

Abstract
Short interfering RNA (siRNA) functions directly in the cytoplasm, where it is assembled into an RNA‐induced silencing complex (RISC). The localized delivery of siRNA to a specific site in vivo is highly challenging. There are many disease states in which a systemic effect of RNAi may be desirable; some examples include non‐localized cancers, HIV, neurodegenerative diseases, respiratory viruses, and heart and vascular disease. In this Concept, we will focus on the localized delivery of siRNA to a target site using various delivery modalities. In certain tissues, such as the eye, central nervous system and lung, it has been demonstrated that a simple injection of naked siRNA will silence gene expression specifically in that tissue. To achieve local gene silencing in other tissues, a variety of approaches have been pursued to help stabilize the siRNA and facilitate uptake; they include chemical modification of the siRNA or complexation within liposomes or polymers to form nanoparticles. Recently, the use of macroscopic biomaterial scaffolds for siRNA delivery has been reported, and although there is still significant work to be done in this area to optimize the delivery systems, it is an important area of research that offers the potential for having great impact on the field of siRNA delivery.
Keywords: biomaterials, biotechnology, gene silencing, localized delivery, siRNA, therapeutics
Tackling the problem of localized siRNA delivery: RNA interference is a powerful gene‐silencing mechanism that inhibits gene expression by the targeted destruction of specific mRNA molecules. It has the potential to revolutionize disease treatment and aid in the functional repair of damaged tissue by decreasing the expression of specific proteins. However, effective delivery of short interfering RNA (siRNA) to target cells in vivo remains a significant challenge. This article describes current research towards the localized and sustained delivery of siRNA.
Introduction
Gene expression can be silenced by the targeted destruction of specific mRNA molecules in a highly conserved process known as RNA interference (RNAi).1 Short interfering RNA (siRNA) functions directly in the cytoplasm, where it is assembled into an RNA‐induced silencing complex (RISC) comprising several proteins and the siRNA (Figure 1). When this complex is activated by ATP, the double‐stranded siRNA becomes single stranded, and the complex is now able to bind to complementary mRNA.2 When a complementary mRNA sequence is found, the nuclease activity of the RISC destroys the mRNA.3 In lieu of delivering siRNA into a cell, it is also possible to deliver long double‐stranded RNA (dsRNA) or a plasmid or virus that encodes for short hairpin RNA (shRNA). The longer dsRNA or shRNA molecules are cleaved by an enzyme called Dicer into the short siRNA sequences that then act via the RISC complex.4 However, the longer double‐stranded RNA molecules can induce a cellular immune response via the interferon system, which is a major obstacle to effectively employing these methods.5
Figure 1.

Schematic of RNA interference mechanism.
RNAi has the potential to treat diseases that can be corrected by the decreased expression of specific proteins, for example in cancer therapeutics6 or in tissue regeneration.7 Despite the enormous potential, the use of siRNA has not yet been successful clinically, largely due to difficulties providing effective delivery of the siRNA to the desired cells in vivo.8 Similar to any RNA molecule, siRNA is highly prone to degradation by ribonucleases that are found throughout the body and in the environment.8b, 9 Additionally, in rapidly dividing cells, the silencing effect that the siRNA provides may only last a few days as the concentration of siRNA is diluted by cell divisions.1b Furthermore, the localized delivery of the siRNA to a specific site in vivo is highly challenging.10 In fact, it has been reported that “the three biggest problems with RNAi therapeutics remain ‘delivery, delivery, and delivery.′”11
In the field of gene delivery, there has been significant research in the delivery of plasmid DNA. However, many of these results do not necessarily translate to the delivery of siRNA, as DNA and siRNA have very different properties. Where DNA functions in the nucleus, siRNA has activity in the cytoplasm. RNA is more prone to degradation by nucleases than DNA because of the hydroxyl group in its 2’ position. Due to its small size, approximately 20–30 nucleotides in length, siRNA is a stiffer molecule than DNA, behaving as a rigid rod. As such, it may not further condense when interacting with cationic agents for delivery, and it has been suggested that the resultant nanoparticles can become too large for endocytosis or provide incomplete encapsulation.12 These differences and their effect on delivery of these molecules have been recently reviewed by Gary, et al.8b
When considering the delivery of siRNA, there are many disease states in which a systemic effect of RNAi may be desirable; some examples include non‐localized cancers, HIV, neurodegenerative diseases, respiratory viruses, and heart and vascular disease. However, in other instances, silencing the expression of the target gene throughout the body could be detrimental if only a localized area needs treatment. Examples of this include non‐malignant tumors or tumor resection sites and also possibly for aiding in the regeneration of diseased or damaged tissue at a defect site. In this Concept, we will focus on the localized delivery of siRNA to a target site.
