

Abstract
There is renewed interest in the role of respiratory virus infections in the pathogenesis of asthma and in the development of exacerbations in pre‐existing disease. This is due to the availability of new molecular and experimental tools. Circumstantial evidence points towards a potentially causative role as well as to possibly protective effects of certain respiratory viruses in the cause of allergic asthma during early childhood. In addition, it now has become clear that exacerbations of asthma, in children as well as adults, are mostly associated with respiratory virus infections, with a predominant role of the common cold virus: rhinovirus. Careful human in vitro and in vivo experiments have shown that rhinovirus can potentially stimulate bronchial epithelial cells to produce pro‐inflammatory chemokines and cytokines, may activate cholinergic‐ or noncholinergic nerves, increase epithelial‐derived nitric oxide synthesis, upregulate local ICAM‐1 expression, and can lead to nonspecific T‐cell responses and/or virus‐specific T‐cell proliferation. Experimental rhinovirus infections in patients with asthma demonstrate features of exacerbation, such as lower airway symptoms, variable airways obstruction, and bronchial hyperresponsiveness, the latter being associated with eosinophil counts and eosinophilic cationic protein levels in induced sputum. This suggests that multiple cellular pathways can be involved in rhinovirus‐induced asthma exacerbations. It is still unknown whether these mechanisms are a distinguishing characteristic of asthma. Because of the limited effects of inhaled steroids during asthma exacerbations, new therapeutic interventions need to be developed based on the increasing pathophysiological knowledge about the role of viruses in asthma.
Keywords: airway hyperresponsiveness, asthma exacerbation, common cold, immune responses, respiratory virus infection
Introduction
Considering the rapid progress in the present research on the pathogenesis, pathophysiology, and immunopathology of asthma and chronic obstructive pulmonary disease (COPD), it is remarkable that the understanding of the potential involvement of respiratory virus infections in these diseases is far from settled. This seems to be due to the only recent availability of data from (i) epidemiological studies using modern virological techniques, and (ii) experimental studies in animals as well as in man. These approaches are increasing our knowledge on the potential role of respiratory viruses in the onset of asthma and COPD as well as in inducing exacerbations in pre‐existing disease.
Respiratory viruses in asthma and COPD
In general, the information on the complex involvement of respiratory virus infections in the pathogenesis of asthma is increasing. It shows circumstantial evidence of a broad spectrum of effects: between a potentially causative or facilitatory role [1, 2] and an inhibitory or protective influence [3, 4], probably depending on the type and age of infection. With regard to the importance of respiratory virus infections in exacerbations of pre‐existing asthma, research during the past decade has provided clear positive evidence, from both epidemiological and experimental studies [5, 6]. This is less clear in COPD. Latent virus infections have been implicated in its pathogenesis [7], but this still needs confirmation. Furthermore, there are only very recent data on a potentially (limited?) role of respiratory viruses in causing exacerbations of pre‐existing COPD [8].
It is likely that respiratory viruses can have pro‐inflammatory and immunomodulatory effects within the airways [9]. This has most extensively been studied for respiratory syncytial virus (RSV) [10, 11] and for human rhinovirus [5, 6, 12]. This review will focus on the latter, and will particularly discuss the association between the pro‐inflammatory and pathophysiological effects of rhinoviruses in patients with asthma.
Rhinovirus and asthma exacerbations
There is little doubt that rhinoviruses are important as a trigger of asthma exacerbations. Several clinical and epidemiological studies have described a close temporal association of respiratory virus infections with exacerbations in patients with asthma [13]. Respiratory viruses can be identified in 10%–44% of the asthma exacerbations in adults [14, 15], whilst in children identification rates vary from 26% to 83% [16, 17, 18, –19]. The use of the sensitive polymerase chain reaction technique to detect rhinovirus and coronavirus in the two most recent studies has resulted in the highest identification rates so far [15, 19]. Among the various respiratory viruses identified, rhinovirus predominates in most of these studies, accounting for about 50% of the detected viruses [15, 16, 17, 18, –19]. The incidence of rhinovirus infections may even be higher in asthmatic patients as compared with nonasthmatic subjects [13, 20], although rhinovirus shedding in the absence of cold symptoms does not seem to be associated with clinical worsening of asthma [17]. Taken together, these findings indicate that rhinovirus infections may have a causal role in exacerbations of asthma. Preliminary data confirm that this also occurs in exacerbations of COPD, but to a lesser degree [8].
Experimental rhinovirus infections in asthma
The observation that common colds are important in induc‐ing exacerbations in asthma, offers the perspective of experimental studies that can be safely performed in humans in vivo [21]. Indeed, experimental infections have been shown to be a useful tool for investigating the effects of rhinovirus infections in allergic disease or asthma [22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, –35]. Such a model allows careful patient selection and monitoring, and intensive assessment of the rhinovirus‐induced effects, under controlled circumstances and timing. Thus, the effects of a rhinovirus infection can be assessed at the level of asthma symptoms, changes in use of asthma medication, lung function, and airway hyperresponsiveness, as well as the underlying airway pathology.
Physiological effects
So far, experimental rhinovirus infections in patients with asthma and/or atopic rhinitis have not been shown to induce a significant change in lung function, e.g. FEV1, when measured during laboratory visits [22,24, 25, –26,28, 29, 31, 35]. This has been considered to be reassuring in terms of patient‐safety of the experimental model; however, recent results from our laboratory are demonstrating that frequent home‐recordings of FEV1 (three times daily) do decrease in atopic asthmatic patients in the acute phase of an experimental RV16 infection [27]. Furthermore, the maximal decrease in FEV1 in the acute phase of the infection, expressed as percentage of the recent personal best, correlated significantly with the observed increase in airways hyperresponsiveness [27]. This suggests that there is transient worsening of airways obstruction after rhinovirus infection in asthma, which may improve spontaneously during the day, or as a consequence of repeated deep‐breath manoeuvres as are being performed in the lung function laboratory. This points at either increased sensitivity to bronchoconstrictive stimuli and/or a reduced bronchodilating effect of a deep breath [36].
