

Abstract
Described is the synthesis of novel arylated and alkynylated 2,4′‐diphenyl sulfones based on what are, to the best of our knowledge, the first palladium(0)‐catalyzed cross‐coupling reactions of bis(triflates) of 2,4′‐bis(hydroxy)diphenyl sulfone. The reactions proceed with very good site‐selectivity.
Keywords: catalysis, palladium, site‐selectivity, Sonogashira reaction, sulfones, Suzuki–Miyaura reaction
A Eurasian collaboration: Suzuki–Miyaura and Sonogashira reactions of the bis(triflate) of 2,4′‐bis(hydroxy)diphenyl sulfone proceeded with excellent site‐selectivity in favor of position C‐4′.

Functionalized arylated sulfones are important structural motifs in medicinal chemistry.1–14 Pharmacological properties include inhibition of the enzymes phospholipidase A2,1, 2 catechol O‐methyltransferase,3 dihydropteroate synthase of Escherichia coli and the main protease of the recombinant SARS coronavirus.4 Biological activities like antibacterial activity,1 hypolipidemic,5 cytotoxic against HeLa cells and the antipicornavirus,[6 ]neuropeptide Y1 receptor binding,7 anti‐HIV,8 anticholesteremic,9 binding to human muscarinic M1 and M2 receptors,10 histamine H3‐receptor antagonistic,11 antiprotozoal,12 and binding to the human cannabinoid CB1 receptor13 activities have also been reported. Alkynylated sulfones are potential candidates for liquid crystalline materials. Besides a vast range of bioactivities, conjugated enynes are also important synthetic intermediates.14
Known syntheses of diaryl sulfones include the oxidation of diaryl sulfides,15 acylation of electron rich benzene derivatives with phenylsulfonyl chloride and benzenesulfonic acid.16 Despite the usefulness of these methods, limitations related to scope and low regioselectivities exist, owing to the harsh reaction conditions. A number of mild transition metal‐mediated approaches to diaryl sulfones have been reported. These include the CuI/proline‐mediated reaction of sodium benzenesulfinate with aryl iodides17a and Cu(OAc)2‐catalyzed reaction sodium benzenesulfinate with 4‐methoxybenzeneboronic acid.17b In addition, Suzuki reactions of phenylsulfonic acid chloride with arylboronic acids have been reported.18 A different approach to diaryl sulfones relies on cyclization reactions of building blocks containing a sulfone moiety.19 In recent years, site selective palladium catalyzed cross coupling reactions have been intensively studied. The selectivity of such reactions is generally controlled by electronic and steric parameters. The first attack usually occurs at the electronically more deficient or sterically less hindered position.20–22 Recently, we reported23 the synthesis of bis(diaryl) sulfones by site‐selective Suzuki–Miyaura reactions of the bis(triflate) of 2,4′‐bis(dihydroxy)diphenyl sulfone. Herein, we report full details of these studies. With regard to our preliminary communication, we have considerably extended the scope and report, for the first time, Sonogashira reactions which also proceed with excellent site‐selectivity. This extension of synthetic scope could be of worth and interest because the regioselectivity in Sonogashira alkynylation reactions remains a challenge, owing to the low steric demand of alkynyl cuprates as reaction partners, Glaser homo‐coupling as a side‐reaction, and increased reactivity of the substrate after the first alkynylation step (owing to the electronic influence of the alkynyl group).1c Indeed, the product distribution of Sonogashira reactions of polyhalogenated substrates or polytriflates is not necessarily the same, as for Suzuki reactions of the same substrates, and the degree of regioselectivity of Sonogashira reactions is also not predictable, based on the results of the analogous Suzuki reactions. The products reported herein, arylated and alkynylated diaryl sulfones are of potential pharmacological relevance and have, to the best of our knowledge, not been reported so far. It can be expected that they are not readily available by other methods.
Results and Discussion
Bis(triflate) 2 was prepared in 81 % yield by reaction of commercially available 2,4′‐bis(hydroxy)‐diphenyl sulfone (1) with triflic anhydride (Scheme 1).
Scheme 1.

Synthesis of 2. Reagents and conditions: i) CH2Cl2, 1 (1.0 equiv), −78 °C, pyridine (4.0 equiv), Tf2O (2.4 equiv), −78→0 °C, 4 h.
Suzuki–Miyaura reactions
2,4′‐Bis(aryl)diphenyl sulfones 4 a–i were prepared in 55–75 % yields by Suzuki–Miyaura reaction24 of 2 with 2.6 equiv of arylboronic acids 3 a–i (Scheme 2, Table 1). The best yields were achieved when the reactions were carried out using [Pd(PPh3)4] (6 mol %) as the catalyst, whereas employment of other catalysts, such as [Pd(OAc)2]/XPhos, resulted in a decrease of the yield. Similar conditions were previously used in Suzuki reactions of related bis(triflates).22 The use of potassium phosphate (K3PO4) as the base and 1,4‐dioxane (110 °C, 4 h) gave optimal yields. The best yield was obtained for the reaction of simple phenylboronic acid. The lowest yield was obtained for 4‐fluorophenylboronic acid, which might be attributed to its low nucleophilicity (owing to the electron‐withdrawing fluorine atom). The structure of 4 e was independently confirmed by X‐ray crystal structure analysis (Figure 1).25
Scheme 2.

Synthesis of 4 a–i. Reagents and conditions: i) 2 (1.0 equiv), 3 a–i (2.6 equiv), K3PO4 (3.0 equiv), [Pd(PPh3)4] (6 mol %), 1,4‐dioxane (5 mL per 1 mmol of 2), 110 °C, 4 h.
Table 1.
The publisher did not receive permission from the copyright owner to include this object in this version of this product. Please refer either to the publisher's own online version of this product or the printed product where one exists.
Figure 1.

Crystal structure of 4 e.