Delivery of Naked siRNA
“Naked” siRNA delivery is the direct injection of a saline or excipient solution containing non‐complexed siRNA sequences to a target site. Generally, naked siRNA injected into the body has a very short half life, on the order of minutes, limiting its usefulness.13 However, it has been found that some tissues are able to uptake naked siRNA to a much higher degree than other tissues (for instance the eye, central nervous system, and lung) making localized delivery by direct injection of siRNA to these sites a possibility. One of the largest complications leading to loss of sight is uncontrolled retinal neovascularization in patients that have ischemic retinal disorders.14 Inhibition of VEGF by antibodies has been demonstrated to reduce ocular neovascularization by about 50 %, although further reduction could not be achieved due to difficulties in delivering the antibodies throughout the retinal tissue.14 Thus, other groups have examined the direct delivery to the eye of naked siRNA targeting VEGF to reduce this neovascularization using RNA interference. Ocular neovascularization was decreased by direct injection of naked siRNA against VEGF into the retina of mice.15 Also, the delivery of siRNA targeting VEGF receptor 1 (VEGFR1) resulted in decreased ocular neovascularization with siRNA remaining present in these cells for at least five days.16 There are currently two clinical trials underway exploring the delivery of siRNA against VEGF or VEGFR1 to treat age‐related macular degeneration.17
There have been several reports on the local delivery of naked siRNA to the nervous system. The intrathecal injection of naked siRNA complementary to a pain‐related ion channel resulted in diminished pain response.18 The silencing of agouti‐related peptide by direct injection of siRNA into the hypothalamus of mice led to decreased body weight and increased metabolism, which could impact obesity.19 The intracerebroventricular injection of naked siRNA has also been shown to provide gene silencing that is confined to the brain. Using this method, dopamine and serotonin transporters have been silenced, affecting the locomotive behaviors of mice.20
Studies have also demonstrated the ability to deliver naked siRNA to the lungs, typically by an intranasal route. siRNA‐targeting heme oxygenase‐1 was delivered intranasally to modulate cell apoptosis, and the silencing of this gene was shown to be specific to the lung.21 Intranasal delivery of naked siRNA targeting the genome of the severe acute respiratory syndrome (SARS) coronavirus, delivered either before or after infection with the SARS virus, was found to decrease the virus levels in the lungs of rhesus macaques and substantially reduce the symptoms of the disease.22 Two other viruses that infect the respiratory system and for which vaccines or other antiviral treatments are currently not available include respiratory syncytial virus (RSV) and parainfluenza virus. These RNA viruses encode in part for RNA polymerases to aid in the replication of the viral genome. It has been demonstrated that the intranasal delivery of naked siRNA targeted to a subunit of the viral polymerases was able to prevent and treat both of these viruses in mice.23 There is currently a clinical trial underway to examine the efficacy of this treatment in humans suffering from RSV infection.17 Intratracheal instillation, where a material is introduced directly into the lungs via a catheter or needle placed in the trachea,24 has also been used to deliver unmodified naked siRNA to the lungs to silence the expression of macrophage inflammatory protein 2, which helped to reduce the migration of neutrophils to the lung after acute lung injury.26
Chemical Conjugation of siRNA
Efforts to increase the activity of siRNA in vivo have been pursued by conjugating the siRNA molecule with a chemical entity to help enhance the cellular uptake of the siRNA and/or help improve its pharmacokinetics. Various chemical modifications of siRNA have been reviewed extensively.26 Frequently, lipophilic moieties such as cholesterol are covalently bound to the siRNA as they have been shown to provide greater in vivo stability by associating with serum proteins in the blood thereby preventing plasma clearance, and also to promote increased cell uptake due to increased cell membrane permeability.27 Localized delivery using chemically modified siRNA has focused on cholesterol‐conjugated siRNA. Cholesterol has been conjugated to siRNA complementary to the huntingtin gene, found in patients with Huntington’s disease, and when injected directly into the striatum of mice was found to locally silence gene expression and decrease the pathology of Huntington’s disease in these mice.28 Also, the delivery of cholesterol‐conjugated siRNA targeted to herpes viral proteins was able to protect mice from herpes simplex virus 2 when the siRNA was applied directly to the vaginal mucosa.29 Additionally, cholesterol‐conjugated siRNA molecules infused directly into the central nervous system in rats were uptaken by oligodendrocytes, a cell type that is difficult to transfect.30 A recent study demonstrated that the modification of siRNA with poly(ethylene glycol) (PEG) and either mannose‐6‐phosphate or galactose efficiently delivered siRNA into hepatocytes in vitro.31 Their previous study demonstrated specific uptake after intravenous injection by hepatocytes in vivo when a similar modification was used for antisense oligonucleotides, and although not demonstrated in this study, it was hypothesized that similar results will be obtained with this modified siRNA.
siRNA‐Laden Particles
Nano‐ or microparticles with siRNA encapsulated in them can be fabricated from a variety of natural and synthetic polymers. The use of siRNA‐laden particles has been widely researched, as the siRNA can be protected from environmental factors which may degrade it and is typically uptaken by cells with a higher efficiency.32 The particles can be uptaken by cells through endocytosis, with the siRNA subsequently released into the cytoplasm where it has activity. This process can be nonspecific, with any type of cell able to uptake the particles (Figure 2 a), or it can be targeted to specific cell receptors so that only the desired cell population uptakes the particles (Figure 2 b). Again, there is much work in the systemic delivery of siRNA particles, but here the focus will be on technologies that have demonstrated the ability to deliver siRNA locally.