The effects of experimental rhinovirus infection on airway responsiveness vary among different studies, using different rhinovirus serotypes. Lemanske et al. and Gern et al. have demonstrated an enhanced hypersensitivity to histamine and allergen challenge after experimental rhinovirus 16 (RV16) infection in nonasthmatic patients with atopic rhinitis [24, 34], which was significantly different from the lack of response in normals [34]. Others, however, have not observed such an effect when using rhinovirus 39 [35]. In asthmatic subjects, Halperin et al. has found increased hypersensitivity to histamine in only four of 22 subjects after experimental rhinovirus (serotype 39 and HH strain) infection [22]. More recently, in our laboratory Cheung et al. have shown that RV16 increases asthma symptoms, coinciding an increase in the maximal bronchoconstrictive response to methacholine up to 15 days after infection [25], pointing towards the development of excessive airway narrowing due to a rhinovirus infection. In a similar design, we have shown a significant increase in airway sensitivity to histamine in asthmatic subjects after RV16 infection, which was most pronounced in those subjects who had severe cold symptoms [28]. Taken together, these data indicate that patients with asthma and/or atopic rhinitis may suffer more prominent pathophysiological consequences from a rhinovirus infection as compared with nonatopic nonasthmatic subjects, suggesting an interactive effect of virus‐induced airways inflammation with features of the underlying disease, such as altered airway geometry [37, 38] and/or pre‐existing allergic sensitization/inflammation [39].
Animal studies
Experimental animal studies have provided strong evidence of the induction of airways inflammation by respiratory viruses, as is apparent from epithelial shedding and morphological changes after, for example, parainfluenza infection in guinea pigs [40]. It has not, however, been resolved to what extent such induced inflammation contributes to the accompanying physiological changes. Initial findings suggested a major role of inflammatory cells, such as T cells, basophils, and their mediators [41]. And indeed, virus‐induced airway hyperresponsiveness could be transferred by bronchoalveolar lavage cells recovered after viral infection in guinea pigs [42]. However, more recent results obtained by using antibodies against pro‐inflammatory cytokines, such as IL‐5, are indicative of (partly) independent induction of airways hyperresponsiveness and eosinophilic airways inflammation by respiratory viruses in guinea pigs in vivo [43].
Apparently, neurogenic mechanisms can also be responsible for virus‐induced hyperresponsiveness in animals. Interestingly, this seems to be mainly due to impaired inhibitory mechanisms [44]. Such impaired neurogenic protection might arise from reduced production or activity of the neuropeptide degrading enzyme neutral endopeptidasen [45], or to dysfunction of inhibitory cholinergic M2‐receptors [46]. In addition, there is evidence that virus‐induced airways hyperresponsiveness in guinea pigs can be caused by impaired synthesis of nitric oxide (NO) [47]. This is potentially important, since it has recently been shown that rhinovirus can induce NO‐synthase expression in human primary bronchial epithelial cells in vitro [48], and that such NO can reduce rhinovirus replication and the induced inflammatory cytokine release by epithelial cells [49]. Interestingly, our latest data in asthmatics in vivo are in keeping with such a protective mechanism by NO after rhinovirus infection (Fig. 1) [50], but this hypothesis remains to be tested using appropriate pharmacological blocking agents in asthmatics in vivo.
Figure 1.
. Relationship between the change in exhaled NO and the change in airway hyperresponsiveness to histamine (PC20) during the first week after experimental infection with RV16 in patients with asthma in vivo. The patients with the greatest increase in airway hyperresponsiveness (reduction in PC20) are showing the lowest increase in exhaled NO and vice versa. The regresssion line has been drawn. Rs, Spearman rank correlation coefficient. Duplicated with permission from [50].
Inflammatory response in humans
What do we know about the inflammatory response to a rhinovirus infection in asthmatic and/or atopic rhinitis patients? It is likely that epithelial cells are playing an active and major role in this. In vitro, there is evidence that infection with rhinovirus in pulmonary epithelial cells and fibroblasts induces de novo synthesis of pro‐inflammatory cytokines, such as GM‐CSF, IL‐11, and IL‐6, and the chemokines IL‐8 and RANTES [51, 52, 53, 54, –55] by activating transcription factors such as NF‐κB [53, 56, 57]. Such epithelial chemokine release appears to be prolonged until 5 days after in vitro infection [58]. Indeed, during common colds in vivo, these mediators and others were found to be elevated in nasal secretions [28, 57,59, 60, 61, 62, 63, 64, 65, 66, –67] as well as in sputum (IL‐8, IL‐6) [30, 68].
In view of the chemotactic properties of the above‐mentioned chemokines on lymphocytes, basophils, neutrophils, and (primed) eosinophils, one can postulate that such chemokines drive the secondary recruitment of inflammatory cells [69]. Indeed, a rhinovirus infection is known to lead to infiltration of leucocytes, particularly neutrophils and mononuclear cells, into nasal secretions [68,70, 71, –72] rather than the nasal mucosa [59, 70, 73]. Both in normals and asthmatic subjects this occurs in conjunction with an increase in the number of neutrophils in peripheral blood, whereas the number of circulating lymphocytes falls in the acute phase of infection [25, 26, 28, 74]. These effects, as well as the decrease in PC20 in asthmatic subjects, are significantly related to the severity of the cold, as reflected by the cold score [28, 74] and the increase in IL‐8 in nasal lavage [28], underlining the relationship between the severity of the cold and the pathophysiological consequences [17, 28].
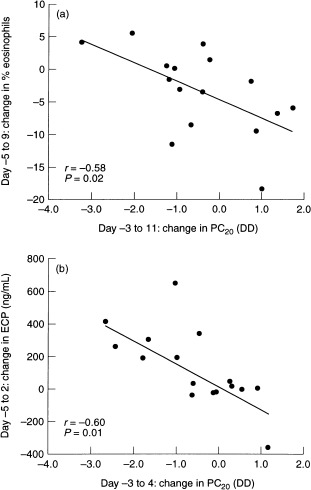
In contrast to the findings in the nose, the only available study so far on the effects of a common cold on intrapulmonary airways inflammation showed that in the bronchial biopsies the numbers of T lymphocytes in the submucosa and eosinophils in the epithelium were elevated both in normal and/or in atopic asthmatic subjects following RV16 infection [26]. Interestingly, the numbers of eosinophils remained elevated up to 6 weeks after infection in the six asthmatic subjects [26]. This is in keeping with the increased levels of eosinophilic cationic protein (ECP) in sputum that we have observed after experimental RV16 infection in asthmatics, which correlated significantly with the increase in airway sensitivity to histamine (Fig. 2) [30]. One could speculate that local production of eosinophil chemotactic chemokines contributes to virus‐induced eosinophilic airways inflammation in asthma.
