

Introduction
Peptidases are enzymes capable of cleaving, and thereby often inactivating, small peptides. They are widely distributed on the surface of many different cell types, with the catalytic site exposed only at the external surface. Peptidases are involved in a variety of processes, including peptide‐mediated inflammatory responses, stromal cell‐dependent B lymphopoiesis, and T‐cell activation. In addition, some peptidases may have functions that are not based on their enzymatic activity.
Peptidases are classified according to the location of the cleavage site in the putative substrate ( Table 1). Endopeptidases recognize specific amino acids in the middle of the peptide, whereas exopeptidases recognize one or two terminal amino acids. Exopeptidases that attack peptides from the N‐terminus (removing either single amino acids or a dipeptide) are termed (dipeptidyl) aminopeptidases, whereas peptidases attacking the C‐terminus are termed carboxypeptidases.
Table 1.
. Peptidases and subtrates * The peptidase cleaves peptides in which the open circle represents (one of) the mentioned amino acids. The closed circle can be any amino acid. The cleaved bond is represented by ‘ −: ’. Peptidases: ACE, angiotensin‐converting enzyme; APA, aminopeptidase A; APN, aminopeptidase N; APP, aminopeptidase P; CPN, carboxypeptidase N; DPP IV, dipeptidyl(amino)peptidase IV; NEP, neprilysin. Substrates: BK, bradykinin; ANF, atrial no uretic factor; BLP, bombesin‐like peptides; ET–1, enothelin‐1; fMLP, formyl‐metheonyl‐leucyl‐phenylalanine; IL, interleukin; NKA, neukonin A; NPY, neuropenptide Y; SP, substance P; VIP, vasoactive intestinal peptide.

Neutral endopeptidase 24.11
Characteristics
Biochemical and molecular characterization
Neutral endopeptidase (NEP, neprilysin, EC 3.4.24.11) was first characterized from rabbit kidney brush border. It soon became apparent that NEP was similar to enkephalinase, originally discovered in the brain. Furthermore, cloning of the NEP gene and subsequent cloning of the common acute lymphoblastic leukaemia antigen (CALLA, CD10) showed that both sequences were similar [ 1].
NEP is a glycoprotein of 750 amino acids, with a single 24 amino acid hydrophobic segment that functions as both a transmembrane region and a signal peptide [ 1]. The C‐terminal 700 amino acids compose the extracellular domain, whereas the 25 N‐terminal amino acids form the cytoplasmic tail. The extracellular domain contains six potential N‐glycosylation sites. Tissue‐specific glycosylation may result in different molecular masses, ranging from approximately 90–110 kDa. The extracellular domain contains the pentapeptide consensus sequence (His‐Glu‐[Ile, Leu, Met]‐X‐His) of zinc‐binding metalloproteases, in which the two histidines co‐ordinating zinc and the glutamic acid residue, together with an aspartic acid residue, are critically involved in the catalytic process.
Gene structure
Characterization of the human NEP gene, which is located at chromosome 3 (q21‐q27), showed that it spans more than 80 kb and is composed of 25 exons [ 2]. Exons 1, 1bis, and 2 encode 5′ untranslated sequences; exon 3 encodes the initiation codon and the transmembrane and cytoplasmic domain; 20 short exons (exons 4–23) encode most of the extracellular region; and exon 24 encodes the C‐terminal 32 amino acids of the protein and contains the entire 3′ untranslated region (UTR). Within exon 24 are five poly (A) addition signals. Alternative splicing of exon 1, exon 1bis, exon 2 (2a), or part of exon 2 (2b) to the common exon 3, resulting in four different transcripts, may be the origin of the tissue‐specific or stage of development‐specific expression of NEP [ 3]. Indeed, two separate regulatory elements have been found in the NEP promoter region and these elements may be regulated by the transcription factor CBF/NF‐Y in a tissue‐specific manner. A cDNA clone lacking the complete exon 16 has been isolated from human lung tissue [ 4]. Deletion of this 27 amino acid segment was shown to reduce enzyme activity to barely detectable levels. However, the physiological relevance of this truncated enzyme remains to be determined. In the rat, an exon 5–18 deletion has been described, but no evidence was found to support the expression of this truncated variant in the human lung.
Distribution
NEP is expressed by a variety of haematopoietic and non‐hematopoietic cells [ 5]. NEP is abundantly present in renal proximal tubular epithelial cells, small intestinal epithelium, and biliary canaliculae. In addition, NEP can be found in synaptic membranes of the central nervous system, bone marrow stromal cells, fibroblasts, placenta, lymphoid progenitors, and neutrophils. Given the expression of NEP on lymphoid progenitors, expression of NEP is used as a diagnostic marker for several lymphoid malignancies, including Burkitt's lymphomas and certain myelomas.
In the human lung, NEP is expressed by bronchial epithelial cells, submucosal glands, bronchial smooth muscle, and endothelium [ 6, 7]. In addition, NEP can be found on alveolar epithelial cells.
Enzymatic activity and biological functions
NEP is able to hydrolyse peptide bonds on the N‐terminal site of hydrophobic amino acids, including Phe, Leu, Ile, Val, Tyr, Ala, and Trp ( Table 1). However, sub‐site interactions and conformational factors greatly influence the efficiency of hydrolysis. Among the possible substrates of NEP are substance P (SP), neurokinin A (NKA), formyl‐metheonyl‐leucyl‐phenylalanine (fMLP), atrial natriuretic factor (ANF), endothelin‐1 (ET‐1), bombesin‐like peptides (BLP), angiotensins, vasoactive intestinal peptide (VIP), neuropeptide Y (NPY), bradykinin (BK), enkephalins, cholecystokinin, and neurotensin. Although NEP predominantly cleaves simple peptides, it has been reported that NEP may also be able to hydrolyse certain larger substrates, including cytokines such as interleukin (IL)‐1β and IL‐6.
The general biological function of NEP is to reduce cellular responses to peptide hormones. Target cells express both NEP and the peptide‐receptor; by degrading the peptide substrate, NEP reduces the local concentration of the peptide available for binding to the receptor. For example, NEP reduces ANF‐mediated hypotension, fMLP‐mediated chemotaxis of neutrophils, and enkephalin‐mediated analgesia. Targeted disruption of the NEP locus in mice results in enhanced lethality to endotoxin, indicating an important protective role for NEP in septic shock. A role for NEP in lymphoid development has been suggested by studies showing that inhibition of NEP results in increased proliferation and maturation of B cells, both in vitro and in vivo [ 8]. Therefore, it has been suggested that NEP functions to regulate B‐cell development by inactivating a peptide that stimulates B‐cell proliferation and differentiation. Alternatively, NEP may activate a pro‐peptide that inhibits proliferation and differentiation of B cells. The role of NEP in the regulation of cellular proliferation and differentiation in the lung is discussed next in more detail. The role of NEP in the modulation of neurogenic inflammation will be discussed further on.