2‐Trifluoromethanesulfonyloxy‐4′‐(aryl)diphenyl sulfones 5 a–d were prepared in 62–76 % yield by Suzuki–Miyaura reaction of 2 with arylboronic acids 3 b, f, j, k (1.1 equiv) in the presence of [Pd(PPh3)4] (3 mol %). The reactions proceeded with very good site‐selectivity in favor of position C‐4′ (Scheme 3, Table 2). In most cases, a small amount of the bis‐coupled product could be detected in the crude product before chromatography (by 1H NMR spectroscopy and GC–MS). Therefore, the yields of mono‐substituted products 5 are often less than the yields of the corresponding disubstituted products 4, because of practical difficulties during the chromatographic purification of products 5 and loss of material (because the R f values of impurities were close the R f value of the desired product. In case of the synthesis of products 4, the reactions were much cleaner and the chromatographic purifications much easier without much loss of material. All products were isolated in pure form by chromatographic purification. During the optimization, the temperature played a crucial role. It proved to be important to perform the reaction at 70 instead of 110 °C to induce a good selectivity.
Scheme 3.

Synthesis of 5 a–d. Reagents and conditions: i) 2 (1.0 equiv), 3 b, f, j, k (1.1 equiv), K3PO4 (1.5 equiv), [Pd(PPh3)4] (3 mol %), 1,4‐dioxane (5 mL per 1 mmol of 2), 70 °C, 4 h.
Table 2.
The publisher did not receive permission from the copyright owner to include this object in this version of this product. Please refer either to the publisher's own online version of this product or the printed product where one exists.
The structure of 5 b was confirmed by 2D NMR spectroscopy (Figure 2). Assignments of chemical shifts of C and H were completed with the help of 1H NMR, HMQC and COSY experiments. The NOESY correlation of CH2 with H‐3′′/H‐5′′ and H‐2′′/H‐6′′ with H‐3′/H‐5′ provided the information that the aryl group is connected to carbon C‐4′. The structure was further confirmed by strong HMBC correlations of H‐2′′/H‐6′′ with C‐1′′ & C‐4′ and HMBC correlation from the other ring H‐3′/H‐5′ to C‐1′′. These careful and clear correlations proved, unambiguously, the connectivity of the first aryl group to C‐4′.
Figure 2.

2D NMR correlations (NOESY and HMBC) of 5 b.
The reaction of 5 a and 5 c with arylboronic acids 3 a, b, e, j, k (1.1 equiv) gave 2,4′‐bis(aryl)diphenyl sulfones 6 a–e containing two different aryl groups in 50–64 % yield (Scheme 4, Table 3). Interestingly, the yield of 6 d derived from the electron poor boronic acid 3 e proved to be the highest.
Scheme 4.
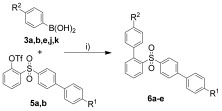
Synthesis of 6 a–e. Reagents and conditions: i) 5 (1.0 equiv), 3 a, b, e, j–k (1.1 equiv), K3PO4 (1.5 equiv), [Pd(PPh3)4] (3 mol %), 1,4‐dioxane (5 mL per 1 mmol of 2), 70 °C, 4 h.
Table 3.
The publisher did not receive permission from the copyright owner to include this object in this version of this product. Please refer either to the publisher's own online version of this product or the printed product where one exists.
Sonogashira reactions
Recently, Takahashi et al. described the formation of highly substituted enynes by coupling reaction of alkenylzirconium compounds with alkynyl halides.26 Gimeno et al. reported the stereoselective synthesis of chiral terminal (E)‐1,3‐enynes derived from optically active aldehydes.27 The stereoselective synthesis of 1,3‐enynylsulfides,28 1,3‐enynylselenides,29 1,3‐enynyltellurides,30 1,3‐enynylsilanes,31 1,3‐enynylstannanes32 and fluoro or CF3‐substituted 1,3‐enyne has previously been described in the literature. However, the synthesis of enynyl sulfones has received less attention33 and enynyl sulfones have, to the best of our knowledge, not been reported so far.
The Sonogashira reaction34 of 2 with alkynes 7 a–e (2.2 equiv) gave the 2,4′‐bis(alkynyl)diphenyl sulfones 8 a–e containing two different aryl groups in 85–92 % yield (Scheme 5, Table 4). The reaction was carried using catalytic amounts of CuI (10 mol %), [Pd(PPh3)2Cl2] (2.5 mol %), and (Bu)4NI (15 mol %). In the reaction Et3N (1.25 equiv) was used as the base and DMF as the solvent (60 °C, 1 h). The structure of 8 b was independently confirmed by X‐ray crystal structure analysis (Figure 3).25 Copper‐free Sonogashira reactions have been previously reported.35 The application of copper free conditions to the synthesis of product 8 a failed (no conversion).
Scheme 5.

Synthesis of 8 a–e. Reagents and conditions: i) 2 (1.0 equiv), 7 a–e (2.2 equiv), (Bu)4NI (15 mol %), CuI (10 mol %), [Pd(PPh3)2Cl2] (5 mol %), Et3N (2.5 equiv), DMF, 80 °C, 4 h.
Table 4.
The publisher did not receive permission from the copyright owner to include this object in this version of this product. Please refer either to the publisher's own online version of this product or the printed product where one exists.
Figure 3.

Ortep plot of 8 b (50 % probability level).
2‐Trifluoromethanesulfonyloxy‐4′‐(alkynyl)diphenyl sulfones 9 a–d were prepared in 83–91 % yield by Sonogashira reaction of 2 with alkynes 7 b, d, e, g (1.1 equiv) in the presence of 2.5 mol % of [Pd(PPh3)2Cl2]. The reactions proceeded with very good site‐selectivity in favor of position C‐4′ (Scheme 6, Table 5). In most cases, a small amount of the bis‐coupled product could be detected in the crude product before chromatography (by 1H NMR spectroscopy and GC–MS). All products were isolated in pure form by chromatographic purification. Therefore, similar to the synthesis of mono‐Suzuki products 5, the yields of mono‐Sonogashira products 9 are often less than the yields of the corresponding disubstituted products 8, because of difficulties during the chromatographic purification of products 9 and loss of material. During the optimization, the rate of addition of the acetylene played a crucial role. It proved to be important to perform the reaction at 70 rather than 110 °C with drop‐wise addition of the acetylene to achieve a good site‐selectivity.
Scheme 6.

