

Summary
Dendritic cells (DCs) act not only as sentinels for detection of, but also as target cells for viruses, and this can be important for viral transport and spread. All subsets of DCs are equipped with a battery of receptors recognizing virus‐associated molecular signatures, and recognition of those launches a maturation programme that results in substantial alterations of morphology, motility and the DCs' interactive properties with the extracellular matrix and scanning T cells. In addition to being sensed, viruses are internalized into DCs and, for the major proportion, processed into peptides that are subsequently presented by major histocompatibility complex (MHC) molecules. Transmission of virus to T cells can occur after completion of their replication cycle if the intracellular milieu of the DC permits that. Alternatively, viruses can remain protected from degradation following entrapment by pattern recognition receptors in intracellular compartments, also referred to as virosomes, which translocate towards the DC/T cell interface. Most likely, transfer of virus to T cells occurs in these junctions, referred to as infectious synapses. In addition to promoting DC maturation, many viruses are able to downmodulate DC development and functions in order to evade immune recognition or to induce a generalized immunosuppression.
Introduction
In their interaction with viruses dendritic cells (DCs) can be looked upon as any other cell type of the body that supports entry and replication of some (but not all) viruses and is compromised in function and viability to various extent. The interaction of viruses with DCs is, however, of particular pathogenic relevance because these cells are ‘conductors’ of the adaptive immunity, which orchestrate lymphocytes capable of specifically recognizing and handling antigens to do so in the most effective manner. DCs require recognition of and signals by pathogens, including viruses, to start this programme also referred to as maturation. This includes major changes in the expression of their receptor repertoire, acquisition of a migratory phenotype, secretion of soluble mediators to attract and surface accumulation of receptors to interact with T cells to which they present processed antigens (Steinman et al., 1997; , Banchereau and Steinman, 1998; , Steinman, 2003). Because they have been easier to access [directly from blood or skin, or generated in vitro by established protocols from precursors (Inaba et al., 1992; , Sallusto and Lanzavecchia, 1994; , Romani et al., 1996)] DCs of myeloid origin, also referred to as conventional DCs (cDCs), have mainly been used to study viral interactions. In homeostatic conditions, immature cDCs continuously sample antigens from their environment and are thought to induce or maintain tolerance as they can anergize or clonally delete autoreactive T cells (Lutz and Schuler, 2002). They are equipped with a variety of surface and intracellular sensors allowing for detection of pathogen‐associated molecular patterns (PAMPs), referred to as pathogen recognition receptors (PRRs). Some PRRs, such as scavenger (SR) and C‐type lectin receptors (CLRs) are specialized in internalization of pathogens for subsequent processing and presentation, while others trigger DC maturation in response to PAMP recognition (see below). In immature cDCs, mainly residing in peripheral, especially mucosal tissues, this results in the acquisition of a migratory phenotype that allows for tissue exit and homing to T cell‐rich areas of the secondary lymphatics. Maturation of cDC is generally associated with a major switch from endocytotic to antigen processing and presentation activity as characterized e.g. by downregulation of PRRs, activation of antigen‐processing pathways and lastly displaying and presenting loaded major histocompatibility complex (MHC) class II molecules at their surface (Banchereau and Steinman, 1998). In addition, lipid antigens are diplayed to T cells by CD1 molecules (Thurnher, 2007). Activation of MHC class I‐restricted CD8+ T cells by cDCs relies on viral replication, or cross‐priming after uptake of cell‐bound or cell‐free antigens by specific receptors including Fcγ‐receptors, CD91 and scavenger receptor A (SR‐A), which also enables acquisition of antigens from live cells by nibbling [for review see Wilson and Villadangos (2005)]. T cell scanning is facilitated by extensive morphing of the maturing cDC, which finally reveals characteristic veils to provide contact planes (Shutt et al., 2000). As they are the only antigen presenting cell (APC) subset licensed to activate naïve T cells, cDCs both initiate clonal selection and shape the quality of the ensuing adaptive response by regulated expression of co‐stimulatory molecules and directed release of cytokines. Mature DCs do not leave but rather die in lymphoid tissues thereafter and are not found in the efferent lymph (Banchereau and Steinman, 1998).
Multiple cDC subtypes are defined, which differ in phenotype, localization and immune function (Banchereau and Steinman, 1998; , Shortman and Liu, 2002), and this has been enlarged by the phenotypic and functional identification of plasmacytoid DCs (pDCs) (Liu, 2005). Although their functional distinction in response to PAMP recognition and antigen capture and uptake is not absolute, cDCs and pDCs may have evolved to preferentially controlling bacterial and/or viral infections respectively. Phenotypically, this is indicated by their distinct repertoire of PRRs and transcription factors. These coin cDCs as major producers of inflammatory cytokines such as IL‐12, TNF‐α, IL‐6 and IL‐1α/β as important for effector cell activation, and pDCs as early effector cells of the innate immune system particularly in viral infections, because they readily produce high amounts of type I IFN (Asselin‐Paturel and Trinchieri, 2005). pDCs are mainly found in blood from where they most likely directly access secondary lymphatic tissues (Liu, 2005). They also mature after pathogen encounter, but their ability to present antigen is limited compared with that of cDCs and thus, their physiological role in pathogen defence remains controversial (see below). Interestingly, pDCs and cDCs share a common programme for chemokine induction after virus encounter, which allows for a co‐ordinated attraction of the different immune effectors in response to viral infection (Piqueras et al., 2006).
Interaction of DCs (of either origin and subset) with pathogens has been extensively studied with regard to their activation and subsequent role in initiating and shaping adaptive immunity. For this, model PAMPs have often been used such as lipopolysaccharide (LPS), macrophage activating lipopeptide, 2 kD (MALP‐2) and CpG DNA and compared with endogenous signals provided by inflammatory signals (e.g. TNF‐α) or receptors (e.g. CD40L) and large‐scale alterations of gene expression were determined by microarray analyses. These allowed for compiling alterations generally associated with pathogen encounter, specific for groups of pathogens such as viruses or even a given virus (e.g. Huang et al., 2001). This review will rather attempt to follow interaction of viruses with DCs from a viral angle, from being detected and taken up, subsequently replicated, processed or retained, to being transmitted to T cells. Lastly, we will summarize consequences of viral interaction on DC functions and viability associated with immune evasion or suppression.
Sensing of PAMPs: TLRs and their relative role in type I IFN induction
Pathogen recognition receptors are specialized either on inducing DC maturation [e.g. Toll‐like receptors (TLRs) and helicases such as retinoic acid inducible gene 1 and melanoma differentiation‐associated gene 5 (RIG‐I and Mda‐5)] or on internalization of pathogens for processing and subsequent presentation. Both systems can, however, functionally cooperate as for example SRs can modulate access of TLR ligands to their receptors (Hoebe et al., 2005), and ligation of the CLR DC‐SIGN modulates the signalling activity of TLRs (van Kooyk et al., 2003).