Role of NEP in cellular differentiation and proliferation in the lung
NEP plays an important role in the cellular differentiation and proliferation of bronchial epithelial cells by inactivating BLP [ 9]. BLP are potent growth factors for bronchial epithelial cells and are involved in lung development. The temporal and cellular patterns of NEP expression implicate the enzyme in the regulation of BLP‐mediated fetal lung development. Indeed, both in vitro and in vivo, it was shown that inhibition of NEP resulted in increased maturation of the developing fetal lung [ 10]. Reduced NEP activity may also promote BLP‐mediated proliferation of bronchial epithelial cells. Indeed, the growth and proliferation of BLP‐dependent carcinomas is inhibited by NEP and potentiated by NEP inhibition [ 9]. NEP expression by epithelial cells is inversely correlated with cellular proliferation. Therefore, reduced NEP activity may promote BLP‐mediated proliferation and facilitate the development of small‐cell carcinomas of the lung [ 9]. A role for NEP in the regulation of tumour cell proliferation is also supported by studies using a human T‐cell line (Jurkat). In these cells, NEP is required for phorbol ester‐induced growth arrest.
Aminopeptidase N
Characteristics
Biochemical and molecular characterization
Aminopeptidase N (APN, EC 3.4.11.2) is a widely studied peptidase, which is known under a variety of names, including aminopeptidase M, alanine aminopeptidase, arylamidase, and microsomal α‐aminoacyl‐peptide hydrolase.
APN is a glycoprotein of 967 amino acids with 11 potential sites of asparagine‐linked oligosaccharide addition [ 11]. The unglycosylated protein has a molecular size of 110 kDa; post‐translational modification results in the 130‐kDa precursor (gp130) and the 150‐kDa mature protein (gp150) [ 12]. The 23‐amino acid retained signal also functions as the membrane‐spanning segment, orientating the APN N‐terminus inside and the C‐terminus outside the cell (thereby defining APN as a type II integral membrane protein) [ 11]. The intracellular domain of APN is only nine amino acids long, whereas the extracellular domain contains 935 amino acids. Similar to NEP, the extracellular domain contains a pentapeptide consensus sequence characteristic of members of the zinc‐binding metalloprotease family. On the surface of cells, APN is expressed as a non‐covalently bound homodimer. Cloning of the APN cDNA revealed that its sequence was identical to the myeloid marker CD13 [ 11, 12].
Gene structure
The APN gene is located on the long arm of chromosome 15 (q25–26) and exists of 20 exons [ 13]. Northern blot analysis of RNA extracted from several tissues revealed two distinct APN transcripts: a 3.7‐kb transcript expressed by monocytes, myeloid leukaemia cells, and fibroblasts, and a 3.4‐kb transcript expressed by intestinal epithelium and kidney cells [ 14]. In epithelial cells, transcripts originate 47 bp upstream from the initiation codon and 22 bp downstream from a TATA box. In contrast, the longer transcripts found in myeloid cells and fibroblasts originated from several sites clustered in an upstream exon located 8 kb from the exon containing the initiation codon. Nevertheless, both transcripts encode the same protein, indicating that separate promoters control the tissue‐specific expression of the APN gene [ 14]. In addition, a 300‐bp region with enhancer activity, located 2.7 kb upstream of the transcriptional start site which is used in epithelial cells, may also be important for the tissue‐specific expression.
Distribution
The non‐hematopoietic distribution of APN shows a pattern comparable with NEP [ 5, 7]. Thus, APN is expressed on renal proximal tubular epithelial cells, small intestinal epithelium, biliary canaliculae, synaptic membranes of the central nervous system, bone marrow stromal cells, fibroblasts, osteoclasts, placenta, and granulocytes [ 5]. In contrast with NEP, APN is also expressed on monocytes and all myeloid progenitors. Expression of APN may be used as a marker for myeloid leukaemia. Mast cells may also express APN, whereas peripheral blood lymphocytes do not express this enzyme. However, expression of APN on lymphocytes can be induced after mitogenic stimulation or after adhesion to fibroblast‐like synoviocytes, endothelial cells, epithelial cells and monocytes/macrophages. In the human bronchus, APN is present on blood vessels, connective tissue, glandular ducts, nerves, perichondrium, and leucocytes (predominantly mononuclear phagocytes, dendritic cells, and granulocytes) [ 7].
Enzymatic activity and biological function
APN is a peptidase which hydrolyses preferentially natural or synthetic substrates with an N‐terminal alanine residue ( Table 1). Other amino acids, especially neutral ones, may also be removed hydrolitically, with the exception of proline. Natural APN substrates appear to be small peptides rather than larger proteins, although the enzyme is more effective in removing residues from oligopeptides than dipeptides. Among the possible substrates for APN are enkephalins, tachykinins, bradykinin, fMLP, and possibly cytokines such as IL‐1β, IL‐6, and IL‐8. However, in certain cases initial cleavage by endopeptidases (like NEP) may be required.
Several functions of APN have been described [ 5]. First, APN expressed on the brush border of the intestine may be involved in the final stages of digestion of small peptides. Second, comparable with NEP and often in collaboration with NEP, APN may function to reduce cellular responses to peptide hormones. Third, a recent report implicates APN in the processing of peptides presented by the major histocompatibility complex class II molecule [ 15]. Fourth, APN may be involved in tumour invasion and metastasis by degradation of collagen type IV. Finally, APN serves as a receptor for coronaviruses, which are RNA viruses that cause respiratory disease in humans.
Dipeptidyl peptidase IV
Characteristics
Biochemical and molecular characterization
Dipeptidyl (amino)peptidase IV (DPP IV; EC 3.4.14.5) is an atypical serine protease of 766 amino acids with type II membrane topology [ 16]. It contains a short, highly conserved intracellular domain of six amino acids, a 22‐amino acid hydrophobic transmembrane region (which also functions as signal peptide), and a 738‐amino acid extracellular domain. The extracellular domain, which contains nine potential glycosylation sites, can be divided into three regions: an N‐terminal glycosylated region containing seven glycosylation sites and starting with a 20‐amino acid flexible ‘stalk region’; a cysteine‐rich region; and a 260‐amino acid C‐terminal domain containing the putative catalytic sequence. On the surface of cells, DPP IV probably is present as a homodimer comprising two identical subunits of approximately 110‐kDa molecular mass. Recent studies indicate that several isoforms of DPP IV can be found [ 17].
In contrast with NEP and APN, DPP IV does not contain zinc in its catalytic centre. Based upon its structural homology with other non‐classic serine proteases, DPP IV is assigned to the prolyl oligopeptidase family. Members of this family share a catalytic site in which the essential residues are arranged in the unique sequence Ser‐Asp‐His. Cloning of the DPP IV cDNA revealed that its sequence was identical to the T‐cell activation antigen CD26 [ 16].