Synthesis of 9 a–d. Reagents and conditions: i) 2 (1.0 equiv), 7 b, d, e, g (1.0 equiv), (Bu)4NI (15 mol %), CuI (10 mol %), [Pd(PPh3)2Cl2] (2.5 mol %), Et3N (1.25 equiv), DMF, 60 °C, 1 h.
Table 5.
The publisher did not receive permission from the copyright owner to include this object in this version of this product. Please refer either to the publisher's own online version of this product or the printed product where one exists.
The Sonogashira cross‐coupling reaction of monoalkynylated diaryl sulfones 9 c, d with terminal alkynes (1.0 equiv), in the presence of [Pd(PPh3)2Cl2] (2.5 mol %), CuI (10 mol %), (Bu)4NI (15 mol %), Et3N (2.5 equiv) (DMF, 80 °C, 4 h), gave products 10 a–c in 79–87 % yield (Scheme 7, Table 6).
Scheme 7.

Synthesis of 10 a–c. Reagents and conditions: i) 9 c, d (1.0 equiv), 7 a, g, h (1.0 equiv), (Bu)4NI (15 mol %), CuI (10 mol %), [Pd(PPh3)2Cl2] (2.5 mol %), Et3N (1.25 equiv), DMF, 60 °C, 4 h.
Table 6.
The publisher did not receive permission from the copyright owner to include this object in this version of this product. Please refer either to the publisher's own online version of this product or the printed product where one exists.
The Suzuki–Miyaura reaction of 9 a, d with arylboronic acids 3 a, e, j, k in the presence of [Pd(PPh3)4] and K3PO4, gave sulfones 11 a–d (Scheme 8). All these reactions proceeded in good yields (80–91 %) (Scheme 8, Table 7).
Scheme 8.

Synthesis of 11 a–d. Reagents and conditions: i) 9 a, d (1.0 equiv), ArB(OH)2 (1.1 equiv), [Pd(PPh3)4] (2.5 mol %), K3PO4 (1.5 equiv), Dioxane, 100 °C, 6 h.
Table 7.
The publisher did not receive permission from the copyright owner to include this object in this version of this product. Please refer either to the publisher's own online version of this product or the printed product where one exists.
Conclusions
We have reported the synthesis of novel arylated and alkynylated 2,4′‐diphenyl sulfones based on what are, to the best of our knowledge, the first palladium(0)‐catalyzed cross‐coupling reactions of bis(triflates) of 2,4′‐bis(hydroxy)diphenyl sulfone. The reactions proceed with very good site‐selectivity.
The oxidative addition of palladium usually occurs first at the most electron deficient carbon atom. Carbon atoms C‐2 and C‐4′ of bis(triflate) 2 are expected to be equally electron deficient. The site‐selective formation of 5 a–d and 9 a–d can be explained by the fact that carbon atom C‐4′ is less sterically hindered. Interestingly, excellent site‐selectivities could be obtained not only for Suzuki–Miyaura reactions, but also for Sonogashira reactions (which rely on the use of sterically undemanding alkynes).
Experimental Section
General procedure A for the synthesis of 4 a–i, 5 a–d 6 a–e and 11 a–d
In a pressure tube, 1,4‐dioxane solution of arylboronic acid, K3PO4, [Pd(PPh3)4] and 2 or 5 was stirred at 110 °C for 4 h under an argon atmosphere. After cooling to 20 °C, a saturated aqueous solution of NH4Cl was added. The organic and the aqueous layers were separated and the latter was extracted with CH2Cl2. The combined organic layers were dried (Na2SO4), filtered and the filtrate was concentrated in vacuo. The residue was purified by using column chromatography.
2‐(Biphenyl‐4‐ylsulfonyl)biphenyl (4 a)
Starting with 2 (205 mg, 0.40 mmol), K3PO4 (254 mg, 1.2 mmol), [Pd(PPh3)4] (28 mg, 6 mol %), phenylboronic acid (122 mg, 1.0 mmol) and 1,4‐dioxane (2 mL), following general procedure A, 4 a was isolated as a white solid (111 mg, 75 %). M.p. 131 °C; 1H NMR (300 mhz, CDCl3): δ=6.86–6.93 (m, 3 H), 7.31–7.39 (m, 5 H), 7.44–7.48 (m, 3 H), 7.59–7.63 (m, 4 H), 7.88–7.91 (m, 2 H), 8.35–8.38 ppm (m, 1 H); 13C NMR (75.5 mhz, CDCl3): δ=126.0, 126.5, 126.9, 127.5, 128.3, 128.5, 128.7, 129.1, 129.9, 131.1, 132.7, 132.9 (CH), 135.2, 139.2, 140.1, 145.0, 150.8, 151.8 ppm (C); IR (KBr):  =3066, 2923, 2852, 1595, 1466 (w), 1317 (m), 1291 (w), 1195 (m), 1126 (s), 1091, 1003 (m), 834, 759, 690 (s), 732, 572 cm−1 (m); GC–MS (EI, 70 eV): m/z (%): 370 ([M]+, 100), 305 (43), 291 (11), 289 (40), 215 (11), 207 (49), 201 (17), 184 (10), 169 (26), 152 (95), 141 (15), 127 (11), 115 (13), 44 (17); HRMS (EI, 70 eV): calcd for C24H18O2S [M]+: 370.10275; found 370.102731.
=3066, 2923, 2852, 1595, 1466 (w), 1317 (m), 1291 (w), 1195 (m), 1126 (s), 1091, 1003 (m), 834, 759, 690 (s), 732, 572 cm−1 (m); GC–MS (EI, 70 eV): m/z (%): 370 ([M]+, 100), 305 (43), 291 (11), 289 (40), 215 (11), 207 (49), 201 (17), 184 (10), 169 (26), 152 (95), 141 (15), 127 (11), 115 (13), 44 (17); HRMS (EI, 70 eV): calcd for C24H18O2S [M]+: 370.10275; found 370.102731.