Figure 2.

. Relationships between the change in precentage of eosinophils in induced sputum (a) or in levels of ECP in supernatant of induced sputum (b) and the change in airway hyperresponsiveness to histamine (PC20) in the first week after experimental RV16 infection in patients with asthma in vivo. Even though there were no significant virus‐induced changes in percentage eosinophils or ECP levels, the observed changes in these variables were significantly related to those in PC20: the greater the development of airways hyperresponsiveness (decrease in PC20), the greater the increase in percentage of sputum eosinophils or ECP levels. The regression lines have been drawn; r, correlation coefficient. Duplicated with permission from [30].
Involvement of intercellular adhesion molecule‐1 (ICAM‐1)
ICAM‐1 has been the focus of much attention in rhinovirus‐induced inflammation because of its dual role as (i) an adhesion molecule on endothelial cells or epithelial cells for binding of infiltrating leucocytes such as eosinophils and lymphocyte subsets [75, 76, 77, –78], and (ii) as the major rhinovirus receptor [79, 80]. It appears that ICAM‐1 expression is up‐regulated upon stimulation with rhinovirus in pulmonary epithelial cell lines in vitro [55, 56]. This has now been confirmed in vivo in a recent study from our laboratory demonstrating that experimental rhinovirus colds in asthmatics increase epithelial ICAM‐1 expression in bronchial biopsy specimens [81]. Hence, up‐regulation of ICAM‐1 by secondarily released cytokines such as IL‐1β and TNF‐α, may facilitate rhinovirus infection in susceptible cell types [52, 55, 82].
ICAM‐1‐binding to its ligands [77], lymphocyte function associated antigen (LFA‐1, CD11a/CD18), and macrophage‐1 antigen (MAC‐1, CD11b/CD18), is involved in the process of leucocyte adherence and migration, and in addition, may provide costimulatory signals for CD4 cell and lymphokine‐activated killer cell activation, T‐cell mediated cytotoxicity, and T‐cell dependent B‐cell activation. It has been postulated that cytokines such as TNFα, IL‐1β, IFN‐γ, and IL‐4 may alter the expression, function, or configuration of ICAM‐1, thereby specifically promoting (TNFα, IFN‐γ, IL‐4) or impeding (IL‐6, IL‐1) lymphocyte adhesion to, for example, cytokine‐pretreated fibroblasts or primary tracheal epithelial cells [52, 83, 84]. Rhinovirus binding, however, does not seem to be affected by such cytokine‐induced functional changes [52, 83, 84], suggesting that virus‐induced pro‐inflammatory cytokines may facilitate infection, whilst promoting or inhibiting the interaction between epithelial cells and lymphocytes.
Immune response
What are the immunological changes following rhinovirus infection? Incubation of either infectious or inactivated rhinovirus particles with a mixture of antigen presenting cells (APC; such as monocytes and also eosinophils [78]) and lymphocytes can induce a proliferative response to rhinovirus [85], while concomitantly hampering the monocytes‐induced, ICAM‐1‐mediated, specific T‐cell proliferation and cytotoxicity to other antigens [86]. This indicates that binding of rhinovirus to ICAM‐1 may interfere with ICAM‐1/LFA‐1 interaction [86]. Indeed, the ICAM‐1 binding sites for LFA‐1 and rhinovirus overlap partially [87]. Natural killer cell activation and cytotoxicity, which is less dependent on ICAM‐1/LFA‐1 interaction, was not shown to be affected, or even increased after in vitro rhinovirus inoculation [86, 88, 89].
Rhinovirus does not replicate inside monocytes and airway macrophages [90]; however, the uptake of infectious rhinovirus particles (either mediated or not mediated by ICAM‐1) by APCs may increase nonspecific T cells activation [91]. This is reflected by the increased expression of the activation marker CD69, but not CD25 on the cell membrane [91], and spontaneous [85] or mitogen‐induced secretion of IL‐2 and IFN‐γ in normal subjects [88, 91]. In addition, from 3 weeks after infection onward, antigen‐specific lymphocyte proliferation could be demonstrated [88, 92, 93], indicating a cell‐mediated immune response to the infection [85]. Such CD4+ cells may be specific to either serotype‐restricted or serotype‐shared viral epitopes [85, 94, 95]. A recent report suggests that eosinophils, when expressing ICAM‐1, may function as APCs, in that they induce T‐cell proliferation when incubated with rhinovirus [78]. This is associated with an increase in CD18 expression (an ICAM‐1 ligand), thus providing evidence for a possible positive feedback loop in rhinovirus‐induced eosinophil activation within the airways.
In a comparative study, using in vivo experimental RV39 infection, patients with allergic rhinitis appeared to have lower numbers of T‐helper cells (either activated or not activated) and RV39‐induced peripheral blood mononuclear cell proliferation as compared with normals [93], while only in the rhinitis patients was an increase in proliferation to RV39 noted as early as during the acute phase of the infection. Furthermore, the decrease in NK‐activity was less marked in the rhinitis patients as compared with the normals [93]. This suggests that there may be disease‐related differences in cell‐mediated immunity, although it is still unclear as to how such differences might be interpreted in terms of mechanisms of rhinovirus‐induced exacerbations of asthma.
Infection of the lower airways?
Is rhinovirus actually present within the lower airways? In the nasopharynx, rhinovirus replicates in the adenoid tissue and the epithelium [96], and can readily be detected in tissue culture or by reverse transcriptase‐polymerase chain reaction [97]. Evidence of rhinovirus infection of the intrapulmonary airways is more difficult to obtain. Although rhinoviruses can occasionally be cultured from sputum [98], tracheal brushings [31], and bronchoalveolar lavage (BAL) fluid [33, 99], possible nasopharyngeal contamination precludes definite conclusions as to virus infection of the lower airways. The use of reverse transcriptase‐ polymerase chain reaction on BAL cells, rather than BAL fluid, has increased the likelihood of detecting lower airways infection, and has led to a detection rate of 80%, while the detection rate in nasal lavage fluid in the same subjects was 100% after experimental RV16 infection [100].