Figure 2.

Uptake of siRNA particles into cell by endocytosis by either a) nonspecific means or b) receptor‐ligand targeting.
siRNA Incorporation into Liposomes and Lipoplexes
To increase the half‐life of siRNA in vivo, it can be encapsulated within liposomes or complexed with cationic lipids to form siRNA‐laden nanoparticles. Liposomes typically consist of a phospholipid bilayer surrounding an aqueous core, and it is within this core that siRNA (or other nucleotides, proteins, or drugs) is contained. Recently, liposomes containing siRNA were modified with a peptide that targeted MCF‐7 breast cancer cells, and were shown to effectively silence the expression of the PDMR14 gene that plays a role in breast cancer carcinogenesis, with minimal uptake and silencing effect in other non‐cancerous cells.33
A more popular alternative to liposomes is the use of lipoplexes, where cationic lipids are mixed with the anionic siRNA to form complexes based on electrostatic interactions. However, these complexes tend to be more unstable in solution and may aggregate over time.34 Additionally, due to their charge, they can be cytotoxic or induce an inflammatory response.35 Complexes of siRNA‐targeting red fluorescent protein (RFP) with the cationic lipid N′,N′′‐dioleylglutamide were demonstrated to silence the expression of RFP when injected locally into a tumor expressing RFP that was formed subcutaneously.36 Another group demonstrated the uptake of anti‐VEGF siRNA in a subcutaneous tumor when siRNA complexed with a cholesterol derivative, cholesteryl oligo‐d‐arginine, was injected locally into the tumor.37
siRNA incorporated into lipoplexes has also been found to be uptaken at a high efficiency in vaginal mucosa when directly applied to the area. This has been shown to effectively silence the expression of herpes viral proteins to protect mice from being infected with herpes simplex virus 2.38 siRNA against lamin A/C was incorporated into lipoplexes with localized silencing found in the vaginal mucosa.39 Additionally, this group demonstrated local delivery of siRNA lipoplexes to the colon, another mucosal surface. By silencing the expression of TNF‐α in the colon, inflammation was reduced in mice with inflammatory bowel disease.39
Delivery of siRNA to hepatic tissue in both rodents and primates has been demonstrated by using a modification of the lipid 1,2‐dilinoleyloxy‐3‐dimethylaminopropane.40 The hepatic gene transthyretin was targeted in primates using this formulation, and at the highest concentration of siRNA (1 mg kg−1) approximately 70 % reduction in gene expression was observed 48 h after intravenous delivery.
An interesting approach to the localized delivery of siRNA involved the synthesis of oleic acid‐based liposomes with a magnetic core comprising a colloidal suspension of magnetic nanocrystals. siRNA was able to interact with these liposomes by electrostatic interaction of the siRNA with the oleic acid, thus essentially forming lipoplexes. When these magnetic liposomal particles carrying siRNA targeted to epidermal growth factor receptor were injected intravenously and allowed to travel systemically in mice, they were found to accumulate in high amounts locally in a tumor to which an external magnetic field was focused.41 Some particles, however, still made their way into other tissues, including the lung, liver, and spleen.