More than 10 mammalian TLRs are known, which are differentially expressed mainly on professional APC. Some TLRs are located at the cell surface and sense – in addition to a variety of bacterial and fungal PAMPs – envelope proteins of respiratory syncytial virus (RSV), measles virus (MV), human cytomegalovirus (HCMV) or mouse mammary tumour virus (MMTV) (Kawai and Akira, 2006a). In contrast, other TLRs (TLR3, TLR7/8 and TLR9) signal from low‐pH endosomal compartments after recognition of nucleic acid PAMPs including viral ssRNAs (in a species‐specific manner), dsDNAs and dsRNAs (Diebold et al., 2004; Heil et al., 2004; Krug et al., 2004a,, b) (Fig. 1). Myeloid differentiation factor 88 (MyD88) is a general adaptor molecule for TLR signalling important for production of pro‐inflammatory cytokines (Akira and Takeda, 2004). TLR3 and TLR4, however, are able to signal also MyD88‐independently by coupling to another adaptor molecule, referred to as Toll/interleukin‐1 receptor (TIR) domain‐containing adaptor inducing IFN‐β (TRIF), which, by name, implies particular relevance for antiviral activity. In addition to sensing viral nucleic acids by TLR3 or members of the TLR9 family (TLRs 7–9), DCs, as other cell types have additional intracellular PRRs detecting dsRNA such as RIG‐I, Mda‐5 or protein kinase R (PKR), which trigger signalling cascades leading to activation of interferon response factor (IRF) transcription factors (Kawai and Akira, 2006b; Meylan and Tschopp, 2006) (Fig. 1). Depending on the availability of PRRs, their relative contribution in both DC maturation and antiviral defence is DC subset‐specific. Thus, recognition of and activation by nucleic acids occur by members of the TLR9 family on pDCs, which selectively express these proteins, while cDCs rather rely on TLR3 or RIG‐I‐dependent detection (Kato et al., 2005) (Fig. 1). In response to viral encounter, pDCs produce type I IFN at 100‐ to 1000‐fold higher levels than any other cell type dedicating up to 50% of their transcription to synthesizing IFN‐specific mRNAs (Asselin‐Paturel and Trinchieri, 2005). IFN induction in pDCs is unique in its spatiotemporal regulation and dependent on MyD88 and IRF‐7, but not IRF‐3, which is different from that in cDCs, which signal both IRF‐3‐ and IRF‐7‐dependently, but MyD88‐independently (2005a, 2005b; Tailor et al., 2006). In vivo, however, TLR3/4‐dependent and ‐independent pathways can take over in conferring protective viral immunity if signalling via the TLR9 family members is ablated, thus questioning the role of pDCs in this context (Yang et al., 2005). In addition to its antiviral activity, type I IFN is also a maturation factor of DCs as it induces upregulation of MHC, co‐stimulatory molecules and also TLRs and impacts on the cytokine pattern released from matured DCs and their activity (Tough, 2004; Lopez et al., 2006). Thus, virally induced type I IFN promotes TRAIL‐mediated cytotoxicity in both cDCs and, very early, in pDCs (see also Fig. 3) and this could be an important factor in the contraction phase of the antiviral immune response (Vidalain et al., 2001; Chaperot et al., 2006). On the other hand, early exposure to type I IFNs can limit development and expansion of cDCs and this was suggested to contribute to viral immune evasion (Hahm et al., 2005) (see also Fig. 3). While type I IFN acts as an adjuvant in DC maturation and subsequent development of adaptive immunity, it also has antiviral activity and therefore can limit viral spread. It is therefore not surprising that most viruses encode for gene products that interfere with the induction and/or the auto‐ or paracrine activity of IFNs. These and their individual strategies to block IFN induction and/or signalling (most of them target stability or activation of transcription factors such as IRF‐3 or STAT1/2 respectively) (Fig. 1) were mainly analysed in fibroblasts and are extensively reviewed (Horvath, 2004; Takaoka and Yanai, 2006). There is no reason to believe they would act differently in DCs, provided the intracellular milieu in these cells supports viral replication at least to an extent that permits their accumulation. Thus, MV or RSV infection in pDCs block TLR7/9‐dependent and ‐independent IFN induction (Schlender et al., 2005), and the ability of the influenza NS1 protein to specifically suppress DC maturation, migration and T cell stimulatory activity of cDC was recently documented (Fernandez‐Sesma et al., 2006). Because, however, viral IFN antagonists are often non‐structural proteins, their availability depends on the ability of the virus to complete at least early steps of its life cycle. As little is known about viral infection of pDCs, we will further focus on the interaction of viruses with cDCs only.
Figure 1.
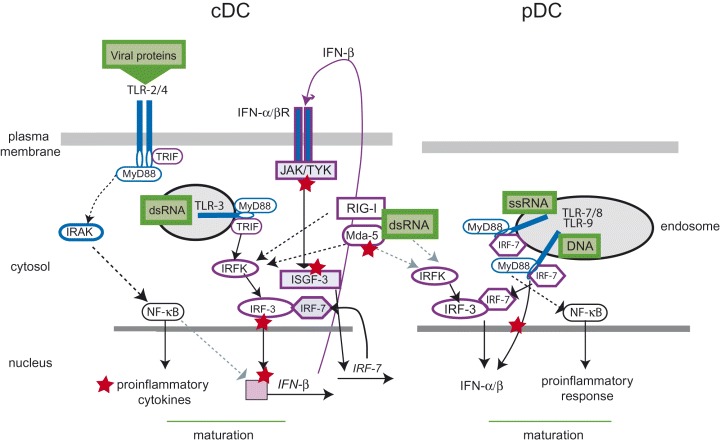
Activation of pro‐inflammatory and antiviral responses in cDCs and pDCs and viral interference. cDCs (left) sense viral proteins by surface TLRs (TLR2 or TLR4) and via their cytoplasmic domains recruit adaptor molecules [MyD88 for both, and in addition, TRIF (TIR domain containing adaptor inducing IFN‐β) for TLR4]. The MyD88‐dependent cascade leading to activation of NF‐κB involves, among other components, the IL‐1R‐associated kinase (IRAK) and is important for the induction of the pro‐inflammatory response. In common with TLR‐4, TLR‐3 (after recognition of viral dsRNA) located in the endosomal compartment, recruits both MyD88 and TRIF and this leads to activation of a cascade including interferon response factor (IRF) kinases (IRFK), which promote phosphorylation of IRF‐3 as required for its dimerization and nuclear translocation. Among other genes, IFN‐β transcription is induced, which, after translation, binds to and activates IFN‐α/β receptor signalling (involving JAK/TYK receptor proximal and the ISGF‐3 complex receptor distal). One of the genes activated by this cascade is IRF‐7, whose gene product, together with IRF‐3, mediates induction of IFN‐α. In pDCs (right), MyD88 and IRF‐7 are constitutively associated with TLR‐7/8 and TLR‐9 (recognizing viral ssRNA or DNA respectively) and this allows for direct induction of IFN‐α/β. In addition to TLRs, intracellular sensors such as mda‐5 and RIG‐I recognize dsRNA (most likely both in cDCs and pDCs) and by that, IRF‐3 can be activated. Many viruses encode for proteins that interfere at various levels with interferon induction or response in cDCs and pDCs (indicated as red asterisks).