In view of the difficulties in detecting rhinoviruses in the lungs so far, and the relative fastidiousness of rhinoviruses for culture condition (particularly the relatively low optimal culture temperature of 33 °C), a high grade infection of the lower airways may not be very likely. One could speculate, however, that host factors such as increased ICAM‐1 expression in the nasal epithelium [101] might increase the susceptibility of asthmatic and/or atopic patients to symptomatic rhinovirus colds [20], even after repeated infection with the same serotype [23, 28, 102]. Similarly, increased expression of ICAM‐1 in the lungs [56, 75] could promote lower airways infection, and its pathophysiological consequences. Apparently, in situ techniques to detect the viral genome [103, 104, –105] will need to be applied in order to conclusively assess viral contamination and/or infection within the lower airways. Preliminary results using in situ hybridization on bronchial biopsy specimens obtained from normals and asthmatics after experimental infection may indeed be suggestive of the possibility that rhinovirus can infect the lower airways [106]; however, confirmation of these results has to be awaited.
Conclusions
There is little doubt that respiratory virus infections can have pro‐inflammatory effects within the airways. Whilst it is still unresolved as to whether this contributes to the onset of asthma, there is convincing evidence that rhinovirus infections in particular are associated with exacerbations of the disease. Rhinovirus infection is able to activate bronchial epithelial cells, leading to the release of pro‐inflammatory cytokines (chemokines) as well as of modulating substances (such as NO). This can be accompanied by virus‐specific T‐cell proliferation weeks after infection, at early stages preceded by nonspecific T‐cell responses that could contribute to virus‐induced airways inflammation. By up‐regulating ICAM‐1 expression on, for example, epithelial cells and eosinophils, there is the possibility of positive feedback loops between rhinovirus infection and the inflammatory response.
Some of these events can be observed following experimental rhinovirus infection in patients in vivo. These are accompanied by clinical symptoms, airways obstruction, and worsened airway hyperresponsiveness. The latter is associated with changes in eosinophil counts and levels of ECP in induced sputum. This suggests, but does not prove, that eosinophilic inflammation can be involved in rhinovirus‐induced airways inflammation and the associated exacerbations of asthma.
In general, the clinical, physiological, and cellular responses to experimental rhinovirus infections in patients with asthma are relatively mild. This underlines the safety of this procedure, but may not adequately mimic the events occurring after a natural common cold. It can be postulated that this requires more complex experimental models, such as those with a pre‐existing flare‐up of allergic inflammation with, for example, up‐regulated ICAM‐1 [107]. This might be obtained by a design in which rhinovirus inoculation follows repeated low‐dose allergen exposure [108, 109]. This may mimic the course of events that occur during spontaneous exacerbations more accurately [110]. It is hoped that such experiments will bring us a therapy for virus‐induced acute exacerbations of asthma that is more successful than the ones currently available [111, 112].
References
- 1. Welliver RC. RSV and chronic asthma. Lancet 1995; 34:789 – 90. [DOI] [PubMed] [Google Scholar]
- 2. Macek V, Sorli J, Kopriva S, Marin J. Persistent adenoviral infection and chronic airway obstruction in children. Am J Respir Crit Care Med 1994; 150:7 – 10. [DOI] [PubMed] [Google Scholar]
- 3. Martinez FD. Role of viral infections in the inception of asthma and allergies during childhood: could they be protective? Thorax 1994; 49:1189 – 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shaheen SO, Aaby P, Hall AJ. Measles and atopy in Guinea‐Bissau. Lancet 1997; 347:1792 – 6. [DOI] [PubMed] [Google Scholar]
- 5. Corne JM, Holgate ST. Mechanisms of virus induced exacerbations of asthma. Thorax 1997; 52:380 – 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Folkerts G, Busse WW, Nijkamp FP, Sorkness R, Gern JE. Virus‐induced airway hyperresponsiveness and asthma. Am J Respir Crit Care Med 1998; 157:1708 – 20. [DOI] [PubMed] [Google Scholar]
- 7. Matsuse T, Hayashi S, Kuwano K. Latent adenoviral infection in the pathogenesis of chronic airways obstruction. Am Rev Respir Dis 1992; 146:177 – 84. [DOI] [PubMed] [Google Scholar]
- 8. Seemungal T, Donaldson GC, Breuer J. Rhinoviruses are associated with exacerbations in COPD. Am J Respir Crit Care Med 1998; 157:A173. [Google Scholar]
- 9. Neuzil KM, Graham BS. Cytokine release and innate immunity in respiratory viral infection. Semin Virol 1996; 7:255 – 64. [Google Scholar]
- 10. Schwarze J, Hamelmann E, Bradley KL, Takeda K, Gelfand EW. Respiratory syncytial virus infection results in airway hyperresponsiveness and enhanced airway sensitization to allergen. J Clin Invest 1997; 99:226 – 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Roman M, Calhoun WJ, Hinton KL. Respiratory syncytial virus infection in infants is associated with predominant Th‐2‐like response. Am J Respir Crit Care Med 1997; 156:190 – 5. [DOI] [PubMed] [Google Scholar]
- 12. Gern JE, Busse WW. The effects of rhinovirus infections on allergic airway responses. Am J Respir Crit Care Med 1995; 152:540 – 3. [DOI] [PubMed] [Google Scholar]
- 13. Pattemore PK, Johnston SL, Bardin PG. Viruses as precipitants of asthma symptoms. I. Epidemiology. Clin Exp Allergy 1992; 22:325 – 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Beasley R, Coleman ED, Hermon Y. Viral respiratory tract infection and exacerbations of asthma in adult patients. Thorax 1988; 43:679 – 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nicholson KG, Kent J, Ireland DC. Respiratory viruses and exacerbations of asthma in adults. BMJ 1993; 307:982 – 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. McIntosh K, Ellis EF, Hoffman LS. The association of viral and bacterial respiratory infections with exacerbations of wheezing in young asthmatic children. J Pediatr 1973; 82:578 – 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Minor TE, Dick EC, DeMeo AN. Viruses as precipitants of asthmatic attacks in children. JAMA 1974; 227:292 – 8. [PubMed] [Google Scholar]