Polymeric Nanoparticles Containing siRNA
Frequently, cationic polymers are used to form nanoscale siRNA complexes based on electrostatic interaction of the negatively charged siRNA with the positively charged polymer. Poly(ethyleneimine) (PEI) is one such polymer. PEI is a synthetic polymer that can be either linear or branched, and with a high percentage of free amine groups which are positively charged. PEI complexed with siRNA targeting VEGF has been injected directly into subcutaneous tumors, and the silencing of VEGF and subsequent diminishment of tumor growth was confirmed.42 PEI‐siRNA was also delivered intratumorally with siRNA complementary to STAT3 to promote the apoptosis of the tumor cells, resulting in reduced tumor weight and size in mice in vivo.43
To increase the circulation time of PEI‐RNA particles, the PEI can be modified with PEG, as this hydrophilic molecule does not adsorb serum proteins. The drawback of adding PEG to PEI is that it will reduce the positive charge of the polymer, thus lessening its interaction with cell membranes, which leads to decreased uptake of the PEI particles. siRNA complexed with PEG‐PEI was successfully delivered by tracheal intubation to the lungs, with the majority of the particles ending up in the lungs for gene silencing, although some siRNA complexes were found in other tissues.44
Chitosan, a natural polysaccharide derived from the shells of crustaceans, is another polycation that can be used for siRNA complexation. Chitosan–siRNA nanocomplexes with siRNA targeted to enhanced green fluorescent protein (eGFP) have been delivered to the lungs of eGFP transgenic mice by nasal administration and were shown to silence eGFP expression by approximately 40 % at six days when delivered daily at 30 μg per day.45 Chitosan complexed with siRNA against red fluorescent protein (RFP) has been delivered intratumorally into a subcutaneous tumor expressing RFP, and was shown to reduce RFP expression by approximately 83 %.46
Thioketal nanoparticles have been demonstrated to protect siRNA delivered orally so that it can reach the intestinal tract and be effectively uptaken there.47 These nanoparticles were formulated from the polymer poly(1,4‐phenyleneacetone dimethylene thioketal), which degrades in response to the high levels of reactive oxygen species found at sites of intestinal inflammation. Using these nanoparticles, the silencing of TNF‐α in the colon was shown to protect mice from ulcerative colitis.
Nanoparticles can also be modified with a targeting ligand to increase uptake by only the desired cell population, even if the nanoparticle–siRNA formulation is delivered systemically. For instance, EGFR siRNA‐laden nanogels composed primarily of N‐isopropylmethacrylamide were conjugated with a peptide that targeted the EphA2 receptor.48 Although these have not yet been tested in vivo, the silencing of EGFR was seen in a cell line expressing EphA2 receptor, with no silencing found in a cell line that does not express this receptor. Poly(propyleneimine) (PPI) dendrimers have also been used to create siRNA‐containing nanoparticles; these particles were modified with a synthetic analogue luteinizing hormone‐releasing hormone (LHRH) peptide for increased uptake by cancer cells that overexpress the LHRH receptor.49 This group demonstrated that the siRNA particles were indeed uptaken to a much higher degree in vivo by the tumor cells. Notably, a recent clinical trial examined the delivery of siRNA‐laden cyclodextrin‐based polymeric nanoparticles modified with a targeting ligand for human transferrin protein receptors, which are upregulated in tumors, to humans in vivo.50 Although these nanoparticles were delivered systemically, they were found to localize in the solid tumors of the patients. The siRNA was targeted to the M2 subunit of ribonucleotide reductase (RRM2), an established anti‐cancer target. Both mRNA and protein levels of RRM2 in tumor biopsies were found to be reduced. This system is very promising for the targeted treatment of solid tumors.
Alternatively, “smart” nanoparticles can be designed such that they do not release their payload until they receive a physical signal to do so. One example of this is the use of gold nanospheres to which thiolated siRNA complementary to NF‐kB is bound.51 The gold nanoparticles exposed to near‐infrared light absorb the light and experience a change in shape, which releases the siRNA. Thus when these nanoparticles are uptaken by cells, only particles in cells exposed to near‐infrared light (which can penetrate deep into tissues) will release the siRNA into the cytoplasm. This group demonstrated targeted knockdown of NF‐kB in tumor tissue, which increased the susceptibility of the tumor to chemotherapeutic drugs.
Polymeric microparticles can also be used for the delivery of siRNA, although there are few reports on this. Poly(lactic‐co‐glycolic acid) (PLGA), a biodegradable, FDA‐approved synthetic polymer, has been used to encapsulate anti‐VEGF siRNA in PLGA microspheres of about 40 mm in diameter, and these microspheres were then injected adjacent to a subcutaneous tumor, resulting in decreased tumor growth over the course of three weeks.52
Macroscopic Biomaterial Scaffolds with Encapsulated siRNA
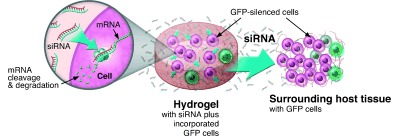
As described in the sections above, many of the current technologies being examined for siRNA delivery rely on the formation of nanoscale siRNA complexes and in some cases microscale complexes. A major disadvantage of using these techniques for local delivery is that they can be rapidly dispersed from the delivery site due to their small size. Therefore, the challenge of getting a sufficient amount of siRNA to the desired location during a desired period of time still remains. There is a paucity of work examining the use of macroscopic biomaterial scaffolds for the local delivery of siRNA. Biomaterial scaffolds are frequently used in tissue engineering to encourage cell migration and proliferation within the material, which is placed at a tissue defect site. It is often possible to incorporate bioactive factors into these scaffolds to encourage the tissue regeneration process, as these factors can be released from the scaffold over time to incorporated cells and the