Figure 3.
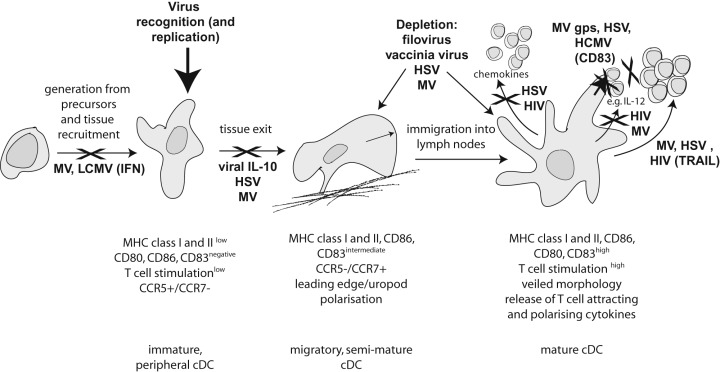
Viral interference with cDC maturation and function. Important steps in the life/maturation cycle of a cDC are schematically depicted including generation from precursors and stimulation‐dependent maturation from a tissue resident immature to a migratory semi‐mature (interacting with extracellular matrix components) to a fully mature DC in the lymph node. Viruses can interfere at virtually any stage of this programme and only some of them or strategies employed are shown.
Virus interaction with DCs: from uptake to assembly
The role of surface receptors in viral uptake and routing to subcellular compartments
While interaction of viruses with TLRs is important in conveying differentiation signals, these do not support viral uptake. For DCs, ‘cis’‐infection and ‘trans’‐infection are distinguished, the first describing DC infection, while the second refers to the ability of DCs to transmit virus, often independently of cis‐infection, to contacting T cells. Cis‐infection of DCs relies on the availability of receptors allowing for attachment and fusion, the latter either at the cell surface or, following internalization, mostly after acidification in late endosomal/lysosomal compartments (Fig. 2). Cis‐infection of immature DCs has best been studied for HIV‐1, which can enter into these cells because they express the attachment receptor CD4 together with CCR5, which serves as co‐receptor of R5 and dual tropic R4/R5 strains (Rinaldo and Piazza, 2004). Immature DCs also express CXCR4 at low levels, yet support cis‐replication of R5 strains only, unless they are exposed to IL‐10 indicating that the intracellular milieu essentially contributes to permissivity. In line with this hypothesis, neither R5 nor R4 strains are able to replicate efficiently in mature DCs in vitro (Rinaldo and Piazza, 2004). The latter, however, efficiently supports trans‐infection of both R4 and R5 tropic strains to T cells (Granelli‐Piperno et al., 1998). Trans‐infection relies on the interaction of HIV‐1 with CLRs such as Langerin (on Langerhans cells, LCs) and DC‐SIGN abundantly expressed on immature DCs, which serves as both adhesion (for ICAM‐2 on endothelial cells, ICAM‐3 on T cells and Mac‐1 on neutrophils) and carbohydrate PAMP receptor (Geijtenbeek and van Kooyk, 2003). DC‐SIGN bears an internalization motif in its cytoplasmic tail and endocytoses after pathogen binding‐driven multimerization thereby usually routing bound antigens into compartments specialized for antigen processing and MHC loading. HIV‐1 was the first virus found to bind to DC‐SIGN at the cell surface with high affinity (Geijtenbeek et al., 2000; , 2002).
Figure 2.
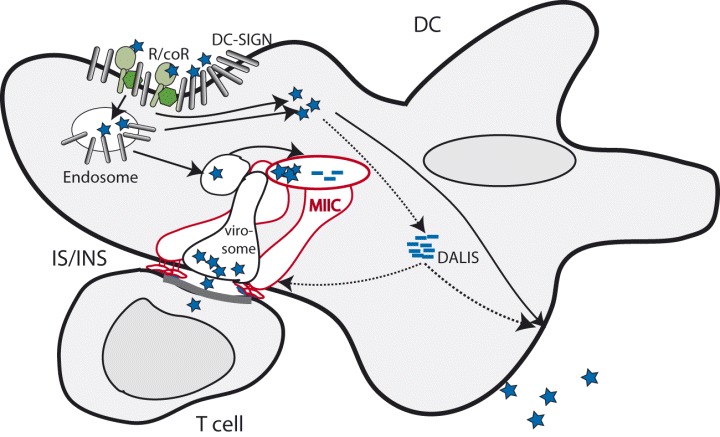
Viral entry into and transmission from DCs. Viruses (indicated as blue asterisks) can enter DCs using specific receptor/co‐receptor (R/coR) complexes (in green) or, following capture by DC‐SIGN, internalization. Entry can be followed by viral replication leading to production of progeny virus, and some viral proteins or defective ribosomal products (DRiPs) may accumulate in DALIS for subsequent trafficking or proteasomal processing. Viral release from endolysosomal compartments can be promoted by low pH, and replication will ensue, in case the intracellar environment in the DC is permissive. The majority of internalized virus will proceed from endolysosomal compartments for processing (peptides indicated as blue bars) and loading onto MHC class II molecules in MHC class II compartments (MIICs and MHC II molecules in red), which will be transported to the surface and presented to T cells in mature DCs. A fraction of internalized virus can remain protected from degradation in compartments referred to as ‘virosomes’, which are similar but maybe not identical to MIICs. In common with the latter, virosomes relocate towards the DC/T cell interface, commonly referred to as immunological synapse (IS). As virus is transmitted at this interface, this is also called infectious synapse (INS). On the T cell side, IS/INS formation (as indicated by the bold line) requires profound cytoskeletal rearrangements and receptor clustering.
After binding to the cell surface, HIV can follow different routes. Sorting of virus can take place simultaneously into the cytoplasm and proceed to cis‐infection or into an endolysosomal compartment after capturing by DC‐SIGN from where (i) it can proceed to degradation and antigen presentation (for the majority of particles); and (ii) it can escape after acidification into the cytoplasm or (iii) where it is maintained in an infectious form (Fackler and Peterlin, 2000; , Moris et al., 2004; , Kramer et al., 2005) (Fig. 2). In macrophages, this ‘storage compartment’, also referred to as virosome, shares some, but not all markers with late endosomes, and in DCs with tetraspanin‐rich MHC class II loading compartments (MIICs) (Garcia et al., 2005). Subsequent trans‐infection of T cells involves fusion of the tubulizing compartment with the cell membrane in response to as yet undefined signals elicited by DC/T cell interaction (Fig. 2). Its role in HIV‐1 uptake and transmission has been underlined by the finding that ablation of DC‐SIGN on immature DCs impaired transport of HIV‐1 to and formation of an infectious synapse at the DC/T cell interface (Arrighi et al., 2004) (see below). The relative role of DC‐SIGN‐mediated trans‐infection may, however, vary dependent on the DC maturation state (Granelli‐Piperno et al., 1998); moreover, long‐term carriage of HIV‐1 in DCs is via low level of infection rather than captured virus (Turville et al., 2004). Thus, viral transfer can be dependent or independent of DC cis‐infection or DC‐SIGN. Other viruses were also found to interact with DC‐SIGN, and this can, probably by concentrating viral receptors at the site of entry, enhance cis‐infection of DCs and mediate trans‐infection of target cells as shown for Dengue virus, HCMV, hepatitis C virus, Ebola virus, SARS corona virus and MV (Alvarez et al., 2002; Halary et al., 2002; , Tassaneetrithep et al., 2003; , Lozach et al., 2004; , de Witte et al., 2006).