- 18. Horn MEC, Brain EA, Gregg I. Respiratory virus infection and wheezy bronchitis in childhood. Thorax 1979; 34:23 – 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Johnston SL, Pattemore PK, Sanderson G. Community study of role of viral infections in exacerbations of asthma in 9–11 year old children. BMJ 1995; 310:1225 – 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Minor TE, Baker JW, Dick EC. Greater frequency of viral respiratory infections in asthmatic children as compared with their nonasthmatic siblings. J Pediatr 1974; 85:472 – 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gwaltney JM Jr, Hendley O, Hayden FG. Updated recommendations for safety‐testing of viral inocula used in volunteer experiments on rhinovirus colds. Prog Med Virol 1992; 39:256 – 63. [PubMed] [Google Scholar]
- 22. Halperin SA, Eggleston PA, Beasley P. Exacerbations of asthma in adults during experimental rhinovirus infection. Am Rev Respir Dis 1985; 132:976 – 80. [DOI] [PubMed] [Google Scholar]
- 23. Bardin PG, Fraenkel DJ, Sanderson G. Amplified rhinovirus colds in atopic subjects. Clin Exp Allergy 1994; 24:457 – 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lemanske RF Jr, Dick EC, Swenson CA, Vrtis RF, Busse WW. Rhinovirus upper respiratory infection increases airway hyperreactivity and late asthmatic reactions. J Clin Invest 1989; 83:1 – 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cheung D, Dick EC, Timmers MC. Rhinovirus inhalation causes long‐lasting excessive airway narrowing in response to methacholine in asthmatic subjects in vivo . Am J Respir Crit Care Med 1995; 152:1490 – 6. [DOI] [PubMed] [Google Scholar]
- 26. Fraenkel DJ, Bardin PG, Sanderson G. Lower airways inflammation during rhinovirus colds in normal and in asthmatic subjects. Am J Respir Crit Care Med 1995; 151:879 – 86. [DOI] [PubMed] [Google Scholar]
- 27. Grünberg K, Timmers MC, De Klerk EPA. Experimental rhinovirus 16 (RV16) infection decreases home‐recordings of FEV1 in asthmatics. Eur Respir J 1997; 10:9s. [Google Scholar]
- 28. Grünberg K, Timmers MC, Smits HH. Effects of experimental rhinovirus 16 colds on airway hyperresponsiveness to histamine and interleukin‐8 in nasal lavage in asthmatic subjects in vivo . Clin Exp Allergy 1997; 27:36 – 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Grünberg K, Kuijpers EAP, De Klerk EPA. Effects of experimental rhinovirus 16 (RV16) infection on airway hyperresponsiveness to bradykinin in asthmatic subjects in vivo . Am J Respir Crit Care Med 1997; 155:833 – 8. [DOI] [PubMed] [Google Scholar]
- 30. Grünberg K, Smits HH, Timmers MC. Experimental rhinovirus 16 infection: effects on cell differentials and soluble markers in sputum in asthmatic subjects. Am J Respir Crit Care Med 1997; 156:609 – 16. [DOI] [PubMed] [Google Scholar]
- 31. Halperin SA, Eggleston PA, Hendley JO. Pathogenesis of lower respiratory tract symptoms in experimental rhinovirus infection. Am Rev Respir Dis 1983; 128:806 – 10. [DOI] [PubMed] [Google Scholar]
- 32. Calhoun WJ, Swenson CA, Dick EC. Experimental rhinovirus 16 infection potentiates histamine release after antigen bronchoprovocation in allergic subjects. Am Rev Respir Dis 1991; 144:1267 – 73. [DOI] [PubMed] [Google Scholar]
- 33. Calhoun WJ, Dick EC, Schwartz LB, Busse WW. A common cold virus, rhinovirus 16, potentiates airway inflammation after segmental antigen bronchoprovocation in allergic subjects. J Clin Invest 1994; 94:2200 – 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gern JE, Calhoun WJ, Swenson CA, Shen G, Busse WW. Rhinovirus infection preferentially increases lower airway responsiveness in allergic subjects. Am J Respir Crit Care Med 1997; 155:1872 – 6. [DOI] [PubMed] [Google Scholar]
- 35. Skoner DP, Doyle WJ, Seroky J, Van Deusen MA, Fireman P. Lower airway responses to rhinovirus 39 in healthy allergic and nonallergic subjects. Eur Respir J 1996; 9:1402 – 6. [DOI] [PubMed] [Google Scholar]
- 36. Skloot G, Permutt S, Togias A. Airway hyperresponsiveness in asthma: a problem of limited smooth muscle relaxation with inspiration. J Clin Invest 1995; 96:2393 – 403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hogg JC. Pathology of asthma. J Allergy Clin Immunol 1993; 92:1 – 5. [DOI] [PubMed] [Google Scholar]
- 38. Redington AE, Howarth PH. Airway remodelling in asthma. Thorax 1997; 52:310 – 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Robinson PJ, Hegele RG, Schellenberg RR. Allergic sensitization increases airway reactivity in guinea pigs with respiratory syncytial virus bronchiolitis. J Allergy Clin Immunol 1997; 100:492 – 8. [DOI] [PubMed] [Google Scholar]
- 40. Folkerts G, Verheyen KCP, Geuens GMA, Folkerts HF, Nijkamp FP. Virus‐induced changes in airway responsiveness, morphology, and histamine levels in guinea pigs. Am Rev Respir Dis 1993; 147:1569 – 57. [DOI] [PubMed] [Google Scholar]
- 41. Huftel MA, Swensen CA, Borcherding WR. The effect of T‐cell depletion on enhanced basophil histamine release after in vitro incubation with live influenza A virus. Am J Respir Cell Mol Biol 1992; 7:434 – 40. [DOI] [PubMed] [Google Scholar]
- 42. Folkerts G, Verheyen A, Janssen M, Nijkamp FP. Virus‐induced airway hyperresponsiveness in the guinea pig can be transferred by bronchoalveolar cells. J Allergy Clin Immunol 1992; 90:364 – 72. [DOI] [PubMed] [Google Scholar]
- 43. Van Oosterhout AJM, Van Ark I, Folkers G. Antibody to interleukin‐5 inhibits virus‐induced airway hyperresponsiveness to histamine in guinea pigs. Am J Respir Crit Care Med 1995; 151:177 – 83. [DOI] [PubMed] [Google Scholar]
- 44. Jacoby DB. Virus‐induced bronchial hyperreactivity. In: Raeburn D, Giembycz MA (eds). Airways smooth muscle modelling the asthmatic response in vivo Basel: Birkhäuser‐Verlag 1996:121 – 45.