2‐(4′‐Methylbiphenyl‐4‐ylsulfonyl)phenyl trifluoromethanesulfonate (5 a)
Starting with with 2 (205 mg, 0.4 mmol), K3PO4 (127 mg, 0.60 mmol), [Pd(PPh3)4] (14 mg, 3 mol %), 4‐methylphenylboronic acid (60 mg, 0.44 mmol) and 1,4‐dioxane (2 mL), following general procedure A, 5 a was isolated as a white solid (113 mg, 62 %). M.p. 91 °C; 1H NMR (300 mhz, CDCl3): δ=2.33 (s, 3 H, CH3), 7.19–7.22 (d, J=8.0 Hz, 2 H, ArH), 7.32–7.35 (m, 1 H, ArH), 7.41 (d, J=8.2 Hz, 2 H, ArH), 7.47–7.53 (m, 1 H, ArH), 7.58–7.61 (m, 1 H, ArH), 7.63–7.66 (m, 2 H, ArH), 7.95 (d, J=8.6 Hz, 2 H, ArH), 8.23–8.27 ppm (m, 1 H, ArH); 19F NMR (282 mhz, CDCl3): δ=−73.1; 13C NMR (62.9 mhz, CDCl3): δ=21.1 (CH3), 122.2 (CH), 124.0 (q, J
F,C
=321.7 Hz, CF3), 127.2, 127.5, 128.5, 128.9, 129.7, 130.9 (CH), 134.4 (C), 135.5 (CH), 136.0, 138.3, 138.8, 146.5, 146.8 ppm (C); IR (KBr):  =2959, 2928, 2869 (w), 1590, 1514, 1467, 1392 (m), 1313, 1151 (s), 1093, 1003 (m), 820 (s), 752, 623, 565 cm−1 (m); GC–MS (EI, 70 eV): m/z (%): 456 ([M]+, 100), 332 (21), 304 (14), 198 (28), 259 (10), 244 (26), 215 (21), 183 (16), 165 (15), 152 (18); HRMS (EI, 70 eV): calcd for C20H15F3O5S2 [M]+: 456.03075; found 456.030612.
=2959, 2928, 2869 (w), 1590, 1514, 1467, 1392 (m), 1313, 1151 (s), 1093, 1003 (m), 820 (s), 752, 623, 565 cm−1 (m); GC–MS (EI, 70 eV): m/z (%): 456 ([M]+, 100), 332 (21), 304 (14), 198 (28), 259 (10), 244 (26), 215 (21), 183 (16), 165 (15), 152 (18); HRMS (EI, 70 eV): calcd for C20H15F3O5S2 [M]+: 456.03075; found 456.030612.
2‐(4′‐Methylbiphenyl‐4‐ylsulfonyl)biphenyl (6 a)
Starting with 5 a (182 mg, 0.40 mmol), K3PO4 (127 mg, 0.6 mmol), [Pd(PPh3)4] (14 mg, 3 mol %), phenylboronic acid (53 mg, 0.44 mmol) and 1,4‐dioxane (2 mL), following the general procedure A,6 a was isolated as a solid (87 mg, 57 %). M.p. 146 °C; 1H NMR (300 mhz, CDCl3): δ=2.32 (s, 3 H, CH3), 6.91 (d, J=7.0 Hz, 2 H, ArH), 7.08–7.14 (m, 3 H, ArH), 7.16–7.20 (m, 4 H, ArH), 7.29–7.33 (m, 1 H, ArH), 7.28 (d, J=8.0 Hz, 2 H, ArH), 7.34 (d, J=8.2 Hz, 2 H, ArH), 7.49–7.53 (m, 2 H, ArH), 8.36–8.39 ppm (m, 1 H, ArH); 13C NMR (62.9 mhz, CDCl3): δ=21.1 (CH3), 126.6, 127.0, 127.2, 127.5, 127.6, 128.1, 128.4, 129.7, 130.0, 132.6, 132.8 (CH), 136.4, 138.1, 138.5, 139.0, 139.9, 142.1, 145.2 ppm (C); IR (KBr):  =3052, 2918, 2852 (w), 1591, 1464 (m), 1297, 1147 (s), 1090, 1005, 813, 757, 702, 690, 625, 582 (s), 541 cm−1 (m); GC–MS (EI, 70 eV): m/z (%): 384 ([M]+, 100), 320 (28), 304 (19), 289 (19), 215 (13), 183 (43), 165 (26), 152 (75); HRMS (EI, 70 eV): calcd for C25H20O2S [M]+: 384.11785; found 384.117575.
=3052, 2918, 2852 (w), 1591, 1464 (m), 1297, 1147 (s), 1090, 1005, 813, 757, 702, 690, 625, 582 (s), 541 cm−1 (m); GC–MS (EI, 70 eV): m/z (%): 384 ([M]+, 100), 320 (28), 304 (19), 289 (19), 215 (13), 183 (43), 165 (26), 152 (75); HRMS (EI, 70 eV): calcd for C25H20O2S [M]+: 384.11785; found 384.117575.
General procedure B for the synthesis of (8 a–e), (9 a–d) and (10 a–c)
In a pressure tube (glass bomb) a suspension of [Pd(PPh3)2Cl2] (2.5–5.0 mol %), 2 (257 mg, 0.5 mmol), alkyne (0.50–1.10 mmol), (Bu)4NI (27 mg, 15 mol %), CuI (10 mol %), Et3N (0.62–1.25 mmol) in DMF (5 mL) was purged with argon and stirred at 20 °C for 10 min. The reaction mixture was stirred at 60 −80 °C for 2–4 h. The solution was cooled to 20 °C, poured into H2O and CH2Cl2 (25 mL each), and the organic and the aqueous layer were separated. The latter was extracted with CH2Cl2 (3×25 mL). The combined organic layers were washed with H2O (3×20 mL), dried (Na2SO4), concentrated in vacuo and the residue was purified by chromatography (flash silica gel, heptanes/EtOAc) to give 8 a–e, 9 a–e and 10 a–c.