Intracellular permissivity of DC to viral infection, viral assembly and propagation
Subsequent to entry, the intracellular milieu is an important viral permissivity determinant in DCs. Their ability to support viral replication is virus‐dependent, if not even strain‐dependent, often changes with maturation and can be modulated in response to external stimulation. In general, immature DCs are more permissive for almost any virus that has been studied than those already matured (Rinaldo and Piazza, 2004; , Pollara et al., 2005). Immature DCs are, for instance, fully permissive to infection with herpesviruses such as herpes simplex virus (HSV) and endothelial cell‐adapted (E‐strains), but not with fibroblast‐adapted HCMV, while in mature DCs, production of viral proteins and particles as well as cytopathic changes are greatly restricted (Bosnjak et al., 2005). For HIV‐1, a post‐entry block of replication in mature DCs has been linked to restrictions of viral transcription occurring prior and post integration (Bakri et al., 2001; , Turville et al., 2004). Mostly, however, viruses may encounter immature DCs and thus, the intracellular milieu of the infected DC changes in response to viral infection, inflammatory signals and, lastly, endogenous ligands such as CD40. The ability to modulate induction or activity of type I IFNs is certainly one of these determinants and has been referred to above (Fernandez‐Sesma et al., 2006). Viral IL‐10, as encoded by HCMV, is known to inhibit DC maturation, and thus, the virus can sustain its preferred DC maturation stage. However, at the same time it promotes DC apoptosis, which in turn limits viral replication (Raftery et al., 2004). Sorting of viral proteins as required for the formation of infectious particles would also be an attractive means of regulating viral infection in DCs. Maturing DCs and macrophages can accumulate newly synthesized proteins in large cytosolic structures referred to as DC aggresome‐like induced structures (DALIS) (Lelouard et al., 2002; , 2004; Canadien et al., 2005). There, proteins often representing ubiqutinated defective ribosomal products (DRiPs) are stored for subsequent proteasomal degradation and MHC class I presentation. Recently, targeting of viral proteins into DALIS has been directly documented for influenza virus nucleoprotein (Herter et al., 2005), and this may regulate not only degradation but also availability of this protein for replication (Fig. 2).
While some viruses including filoviruses and hantavirus productively replicate in immature DCs to titres not different from or even exceeding those in standard cell lines (Raftery et al., 2002; , Mahanty et al., 2003), replication of others such as MV and HIV‐1 is restricted, cell differentiation‐ and sometimes even virus strain‐dependent (Schneider‐Schaulies et al., 2003; , Rinaldo and Piazza, 2004). For both viruses, the efficiency of viral replication in DCs was found to be greatly enhanced upon co‐culture with activated T cells (Fugier‐Vivier et al., 1997; , Frank and Pope, 2002). In MV‐infected DCs, CD40 ligation was found to provide this stimulatory signal (Servet‐Delprat et al., 2000a;, 2003). Cis‐infection of DCs may also be augmented in response to T cell contact, but the effect of cellular interaction on viral transmission may predominate in the explosive HIV‐1 replication in DC/T cell conjugates. As indicated above, HIV‐1 can access intracellular compartments in DCs and macrophages that redistribute or tubulize back towards the surface, which may give rise to an infectious synapse (Turville et al., 2004; , Garcia et al., 2005). The latter, so far been characterized for transfer of retroviruses between T cells and in DC/T cell conjugates, is associated with cytoskeletal reorganizations and receptor clustering in both donor and acceptor cells, which favour fusion with and entry into the target cells (Arrighi et al., 2004; Piguet and Sattentau, 2004; , Fackler and Krausslich, 2006). Suggesting that this occurs by efficient trans‐infection, accumulation of CD4, CCR5 and CXCR4 molecules at the site of contact has been found essential for HIV‐1 transmission in conjugates (McDonald et al., 2003; , Jolly and Sattentau, 2004). Unlike that of immunological synapses, formation of infectious synapses is not triggered by T cell receptor‐mediated antigen recognition (Piguet and Sattentau, 2004). At present it is unknown to what extent infectious synapses formed in DC/T cell conjugates may differ from stable immunological synapses formed after antigen recognition. Importantly, the latter has been associated with surface‐orientated tubulation of MHC class II containing intracellular compartments towards the interface (Boes et al., 2003; , Boes and Ploegh, 2004). Though transmission via infectious synapses was described only for retroviruses as yet, it is likely that this is an important, if not the predominant, mode of transfer from DC to T cells for other viruses as well (Fig. 2).
Interference of viral interaction with DC function and maturation
Most studies addressing the interaction of viruses with DCs have focused on their ability to modulate immune responses. Much has been learned about to what extent individual viruses promote DC maturation and thereby prime, activate and shape immunity and this has not only increased our knowledge of immune activation in general but also provided the basis for exploiting their ability to serve as adjuvants for preventive and therapeutical interventions. Interferences with DC development, viability, maturation and function are, however, considered as important strategies for viral immune evasion and/or immunosuppression (some of which are summarized in Fig. 3). At a first level, viruses could modulate the frequency of DC subsets. This has been seen after experimental infection of mice with MV and lymphocytic choriomeningitis virus (LCMV), where interference with DC development and expansion in vivo and in vitro has been linked to type I IFN production (Hahm et al., 2005). Though frequencies of both pDC and cDC were found to be decreased in blood with AIDS progression (Pacanowski et al., 2001), those in secondary lymphoid tissues of experimentally simian immunodeficiency virus (SIV)‐infected animals were comparable to healthy animals (Teleshova et al., 2006), suggesting that active DC depletion or aberrant trafficking in the periphery may contribute. DC depletion could also result from apoptosis or cytolysis as induced by many viruses such as filoviruses, Vaccinia virus, HSV and MV in vitro (Engelmayer et al., 1999; , Servet‐Delprat et al., 2000b; , Mahanty et al., 2003; , Muller et al., 2004). Secondly, trafficking of DCs could be affected by viral encounter. There is evidence that infection of immature DCs impairs chemokine receptor switching as required for tissue exit and recruitment into lymphatics. Thus, HSV‐ or HCMV‐infection interferes with upregulation of CCR7 and, consequently DC mobilization in response to CCL19, which is constitutively produced at the luminal sites of high endothelial venules and in the T cell‐rich areas of secondary lymphoid tissues by mature interdigitating DCs or stromal cells (Caux et al., 2002; , Moutaftsi et al., 2004; , Prechtel et al., 2005). In addition, IL‐10, as produced for instance upon infection with HCMV (Raftery et al., 2004), can uncouple signalling via chemokine receptors and this would be expected to greatly interfere with DC trafficking (D′Amico et al., 2000).