Gene structure
The human DPP IV gene, located on chromosome 2 (q24.3), spans approximately 70 kb and contains 26 exons [ 18]. The serine recognition site is split across two exons, the first half Gly Trp is in exon 21 and the second half Ser‐Tyr‐Gly is in exon 22. The three residues comprising the catalytic site are each present in a distinct exon: Ser in exon 22, Asp in exon 24, and His in exon 26. This latter exon also contains the stop codon and the 3′ untranslated region of the gene. The 5′ flanking domain of the DPP IV gene contains neither a TATA box nor a CAAT box, but a 300 bp region extremely rich in C and G contains potential binding sites for several transcription factors, including Sp‐1 and activator protein (AP)‐1. The human DPP IV gene encodes two RNA transcripts of approximately 4.2 and 2.8 kb, which differ in sequence only at the 3′ untranslated region [ 18]. Probably, the two mRNA arise from the use of different polyadenylation sites in the last exon of the DPP IV gene.
Distribution
In many respects, the non‐hematopoietic tissue distribution of DPP IV resembles that of NEP and APN. DPP IV is constitutively expressed on renal proximal tubular epithelial cells, epithelial cells in the small intestine, and biliary canaliculae, but can also be found on alveolar pneumocytes and endothelia [ 5]. In the human bronchus, DPP IV is strongly present in serosal submucosal glands [ 7]. The expression of DPP IV on haematopoietic cells is regulated stringently. DPP IV is absent from the majority of human resting peripheral blood T lymphocytes, but some subsets of resting peripheral blood T cells weakly express the molecule. DPP IV expression on T lymphocytes is increased after T‐cell activation. Thus, DPP IV is a suitable marker for T cells activated in vivo. Recent data indicate that DPP IV expression on T cells may correlate with T‐helper (TH) subsets [ 19]. High DPP IV expression was found on TH1 and TH0 cells, whereas TH2 cells displayed lower expression of DPP IV. The amount of IL‐4 secretion was responsible for this correlation [ 19]. Memory T cells have been reported to reside in the DPP IV‐positive T‐cell fraction, although this was not found in another study. DPP IV is also expressed by medullary thymocytes in humans and can be induced on activated natural killer cells.
Enzymatic activity and biological functions
DPP IV is a serine peptidase with a unique specificity: it cleaves dipeptides from the N‐terminus of polypeptides if proline is at the penultimate position. Peptides with alanine in the penultimate position may also be cleaved, although with a much lower efficiency. Since N‐termini‐containing X‐Pro are not easily cleaved by other peptidases, the action of DPP IV is a rate‐limiting step in the degradation of such peptides. Several biologically active peptides have the X‐Pro sequence at their N‐terminus and therefore DPP IV may play an important role in modulating their action. These peptides include SP and bradykinin. Hydrolysis of SP by DPP IV yields two products (SP3–11 and SP5–11) which both are more potent bronchoconstrictors than intact SP1–11. Both products can rapidly be inactivated by APN. A proline residue is also present at the penultimate position of several cytokines and chemokines, like IL‐1β, IL‐2, tumour necrosis factor (TNF)‐β, RANTES, and granulocyte‐colony‐stimulating factor (G‐CSF).
DPP IV may have several functions, dependent upon the tissue in which it is expressed. DPP IV plays an obligatory role in the renal transport and intestinal digestion of proline‐containing polypeptides. However, most attention has been given to the function of DPP IV on T lymphocytes.
Role of DPP IV on T lymphocytes
Although the role of DPP IV on activated T cells is not completely understood, recent studies indicate that it may act as a costimulatory molecule that can up‐regulate the signal transducing properties of the T‐cell receptor (TCR) [ 20, 21]. Stimulation of DPP IV (using monoclonal antibodies) leads to the activation of all functional programs of the T cells, including cytotoxicity and production of IL‐2. This activation requires the expression of the TCR and DPP IV enzymatic activity. Furthermore, antibody‐induced cross‐linking of DPP IV‐induced tyrosine phosphorylation of several intracellular proteins with a similar pattern to that seen after TCR/CD3 stimulation [ 20]. Co‐cross‐linking of DPP IV and CD3 antigens induced prolonged and increased tyrosine phosphorylation in comparison with CD3 alone, indicating that DPP IV is a true costimulatory entity [ 20]. In addition to T‐cell activation, anti‐DPP IV‐stimulated T cells show enhanced proliferative responses, increased CD3 (phosphorylation and increased p56lck activity. One possible mechanism for the enhanced response of T cells to perturbation of DPP IV was suggested by the demonstration that CD45, a tyrosine phosphatase that positively regulates TCR signalling, coprecipitates with DPP IV [ 21]. Thus, DPP IV antibodies may stimulate T‐cell proliferation in part by decreasing CD45‐mediated dephosphorylation of key substrates.
Inhibition of DPP IV activity results in reduced DNA synthesis as well as reduced production of IL‐2, IL‐10, IL‐12, IL‐13, and interferon (IFN)‐γ of pokeweed mitogen (PWM)‐stimulated purified T cells [ 22]. Most importantly, DPP IV inhibition increased mRNA synthesis and secretion of transforming growth factor (TGF)‐β and a neutralizing antibody directed against TGF‐β abolished the DPP IV inhibitor‐induced suppression in cytokine production [ 22]. In a rat study, repeated subcutaneous injection of DPP IV inhibitors reduced serum DPP IV activities to levels less than 30% of the normal [ 23]. When primary, secondary or tertiary immune responses to bovine serum albumin (BSA) were evoked in these animals, they showed reduced anti‐BSA antibody production. In normal rats, immunization with BSA was followed by a temporary decrease in serum DPP IV activity and then by enhanced serum enzyme activity after several days. These results suggest that DPP IV plays an important role in immune responses in vivo.
Memory T cells have been shown to increase their antigen sensitivity gradually with time after restimulation, an effect that is accompanied by increased cell‐surface expression of DPP IV. Using antibodies directed against DPP IV, it has been shown that DPP IV directly contributed to this increased antigen sensitivity of late‐memory T cells. As mentioned above, this effect may be explained by the costimulatory capacity of DPP IV [ 20]. Increasing the antigen‐sensitivity via antigen‐non‐specific molecules may be physiologica mechanism for maintaining T‐cell memory in face of decreasing antigen concentrations, and may ensure preferential activation of memory T cells upon repeated antigen challenge.
DPP IV is also found to be associated with adenosine deaminase (ADA), and this complex is thought to serve as an important immunoregulatory mechanism. Released ADA may bind to cell surface DPP IV, and the DPP IV/ADA complex subsequently binds adenosine, thereby reducing its local concentration.
DPP IV may also function as an auxiliary adhesion factor. DPP IV was found to bind to components of the extracellular matrix, such as fibronectin and collagen. Binding of human CD4‐positive T cells to collagen produced a costimulatory signal in anti‐CD3‐mediated T‐cell activation, resulting in increased proliferation. An anti‐DPP IV antibody inhibited this effect.
Finally, DPP IV may be involved in the pathogenesis of the acquired immunodeficiency syndrome (AIDS). DPP IV may act as one of the coreceptors for human immuno‐deficiency virus (HIV). Furthermore, the HIV Tat antigen has been shown to inhibit the enzymatic activity of DPP IV, resulting in the inhibition of T‐cell responses to antigen and anti‐CD3 antibodies. Thus, the immunosuppressive effects of the HIV‐1 Tat protein may be mediated by DPP IV inhibition.