2‐(Phenylethynyl)‐1‐[4‐(phenylethynyl)benzenesulfonyl]benzene (8 a)
Starting with 2 (257 mg, 0.50 mmol), [Pd(PPh3)2Cl2] (18 mg, 5 mol %), (Bu)4NI (27 mg, 15 mol %), CuI (10 mg, 10 mol %), triethylamine (0.18 mL, 1.25 mmol), ethynylbenzene (0.13 mL, 1.1 mmol) and DMF (5 mL), following the general procedure B, 8 a was isolated as a white solid (184 mg, 88 %). M.p. 87 °C; 1H NMR (300 mhz, CDCl3): δ=7.25–7.27 (m, 4 H, ArH), 7.31–7.34 (m, 3 H, ArH), 7.40–7.56 (m, 8 H, ArH), 7.77–7.80 (m, 2 H, ArH), 8.18 ppm (dd, 1 H, J=1.8, 7.8 Hz, ArH); 13C NMR (62.9 mhz, CDCl3): δ=85.8, 87.8, 93.0, 98.2 (C≡), 122.3, 122.4, 122.7 (C), 128.3, 128.4, 128.5, 129.0, 129.1, 129.2, 131.6, 131.7, 131.8, 132.3, 133.1, 134.8 (CH), 135.2, 139.7, 141.0 ppm (C); IR (KBr):  =3060, 2924, 2853 (w), 1780, 1597, 1587, 1491, 1465, 1441, 1395, 1359 (m), 1319, 1279, 1249, 1220, 1155 (s), 1123, 1086, 916, 834 (m), 746, 688, 673, 622, 583 (s), 548 cm−1 (m); GC–MS (EI, 70 eV): m/z (%): 418 ([M]+, 21), 352 (33), 313 (100), 284 (21), 213 (10); HRMS (EI, 70 eV): calcd for C28H18O2S [M]+: 418.10220; found: 418.102241.
=3060, 2924, 2853 (w), 1780, 1597, 1587, 1491, 1465, 1441, 1395, 1359 (m), 1319, 1279, 1249, 1220, 1155 (s), 1123, 1086, 916, 834 (m), 746, 688, 673, 622, 583 (s), 548 cm−1 (m); GC–MS (EI, 70 eV): m/z (%): 418 ([M]+, 21), 352 (33), 313 (100), 284 (21), 213 (10); HRMS (EI, 70 eV): calcd for C28H18O2S [M]+: 418.10220; found: 418.102241.
2‐[4‐(p‐Tolylethynyl)phenylsulfonyl]phenyl trifluoromethanesulfonate (9 a)
Starting with 2 (257 mg, 0.50 mmol), [Pd(PPh3)2Cl2] (9 mg, 2.5 mol %), (Bu)4NI (27 mg, 15 mol %), CuI (10 mg, 10 mol %), triethylamine (0.09 mL, 0.62 mmol), 1‐ethynyl‐4‐methylbenzene (0.06 mL, 0.5 mmol) and DMF (4 mL), following general procedure B, (9 b) was prepared as a reddish oil (199 mg, 83 %). 1H NMR (300 mhz, CDCl3): δ=2.30 (s, 3 H, CH3), 7.10 (d, J=7.9 Hz, 2 H, ArH), 7.31–7.36 (m, 3 H, ArH), 7.48–7.55 (m, 2 H, ArH), 7.57–7.65 (m, 2 H, ArH), 7.87 (d, J=8.7 Hz, 2 H, ArH), 8.21–8.25 ppm (m, 1 H, ArH); 19FNMR (282.4 mhz, CDCl3): δ=−73.1; 13CNMR (75.5 mhz, CDCl3): δ=21.5 (CH3), 87.1, 94.1 (C≡), 116.3, 120.5 (C), 119.8 (q, J
F,C
=321.0 Hz, CF3), 122.3, 128.3, 128.6, 129.2,130.9, 131.7, 132.0, 135.7 (CH), 129.8, (CH), 135.7, 138.9, 139.5, 146.5 ppm (C); IR (KBr):  =3068, 2919 (w), 1586 (m), 1426, 1332 (s), 1231, 1198, 1157 (m), 1127 (s), 1087 (m), 882 (s), 771, 719, 608 (m), 572 cm−1 (s); GC–MS (EI, 70 eV): m/z (%)=480 ([M]+
, 100), 282 (18), 268 (33), 239 (27), 189 (16); HRMS (EI, 70 eV): calcd for C22H15F3O5S2 [M]+: 480.03075; found: 480.030830.
=3068, 2919 (w), 1586 (m), 1426, 1332 (s), 1231, 1198, 1157 (m), 1127 (s), 1087 (m), 882 (s), 771, 719, 608 (m), 572 cm−1 (s); GC–MS (EI, 70 eV): m/z (%)=480 ([M]+
, 100), 282 (18), 268 (33), 239 (27), 189 (16); HRMS (EI, 70 eV): calcd for C22H15F3O5S2 [M]+: 480.03075; found: 480.030830.
1‐[4‐(Hept‐1‐ynyl)benzenesulfonyl]‐2‐hex‐1‐ynyl‐benzene (10 a)
Starting with 9 c (230 mg, 0.5 mmol), [Pd(PPh3)2Cl2] (9 mg, 2.5 mol %), CuI (10 mg, 10 mol %), triethylamine (0.09 mL, 0.62 mmol), 1‐heptyne (0.06 mL, 0.5 mmol) and DMF (5 mL) following general procedure B, 10 a was isolated as a reddish oil (171 mg, 87 %). 1H NMR (300 mhz, CDCl3): δ=0.77–0.93 (m, 6 H, 2CH3), 1.18–1.24 (m, 2 H, CH2), 1.29–1.61 (m, 8 H, 4CH2), 2.30–2.36 (m, 4 H, 2CH2), 7.36–7.42 (m, 5 H, ArH), 7.74–7.78 (m, 2 H, ArH), 8.16–8.19 ppm (m, 1 H, ArH); 13C NMR (75.4 mhz, CDCl3): δ=13.6, 14.0 (CH3), 19.5, 22.1, 22.6, 28.4, 29.1, 30.2, 31.8 (CH2), 78.1, 79.5, 94.8, 100.4 (C≡), 123.5 (C), 127.6, 128.1, 128.9 (CH), 129.3 (C), 131.4, 132.9, 135.1 (CH), 139.1, 140.7 ppm (C); IR (KBr):  =3063, 2924 (w), 2929, 2859, 1590, 1467 (m), 1318, 1153 (s), 1089, 835, 754, 730 (m), 612, 581 cm−1 (s); GC–MS (EI, 70 eV): m/z (%): 392 ([M]+, 100), 365 (17), 350 (16), 349 (56), 228 (10), 205 (38), 163 (12), 115 (12); HRMS (EI, 70 eV): calcd for C25H28O2S [M]+: 392.18045; found: 392.180237.