While alterations of DC trafficking may just be induced by some viruses, loss of T cell stimulatory activity is a feature shared by DCs infected with most viruses investigated, and the mechanisms and targets of this interference vary between viruses. Adequate expression levels of MHC complexes loaded with correctly processed peptides are important especially for priming of naïve T cells, and inefficient induction or even downmodulation of MHC molecules and/or interference with antigen processing occurs in HSV‐, HCMV‐ or MV‐infected DCs (Servet‐Delprat et al., 2000a; , Raftery et al., 2004; , Novak and Peng, 2005). Secondly, accumulation of co‐stimulatory molecules that engage CD28 and amplify T cell receptor signalling are also targeted by these viruses. Whether or to what extent virus infection of DCs interferes with organization of functional immunological synapses as required for successful T cell activation is unknown as yet. Most recently, the importance of surface‐bound chemokines for capturing and priming T cells for synapse formation has been documented (Friedman et al., 2006). Thus, the ability of viruses to suppress induction of CCR7 expression may, in addition to trafficking and chemoattraction, also directly affect the ability of infected DCs to promote successful interaction with T cells. Virus‐infected DCs can, however, also directly convey inhibitory signals to T cells, one of which may be brought about by upregulation of inhibitory members of the B7 family, which ligate inhibitory receptors such as PD‐1 on T cells (Barber et al., 2006). Another example of active inhibition by cellular proteins is shedding of CD83 by HCMV‐ or HSV‐infected mature DCs, which was found to inhibit expansion of allogenic T cells. Receptor(s) involved remain elusive, but apparently, soluble CD83 acts on both DCs and T cells and prevents efficient formation of DC/T cell conjugates (Lechmann et al., 2001; , Kotzor et al., 2004; Senechal et al., 2004). Lastly, viral proteins expressed by infected DCs can directly silence T cells, and among those, the ability of the MV glycoprotein complex to block S phase entry of T cells by interference with signalling cascades activated in response to CD3/CD28 ligation has been studied in more detail on a molecular level [for a recent review see that by Schneider‐Schaulies and Dittmer (2006)]. In addition to silencing T cells, infected DCs may trigger differentiation and expansion of effector T cells that could support establishment of chronic rather than resolving viral infections. There is, for instance evidence that secretion of IL‐12 from cDCs is suppressed or that of IL‐10 is induced after viral infection (Servet‐Delprat et al., 2000a; , Moutaftsi et al., 2002) and this may promote preferential humoral rather than cellular immunity. Activation of effector T cells can also be efficiently prevented by regulatory T cells, the induction and expansion of which can be triggered upon interaction with immature or partially matured DCs, a phenotype (MHClow, CD86low) sustained upon infection with some viruses (see above) (Mills, 2004; , Robertson and Hasenkrug, 2006; Schneider‐Schaulies and Dittmer, 2006).
Conclusions and outlook
After all, what is so special about the interaction of viruses with DCs? As any other cell type of the body, DCs could just be regarded as hosts more or less permissive for viruses, which provide the metabolic environment for amplification of viral genetic material and subsequent transmission and spread. There are, however, facets of this particular interaction that make it a double‐edged sword for viruses. The fact that many of them target these cells (as for instance reflected by the variety of entry modes into DCs) suggests that this can be very favourable – DCs are perfect ferries that take off as soon as the passenger enters. Especially lymphotropic viruses are transported safely to the desired destination, the secondary lymphatics, from where there are many ways for efficient spread. By scanning their receptor repertoire, viruses can choose DC subsets ideally suited for their individual purpose and they can, by actively modifying the maturation programme, determine for instance the efficiency of migration or if they would or would rather not like to attract T cells for further propagation. The potential to interfere with DC functions thereby avoiding recognition by the immune system is being exploited by many viruses, and this is certainly a highly efficient mode of immune evasion or, at a more general level, immunosuppression, which is of particular pathogenic importance for HIV‐1 and MV in humans. The other side of the coin is, however, that DCs refuse to just serve the virus, but are perfect adjuvants in the induction of immunity – so uptake by these cells is at the expense of being sensed, degraded, presented, and lastly, eliminated by effector cells orchestrated by these sentinels. The interplay between viruses and DCs is apparently delicate and needs fine‐tuning to allow for the survival of both the passenger and the host. Manipulation of this interaction is a highly attractive strategy followed by many laboratories worldwide in order to exploit the ability of the best APC we have to induce protection or immune responses to auto‐ or tumour antigens (often by loading them with recombinant viral vectors). A deeper understanding of these fascinating cells in terms of for instance plasticity and function of subsets and how they are modified by viruses on a cellular (e.g. mechanisms of priming of differential T cell subsets) and particularly cell biological level (how does virus infection modify DC morphing and adhesion, and how efficiently are immunological synapses formed, how do they look like, how stable are they and how are they lastly resolved) will pave the way for major breakthroughs in their rationale preventive and therapeutic applications.
Acknowledgements
We thank Jürgen Schneider‐Schaulies, Alexander Steinkasserer and Bertus K. Rima for constructive critical comments on the manuscript and apologize to all scientists, viruses and DC subsets who did not get the recognition they deserve in this review to keep the list of references manageable. We thank our lab members for their continuous practical support and the Deutsche Forschungsgemeinschaft and the Interdisciplinary Center for Clinical Research of the Medical Faculty of the University of Würzburg for financial support.