Other peptidases
In addition to the three peptidases described above, other peptidases are involved in the degradation of (neuro)peptides. These include angiotensin‐converting enzyme (ACE), endothelin‐converting enzyme (ECE), aminopeptidases, and carboxypeptidases.
Angiotensin‐converting enzyme
ACE, also known as peptidyl peptidase A or kinase II, is a type II integral membrane endopeptidase belonging to the superfamily of metallopeptidases (reviewed in [ 24]). Two isoforms of ACE are present within the human body: a somatic form with a molecular weight around 150 kDa, which is found in endothelial, epithelial and neural cells, and a smaller isoform (90–110 kDa) found in germinal cells. Both forms are transcribed from a single gene by the use of two separate functional promoters, a somatic and a testicular form. The somatic form is composed of two highly homologous domains, probably arising by gene duplication in the course of evolution. The germinal isoform only contains one of the two homologous domains. Somatic ACE comprises 1306 amino acids with 17 potential N‐linked glycosylation sites. Each domain has a catalytic site, containing zinc, which functions independently.
ACE is widely distributed in human tissues: it is present on vascular endothelial cells, in the brush border of absorptive epithelia of the small intestine and the renal proximal tubuli, and in monocytes, macrophages, and T lymphocytes. Nevertheless, its major location is considered to be the vascular endothelial surface of the lung. The enzyme preferentially cleaves peptides containing an aromatic residue in the P1 position ( Table 1), but the enzyme is far less selective than NEP. It is capable of inactivating bradykinin and enkephalins, and hydrolyses angiotensin I to yield the vasoconstrictor peptide angiotensin II. ACE appears to play a major role in controlling blood pressure and water and salt metabolism. In addition, ACE hydrolyses intravascular substance P, but neurokinin A is not a good substrate.
Endothelin‐converting enzyme
ECE is a type II integral membrane protein homologous with NEP [ 25]. Unlike NEP, however, ECE exists as a highly glycosylated disulphide‐linked dimer of subunit molecular weight 120–130 kDa. ECE convert big‐endothelin to its biologically active product ET‐1 ( Table 1), which is a potent broncho‐ and vasoconstrictor that may regulate vascular tone and blood pressure. Three isoforms of ECE can be distinguished: ECE‐1α, ECE‐1β (resulting from alternative splicing of a single gene), and ECE‐2.
In the human lung, ECE has been found in airway epithelium, pulmonary endothelium, airway and vascular smooth muscle, and serous bronchial glands [ 26]. Although ECE may play a role in modulating biologically active peptides, it remains to be determined whether it is involved in the pathogenesis of asthma. Nevertheless, in asthmatic patients increased levels of ET‐1 have been found in bronchoalveolar lavage fluid, plasma, and bronchial epithelial cells compared with healthy controls.
Aminopeptidases
Human tissues contain an array of cytosolic and membrane‐bound aminopeptidases. The best‐characterized, aminopeptidase N, is described above. Other aminopeptidases are aminopeptidase A specific for N‐terminal Glu and Asp residues, and aminopeptidase P, which will release an N‐terminal residue adjacent to a proline ( Table 1). The role of these peptidases in the metabolism of susceptible peptides has been little investigated, but it may be hypothesized that these enzymes are involved in the final hydrolysis of a variety of substrates, with or without initial cleavage by an endopeptidase. A role for aminopeptidase A in modulating the potency of peptides binding to the neurokinin (NK)2 receptor has been suggested. Aminopeptidases may also be involved in the regulation of CC chemokine activities, as deletion of the NH2‐terminal residue converts monocyte chemotactic protein‐1 from an activator of basophil mediator release to an eosinophil chemoattractant.
Carboxypeptidases
Carboxypeptidase N (CPN, kininase I) cleaves the C‐terminal arginine and lysine of peptides such as bradykinin [ 27]. One of the functions of CPN is to protect the body from potent vasoactive and inflammatory peptides containing COOH‐terminal Arg or Lys which are released into the circulation. In the human lung, CPN has been detected in alveolar type I cells, in the glycocalyx of the epithelium, in some vessels, and in gland ducts near the epithelial basement membrane [ 28]. CPN activity in nasal lavage fluid has been shown to be enhanced after histamine challenge. This CPN originated in plasma, suggesting that plasma extravasation and interstitial fluid exudation across the epithelium are the primary processes regulating its appearance in nasal secretions. CPN has also been found in BAL fluid. Since increased CPN activity was found in patients with lung disease (pneumonia or lung cancer), it was hypothesized that CPN activity in BAL fluid may be an indicator of type I cell injury.
Soluble counterparts of membrane‐bound peptidases
Although the above mentioned peptidases are integral membrane glycoproteins, soluble peptidases with comparable enzymatic activity can be detected in body fluids. These soluble counterparts may either be derived from shedding of membrane‐bound peptidases, or may be formed by post‐translational cleavage of the membrane‐bound form. For most peptidases, however, the physiological role of the soluble molecules remains to be elucidated.
Serum neutral endopeptidase activity probably arises from shedding of the membrane‐bound peptidase [ 29]. Increased serum activity of NEP has been observed in underground miners exposed to coal dust particles [ 29] and in patients with adult respiratory distress syndrome (ARDS), rheumatoid arthritis or sarcoidosis. Although the source of the increased NEP levels remains to be determined, it has been suggested that increased NEP levels may reflect local tissue damage with subsequent shedding of membrane‐bound NEP. Furthermore, serum activity of NEP is increased in acute renal graft rejection, in patients with end‐stage renal failure, and in cholestatic liver disease.
Human serum contains an array of aminopeptidase activities, including alanine aminopeptidase and leucine aminopeptidase. Serum alanine aminopeptidase activity predominantly comprises a circulating isoform of CD13 [ 30]. Increased activity of leucine aminopeptidase has been observed in BAL fluid of patients with pulmonary tuberculosis and it was shown that this increase could be attributed to lung tissue damage.
Dipeptidyl peptidase IV is present in several forms in human serum and may enhance antigen‐induced T‐cell proliferation [ 31]. Recent studies indicate that serum DPP IV is a monomer of 175 kDa and that this molecule, which is a potent T‐cell costimulator, is not a breakdown product of membrane‐bound CD26 [ 32]. Furthermore, the 175‐kDa form of DPP IV found in normal serum is identical with a similarly sized molecule, DPPT‐L, found to be rapidly expressed on the surface of activated T cells [ 17]. CD45RO‐ CD4‐positive T cells appeared to be the major source of serum DPP IV activity [ 17]. DPP IV activity in serum is decreased in patients with major depression, and a correlation was observed between DPP IV activity and CD4‐positive T cells in blood of depressed subjects, but not of normal controls. There were no significant relationships between serum DPP IV activity and plasma cortisol or immune‐inflammatory markers, such as serum IL‐6 or soluble IL‐2 receptor (CD25). Reduced serum DPP IV activity has also been described in patients with systemic lupus erythematosus and in oral cancer patients. In the latter study a significant correlation between serum DPP IV activity and peripheral blood lymphocytes or CD26‐positive T cells was found.