surrounding host cells. Although traditionally these bioactive factors have been growth factors or DNA to upregulate the expression of growth or transcription factors, it may be beneficial to use siRNA to aid in the tissue regeneration process, perhaps by inhibiting the production of molecules that hinder the healing process or by influencing cell behaviors such as proliferation or differentiation. The use of RNA interference for regenerative medicine applications has been reviewed by Yao, et al.53 The release of siRNA incorporated into a biomaterial scaffold could be mediated by diffusion through the biopolymer pores, the degradation of the polymer, or through ionic or affinity interactions with the scaffold. The released siRNA would then act locally, and the scaffold itself could aid in tissue regeneration by providing a conductive or inductive environment for cellular growth within the defect.54 Work in our laboratory was the first to demonstrate the potential of such a system.55 Ionically crosslinked alginate, photocrosslinked alginate, and collagen hydrogels were shown to provide varying siRNA release profiles, and in all cases the siRNA released was bioactive and able to silence protein expression in cells both surrounding the scaffolds and incorporated within the scaffolds (schematic shown in Figure 3). The use of collagen scaffolds to deliver siRNA‐dendrimer nanoparticles has also recently been demonstrated, although it was found that approximately 60 % of the siRNA was released in the first two days with little subsequent release.56 However, the nanoparticles delivered from these scaffolds were shown to reduce protein expression for up to seven days when cells were cultured directly on the scaffolds. Singh, et al. incorporated DNA, siRNA, and chemokines into PLGA microparticles, which were then incorporated into hydrogels made either of dextran vinyl sulfone and poly(ethylene glycol) (PEG) or PEG alone.57 The chemokines were released from the hydrogels to promote the migration of antigen‐presenting cells into the hydrogels, and these cells were then able to uptake the siRNA and DNA released from the PLGA microparticles. Further developing these biomaterial scaffold systems and others that incorporate siRNA offers exciting possibilities for the sustained and targeted delivery of siRNA to a specific site in vivo.
Figure 3.

Schematic of hydrogel formation for delivery of siRNA and subsequent inhibition of gene expression in incorporated and neighboring cells. Biomaterial solutions of alginate, photo alginate, or collagen are mixed with siRNA and cells, and hydrogels are then formed by ionic crosslinking, photocrosslinking, or thermogelling, respectively. The siRNA diffuses through the hydrogel to affect incorporated cells, and it is also released from the hydrogel to locally affect surrounding cells. (Reproduced from reference 55 with permission from Journal of the American Chemical Society.)
Summary and Outlook
RNA interference offers great promise for treating various disease states or helping to repair damaged tissue. However, there are significant challenges that need to be overcome before this technology can be used in the clinical setting. In particular, for applications that will benefit from the local delivery of the siRNA, it will be important to develop systems that are capable of providing bioactive siRNA to a specific site rather than systemically. As highlighted in this article, many groups have examined the local delivery of siRNA using various delivery modalities (Table 1). In certain tissues, it has been demonstrated that a simple injection of naked siRNA will silence gene expression specifically in that tissue. However, this technique for local delivery is unfortunately limited to only a few tissue types including the eye, central nervous system, and lung. To achieve local gene silencing in other tissues, a variety of approaches have been pursued to help stabilize the siRNA and to encourage cellular uptake by administration to the site of interest; they include chemical modification of the siRNA or complexation within liposomes or polymers to form nanoparticles. Overall, the field of siRNA delivery has been very focused on the use of nanoparticles. While these may promote the stability and uptake of the siRNA, they do not necessarily provide localization as they may be readily carried away from the site of interest. Only recently has the use of macroscopic biomaterial scaffolds for siRNA delivery been examined and reported. There is still significant work to be done in this area to optimize the delivery systems and ensure longer‐term release of bioactive siRNA at the site of interest. However, this is an important area of research that warrants further study and offers the potential for having great impact on the field of siRNA delivery.
Table 1.
The publisher did not receive permission from the copyright owner to include this object in this version of this product. Please refer either to the publisher's own online version of this product or the printed product where one exists.
Acknowledgements
We would like to thank Chirag Dhami for his assistance in creating some of the figures.
The frontispiece graphic and table of contents entry was reproduced from reference 55 with permission from Journal of the American Chemical Society.
References
- 1.
- 1a. Fire A., Xu S., Montgomery M. K., Kostas S. A., Driver S. E., Mello C. C., Nature 1998, 391, 806–811; [DOI] [PubMed] [Google Scholar]
- 1b. Dykxhoorn D. M., Palliser D., Lieberman J., Gene Ther. 2006, 13, 541–552. [DOI] [PubMed] [Google Scholar]
- 2. Nykänen A., Haley B., Zamore P. D., Cell 2001, 107, 309–321. [DOI] [PubMed] [Google Scholar]
- 3. Hammond S. M., Bernstein E., Beach D., Hannon G. J., Nature 2000, 404, 293–296. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Bernstein E., Caudy A. A., Hammond S. M., Hannon G. J., Nature 2001, 409, 363–366; [DOI] [PubMed] [Google Scholar]
- 4b. Collins R. E., Cheng X., FEBS Lett. 2005, 579, 5841–5849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Aigner A., Appl. Microbiol. Biotechnol. 2007, 76, 9–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chen Y., Huang L., Expert Opin. Drug Delivery 2008, 5, 1301–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.