References
- Akira, S. , and Takeda, K. (2004) Toll‐like receptor signalling. Nat Rev Immunol 4: 499–511. [DOI] [PubMed] [Google Scholar]
- Alvarez, C.P. , Lasala, F. , Carrillo, J. , Muniz, O. , Corbi, A.L. , and Delgado, R. (2002) C‐type lectins DC‐SIGN and L‐SIGN mediate cellular entry by Ebola virus in cis and in trans . J Virol 76: 6841–6844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrighi, J.F. , Pion, M. , Wiznerowicz, M. , Geijtenbeek, T.B. , Garcia, E. , Abraham, S. , et al. (2004) Lentivirus‐mediated RNA interference of DC‐SIGN expression inhibits human immunodeficiency virus transmission from dendritic cells to T cells. J Virol 78: 10848–10855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asselin‐Paturel, C. , and Trinchieri, G. (2005) Production of type I interferons: plasmacytoid dendritic cells and beyond. J Exp Med 202: 461–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakri, Y. , Schiffer, C. , Zennou, V. , Charneau, P. , Kahn, E. , Benjouad, A. , et al. (2001) The maturation of dendritic cells results in postintegration inhibition of HIV‐1 replication. J Immunol 166: 3780–3788. [DOI] [PubMed] [Google Scholar]
- Banchereau, J. , and Steinman, R.M. (1998) Dendritic cells and the control of immunity. Nature 392: 245–252. [DOI] [PubMed] [Google Scholar]
- Barber, D.L. , Wherry, E.J. , Masopust, D. , Zhu, B. , Allison, J.P. , Sharpe, A.H. , et al. (2006) Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 439: 682–687. [DOI] [PubMed] [Google Scholar]
- Boes, M. , and Ploegh, H.L. (2004) Translating cell biology in vitro to immunity in vivo . Nature 430: 264–271. [DOI] [PubMed] [Google Scholar]
- Boes, M. , Bertho, N. , Cerny, J. , Op den Brouw, M. , Kirchhausen, T. , and Ploegh, H. (2003) T cells induce extended class II MHC compartments in dendritic cells in a Toll‐like receptor‐dependent manner. J Immunol 171: 4081–4088. [DOI] [PubMed] [Google Scholar]
- Bosnjak, L. , Jones, C.A. , Abendroth, A. , and Cunningham, A.L. (2005) Dendritic cell biology in herpesvirus infections. Viral Immunol 18: 419–433. [DOI] [PubMed] [Google Scholar]
- Canadien, V. , Tan, T. , Zilber, R. , Szeto, J. , Perrin, A.J. , and Brumell, J.H. (2005) Cutting edge: microbial products elicit formation of dendritic cell aggresome‐like induced structures in macrophages. J Immunol 174: 2471–2475. [DOI] [PubMed] [Google Scholar]
- Caux, C. , Vanbervliet, B. , Massacrier, C. , Ait‐Yahia, S. , Vaure, C. , Chemin, K. , et al. (2002) Regulation of dendritic cell recruitment by chemokines. Transplantation 73: S7–S11. [DOI] [PubMed] [Google Scholar]
- Chaperot, L. , Blum, A. , Manches, O. , Lui, G. , Angel, J. , Molens, J.P. , and Plumas, J. (2006) Virus or TLR agonists induce TRAIL‐mediated cytotoxic activity of plasmacytoid dendritic cells. J Immunol 176: 248–255. [DOI] [PubMed] [Google Scholar]
- D'Amico, G. , Frascaroli, G. , Bianchi, G. , Transidico, P. , Doni, A. , Vecchi, A. , et al. (2000) Uncoupling of inflammatory chemokine receptors by IL‐10: generation of functional decoys. Nat Immunol 1: 387–391. [DOI] [PubMed] [Google Scholar]
- Diebold, S.S. , Kaisho, T. , Hemmi, H. , Akira, S. , and Reis e Sousa, C. (2004) Innate antiviral responses by means of TLR7‐mediated recognition of single‐stranded RNA. Science 303: 1529–1531. [DOI] [PubMed] [Google Scholar]
- Engelmayer, J. , Larsson, M. , Subklewe, M. , Chahroudi, A. , Cox, W.I. , Steinman, R.M. , and Bhardwaj, N. (1999) Vaccinia virus inhibits the maturation of human dendritic cells: a novel mechanism of immune evasion. J Immunol 163: 6762–6768. [PubMed] [Google Scholar]
- Fackler, O.T. , and Krausslich, H.G. (2006) Interactions of human retroviruses with the host cell cytoskeleton. Curr Opin Microbiol 9: 409–415. [DOI] [PubMed] [Google Scholar]
- Fackler, O.T. , and Peterlin, B.M. (2000) Endocytic entry of HIV‐1. Curr Biol 10: 1005–1008. [DOI] [PubMed] [Google Scholar]
- Fernandez‐Sesma, A. , Marukian, S. , Ebersole, B.J. , Kaminski, D. , Park, M.S. , Yuen, T. , et al. (2006) Influenza virus evades innate and adaptive immunity via the NS1 protein. J Virol 80: 6295–6304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank, I. , and Pope, M. (2002) The enigma of dendritic cell‐immunodeficiency virus interplay. Curr Mol Med 2: 229–248. [DOI] [PubMed] [Google Scholar]
- Friedman, R.S. , Jacobelli, J. , and Krummel, M.F. (2006) Surface‐bound chemokines capture and prime T cells for synapse formation. Nat Immunol 7: 1101–1108. [DOI] [PubMed] [Google Scholar]
- Fugier‐Vivier, I. , Servet‐Delprat, C. , Rivailler, P. , Rissoan, M.C. , Liu, Y.J. , and Rabourdin‐Combe, C. (1997) Measles virus suppresses cell‐mediated immunity by interfering with the survival and functions of dendritic and T cells. J Exp Med 186: 813–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia, E. , Pion, M. , Pelchen‐Matthews, A. , Collinson, L. , Arrighi, J.F. , Blot, G. , et al. (2005) HIV‐1 trafficking to the dendritic cell–T‐cell infectious synapse uses a pathway of tetraspanin sorting to the immunological synapse. Traffic 6: 488–501. [DOI] [PubMed] [Google Scholar]
- Geijtenbeek, T.B. , and Van Kooyk, Y. (2003) Pathogens target DC‐SIGN to influence their fate DC‐SIGN functions as a pathogen receptor with broad specificity. APMIS 111: 698–714. [DOI] [PubMed] [Google Scholar]
- Geijtenbeek, T.B. , Kwon, D.S. , Torensma, R. , Van Vliet, S.J. , Van Duijnhoven, G.C. , Middel, J. , et al. (2000) DC‐SIGN, a dendritic cell‐specific HIV‐1‐binding protein that enhances trans‐infection of T cells. Cell 100: 587–597. [DOI] [PubMed] [Google Scholar]
- Geijtenbeek, T.B. , Van Duijnhoven, G.C. , Van Vliet, S.J. , Krieger, E. , Vriend, G. , Figdor, C.G. , and Van Kooyk, Y. (2002) Identification of different binding sites in the dendritic cell‐specific receptor DC‐SIGN for intercellular adhesion molecule 3 and HIV‐1. J Biol Chem 277: 11314–11320. [DOI] [PubMed] [Google Scholar]
- Granelli‐Piperno, A. , Delgado, E. , Finkel, V. , Paxton, W. , and Steinman, R.M. (1998) Immature dendritic cells selectively replicate macrophagetropic (M‐tropic) human immunodeficiency virus type 1, while mature cells efficiently transmit both M‐ and T‐tropic virus to T cells. J Virol 72: 2733–2737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahm, B. , Trifilo, M.J. , Zuniga, E.I. , and Oldstone, M.B. (2005) Viruses evade the immune system through type I interferon‐mediated STAT2‐dependent, but STAT1‐independent, signaling. Immunity 22: 247–257. [DOI] [PubMed] [Google Scholar]