Modulation of (neurogenic) inflammation
In addition to the two well‐known autonomic nervous systems (parasympathetic and sympathetic) that innervate the airways, a non‐adrenergic non‐cholinergic (NANC) neural pathway is present. While inhibitory NANC (i‐NANC) effects are bronchodilatory through the activity of VIP and nitric oxide (NO) released from cholinergic nerves, excitatory NANC (e‐NANC) effects are bronchoconstrictor and mediated through the release of neuropeptides (especially tachykinins and calcitonin gene‐related peptide [CGRP]) from sensory nerves [ 33]. Stimulation of sensory nerves, either by chemical or physical triggers, results in an axon reflex and subsequent release of neuropeptides from the peripheral endings of the sensory nerves. Following release, these neuropeptides exert a variety of effects through activation of specific neurokinin receptors, including vasodilation, increased microvascular permeability, leucocyte recruitment and adhesion, submucosal gland secretion, smooth muscle contraction, cough, and facilitation of cholinergic neurotransmission. This sequence of events is now known as ‘neurogenic inflammation’ [ 34]. Since the neurogenic inflammatory response mimics many of the pathophysiological features of asthma, a role for neuropeptides in the pathogenesis of asthma has been implicated. In the asthmatic airways, the effects of bronchoconstrictor peptides (including tachykinins and bradykinin) may be enhanced, whereas the effects of bronchodilator peptides (including VIP) may be reduced.
After it became apparent that neuropeptides were responsible for the neurogenic inflammatory responses, it was hypothesized that degradative mechanisms existed which may limit the effects of neuropeptides, comparable with the role of cholinesterase in limiting the effects of acetylcholine. Several studies now have demonstrated that peptidases play a major role in the modulation of peptide‐mediated effects in the airways (reviewed in [ 35]). Much research has focused on the degradation of the tachykinins, like SP and NKA, and the enzyme NEP.
The physiological relevance of tachykinin inactivation by enzymatic hydrolysis has been deduced from studies of the effects of enzyme inhibition on the physiological action of exogenously administered or endogenously released peptides. In the first study, it was shown that selective inhibition of NEP potentiated the secretagogue effect of SP on submucosal gland secretion in the ferret trachea in vivo. Several other reports subsequently demonstrated that inhibition of NEP potentiated the effects of SP on cough, vascular permeability, cholinergic neurotransmission, and smooth muscle contraction. In guinea‐pigs, it was shown that both NEP and ACE participate in the metabolism of SP when administered intravascularly, whilst SP administered by aerosol was degraded by NEP only. In addition, the ACE inhibitor captopril did not affect TK‐induced bronchial smooth muscle contraction in man. Therefore, ACE is thought to play an important role in modulating the biological activity of intravascular peptides, whereas NEP is also involved in the hydrolysis of peptides present within lung tissue or within the bronchial lumen. The importance of NEP in modulating tachykinin‐mediated effects is further supported by the observation that administration of other peptidase inhibitors (including inhibitors of aminopeptidases, serine proteases, and carboxypeptidases), did not potentiate tachykinin‐induced effects in the airways. The involvement of NEP in the breakdown of tachykinins has also been shown in in vivo studies in humans. These studies showed that both NKA‐ and SP‐induced bronchoconstriction could be potentiated by NEP inhibition [ 36, 37]. Furthermore, these studies indicated that SP, but not NKA, increased the airway responsiveness to methacholine, suggesting that inflammatory processes are contributing to SP‐induced airway narrowing [ 38].
In contrast with the studies above, in which the effects of neuropeptides were increased due to the inhibition of peptidases, some studies have shown that administration of recombinant NEP may prevent neurogenic inflammation. Thus, administration of aerosolized NEP inhibited the SP‐induced cough and ozone‐induced hyperreactivity to SP in guinea‐pigs [ 34].
Biochemical and immunohistochemical studies have shown that NEP is present on airway epithelial cells [ 6]. Removal of the epithelium was further shown to result in increased responses to exogenously applied or endogenously released tachykinins [ 39]. However, NEP is also present at other sites within the airways, and also after removal of the epithelium NEP inhibitors potentiate tachykinin‐mediated effects [ 6]. Nevertheless, NEP expressed by epithelial cells may more easily be modulated by inhaled agents than NEP located at other sites.
Several environmental agents may modulate peptidase activity, thereby exaggerating responses to tachykinins (and other peptides) and increasing airway inflammation. These agents include viruses, ozone, cigarette smoke, chemical irritants, and possibly antigen challenge. In contrast, inhaled steroids may exert their anti‐inflammatory actions in part by up‐regulating NEP activity.
Viruses
Viral infections may potentiate neurogenic inflammatory responses through inhibition of NEP activity. In laboratory animals, infection with influenza virus or Sendai virus was shown to result in enhanced bronchoconstrictor responses to tachykinins, an effect that was mediated by decreased epithelial NEP activity [ 40].
Ozone
In humans, guinea‐pigs and many other species, exposure to ozone results in the recruitment of neutrophils to the airways and increased responsiveness to inhaled bronchoconstrictor agents. Ozone‐induced airway hyperreactivity can be blocked by capsaicin‐pretreatment, which depletes TK from sensory nerves. Exposure to ozone also results in increased responsiveness for SP, and this effect could not be enhanced by inhibition of NEP [ 41]. This suggests that ozone exposure inactivated NEP, which is supported by the observation that the tracheal NEP activity in ozone‐exposed animals was significantly lower than the NEP activity in air‐exposed animals [ 41].
Toluene diisocyanate
Toluene diisocyanate (TDI) is a widely used plasticizer that may cause occupational asthma. In guinea‐pigs it was shown that TDI, albeit at rather unrealistic doses, increased airway responsiveness to SP and decreased airway neutral endopeptidase [ 42].
Cigarette smoke
Inhalation of cigarette smoke enhances the bronchoconstrictor response to inhaled SP in guinea‐pigs [ 43]. Inhibition of NEP by phosphoramidon increased the bronchoconstriction induced by SP in control animals, but not in animals exposed to cigarette smoke. NEP activity in homogenates of guinea‐pig trachea was inhibited by cigarette smoke. However, in another study no effect of cigarette smoke on airway NEP activity in vivo could be observed. A possible explanation for this discrepancy may be that the NEP inhibited by cigarette smoke only represents a small fraction of the total amount of NEP in the airways.
Cigarette smoke is an important factor contributing to the development of small‐cell lung carcinomas of the lung. As already mentioned, NEP activity is decreased in lung cancers [ 9]. Therefore, one may speculate that cigarette smoke contributes to the development of lung cancers in part by inhibiting NEP, thereby enhancing the mitogenic effects of peptides (e.g. SP and BLP) on bronchial epithelial cells.