=3063, 2924 (w), 2929, 2859, 1590, 1467 (m), 1318, 1153 (s), 1089, 835, 754, 730 (m), 612, 581 cm−1 (s); GC–MS (EI, 70 eV): m/z (%): 392 ([M]+, 100), 365 (17), 350 (16), 349 (56), 228 (10), 205 (38), 163 (12), 115 (12); HRMS (EI, 70 eV): calcd for C25H28O2S [M]+: 392.18045; found: 392.180237.
Supporting information
Detailed facts of importance to specialist readers are published as ”Supporting Information”. Such documents are peer‐reviewed, but not copy‐edited or typeset. They are made available as submitted by the authors.
miscellaneous_information
Acknowledgements
Financial support by the DAAD (scholarships for R. A. K., A. A., and M. F. I.), by the Friedrich‐Irmgard‐Harms‐Stiftung (scholarships for A. A. and R. A. K.), by the State of Pakistan (HEC scholarship for R. A. K. and M. H.), (HEC research grant under IPFP for R. A. K. by state of Pakistan) and by the State of Mecklenburg‐Vorpommern (scholarship for M. H.) is gratefully acknowledged.
References
- 1.
- 1a. Shrimali S. S., Joshi B. C., Kishore D., J. Indian Chem. Soc. 1988, 65, 438; [Google Scholar]
- 1b. Upadhyay P. S., Vansdadia R. N., Baxi A. J., Indian J. Chem. Sect. B 1990, 29, 793. [Google Scholar]
- 2. Teshirogi I., Matsutani S., Shirahase K., Fujii Y., Yoshida T., J. Med. Chem. 1996, 39, 5183. [DOI] [PubMed] [Google Scholar]
- 3. Paulini R., Lerner C., Diederich F., Jakob‐Roetne R., Zuercher G., Borroni E., Helv. Chim. Acta 2006, 89, 1856. [Google Scholar]
- 4.
- 4a. de Benedetti P. G., Iarossi D., Menziani C., Caiolfa V., Frassineti C., Cennamo C., J. Med. Chem. 1987, 30, 459; [DOI] [PubMed] [Google Scholar]
- 4b. de Benedetti P. G., Iarossi D., Folli U., Frassineti C., Menziani M. C., Cennamo C., J. Med. Chem. 1989, 32, 2396. [DOI] [PubMed] [Google Scholar]
- 5. Sircar I., Hoefle M., Maxwell R. E., J. Med. Chem. 1983, 26, 1020. [DOI] [PubMed] [Google Scholar]
- 6. Markley L. D., Tong Y. C., Dulworth J. K., Steward D. L., Goralski C. T., J. Med. Chem. 1986, 29, 427. [DOI] [PubMed] [Google Scholar]
- 7. Wright J., Bolton G., Creswell M., Downing D., Georgic L., Bioorg. Med. Chem. Lett. 1996, 6, 1809. [Google Scholar]
- 8.
- 8a. Neamati N., Mazumder A., Zhao H., Sunder S., T. R. Burke, Jr. , Schultz R. J., Pommier Y., Antimicrob. Agents Chemother. 1997, 41, 385; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8b. Chan J. H., Hong J. S., Hunter R. N., Orr G. F., Cowan J. R., Sherman D. B., Sparks S. M., Reitter B. E., Andrews C. W., Hazen, Richard J., Clair M. S., J. Med. Chem. 2001, 44, 1866; [DOI] [PubMed] [Google Scholar]
- 8c. Tagat J. R., McCombie S. W., Steensma R. W., Lin S.‐I., Nazareno D. V., Baroudy B., Vantuno N., Xu S., Liu J., Bioorg. Med. Chem. Lett. 2001, 11, 2143. [DOI] [PubMed] [Google Scholar]
- 9. Stanton J. L., Cahill E., Dotson R., Tan J., Tomaselli H. C., Wasvary J. M., Stephan Z. F., Steele R. E., Bioorg. Med. Chem. Lett. 2000, 10, 1661. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Kozlowski J. A., Zhou G., Tagat J. R., Lin S.‐I., McCombie S. W., Ruperto V. B., Duffy R. A., McQuade R. A., Crosby G., Taylor L. A., Billard W., Bioorg. Med. Chem. Lett. 2002, 12, 791; [DOI] [PubMed] [Google Scholar]
- 10b. Wang Y., Chackalamannil S., Hu Z., Clader J. W., Greenlee W., Billard W., Binch H., Crosby G., Ruperto V., Duffy R. A., McQuade R., Lachowicz J. E., Bioorg. Med. Chem. Lett. 2000, 10, 2247; [DOI] [PubMed] [Google Scholar]
- 10c. Boyle C. D., Chackalamannil S., Chen L.‐Y., Dugar S., Pushpavanam P., Billard W., Binch H., Crosby G., Cohen‐Williams M., Coffin V. L., Duffy R. A., Bioorg. Med. Chem. Lett. 2000, 10, 2727; [DOI] [PubMed] [Google Scholar]
- 10d. Wang Y., Chackalamannil S., Chang W., Greenlee W., Ruperto V., Duffy R. A., McQuade R., Lachowicz J. E., Bioorg. Med. Chem. Lett. 2001, 11, 891; [DOI] [PubMed] [Google Scholar]
- 10e. Tagat J. R., McCombie S. W., Steensma R. W., Lin S.‐I., Nazareno D. V., Baroudy B., Vantuno N., Xu S., Liu J., Bioorg. Med. Chem. Lett. 2001, 11, 2143; [DOI] [PubMed] [Google Scholar]
- 10f. Boyle C. D., Chackalamannil S., Clader J. W., Greenlee W. J., Josien H. B., Kaminski J. J., Kozlowski J. A., McCombie S. W., Nazareno D. V., Tagat J. R., Wang Y., Bioorg. Med. Chem. Lett. 2001, 11, 2311; [DOI] [PubMed] [Google Scholar]
- 10g. Boyle C. D., Vice S. F., Campion J., Chackalamannil S., Lankin C. M., McCombie S. W., Billard W., Binch H., Crosby G., Williams M.‐C., Coffin V. L., Bioorg. Med. Chem. Lett. 2002, 12, 3479. [DOI] [PubMed] [Google Scholar]
- 11. Sasse A., Ligneau X., Sadek B., Elz S., Pertz H. H., Ganellin C. R., Arrang J.‐M., Schwartz J.‐C., Schunack W., Stark H., Arch. Pharm. 2001, 334, 45. [DOI] [PubMed] [Google Scholar]
- 12. Langler R. F., Paddock R. L., Thompson D. B., Crandall I., Ciach M., Kain K. C., Aust. J. Chem. 2003, 56, 1127. [Google Scholar]
- 13. Lavey B. J., Kozlowski J. A., Hipkin R. W., Gonsiorek W., Lundell D. J., Piwinski J. J., Narula S., Lunn C. A., Bioorg. Med. Chem. Lett. 2005, 15, 783. [DOI] [PubMed] [Google Scholar]
- 14.