- Halary, F. , Amara, A. , Lortat‐Jacob, H. , Messerle, M. , Delaunay, T. , Houles, C. , et al. (2002) Human cytomegalovirus binding to DC‐SIGN is required for dendritic cell infection and target cell trans‐infection. Immunity 17: 653–664. [DOI] [PubMed] [Google Scholar]
- Heil, F. , Hemmi, H. , Hochrein, H. , Ampenberger, F. , Kirschning, C. , Akira, S. , et al. (2004) Species‐specific recognition of single‐stranded RNA via toll‐like receptor 7 and 8. Science 303: 1526–1529. [DOI] [PubMed] [Google Scholar]
- Herter, S. , Osterloh, P. , Hilf, N. , Rechtsteiner, G. , Hohfeld, J. , Rammensee, H.G. , and Schild, H. (2005) Dendritic cell aggresome‐like‐induced structure formation and delayed antigen presentation coincide in influenza virus‐infected dendritic cells. J Immunol 175: 891–898. [DOI] [PubMed] [Google Scholar]
- Hoebe, K. , Georgel, P. , Rutschmann, S. , Du, X. , Mudd, S. , Crozat, K. , et al. (2005) CD36 is a sensor of diacylglycerides. Nature 433: 523–527. [DOI] [PubMed] [Google Scholar]
- Honda, K. , Ohba, Y. , Yanai, H. , Negishi, H. , Mizutani, T. , Takaoka, A. , et al. (2005a) Spatiotemporal regulation of MyD88‐IRF‐7 signalling for robust type‐I interferon induction. Nature 434: 1035–1040. [DOI] [PubMed] [Google Scholar]
- Honda, K. , Yanai, H. , Negishi, H. , Asagiri, M. , Sato, M. , Mizutani, T. , et al. (2005b) IRF‐7 is the master regulator of type‐I interferon‐dependent immune responses. Nature 434: 772–777. [DOI] [PubMed] [Google Scholar]
- Horvath, C.M. (2004) Silencing STATs: lessons from paramyxovirus interferon evasion. Cytokine Growth Factor Rev 15: 117–127. [DOI] [PubMed] [Google Scholar]
- Huang, Q. , Liu, D. , Majewski, P. , Schulte, L.C. , Korn, J.M. , Young, R.A. , et al. (2001) The plasticity of dendritic cell responses to pathogens and their components. Science 294: 870–875. [DOI] [PubMed] [Google Scholar]
- Inaba, K. , Inaba, M. , Romani, N. , Aya, H. , Deguchi, M. , Ikehara, S. , et al. (1992) Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony‐stimulating factor. J Exp Med 176: 1693–1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolly, C. , and Sattentau, Q.J. (2004) Retroviral spread by induction of virological synapses. Traffic 5: 643–650. [DOI] [PubMed] [Google Scholar]
- Kato, H. , Sato, S. , Yoneyama, M. , Yamamoto, M. , Uematsu, S. , Matsui, K. , et al. (2005) Cell type‐specific involvement of RIG‐I in antiviral response. Immunity 23: 19–28. [DOI] [PubMed] [Google Scholar]
- Kawai, T. , and Akira, S. (2006a) Innate immune recognition of viral infection. Nat Immunol 7: 131–137. [DOI] [PubMed] [Google Scholar]
- Kawai, T. , and Akira, S. (2006b) TLR signaling. Cell Death Differ 13: 816–825. [DOI] [PubMed] [Google Scholar]
- Van Kooyk, Y. , Appelmelk, B. , and Geijtenbeek, T.B. (2003) A fatal attraction: Mycobacterium tuberculosis and HIV‐1 target DC‐SIGN to escape immune surveillance. Trends Mol Med 9: 153–159. [DOI] [PubMed] [Google Scholar]
- Kotzor, N. , Lechmann, M. , Zinser, E. , and Steinkasserer, A. (2004) The soluble form of CD83 dramatically changes the cytoskeleton of dendritic cells. Immunobiology 209: 129–140. [DOI] [PubMed] [Google Scholar]
- Kramer, B. , Pelchen‐Matthews, A. , Deneka, M. , Garcia, E. , Piguet, V. , and Marsh, M. (2005) HIV interaction with endosomes in macrophages and dendritic cells. Blood Cells Mol Dis 35: 136–142. [DOI] [PubMed] [Google Scholar]
- Krug, A. , French, A.R. , Barchet, W. , Fischer, J.A. , Dzionek, A. , Pingel, J.T. , et al. (2004a) TLR9‐dependent recognition of MCMV by IPC and DC generates coordinated cytokine responses that activate antiviral NK cell function. Immunity 21: 107–119. [DOI] [PubMed] [Google Scholar]
- Krug, A. , Luker, G.D. , Barchet, W. , Leib, D.A. , Akira, S. , and Colonna, M. (2004b) Herpes simplex virus type 1 activates murine natural interferon‐producing cells through toll‐like receptor 9. Blood 103: 1433–1437. [DOI] [PubMed] [Google Scholar]
- Lechmann, M. , Krooshoop, D.J. , Dudziak, D. , Kremmer, E. , Kuhnt, C. , Figdor, C.G. , et al. (2001) The extracellular domain of CD83 inhibits dendritic cell‐mediated T cell stimulation and binds to a ligand on dendritic cells. J Exp Med 194: 1813–1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lelouard, H. , Gatti, E. , Cappello, F. , Gresser, O. , Camosseto, V. , and Pierre, P. (2002) Transient aggregation of ubiquitinated proteins during dendritic cell maturation. Nature 417: 177–182. [DOI] [PubMed] [Google Scholar]
- Lelouard, H. , Ferrand, V. , Marguet, D. , Bania, J. , Camosseto, V. , David, A. , et al. (2004) Dendritic cell aggresome‐like induced structures are dedicated areas for ubiquitination and storage of newly synthesized defective proteins. J Cell Biol 164: 667–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Y.J. (2005) IPC: professional type 1 interferon‐producing cells and plasmacytoid dendritic cell precursors. Annu Rev Immunol 23: 275–306. [DOI] [PubMed] [Google Scholar]
- Lopez, C.B. , Yount, J.S. , and Moran, T.M. (2006) Toll‐like receptor‐independent triggering of dendritic cell maturation by viruses. J Virol 80: 3128–3134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozach, P.Y. , Amara, A. , Bartosch, B. , Virelizier, J.L. , Arenzana‐Seisdedos, F. , Cosset, F.L. , and Altmeyer, R. (2004) C‐type lectins L‐SIGN and DC‐SIGN capture and transmit infectious hepatitis C virus pseudotype particles. J Biol Chem 279: 32035–32045. [DOI] [PubMed] [Google Scholar]
- Lutz, M.B. , and Schuler, G. (2002) Immature, semi‐mature and fully mature dendritic cells: which signals induce tolerance or immunity? Trends Immunol 23: 445–449. [DOI] [PubMed] [Google Scholar]
- McDonald, D. , Wu, L. , Bohks, S.M. , KewaIRamani, V.N. , Unutmaz, D. , and Hope, T.J. (2003) Recruitment of HIV and its receptors to dendritic cell–T cell junctions. Science 300: 1295–1297. [DOI] [PubMed] [Google Scholar]
- Mahanty, S. , Hutchinson, K. , Agarwal, S. , McRae, M. , Rollin, P.E. , and Pulendran, B. (2003) Cutting edge: impairment of dendritic cells and adaptive immunity by Ebola and Lassa viruses. J Immunol 170: 2797–2801. [DOI] [PubMed] [Google Scholar]
- Meylan, E. , and Tschopp, J. (2006) Toll‐like receptors and RNA helicases: two parallel ways to trigger antiviral responses. Mol Cell 22: 561–569. [DOI] [PubMed] [Google Scholar]