Allergen
Airway inflammation may be linked to the clinical features of asthma by an effect on peptidase activity. In guinea‐pigs, chronic antigen exposure results in airway inflammation and hyperreactivity to SP [ 44]. It was shown that lungs with chronic allergic inflammation were more sensitive to the bronchoconstrictor effects of SP and less sensitive the bronchodilator effects of VIP than lungs from healthy subjects. In addition, the effects of enzyme inhibitors on physiological responses and peptide cleavage profiles were consistent with decreased NEP and enhanced tryptic activity [ 44].
In a recent human in vivo study, no effect of inhaled thiorphan (a NEP inhibitor) on allergen‐induced airway responses in asthmatic subjects was observed [ 45]. This suggests that either neuropeptides do not play a predominant role in allergen‐induced airway responses, or that allergen challenge induces NEP‐dysfunction in humans in vivo. However, in guinea‐pigs, it has been shown that tachykinins contribute to allergen‐induced bronchoconstriction [ 46], an effect that is probably mediated via the release of BK and histamine.
Glucocorticoids
Glucocorticoids have potent anti‐inflammatory effects and therefore are widely used in the treatment of asthma. The anti‐inflammatory effect may be caused, in part, by an up‐regulation of NEP activity, thereby reducing neurogenic inflammatory responses. Indeed, NEP activity by a transformed human tracheal cell line and a bronchial epithelial cell line was shown to be increased after stimulation with glucocorticoids. However, no effect of glucocorticoids was observed in another study using the same bronchial epithelial cell line. In guinea‐pigs, glucocorticoids were shown to reduce capsaicin‐induced microvascular permeability, which might be due to elevated NEP expression. This was supported by the observation that treatment of rats with combined NEP and ACE inhibitors prevented the effect of glucocorticoids. The effect of glucocorticoid treatment in vivo on NEP expression in the human airways has recently been reported [ 47]. In that study it was shown that NEP expression in the bronchial epithelium and lamina propria of steroid‐treated asthmatics was significantly greater than the expression in non‐steroid‐treated asthmatic patients [ 47].
As shown above, many of the agents that lead to exacerbations of asthma appear to reduce the activity of NEP at the airway surface, thus leading to exaggerated responses to tachykinins and neurogenic inflammation ( Fig. 1). However, most of these studies have been performed in laboratory animals, especially the guinea‐pig, and have not been confirmed in humans yet. Furthermore, in many studies the NEP inhibitor phosphoramidon was used. This inhibitor, however, not only inhibits NEP, but later was also shown to inhibit ECE. If it appears that ECE can cleave tachykinins the certainty of the conclusions drawn about NEP from experiments using phosphoramidon is somewhat tempered.
Figure 1.
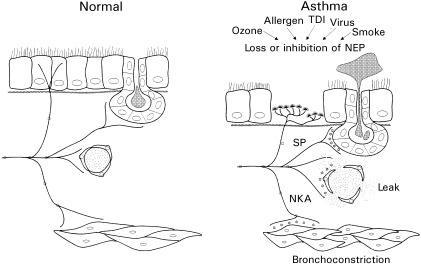
. Neurogenic inflammation in asthmatic airways. Neuropeptides (⋆) released from sensory nerves are normally rapidly degraded by peptidases. Therefore the effects of these neuropeptides are limited. In the asthmatic airways, several factors may result in a decreased peptidase activity, thereby exaggerating the neuropeptide effects. Adapted from reference [ 33].
Neuropeptides and peptidases: important in asthma?
Although neuropeptides and peptidases have been shown to be present in the human airways, their role in asthma still remains to be elucidated. However, several observations may support the hypothesis that neuropeptides and peptidases are involved in the pathogenesis of asthma.
SP and NKA have been shown in several in vivo studies to cause bronchoconstriction, and these effects could be potentiated by inhibition of NEP (reviewed in [ 48]). Furthermore, these studies demonstrated that TK‐mediated bronchoconstriction is greater in allergic asthmatics compared with healthy subjects. However, the thiorphan‐induced leftward shift of the NKA dose response curve was similar in asthmatic patients and healthy subjects, suggesting that the activity of NEP does not differ between both groups [ 36, 37]. Nevertheless, patients used in the latter study were stable asthmatics and it could be argued that reduced NEP activity may occur during exacerbations of asthma.
Increased amounts of SP can be detected in bronchoalveolar lavage fluid of allergic asthmatics and in sputum after allergen challenge, whereas NKA levels do not differ between bronchoalveolar lavage fluid of asthmatics compared with healthy controls. The possibility that tachykinins are endogenously released in vivo has also been supported by the observation that bradykinin‐induced bronchoconstriction in asthmatics can be blocked by a tachykinin receptor antagonist [ 49] (although this could not be confirmed in some other studies) and can be potentiated by NEP inhibition. Bradykinin, which is present in the asthmatic airways and is released after relevant aeroallergen challenge in allergic individuals, can stimulate sensory nerves to induce retrograde release of tachykinins. Increased levels of (neuro)peptides in the airways of asthmatic patients may contribute both to acute and chronic inflammatory abnormalities. In addition to their acute effects, such as the constriction of airway smooth muscle, the secretion of mucus, and vasodilation [ 34], several studies have shown that (neuro)peptides possess a wide number of immunomodulatory effects and may be involved in tissue repair responses [ 50]. Interestingly, several types of leucocytes, including eosinophils and macrophages, are able to produce and release SP themselves, further supporting a role for these molecules in immunological reactions. Thus, dysfunction of peptidases may result in exaggerated immunological responses and thereby contribute to the development and/or maintenance of the inflammatory process.
Inhibition of NEP, either in healthy subjects or asthmatics, has been shown to potentiate the bronchoconstrictor effects of mediators known to be released after allergen challenge (such as LTD4 and bradykinin). However, inhibition of NEP at doses shown to enhance the bronchoconstrictor effect of NKA did not affect the early and late‐phase response in mild asthmatics following allergen challenge [ 45]. This may suggest that endogenously released neuropeptides do not play a role in antigen‐induced airway responses. Alternatively, antigen challenge may result in a dysfunction of NEP activity. To further determine whether peptidases and neuropeptides contribute to asthma, in vivo studies using selective neurokinin receptor antagonists should be performed both in the presence and absence of NEP or other peptidase inhibitors. Neurokinin receptor antagonists should first be tested against tachykinin‐induced bronchoconstriction in order to determine the optimal dose of the antagonists. Second, the effects of these antagonists should be analysed in allergen‐induced bronchoconstriction, both in the absence and in the presence of peptidase inhibitors. Furthermore, it would be interesting to treat allergic asthmatics with appropriate neurokinin receptor antagonists (either intravascular or by inhalation) for a longer period of time, and to determine whether this affects allergen‐induced bronchoconstriction and bronchial inflammation (as determined by analysis of bronchial biopsies and BAL fluid). This will give insight in the contribution of tachykinins to the (chronic) inflammatory process in the airways of asthmatic patients. Finally, the contribution of tachykinins and peptidases in asthma may be demonstrated by treating asthmatic patients with recombinant NEP, and analysing the effects on bronchoconstriction induced by allergens or environmental agents such as cigarette smoke. If it appears that (neuro)peptides and peptidases are indeed involved in the pathogenesis of asthma, selective receptor antagonists or recombinant peptidases may be useful in the therapy of asthma.