- 7a. Natarajan R., Salloum F. N., Fisher B. J., Kukreja R. C., Fowler A. A., Circ. Res. 2005, 98, 133–140; [DOI] [PubMed] [Google Scholar]
- 7b. Gao Z., Wang Z., Shi Y., Lin Z., Jiang H., Hou T., Wang Q., Yuan X., Zhao Y., Wu H., Zin Y., Plast. Reconstr. Surg. 2006, 118, 1328–1337; [DOI] [PubMed] [Google Scholar]
- 7c. Hu G., Kim J., Xu Q., Leng Y., Orkin S. H., Elledge S. J., Genes Dev. 2009, 23, 837–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.
- 8a. Pardridge W. M., Expert Opin. Biol. Ther. 2004, 4, 1103–1113; [DOI] [PubMed] [Google Scholar]
- 8b. Gary D. J., Puri N., Won Y. Y., J. Controlled Release 2007, 121, 64–73. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Rutz S., Scheffold A., Arthritis Res. Ther. 2004, 6, 78; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9b. Ryther R. C., Flynt A. S., Phillips J. A., 3rd, Patton J. G., Gene Ther. 2005, 12, 5–11; [DOI] [PubMed] [Google Scholar]
- 9c. Aigner A., J. Biomed. Biotechnol. 2006, 2006, 71659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Song E., Zhu P., Lee S. K., Chowdhury D., Kussman S., Dykxhoorn D. M., Feng Y., Palliser D., Weiner D. B., Shankar P., Marasco W. A., Lieberman J., Nat. Biotechnol. 2005, 23, 709–717. [DOI] [PubMed] [Google Scholar]
- 11. Perkel J. M., Science 2009, 326, 454–456. [Google Scholar]
- 12. Spagnou S., Miller A. D., Keller M., Biochemistry 2004, 43, 13348–13356. [DOI] [PubMed] [Google Scholar]
- 13. Dykxhoorn D. M., Lieberman J., Cell 2006, 126, 231–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Aiello L. P., Pierce E. A., Foley E. D., Takagi H., Chen H., Riddle L., Ferrara N., King G. L., Smith L. E., Proc. Natl. Acad. Sci. USA 1995, 92, 10457–10461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Reich S. J., Fosnot J., Kuroki A., Tang W., Yang X., Maguire A. M., Bennett J., Tolentino M. J., Mol. Vision 2003, 9, 210–216. [PubMed] [Google Scholar]
- 16. Shen J., Samul R., Silva R. L., Akiyama H., Liu H., Saishin Y., Hackett S. F., Zinnen S., Kossen K., Fosnaugh K., Vargeese C., Gomez A., Bouhana K., Aitchison R., Pavco P., Campochiaro P. A., Gene Ther. 2006, 13, 225–234. [DOI] [PubMed] [Google Scholar]
- 17. Sepp‐Lorenzino L., Ruddy M. K., Clin. Pharmacol. Ther. 2008, 84, 628–632. [DOI] [PubMed] [Google Scholar]
- 18. Dorn G., Patel S., Wotherspoon G., Hemmings‐Mieszczak M., Barclay J., Natt F. J., Martin P., Bevan S., Fox A., Ganju P., Wishart W., Hall J., Nucleic Acids Res. 2004, 32, e49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Makimura H., Mizuno T. M., Mastaitis J. W., Agami R., Mobbs C. V., BMC Neurosci. 2002, 3, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.