- 45. Dusser DJ, Jacoby DB, Djokic TD. Virus induces airway hyperresponsiveness to tachykinins: role of neutral endopeptidase. J Appl Physiol 1989; 67:1504 – 11. [DOI] [PubMed] [Google Scholar]
- 46. Kahn RM, Okaniami OA, Jacoby DB, Fryer AD. Viral infection induces dependence of neuronal M2 muscarinic receptors on cyclooxygenase in guinea pig lung. J Clin Invest 1996; 98:299 – 307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Folkerts G, Van Der Linde HJ, Nijkamp FP. Virus‐induced airway hyperresponsiveness in guinea pigs is related to a deficiency in nitric oxide. J Clin Invest 1995; 95:26 – 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. De Gouw Hwfm, Van Sterkenburg Maja, Van Wetering S. Expression of inducible nitric oxide synthase (iNOS) mRNA in human primary bronchial epithelial cells in vitro: effects of cytokines and rhinovirus‐16. Am J Respir Crit Car Med 1998; 157:A24. [Google Scholar]
- 49. Sanders SP, Siekierski ES, Porter JD, Richards SM, Proud D. Nitric oxide inhibits rhinovirus‐induced cytokine production and viral replication in a human respiratory epithelial cell line. J Virol 1998; 72:934 – 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. De Gouw Hwfm, Grünberg K, Schot R. Relationship between exhaled nitric oxide and airway hyperresponsiveness following experimental rhinovirus infection in asthmatic subjects. Eur Respir J 1998; 11:126 – 32. [DOI] [PubMed] [Google Scholar]
- 51. Johnston SL, Mastronarde JG, Monick MM. Rhinoviruses induce prolonged interleukin‐8 release, increases mRNA and promotor activation in pulmonary epithelial and peripheral blood mononuclear cells. Am J Respir Crit Care Med 1995; 151:A773. [Google Scholar]
- 52. Subauste MC, Jacoby DB, Richards SM, Proud D. Infection of a human respiratory epithelial cell line with rhinovirus. Induction of cytokine release and modulation of susceptibility to infection by cytokine exposure. J Clin Invest 1995; 96:549 – 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhu Z, Tang W, Ray A. Rhinovirus stimulation of interleukin‐6 in vivo and in vitro. Evidence for nuclear factor κB‐dependent transcriptional activation. J Clin Invest 1996; 97:421 – 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Cazemier H, Sterkenburg Maja, Manesse‐Lazeroms SPG. Rhinovirus 16 (RV16) stimulates production and release of the chemokines IL‐8 and RANTES in subcultures of human primary bronchial epithelial cells. Eur Respir J 1996; 9:424s. [Google Scholar]
- 55. Terajima M, Yamaya M, Sekizawa K. Rhinovirus infection of primary cultures of human tracheal epithelium: role of ICAM‐1 and IL‐1β. Am J Physiol 1997; 273:L749 – 59. [DOI] [PubMed] [Google Scholar]
- 56. Papi A, Wilson SJ, Johnston SL. Rhinoviruses increase production of adhesion molecules (CAM) and NF‐κB. Am J Respir Crit Care Med 1996; 153:A866. [Google Scholar]
- 57. Zhu Z, Tang W, Gwaltney JM, Wu Y, Elias JA. Rhinovirus stimulation of interleukin‐8 in vivo and in vitro: role of NF‐κB. Am J Physiol 1997; 273:L814 – 24. [DOI] [PubMed] [Google Scholar]
- 58. Johnston SL, Papi A, Bates PhJ. Low grade rhinovirus infection induces a prolonged release of IL‐8 in pulmonary epithelium. J Immunol 1998; 160:6172 – 81. [PubMed] [Google Scholar]
- 59. Cazemier H, Grünberg K, De Kluijver J. Rhinovirus (RV)‐16 induced nasal inflammation is characterized by an influx of neutrophils and increased chemokine expression in mild asthmatics. Eur Respir J 1997; 10:463s. [Google Scholar]
- 60. Proud D, Naclerio RM, Gwaltney JM, Hendley JO. Kinins are generated in nasal secretions during natural rhinovirus colds. J Infect Dis 1990; 161:120 – 3. [DOI] [PubMed] [Google Scholar]
- 61. Igarashi Y, Skoner DP, Doyle WJ. Analysis of nasal secretions during experimental rhinovirus upper respiratory infections. J Allergy Clin Immunol 1993; 92:722 – 31. [DOI] [PubMed] [Google Scholar]
- 62. Proud D, Gwaltney JM Jr, Hendley JO. Increased levels of interleukin‐1 are detected in nasal secretions of volunteers during experimental rhinovirus colds. J Infect Dis 1994; 169:1007 – 13. [DOI] [PubMed] [Google Scholar]
- 63. Linden M, Greiff L, Andersson M. Nasal cytokines in common cold and allergic rhinitis. Clin Exp Allergy 1995; 25:166 – 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Roseler S, Holtappels G, Wagenmann M, Bachert C. Elevated levels of interleukins IL‐1 beta, IL‐6 and IL‐8 in naturally acquired viral rhinitis. Eur Arch Otorhinolaryngol Suppl 1995; 1:S61 – 3. [DOI] [PubMed] [Google Scholar]
- 65. Teran LM, Johnston SL, Holgate ST. Immunoreactive RANTES and MIP‐1α are increased in the nasal aspirates of children with virus‐associated asthma. Am J Respir Crit Care Med 1995; 151:A385. [Google Scholar]