Acknowledgements
We gratefully acknowledge Mr T. M. van Os for preparing the figures.
References
- 1. Shipp MA, Richardson NE, Sayre PH Molecular cloning of the common acute lymphoblastic leukaemia antigen (CALLA) identifies a type II integral membrane protein. Proc Natl Acad Sci U S A 1988; 85:4819 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. D'Adamio L, Shipp MA, Masteller EL, Reinherz EL. Organization of the gene encoding common acute lymphoblastic leukaemia antigen (neutral endopeptidase 24.11): multiple miniexons and separate 5′ untranslated regions. Proc Natl Acad Sci U S A 1989; 86:7103 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ishimaru F & Shipp MA. Analysis of the human CD10/neutral endopeptidase 24.11 promoter region: two separate regulatory elements. Blood 1995; 85:3199 207. [PubMed] [Google Scholar]
- 4. Iijima H, Gerard NP, Squassoni C, Ewig J, Face D, Drazen JM Exon 16 del: a novel form of human neutral endopeptidase (CALLA). Am J Physiol 1992; 262:L725 L729. [DOI] [PubMed] [Google Scholar]
- 5. Shipp MA & Look AT. Hematopoietic differentiation antigens that are membrane‐associated enzymes: cutting is the key. Blood 1993; 82:1052 70. [PubMed] [Google Scholar]
- 6. Baraniuk JN, Ohkubo K, Kwon OJ Localization of neutral endopeptidase (NEP) mRNA in human bronchi. Eur Respir J 1995; 8:1458 64. [PubMed] [Google Scholar]
- 7. Van Der Velden VHJ, Wierenga‐Wolf AF, Adriaansen‐Soeting PWC Expression of aminopeptidase N and dipeptidyl peptidase IV in the healthy and asthmatic bronchus. Clin Exp Allergy 1998; 28:110 20. [DOI] [PubMed] [Google Scholar]
- 8. Salles G, Rodewald HR, Chin BS, Reinherz EL, Shipp MA. Inhibition of CD10/neutral endopeptidase 24.11 promotes B‐cell reconstitution and maturation in vivo. Proc Natl Acad Sci U S A 1993; 90:7618 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shipp MA, Tarr GE, Chen CY CD10/neutral endopeptidase 24.11 hydrolyses bombesin‐like peptides and regulates the growth of small cell carcinomas of the lung. Proc Natl Acad Sci U S A 1991; 88:10662 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sunday ME, Hua J, Torday JS, Reyes B, Shipp MA. CD10/neutral endopeptidase 24.11 in developing human foetal lung. Patterns of expression and modulation of peptide‐mediated proliferation. J Clin Invest 1992; 90:2517 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Look AT, Ashmun RA, Shapiro LH, Peiper SC. Human myeloid plasma membrane glycoprotein CD13 (gp150) is identical to aminopeptidase N. J Clin Invest 1989; 83:1299 307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Look AT, Peiper SC, Rebentisch MB Transfer and expression of the gene encoding a human myeloid membrane antigen (gp150). J Clin Invest 1985; 75:569 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lerche C, Vogel LK, Shapiro LH, Noren O, Sjostrom H. Human aminopeptidase N is encoded by 20 exons. Mamm Genome 1996; 7:712 3. [DOI] [PubMed] [Google Scholar]
- 14. Shapiro LH, Ashmun RA, Roberts WM, Look AT. Separate promoters control transcription of the human aminopeptidase N gene in myeloid and intestinal epithelial cells. J Biol Chem 1991; 266:11999 2007. [PubMed] [Google Scholar]
- 15. Larsen SL, Pedersen LO, Buus S, Stryhn A. T cell responses affected by aminopeptidase N (CD13)‐mediated major histocompatibility complex class II‐bound peptides. J Exp Med 1996; 184:183 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tanaka T, Camerini D, Seed B, Torimoto Y, Dang NH, Kameoka J Cloning and functional expression of the T cell activation antigen CD26. J Immunol 1992; 149:481 6. [PubMed] [Google Scholar]
- 17. Duke‐Cohan JS, Morimoto C, Rocker JA, Schlossman SF. Serum high molecular weight dipeptidyl peptidase IV (CD26) is similar to a novel antigen DPPT‐L released from activated T cells. J Immunol 1996; 156:1714 21. [PubMed] [Google Scholar]
- 18. Abbott CA, Baker E, Sutherland GR, McCaughan GW. Genomic organization, exact localization, and tissue expression of the human CD26 (dipeptidyl peptidase IV) gene. Immunogenetics 1994; 40:331 8. [DOI] [PubMed] [Google Scholar]
- 19. Willheim M, Ebner C, Baier K Cell surface characterization of T lymphocytes and allergen‐specific T cell clones: correlation of CD26 expression with T (H1) subsets. J Allergy Clin Immunol 1997; 100:348 55. [DOI] [PubMed] [Google Scholar]
- 20. Hegen M, Kameoka J, Dong RP, Schlossman SF, Morimoto C. Cross‐linking of CD26 by antibody induces tyrosine phosphorylation and activation of mitogen‐activated protein kinase. Immunology 1997; 90:257 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Torimoto Y, Dang NH, Vivier E, Tanaka T, Schlossman SF, Morimoto C. Coassociation of CD26 (dipeptidyl peptidase IV) with CD45 on the surface of human T lymphocytes. J Immunol 1991; 147:2514 7. [PubMed] [Google Scholar]
- 22. Reinhold D, Bank U, Buhling F, Lendeckel U, Faust J, Neubert K, Ansorge S. Inhibitors of dipeptidyl peptidase IV induce secretion of transforming growth factor‐beta 1 in PWM‐stimulated PBMC and T cells. Immunology 1997; 91:354 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kubota T, Flentke GR, Bachovchin WW, Stollar BD. Involvement of dipeptidyl peptidase IV in an in vivo immune response. Clin Exp Immunol 1992; 89:192 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. CorVol. P, Michaud A, Soubrier F, Williams TA. Recent advances in knowledge of the structure and function of the angiotensin I converting enzyme. J Hypertens 1995; 13 (Suppl.):S3 S10. [DOI] [PubMed] [Google Scholar]
- 25. Xu D, Emoto N, Giaid A, Slaughter C, Kaw S, DeWit D, Yanagisawa M. ECE‐1: a membrane‐bound metalloprotease that catalyses the proteolytic activation of big endothelin‐1. Cell 1994; 78:473 85. [DOI] [PubMed] [Google Scholar]