- 20a. Hoyer D., Thakker D. R., Natt F., Maier R., Huesken D., Muller M., Flor P., H V. D. P., Schmutz M., Bilbe G., Cryan J. F., J. Recept. Signal Transduction Res. 2006, 26, 527–547; [DOI] [PubMed] [Google Scholar]
- 20b. Thakker D. R., Natt F., Husken D., Maier R., Muller M., van der Putten H., Hoye D., Cryan J. F., Proc. Natl. Acad. Sci. USA 2004, 101, 17270–17275; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20c. Thakker D. R., Natt F., Husken D., van der Putten H., Maier R., Hoyer D., Cryan J. F., Mol. Psychiatry 2005, 10, 782–789, 714. [DOI] [PubMed] [Google Scholar]
- 21. Zhang X., Shan P., Jiang D., Noble P. W., Abraham N. G., Kappas A., Lee P. J., J. Biol. Chem. 2003, 279, 10677–10684. [DOI] [PubMed] [Google Scholar]
- 22. Li B. J., Tang Q., Cheng D., Qin C., Xie F. Y., Wei Q., Xu J., Liu Y., Zheng B. J., Woodle M. C., Zhong N., Lu P. Y., Nat. Med. 2005, 11, 944–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bitko V., Musiyenko A., Shulyayeva O., Barik S., Nat. Med. 2005, 11, 50–55. [DOI] [PubMed] [Google Scholar]
- 24. Driscoll K. E., Costa D. L., Hatch G., Henderson R., Oberdorster G., Salem H., Schlesinger R. B., Toxicol. Sci. 2000, 55, 24–35. [DOI] [PubMed] [Google Scholar]
- 25. Lomas‐Neira J. L., Chung C. S., Wesche D. E., Perl M., Ayala A., J. Leukocyte Biol. 2005, 77, 846–853. [DOI] [PubMed] [Google Scholar]
- 26.
- 26a.G. F. Deleavey, J. K. Watts, M. J. Damha, Curr. Protoc Nucleic Acid Chem. 2009, Chapter 16, Unit 1613; [DOI] [PubMed]
- 26b. Watts J. K., Deleavey G. F., Damha M. J., Drug Discovery Today 2008, 13, 842–855. [DOI] [PubMed] [Google Scholar]
- 27.
- 27a. Lorenz C., Hadwiger P., John M., Vornlocher H. P., Unverzagt C., Bioorg. Med. Chem. Lett. 2004, 14, 4975–4977; [DOI] [PubMed] [Google Scholar]
- 27b. Soutschek J., Akinc A., Bramlage B., Charisse K., Constien R., Donoghue M., Elbashir S., Geick A., Hadwiger P., Harborth J., John M., Kesavan V., Lavine G., Pandey R. K., Racie T., Rajeev K. G., Rohl I., Toudjarska I., Wang G., Wuschko S., Bumcrot D., Koteliansky V., Limmer S., Manoharan M., Vornlocher H. P., Nature 2004, 432, 173–178. [DOI] [PubMed] [Google Scholar]
- 28. DiFiglia M., Sena‐Esteves M., Chase K., Sapp E., Pfister E., Sass M., Yoder J., Reeves P., Pandey R. K., Rajeev K. G., Manoharan M., Sah D. W., Zamore P. D., Aronin N., Proc. Natl. Acad. Sci. USA 2007, 104, 17204–17209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wu Y., Navarro F., Lal A., Basar E., Pandey R. K., Manoharan M., Feng Y., Lee S. J., Lieberman J., Palliser D., Cell Host Microbe 2009, 5, 84–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chen Q., Butler D., Querbes W., Pandey R. K., Ge P., Maier M. A., Zhang L., Rajeev K. G., Nechev L., Kotelianski V., Manoharan M., Sah D. W., J. Control. Release 2010, 144, 227–232. [DOI] [PubMed] [Google Scholar]
- 31. Zhu L., Mahato R. I., Bioconjugate Chem. 2010, 21, 2119–2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang Y., Li Z., Han Y., Liang L. H., Ji A., Curr. Drug Metab. 2010, 11, 182–196. [DOI] [PubMed] [Google Scholar]
- 33.D. Bedi, T. Musacchio, O. A. Fagbohun, J. W. Gillespie, P. Deinnocentes, R. C. Bird, L. Bookbinder, V. P. Torchilin, V. A. Petrenko, Nanomedicine 2011, in press. [DOI] [PMC free article] [PubMed]
- 34. de Fougerolles A. R., Hum. Gene Ther. 2008, 19, 125–132. [DOI] [PubMed] [Google Scholar]
- 35. Filion M. C., Phillips N. C., Biochim. Biophys. Acta Biomembr. 1997, 1329, 345–356. [DOI] [PubMed] [Google Scholar]
- 36. Suh M. S., Shim G., Lee H. Y., Han S. E., Yu Y. H., Choi Y., Kim K., Kwon I. C., Weon K. Y., Kim Y. B., Oh Y. K., J. Controlled Release 2009, 140, 268–276. [DOI] [PubMed] [Google Scholar]
- 37. Kim W. J., Christensen L. V., Jo S., Yockman J. W., Jeong J. H., Kim Y. H., Kim S. W., Mol. Ther. 2006, 14, 343–350. [DOI] [PubMed] [Google Scholar]