- 66. Einarsson O, Geba GP, Zhu Z, Landry M, Elias JA. Interleukin‐11: Stimulation in vivo and in vitro by respiratory viruses and induction of airways hyperresponsiveness. J Clin Invest 1996; 97:915 – 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Teran LM, Johnston SL, Schroder J‐M, Church MK, Holgate ST. Role of nasal interleukin‐8 in neutrophil recruitment and activation in children with virus‐induced asthma. Am J Respir Crit Care Med 1997; 155:1362 – 6. [DOI] [PubMed] [Google Scholar]
- 68. Fahy JV, Kim KW, Liu J, Boushey HA. Prominent neutrophilic inflammation in sputum from subjects with asthma exacerbation. J Allergy Clin Immunol 1995; 95: 843 – 52. [DOI] [PubMed] [Google Scholar]
- 69. Baggiolini M, Dewald B, Moser B. Interleukin‐8 and related chemotactic cytokines − cxc and cc chemokines. Adv Immunol 1994; 55:97 – 179. [PubMed] [Google Scholar]
- 70. Winther B, Farr B, Turner RB. Histopathologic examination and enumeration of polymorphonuclear leukocytes in the nasal mucosa during experimental rhinovirus colds. Acta Otolaryngol (Suppl )(Stockholm) 1984; 413:19 – 24. [DOI] [PubMed] [Google Scholar]
- 71. Levandowski RA, Weaver CW, Jackson GG. Nasal‐secretion leukocyte populations determined by flow cytometry during acute rhinovirus infection. J Med Virol 1988; 25:423 – 32. [DOI] [PubMed] [Google Scholar]
- 72. Naclerio RM, Proud D, Lichtenstein LM. Kinins are generated during experimental rhinovirus colds. J Infect Dis 1988; 157:133 – 42. [DOI] [PubMed] [Google Scholar]
- 73. Fraenkel DJ, Bardin PG, Sanderson G. Immunohistochemical analysis of nasal biopsies during rhinovirus experimental colds. Am J Respir Crit Care Med 1994; 150:1130 – 6. [DOI] [PubMed] [Google Scholar]
- 74. Levandowski RA, Ou DW, Jackson GG. Acute‐phase decrease of T lymphocyte subsets in rhinovirus infection. J Infect Dis 1986; 153:743 – 8. [DOI] [PubMed] [Google Scholar]
- 75. Canonica GW, Ciprandi G, Pesce GP. ICAM‐1 on epithelial cells in allergic subjects: a hallmark of allergic inflammation. Int Arch Allergy Immunol 1995; 107:99 – 102. [DOI] [PubMed] [Google Scholar]
- 76. Winther B, Greve JM, Gwaltney JMJ. Surface expression of intercellular adhesion molecule 1 on epithelial cells in the human adenoid. J Infect Dis 1997; 176:523 – 5. [DOI] [PubMed] [Google Scholar]
- 77. Bloemen PGM, Hendricks PAJ, Nijkamp FP. Cell adhesion and asthma. Clin Exp Allergy 1997; 27:128 – 41. [PubMed] [Google Scholar]
- 78. Handzel ZT, Busse WW, Sedgwick JB. Eosinophils bind rhinovirus and activate virus‐specific T cells. J Immunol 1998; 160:1279 – 84. [PubMed] [Google Scholar]
- 79. Greve JM, Davis G, Meyer AM. The major human rhinovirus receptor is ICAM‐1. Cell 1989; 56:839 – 47. [DOI] [PubMed] [Google Scholar]
- 80. Staunton DE, Merluzzi VJ, Rothlein R. A cell adhesion molecule, ICAM‐1, is the major surface receptor for rhinoviruses. Cell 1989; 56:849 – 53. [DOI] [PubMed] [Google Scholar]
- 81. Sharon RF, Grünberg K, Van Krieken Jhjm. Experimental rhinovirus (RV)‐16 infection enhances ICAM‐1 expression in bronchial mucosal biopsies of mildly asthmatic subjects, regardless of inhaled steroid treatment. Am J Respir Crit Care Med 1998; 157:A22. [Google Scholar]
- 82. Grunert HP, Wolf KU, Langner KD. Internalization of human rhinovirus 14 into HeLa and ICAM‐1‐transfected BHK cells. Med Microbiol Immunol 1997; 186:1 – 9. [DOI] [PubMed] [Google Scholar]
- 83. Piela‐Smith TH, Aneiro L, Korn JH. Binding of human rhinovirus and T cells to intercellular adhesion molecule‐1 on human fibroblasts. Discordance between effects of IL‐1 and IFN‐gamma. J Immunol 1991; 147:1831 – 6. [PubMed] [Google Scholar]
- 84. Piela‐Smith TH, Broketa G, Hand A, Korn JH. Regulation of ICAM‐1 expression and function in human dermal fibroblasts by IL‐4. J Immunol 1992; 148:1375 – 81. [PubMed] [Google Scholar]
- 85. Wimalasundera SS, Katz DR, Chain BM. Characterization of the T cell response to human rhinovirus in children: Implications for understanding the immunopathology of the common cold. J Infect Dis 1997; 176:755 – 9. [DOI] [PubMed] [Google Scholar]
- 86. Gern JE, Joseph B, Galagan DM, Borcherding WR, Dick EC. Rhinovirus inhibits antigen‐specific T cell proliferation through an intercellular adhesion molecule‐1 dependant mechanism. J Infect Dis 1996; 174:1143 – 50. [DOI] [PubMed] [Google Scholar]
- 87. Staunton DE, Dustin ML, Erickson HP, Springer TA. The arrangement of the immunoglobulin‐like domains of ICAM‐1 and the binding sites for LFA‐1 and rhinovirus. Cell 1990; 61:243 – 54. [DOI] [PubMed] [Google Scholar]
- 88. Hsia J, Goldstein AL, Simon GL, Sztein M, Hayden FG. Peripheral blood mononuclear cell interleukin‐2 and interferon‐gamma production, cytotoxicity, and antigen‐stimulated blastogenesis during experimental rhinovirus infection. J Infect Dis 1990; 162:591 – 7. [DOI] [PubMed] [Google Scholar]
- 89. Levandowski RA, Horohov DW. Rhinovirus induces natural killer‐like cytotoxic cells and interferon alpha in mononuclear leukocytes. J Med Virol 1991; 35:116 – 20. [DOI] [PubMed] [Google Scholar]