- 26. Saleh D, Furukawa K, Tsao MS Elevated expression of endothelin‐1 and endothelin‐converting enzyme‐1 in idiopathic pulmonary fibrosis: possible involvement of proinflammatory cytokines. Am J Respir Cell Mol Biol 1997; 16:187 93. [DOI] [PubMed] [Google Scholar]
- 27. Skidgel RA. Basic carboxypeptidases: regulators of peptide hormone activity. Trends Pharmacol Sci 1988; 9: 299 304. [DOI] [PubMed]
- 28. Nagae A, Abe M, Becker RP, Deddish PA, Skidgel RA, Erdos EG. High concentration of carboxypeptidase M in lungs: presence of the enzyme in alveolar type I cells. Am J Respir Cell Mol Biol 1993; 9:221 9. [DOI] [PubMed] [Google Scholar]
- 29. Soleilhac JM, Lafuma C, Porcher JM, Auburtin G, Roques BP. Characterization of a soluble form of neutral endopeptidase‐24.11 (EC 3.4.24.11) in human serum: enhancement of its activity in serum of underground miners exposed to coal dust particles. Eur J Clin Invest 1996; 26:1011 7. [DOI] [PubMed] [Google Scholar]
- 30. Favaloro EJ, Browning T, Nandurkar H. The hepatobiliary disease marker serum alanine aminopeptidase predominantly comprises an isoform of the haematological myeloid differentiation antigen and leukaemia marker CD‐13/gp150. Clin Chim Acta 1993; 220:81 90. [DOI] [PubMed] [Google Scholar]
- 31. Tanaka T, Duke‐Cohan JS, Kameoka J, Yaron A, Lee I, Schlossman SF, Morimoto C. Enhancement of antigen‐induced T‐cell proliferation by soluble CD26/dipeptidyl peptidase IV. Proc Natl Acad Sci U S A 1994; 91:3082 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Duke‐Cohan JS, Morimoto C, Rocker JA, Schlossman SF. A novel form of dipeptidylpeptidase IV found in human serum. Isolation, characterization, and comparison with T lymphocyte membrane dipeptidylpeptidase IV (CD26). J Biol Chem 1995; 270:14107 14. [DOI] [PubMed] [Google Scholar]
- 33. Barnes PJ. Neural control of human airways in health and disease. Am Rev Respir Dis 1986; 134:1289 314. [DOI] [PubMed] [Google Scholar]
- 34. Nadel JA. Neutral endopeptidase modulation of neurogenic inflammation in airways. Eur Respir J 1990; 12 (Suppl.):645S 651S. [PubMed] [Google Scholar]
- 35. Lilly CM, Drazen JM, Shore SA. Peptidase modulation of airway effects of neuropeptides. Proc Soc Exp Biol Med 1993; 203:388 404. [DOI] [PubMed] [Google Scholar]
- 36. Cheung D, Bel EH, Den Hartigh J, Dijkman JH, Sterk PJ. The effect of an inhaled neutral endopeptidase inhibitor, thiorphan, on airway responses to neurokinin A in normal humans in vivo. Am Rev Respir Dis 1992; 145:1275 80. [DOI] [PubMed] [Google Scholar]
- 37. Cheung D, Timmers MC, Zwinderman AH, Den Hartigh J, Dijkman JH, Sterk PJ. Neutral endopeptidase activity and airway hyperresponsiveness to neurokinin A in asthmatic subjects in vivo. Am Rev Respir Dis 1993; 148:1467 73. [DOI] [PubMed] [Google Scholar]
- 38. Cheung D, Van Der Veen H, Den Hartigh J, Dijkman JH, Sterk PJ. Effects of inhaled substance P on airway responsiveness to methacholine in asthmatic subjects in vivo. J Appl Physiol 1994; 77:1325 32. [DOI] [PubMed] [Google Scholar]
- 39. Frossard N, Rhoden KJ, Barnes PJ. Influence of epithelium on guinea pig airway responses to tachykinins: role of endopeptidase and cyclooxygenase. J Pharmacol Expr 1989; 248:292 8. [PubMed] [Google Scholar]
- 40. Dusser DJ, Jacoby DB, Djokic TD, Rubinstein I, Borson DB, Nadel JA. Virus induces airway hyperresponsiveness to tachykinins: role of neutral endopeptidase. J Appl Physiol 1989; 67:1504 11. [DOI] [PubMed] [Google Scholar]
- 41. Murlas CG, Lang Z, Williams GJ, Chodimella V. Aerosolized neutral endopeptidase reverses ozone‐induced airway hyperreactivity to substance P. J Appl Physiol 1992; 72:1133 41. [DOI] [PubMed] [Google Scholar]
- 42. Sheppard D, Thompson JE, Scypinski L, Dusser D, Nadel JA, Borson DB. Toluene diisocyanate increases airway responsiveness to substance P and decreases airway neutral endopeptidase. J Clin Invest 1988; 81:1111 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dusser DJ, Djokic TD, Borson DB, Nadel JA. Cigarette smoke induces bronchoconstrictor hyperresponsiveness to substance P and inactivates airway neutral endopeptidase in the guinea pig. Possible role of free radicals. J Clin Invest 1989; 84:900 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lilly CM, Kobzik L, Hall AE, Drazen JM. Effects of chronic airway inflammation on the activity and enzymatic inactivation of neuropeptides in guinea pig lungs. J Clin Invest 1994; 93:2667 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Diamant Z, Van der Veen H, Kuijpers EA, Bakker PF, Sterk PJ. The effect of inhaled thiorphan on allergen‐induced airway responses in asthmatic subjects. Clin Exp Allergy 1996; 26:525 32. [PubMed] [Google Scholar]
- 46. Kohrogi H, Yamaguchi T, Iwagoe H, Fujii K, Hamamoto J, Kawano O, Ando M. Evidence that allergen‐induced contraction of guinea pig bronchi is mediated in part by the release of tachykinins. Int Arch Allergy Immunol 1997; 112:303 8. [DOI] [PubMed] [Google Scholar]
- 47. Sont JK, Van Krieken JH, Van Klink HC, Roldaan AC, Apap CR, Willems LN, Sterk PJ. Enhanced expression of neutral endopeptidase (NEP) in airway epithelium in biopsies from steroid‐ versus nonsteroid‐treated patients with atopic asthma. Am J Respir Cell Mol Biol 1997; 16:549 56. [DOI] [PubMed] [Google Scholar]
- 48. Joos GF, Germonpre PR, Kips JC, Peleman RA, Pauwels RA. Sensory neuropeptides and the human lower airways: present state and future directions. Eur Respir J 1994; 7:1161 71. [PubMed] [Google Scholar]
- 49. Ichinose M, Nakajima N, Takahashi T, Yamauchi H, Inoue H, Takishima T. Protection against bradykinin‐induced bronchoconstriction in asthmatic patients by neurokinin receptor antagonist. Lancet 1992; 340:1248 51. [DOI] [PubMed] [Google Scholar]
- 50. Maggi CA. The effects of tachykinins on inflammatory and immune cells. Regul Pept 1997; 70:75 90. [DOI] [PubMed] [Google Scholar]