- 38. Palliser D., Chowdhury D., Wang Q. Y., Lee S. J., Bronson R. T., Knipe D. M., Lieberman J., Nature 2006, 439, 89–94. [DOI] [PubMed] [Google Scholar]
- 39. Zhang Y., Cristofaro P., Silbermann R., Pusch O., Boden D., Konkin T., Hovanesian V., Monfils P. R., Resnick M., Moss S. F., Ramratnam B., Mol. Ther. 2006, 14, 336–342. [DOI] [PubMed] [Google Scholar]
- 40. Semple S. C., Akinc A., Chen J., Sandhu A. P., Mui B. L., Cho C. K., Sah D. W., Stebbing D., Crosley E. J., Yaworski E., Hafez I. M., Dorkin J. R., Qin J., Lam K., Rajeev K. G., Wong K. F., Jeffs L. B., Nechev L., Eisenhardt M. L., Jayaraman M., Kazem M., Maier M. A., Srinivasulu M., Weinstein M. J., Chen Q., Alvarez R., Barros S. A., De S., Klimuk S. K., Borland T., Kosovrasti V., Cantley W. L., Tam Y. K., Manoharan M., Ciufolini M. A., Tracy M. A., de Fougerolles A., MacLachlan I., Cullis P. R., Madden T. D., Hope M. J., Nat. Biotechnol. 2010, 28, 172–176. [DOI] [PubMed] [Google Scholar]
- 41. Namiki Y., Namiki T., Yoshida H., Ishii Y., Tsubota A., Koido S., Nariai K., Mitsunaga M., Yanagisawa S., Kashiwagi H., Mabashi Y., Yumoto Y., Hoshina S., Fujise K., Tada N., Nat. Nanotechnol. 2009, 4, 598–606. [DOI] [PubMed] [Google Scholar]
- 42. Kim S. H., Jeong J. H., Lee S. H., Kim S. W., Park T. G., J. Controlled Release 2008, 129, 107–116. [DOI] [PubMed] [Google Scholar]
- 43. Alshamsan A., Hamdy S., Samuel J., El‐Kadi A. O., Lavasanifar A., Uludag H., Biomaterials 2010, 31, 1420–1428. [DOI] [PubMed] [Google Scholar]
- 44. Merkel O. M., Beyerle A., Librizzi D., Pfestroff A., Behr T. M., Sproat B., Barth P. J., Kissel T., Mol. Pharm. 2009, 6, 1246–1260. [DOI] [PubMed] [Google Scholar]
- 45. Howard K. A., Rahbek U. L., Liu X., Damgaard C. K., Glud S. Z., Andersen M. O., Hovgaard M. B., Schmitz A., Nyengaard J. R., Besenbacher F., Kjems J., Mol. Ther. 2006, 14, 476–484. [DOI] [PubMed] [Google Scholar]
- 46. Noh S. M., Park M. O., Shim G., Han S. E., Lee H. Y., Huh J. H., Kim M. S., Choi J. J., Kim K., Kwon I. C., Kim J. S., Baek K. H., Oh Y. K., J. Control. Release 2010, 145, 159–164. [DOI] [PubMed] [Google Scholar]
- 47. Wilson D. S., Dalmasso G., Wang L., Sitaraman S. V., Merlin D., Murthy N., Nat. Mater. 2010, 9, 923–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Dickerson E. B., Blackburn W. H., Smith M. H., Kapa L. B., Lyon L. A., McDonald J. F., BMC Cancer 2010, 10, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Taratula O., Garbuzenko O. B., Kirkpatrick P., Pandya I., Savla R., Pozharov V. P., He H., Minko T., J. Controlled Release 2009, 140, 284–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Davis M. E., Zuckerman J. E., Choi C. H., Seligson D., Tolcher A., Alabi C. A., Yen Y., Heidel J. D., Ribas A., Nature 2010, 464, 1067–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lu W., Zhang G., Zhang R., L. G. Flores II , Huang Q., Gelovani J. G., Li C., Cancer Res. 2010, 70, 3177–3188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Murata N., Takashima Y., Toyoshima K., Yamamoto M., Okada H., J. Controlled Release 2008, 126, 246–254. [DOI] [PubMed] [Google Scholar]
- 53. Yao Y., Wang C., Varshney R. R., Wang D. A., Pharm. Res. 2009, 26, 263–275. [DOI] [PubMed] [Google Scholar]
- 54. Alsberg E., Hill E. E., Mooney D. J., Crit. Rev. Oral Biol. Med. 2001, 12, 64–75. [DOI] [PubMed] [Google Scholar]
- 55. Krebs M. D., Jeon O., Alsberg E., J. Am. Chem. Soc. 2009, 131, 9204–9206. [DOI] [PubMed] [Google Scholar]
- 56. Vinas‐Castells R., Holladay C., di Luca A., Diaz V. M., Pandit A., Bioconjugate Chem. 2009, 20, 2262–2269. [DOI] [PubMed] [Google Scholar]
- 57. Singh A., Suri S., Roy K., Biomaterials 2009, 30, 5187–5200. [DOI] [PMC free article] [PubMed] [Google Scholar]