111, 1605–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Navas-Martin S. R., Weiss S., J. Neurovirol. 2004, 10, 75–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. He Y., Zhou Y., Liu S., Kou Z., Li W., Farzan M., Jiang S., Biochem. Biophys. Res. Commun. 2004, 324, 773–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. O′Neil K. T., Hoess R. H., Jackson S. A., Ramachandran N. S., Mousa S. A., DeGrado W. F., Proteins 1992, 14, 509–515. [DOI] [PubMed] [Google Scholar]
- 17. McLafferty M. A., Kent R. B., Ladner R. C., Markland W., Gene 1993, 128, 29–36. [DOI] [PubMed] [Google Scholar]
- 18. Hipolito C. J., Suga H., Curr. Opin. Chem. Biol. 2012, 16, 196–203. [DOI] [PubMed] [Google Scholar]
- 19. Palei S., Becher K. S., Nienberg C., Jose J., Mootz H. D., ChemBioChem 2019, 20, 72–77. [DOI] [PubMed] [Google Scholar]
- 20. Xiao W., Wang Y., Lau E. Y., Luo J., Yao N., Shi C., Meza L., Tseng H., Maeda Y., Kumaresan P., Liu R., Lightstone F. C., Takada Y., Lam K. S., Mol. Cancer Ther. 2010, 9, 2714–2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang X. S., Chen P. H. C., Hampton J. T., Tharp J. M., Reed C. A., Das S. K., Wang D. S., Hayatshahi H. S., Shen Y., Liu J., Liu W. R. A., Angew. Chem. Int. Ed. 2019, 58, 15904–15909; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 16051–16056. [Google Scholar]
- 22. Hu H., Li L., Kao R. Y., Kou B., Wang Z., Zhang L., Zhang H., Hao Z., Tsui W. H., Ni A., Cui L., Fan B., Guo F., Rao S., Jiang C., Li Q., Sun M., He W., Liu G., J. Comb. Chem. 2005, 7, 648–656. [DOI] [PubMed] [Google Scholar]
- 23. Han D. P., Penn-Nicholson A., Cho M. W., Virology 2006, 350, 15–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kirchdoerfer R. N., Ward A. B., Nat. Commun. 2019, 10, 2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. González R. G., Blackburn B. J., Schleich T., Biochim. Biophys. Acta Nucleic Acids Protein Synth. 1979, 562, 534–545. [DOI] [PubMed] [Google Scholar]
- 26. Cushman M., Wang P., Chang S. H., Wild C., De Clercq E., Schols D., Goldman M. E., Bowen J. A., J. Med. Chem. 1991, 34, 329–337. [DOI] [PubMed] [Google Scholar]
- 27. He R., Adonov A., Traykova-Adonova M., Cao J., Cutts T., Grudesky E., Deschambaul Y., Berry J., Drebot M., Li X., Biochem. Biophys. Res. Commun. 2004, 320, 1199–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yap Y., Zhang X., Andonov A., He R., Comput. Biol. Chem. 2005, 29, 212–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chiou H. E., Liu C. L., Buttrey M. J., Kuo H. P., Liu H. W., Kuo H. T., Lu Y. T., Chest 2005, 128, 263–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Muller M. P., Dresser L., Raboud J., McGeer A., Rea E., Richardson S. E., Mazzulli T., Loeb M., Louie M., Canadian S. R. N., Pharmacotherapy 2007, 27, 494–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Al-Tawfiq J. A., Momattin H., Dib J., Memish Z. A., J. Infect. Dis. 2014, 20, 42–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Smith E. C., Blanc H., Vignuzzi M., Denison M. R., PLoS Pathog. 2013, 9, e1003565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Stockman L. J., Bellamy R., Garner P., PLoS Med. 2006, 3, e343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ferron F., Subissi L., De Morais A. T. S., Le N. T. T., Sevajol M., Gluais L., Decroly E., Vonrhein C., Bricogne G., Canard B., Imbert I., Proc. Natl. Acad. Sci. USA 2018, 115, E162–E171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sheahan T. P., Sims A. C., Graham R. L., Menachery V. D., Gralinski L. E., Case J. B., Leist S. R., Pyrc K., Feng J. Y., Trantcheva I., Bannister R., Park Y., Babusis D., Clarke M. O., Mackman R. L., Spahn J. E., Palmiotti C. A., Siegel D., Ray A. S., Cihlar T., Jordan R., Denison M. R., Baric R. S., Sci. Transl. Med. 2017, 9, eaal3653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Agostini M. L., Andres E. L., Sims A. C., Graham R. L., Sheahan T. P., Lu X., Smith E. C., Case J. B., Feng J. Y., Jordan R., Ray A. S., Cihlar T., Siegel D., Mackman R. L., Clarke M. O., Baric R. S., Denison M. R., mBio 2018, 9, e00221-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Báez-Santos Y. M., John St. S. E., Mesecar A. D., Antiviral Res. 2015, 115, 21–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lee T. W., Cherney M. M., Huitema C., Liu J., James K. E., Powers J. C., Eltis L. D., James M. N., J. Mol. Biol. 2005, 353, 1137–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ratia K., Kilianski A., Baez-Santos Y. M., Baker S. C., Mesecar A., PLoS Pathog. 2014, 10, e1004113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chen X., Chou C. Y., Chang G. G., Antiviral Chem. Chemother. 2009, 19, 151–156. [DOI] [PubMed] [Google Scholar]
- 41. Cheng K. W., Cheng S. C., Chen W. Y., Lin M. H., Chuang S. J., Cheng I. H., Sun C. Y., Chou C. Y., Antiviral Res. 2015, 115, 9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kumar V., Shin J. S., Shie J. J., Ku K. B., Kim C., Go Y. Y., Huang K. F., Kim M., Liang P. H., Antiviral Res. 2017, 141, 101–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Liu W., Zhu H. M., Niu G. J., Shi E. Z., Chen J., Sun B., Chen W. Q., Zhou H. G., Yang C., Bioorg. Med. Chem. 2014, 22, 292–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Park J. Y., Jeong H. J., Kim J. H., Kim Y. M., Park S. J., Kim D., Park K. H., Lee W. S., Ryu Y. B., Biol. Pharm. Bull. 2012, 35, 2036–2042. [DOI] [PubMed] [Google Scholar]
- 45. Park J. Y., Ko J. A., Kim D. W., Kim Y. M., Kwon H. J., Jeong H. J., Kim C. Y., Park K. H., Lee W. S., Ryu Y. B., J. Enzyme Inhib. Med. Chem. 2016, 31, 23–30. [DOI] [PubMed] [Google Scholar]
- 46. Zhou Y., Vedantham P., Lu K., Agudelo J., R. Carrion, Jr. , Nunneley J. W., Barnard D., Pohlmann S., McKerrow J. H., Renslo A. R., Simmons G., Antiviral Res. 2015, 116, 76–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kim Y., Shivanna V., Narayanan S., Prior A. M., Weerasekara S., Hua D. H., Kankanamalage A. C., Groutas W. C., Chang K. O., J. Virol. 2015, 89, 4942–4950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kim Y., Liu H., Galasiti Kankanamalage A. C., Weerasekara S., Hua D. H., Groutas W. C., Chang K. O., Pedersen N. C., PLoS Pathog. 2016, 12, e1005531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Song Y. H., Kim D. W., Curtis-Long M. J., Yuk H. J., Wang Y., Zhuang N., Lee K. H., Jeon K. S., Park K. H., Biol. Pharm. Bull. 2014, 37, 1021–1028. [DOI] [PubMed] [Google Scholar]