- 14a. Lu I.‐L., Mahindroo N., Liang P.‐H., Peng Y.‐H., Kuo C.‐J., Tsai K.‐C., Hsieh H.‐P., Chao Y.‐S., Wu S.‐Y., J. Med. Chem. 2006, 49, 5154; [DOI] [PubMed] [Google Scholar]
- 14b. Magriotis P. A., Kim K. D., J. Am. Chem. Soc. 1993, 115, 2972; [Google Scholar]
- 14c. Miller J. A., Zweifel G., J. Am. Chem. Soc. 1983, 105, 1383; [Google Scholar]
- 14d. Corey E. J., Tramontano A., J. Am. Chem. Soc. 1984, 106, 462; [Google Scholar]
- 14e. Marshall H. R., Chobanian M., Yanik M., Org. Lett. 2001, 3, 4107; [DOI] [PubMed] [Google Scholar]
- 14f. Karatholuvhu M. S., Fuchs P. L., J. Am. Chem. Soc. 2004, 126, 14314; [DOI] [PubMed] [Google Scholar]
- 14g. Yoshida M., Ohsawa Y., Ihara M., Tetrahedron Tetrahedron. 2006, 62, 11218. [Google Scholar]
- 15. Joseph J. K., Jain S. L., Sain B., Synth. Commun. 2006, 36, 2743, and references therein. [Google Scholar]
- 16.
- 16a. Chen D.‐W., Kubiak R. J., Ashley J. A., Janda K. D., J. Chem. Soc. Perkin Trans. 1 2001, 2796; [Google Scholar]
- 16b. Marquie J., Laporterie A., Dubac J., Roques N., Desmurs J.‐R., J. Org. Chem. 2001, 66, 421; [DOI] [PubMed] [Google Scholar]
- 16c. Répichet S., Le Roux C., Dubac J., Tetrahedron Lett. 1999, 40, 9233; [Google Scholar]
- 16d. Hajipour A. R., Zarei A., Khazdooz L., Pourmousavi S. A., Mirjalili B. B. F., Ruoho A. E., Phosphorus Sulfur Silicon Relat. Elem. 2005, 180, 2029; [Google Scholar]
- 16e. Woroshzow V., Kutschkarow V., Zh. Obshch. Khim. 1949, 19, 1943. [Google Scholar]
- 17a. Zhu W., Ma D., J. Org. Chem. 2005, 70, 2696; [DOI] [PubMed] [Google Scholar]
- 17b. Huang F., Batey R. A., Tetrahedron 2007, 63, 7667. [Google Scholar]
- 18. Bandgar B. P., Bettigeri S. V., Phopase J., Org. Lett. 2004, 6, 2105. [DOI] [PubMed] [Google Scholar]
- 19.
- 19a. Erian A. W., Issac Y., Sherif S. M., Mahmoud F. F., J. Chem. Soc. Perkin Trans. 1 2000, 3686; [Google Scholar]
- 19b. Ogura K., Takeda M., Xie J. R., Akazome M., Matsumoto S., Tetrahedron Lett. 2001, 42, 1923; [Google Scholar]
- 19c. Matsumoto S., Kumazawa K., Ogura K., Bull. Chem. Soc. Jpn. 2003, 76, 2179; [Google Scholar]
- 19d. Bianchi L., Dell′Erba C., Maccagno M., Mugnoli A., Novi M., Petrillo G., Sancassan F., Tavani C., J. Org. Chem. 2003, 68, 5254; [DOI] [PubMed] [Google Scholar]
- 19e. Mutsuhiro Y., Watanabe M., Furukawa S., Chem. Pharm. Bull. 1990, 38, 902; [Google Scholar]
- 19f. Padwa A., Gareau Y., Harrison B., Rodriguez A., J. Org. Chem. 1992, 57, 3540; [Google Scholar]
- 19g. Hayakawa K., Nishiyama H., Kanematsu K., J. Org. Chem. 1985, 50, 512; [Google Scholar]
- 19h. Nakayama J., Hirashima A., J. Am. Chem. Soc. 1990, 112, 7648; [Google Scholar]
- 19i. Hu C.‐M., Hong F., Jiang B., Xu Y., J. Fluorine Chem. 1994, 66, 215; [Google Scholar]
- 19j. Antelo B., Castedo L., Delamano J., Gomes A., Lopez C., Tojo G., J. Org. Chem. 1996, 61, 1188; [Google Scholar]
- 19k. Shkoor M., Riahi A., Fatunsin O., Reinke H., Fischer C., Langer P., Synthesis 2009, 2223. [Google Scholar]
- 20.For reviews of cross coupling reactions of polyhalogenated heterocycles, see:
- 20a. Schröter S., Stock C., Bach T., Tetrahedron 2005, 61, 2245; [Google Scholar]
- 20b. Schnürch M., Flasik R., Khan A. F., Spina M., Mihovilovic M. D., Stanetty P., Eur. J. Org. Chem. 2006, 3283. [Google Scholar]
- 21.For reactions of halogenated heterocycles from our group, see:
- 21a. Dang T. T., Dang T. T., Ahmad R., Reinke H., Langer P., Tetrahedron Lett. 2008, 49, 1698; [Google Scholar]
- 21b. Dang T. T., Villinger A., Langer P., Adv. Synth. Catal. 2008, 350, 2109; [Google Scholar]
- 21c. Hussain M., Nguyen T. H., Langer P., Tetrahedron Lett. 2009, 50, 3929; [Google Scholar]
- 21d. Tengho Toguem S.‐M., Hussain M., Malik I., Villinger A., Langer P., Tetrahedron Lett. 2009, 50, 4962; [Google Scholar]