- 90. Gern JE, Dick EC, Ming Lee W. Rhinovirus enters but does not replicate inside monocytes and airway macrophages. J Immunol 1996; 156:621 – 7. [PubMed] [Google Scholar]
- 91. Gern JE, Vrtis RF, Kelly EAB, Dick EC, Busse WW. Rhinovirus produces nonspecific activation of lymphocytes through a monocyte‐dependent mechanism. J Immunol 1996; 157:1605 – 12. [PubMed] [Google Scholar]
- 92. Levandowski RA, Pachucki CT, Rubenis M. Specific mononuclear cell response to rhinovirus. J Infect Dis 1983; 148:1125. [DOI] [PubMed] [Google Scholar]
- 93. Skoner DP, Whiteside TL, Wilson JW. Effect of rhinovirus 39 infection on cellular immune parameters in allergic and nonallergic subjects. J Allergy Clin Immunol 1993; 92:732 – 43. [DOI] [PubMed] [Google Scholar]
- 94. Hastings GZ, Rowlands DJ, Francis MJ. Proliferative responses of T cells primed against human rhinovirus to other rhinovirus serotypes. J Gen Virol 1991; 72:2947 – 52. [DOI] [PubMed] [Google Scholar]
- 95. Gern JE, Dick EC, Kelly EAB, Vrtis RF, Klein B. Rhinovirus‐specific T cells recognize both shared and serotype‐restricted viral epitopes. J Infect Dis 1997; 175:1108 – 14. [DOI] [PubMed] [Google Scholar]
- 96. Winther B, Gwaltney JM Jr, Mygind N, Turner RB, Hendley JO. Sites of rhinovirus recovery after point inoculation of the upper airway. JAMA 1986; 256:1763 – 7. [PubMed] [Google Scholar]
- 97. Johnston SL, Sanderson G, Pattemore PK. Use of polymerase chain reaction for diagnosis of picornavirus infection in subjects with and without respiratory symptoms. J Clin Microbiol 1993; 31:111 – 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Horn MEC, Reed SE, Taylor PM. Role of viruses and bacteria in acute wheezy bronchitis in childhood: a study of sputum. Arch Dis Child 1979; 54:587 – 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Schmidt HJ, Fink RJ. Rhinovirus as a lower respiratory tract pathogen in infants. Pediatr Infect Dis J 1991; 10:700 – 2. [DOI] [PubMed] [Google Scholar]
- 100. Gern JE, Galagan DM, Jarjour NN, Dick EC, Busse WW. Detection of rhinovirus RNA in lower airway cells during experimentally induced infection. Am J Respir Crit Care Med 1997; 155:1159 – 61. [DOI] [PubMed] [Google Scholar]
- 101. Ciprandi G, Pronzato C, Ricca V. Allergen‐specific challenge induces intercellular adhesion molecule 1 (ICAM‐1 or CD54) on nasal epithelial cells in allergic subjects. Am J Respir Crit Care Med 1994; 150:1653 – 9. [DOI] [PubMed] [Google Scholar]
- 102. Alper CM, Doyle WJ, Skoner DP. Prechallenge antibodies: moderators of infection rate, signs, and symptoms in adults experimentally challenged with rhinovirus type 39. Laryngoscope 1996; 106:1298 – 305. [DOI] [PubMed] [Google Scholar]
- 103. Bruce C, Chadwick P, Al‐Nakib W. Detection of rhinovirus RNA in nasal epithelial cells by in situ hybridization. J Virol Meth 1990; 30:115 – 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Arruda E, Boyle TR, Winther B. Localization of human rhinovirus replication in the upper respiratory tract by in situ hybridization. J Infect Dis 1995; 171:1329 – 33. [DOI] [PubMed] [Google Scholar]
- 105. Bardin PG, Johnston SL, Sanderson G. Detection of rhinovirus infection of the nasal mucosa by oligonucleotide in situ hybridization. Am J Respir Cell Mol Biol 1994; 10:207 – 13. [DOI] [PubMed] [Google Scholar]
- 106. Bates PJ, Bardin PG, Fraenkel DJ. Localisation of rhinovirus in the bronchial epithelium of experimentally‐infected human volunteers. Am J Respir Crit Care Med 1998; 157:A25. [Google Scholar]
- 107. Montefort S, Gratziou C, Goulding D. Bronchial biopsy evidence for leukocyte infiltration and upregulation of leukocyte‐endothelial cell adhesion molecules 6 hours after local allergen challenge of sensitized asthmatic airways. J Clin Invest 1994; 93:1411 – 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Sterk PJ. Repeated low dose allergen exposure: a new investigational model of asthma as a persistent disease? Eur Respir J 1998; 11:798 – 800. [DOI] [PubMed] [Google Scholar]
- 109. Arshad S, Hamilton RB, Adkinson NF. Repeated aerosol exposure to small doses of allergen. A model of chronic allergic asthma. Am J Respir Crit Care Med 1998; 157:1900 – 6. [DOI] [PubMed] [Google Scholar]
- 110. Johnston SL, Pattemore PK, Sandeson G. The relationship between upper respiratory infections and hospital admissions for asthma: a time‐trend analysis. Am J Respir Crit Care Med 1996; 154:654 – 60. [DOI] [PubMed] [Google Scholar]
- 111. Doull IJM, Lampe FC, Smith S. Effect of inhaled corticosteroids on episodes of wheezing associated with viral infection in school age children: a randomised double blind placebo controlled trial. BMJ 1997; 315:858 – 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Svedmyr J, Nyberg E, Asbrink‐Nilsson E, Hedlin G. Intermittent treatment with inhaled steroids for deterioration of asthma due to upper respiratory tract infection. Acta Paediatr 1995; 84:884 – 8. [DOI] [PubMed] [Google Scholar]