- 21e. Dang T. T., Dang T. T., Rasool N., Villinger A., Langer P., Adv. Synth. Catal. 2009, 351, 1595; [Google Scholar]
- 21f. Ullah I., Khera R. A., Hussain M., Langer P., Tetrahedron Lett. 2009, 50, 4651. [Google Scholar]
- 22.For reactions of bis(triflates)from our group, see:
- 22a. Nawaz M., Farooq Ibad M., Obaid‐Ur‐Rahman A., Khera R. A., Villinger A., Langer P., Synlett 2010, 150; [Google Scholar]
- 22b. Nawaz M., Khera R. A., Malik I., Ibad M. F., Abid O.‐u. R., Villinger A., Langer P., Synlett 2010, 979; [Google Scholar]
- 22c. Ibad O.‐u.‐R. Abid; M. F., Nawaz M., Sher M., Rama N. H., Villinger A., Langer P., Tetrahedron Lett. 2010, 51, 1541; [Google Scholar]; Ibad M. F., Nawaz M., Sher M., Rama N. H., Villinger A., Langer P., Tetrahedron Lett. 2010, 51, 1541; [Google Scholar]; Mahal A., Villinger A., Langer P., Synlett 2010, 1085. [Google Scholar]
- 23. Ali A., Khera R. A., Ibad M. F., Hussain M., Villinger A., Langer P., Synlett 2010, 731; [Google Scholar]; Hara R., Liu Y., Sun W.‐H., Takahashi T., Tetrahedron Lett. 1997, 38, 4103. [Google Scholar]
- 24.For reviews of the Suzuki reaction, see:
- 24a. Miyaura N., Suzuki A., Chem. Rev. 1995, 95, 2457; [Google Scholar]
- 24b. Suzuki A. in Organopalladium Chemistry for Organic Synthesis (Ed.: E. Negishi), Wiley, 2002, chap. III. 2.2; [Google Scholar]
- 24c. Nicolaou K. C., Bulger P. G., Sarlah D., Angew. Chem. 2005, 117, 4516; [Google Scholar]; Angew. Chem. Int. Ed. 2005, 44, 4442. [Google Scholar]
- 25.CCDC 848539 and CCDC 848540 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif.
- 26. Cadierno V., Gamasa M. P., Gimeno J., J. Organomet. Chem. 2001, 39, 621. [Google Scholar]
- 27.
- 27a. Koreeda M., Yang W., Synlett 1994, 201; [Google Scholar]
- 27b. Cai M. Z., Wang D., Wang P., J. Chem. Res. 2006, 504; [Google Scholar]
- 27c. Jiang J. W., Cai M. Z., Chin. Chem. Lett. 2006, 17, 757. [Google Scholar]
- 28.
- 28a. Braga A. L., Zeni G., Andrade L. H., Silveira C. C., Stefani H. A., Synthesis 1998, 39; [Google Scholar]
- 28b. Ma Y., Huang X., Synth. Commun. 1997, 27, 3441; [Google Scholar]
- 28c. Cai M. Z., Peng C. Y., Zhao H., J. Chem. Res. 2002, 376; [Google Scholar]
- 28d. Zhong P., Huang N., Synth. Commun. 2002, 32, 139. [Google Scholar]
- 29.
- 29a. Huang X., Wang Y. P., Tetrahedron Lett. 1996, 37, 7417; [Google Scholar]
- 29b. Cai M., Chen J. Z., J. Chem. Res. 2004, 840. [Google Scholar]
- 30.
- 30a. Chatani N., Amishiro N., Murai S., J. Am. Chem. Soc. 1991, 113, 7778; [Google Scholar]
- 30b. Cai M. Z., Hao W., Zhao H., Song C., J. Chem. Res. 2003, 485. [Google Scholar]
- 31.
- 31a. Huang X., Zhong P., J. Chem. Soc. Perkin Trans. 1 1999, 1, 1543; [Google Scholar]
- 31b. Cai M. Z., Zhao H., Ye H., Xia J., Song C., J. Chem. Res. 2003, 344. [Google Scholar]
- 32.
- 32a. Yoshida M., Yoshikawa S., Fukuhara T., Yoneda N., Hara S., Tetrahedron 2001, 57, 7143; [Google Scholar]
- 32b. Legros J., Crousse B., Begue J. P., J. Fluorine Chem. 2001, 107, 121. [Google Scholar]
- 33.
- 33a. Yoshimatsu M., Hasegawa J., J. Chem. Soc. Perkin Trans. 1 1997, 211; [Google Scholar]
- 33b. Shen Y., Wang G., Sun J., J. Chem. Soc. Perkin Trans. 1 2001, 519. [Google Scholar]
- 34.For reviews of Sonogashira reactions, see:
- 34a. Sonogashira K. in Metal‐Catalyzed Cross‐Coupling Reactions, Wiley VCH, 1998, p. 203; [Google Scholar]
- 34b. Sonogashira K. in Organopalladium Chemistry for Organic Synthesis (Ed.: E. Negishi), Wiley, 2002, chap. III 2–8, p. 493. [Google Scholar]
- 35. Genet J. P., Blart E., Savignac M., Synlett 1992, 715. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Detailed facts of importance to specialist readers are published as ”Supporting Information”. Such documents are peer‐reviewed, but not copy‐edited or typeset. They are made available as submitted by the authors.
miscellaneous_information