- Mills, K.H. (2004) Regulatory T cells: friend or foe in immunity to infection? Nat Rev Immunol 4: 841–855. [DOI] [PubMed] [Google Scholar]
- Moris, A. , Nobile, C. , Buseyne, F. , Porrot, F. , Abastado, J.P. , and Schwartz, O. (2004) DC‐SIGN promotes exogenous MHC‐I‐restricted HIV‐1 antigen presentation. Blood 103: 2648–2654. [DOI] [PubMed] [Google Scholar]
- Moutaftsi, M. , Mehl, A.M. , Borysiewicz, L.K. , and Tabi, Z. (2002) Human cytomegalovirus inhibits maturation and impairs function of monocyte‐derived dendritic cells. Blood 99: 2913–2921. [DOI] [PubMed] [Google Scholar]
- Moutaftsi, M. , Brennan, P. , Spector, S.A. , and Tabi, Z. (2004) Impaired lymphoid chemokine‐mediated migration due to a block on the chemokine receptor switch in human cytomegalovirus‐infected dendritic cells. J Virol 78: 3046–3054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller, D.B. , Raftery, M.J. , Kather, A. , Giese, T. , and Schonrich, G. (2004) Frontline: induction of apoptosis and modulation of c‐FLIPL and p53 in immature dendritic cells infected with herpes simplex virus. Eur J Immunol 34: 941–951. [DOI] [PubMed] [Google Scholar]
- Novak, N. , and Peng, W.M. (2005) Dancing with the enemy: the interplay of herpes simplex virus with dendritic cells. Clin Exp Immunol 142: 405–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacanowski, J. , Kahi, S. , Baillet, M. , Lebon, P. , Deveau, C. , Goujard, C. , et al. (2001) Reduced blood CD123+ (lymphoid) and CD11c+ (myeloid) dendritic cell numbers in primary HIV‐1 infection. Blood 98: 3016–3021. [DOI] [PubMed] [Google Scholar]
- Piguet, V. , and Sattentau, Q. (2004) Dangerous liaisons at the virological synapse. J Clin Invest 114: 605–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piqueras, B. , Connolly, J. , Freitas, H. , Palucka, A.K. , and Banchereau, J. (2006) Upon viral exposure, myeloid and plasmacytoid dendritic cells produce 3 waves of distinct chemokines to recruit immune effectors. Blood 107: 2613–2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollara, G. , Kwan, A. , Newton, P.J. , Handley, M.E. , Chain, B.M. , and Katz, D.R. (2005) Dendritic cells in viral pathogenesis: protective or defective? Int J Exp Pathol 86: 187–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prechtel, A.T. , Turza, N.M. , Kobelt, D.J. , Eisemann, J.I. , Coffin, R.S. , McGrath, Y. , et al. (2005) Infection of mature dendritic cells with herpes simplex virus type 1 dramatically reduces lymphoid chemokine‐mediated migration. J Gen Virol 86: 1645–1657. [DOI] [PubMed] [Google Scholar]
- Raftery, M.J. , Kraus, A.A. , Ulrich, R. , Kruger, D.H. , and Schonrich, G. (2002) Hantavirus infection of dendritic cells. J Virol 76: 10724–10733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raftery, M.J. , Wieland, D. , Gronewald, S. , Kraus, A.A. , Giese, T. , and Schonrich, G. (2004) Shaping phenotype, function, and survival of dendritic cells by cytomegalovirus‐encoded IL‐10. J Immunol 173: 3383–3391. [DOI] [PubMed] [Google Scholar]
- Rinaldo, C.R. , Jr, and Piazza, P. (2004) Virus infection of dendritic cells: portal for host invasion and host defense. Trends Microbiol 12: 337–345. [DOI] [PubMed] [Google Scholar]
- Robertson, S.J. , and Hasenkrug, K.J. (2006) The role of virus‐induced regulatory T cells in immunopathology. Springer Semin Immunopathol 28: 51–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romani, N. , Reider, D. , Heuer, M. , Ebner, S. , Kampgen, E. , Eibl, B. , et al. (1996) Generation of mature dendritic cells from human blood. An improved method with special regard to clinical applicability. J Immunol Methods 196: 137–151. [DOI] [PubMed] [Google Scholar]
- Sallusto, F. , and Lanzavecchia, A. (1994) Efficient presentation of soluble antigen by cultured human dendritic cells is maintained by granulocyte/macrophage colony‐stimulating factor plus interleukin 4 and downregulated by tumor necrosis factor alpha. J Exp Med 179: 1109–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlender, J. , Hornung, V. , Finke, S. , Gunthner‐Biller, M. , Marozin, S. , Brzozka, K. , et al. (2005) Inhibition of toll‐like receptor 7‐ and 9‐mediated alpha/beta interferon production in human plasmacytoid dendritic cells by respiratory syncytial virus and measles virus. J Virol 79: 5507–5515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider‐Schaulies, S. , and Dittmer, U. (2006) Silencing T cells or T‐cell silencing: concepts in virus‐induced immunosuppression. J Gen Virol 87: 1423–1438. [DOI] [PubMed] [Google Scholar]
- Schneider‐Schaulies, S. , Klagge, I.M. , and Ter Meulen, V. (2003) Dendritic cells and measles virus infection. Curr Top Microbiol Immunol 276: 77–101. [DOI] [PubMed] [Google Scholar]
- Senechal, B. , Boruchov, A.M. , Reagan, J.L. , Hart, D.N. , and Young, J.W. (2004) Infection of mature monocyte‐derived dendritic cells with human cytomegalovirus inhibits stimulation of T‐cell proliferation via the release of soluble CD83. Blood 103: 4207–4215. [DOI] [PubMed] [Google Scholar]
- Servet‐Delprat, C. , Vidalain, P.O. , Bausinger, H. , Manie, S. , Le Deist, F. , Azocar, O. , et al. (2000a) Measles virus induces abnormal differentiation of CD40 ligand‐activated human dendritic cells. J Immunol 164: 1753–1760. [DOI] [PubMed] [Google Scholar]
- Servet‐Delprat, C. , Vidalain, P.O. , Azocar, O. , Le Deist, F. , Fischer, A. , and Rabourdin‐Combe, C. (2000b) Consequences of Fas‐mediated human dendritic cell apoptosis induced by measles virus. J Virol 74: 4387–4393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Servet‐Delprat, C. , Vidalain, P.O. , Valentin, H. , and Rabourdin‐Combe, C. (2003) Measles virus and dendritic cell functions: how specific response cohabits with immunosuppression. Curr Top Microbiol Immunol 276: 103–123. [DOI] [PubMed] [Google Scholar]
- Shortman, K. , and Liu, Y.J. (2002) Mouse and human dendritic cell subtypes. Nat Rev Immunol 2: 151–161. [DOI] [PubMed] [Google Scholar]
- Shutt, D.C. , Daniels, K.J. , Carolan, E.J. , Hill, A.C. , and Soll, D.R. (2000) Changes in the motility, morphology, and F‐actin architecture of human dendritic cells in an in vitro model of dendritic cell development. Cell Motil Cytoskeleton 46: 200–221. [DOI] [PubMed] [Google Scholar]
- Steinman, R.M. (2003) The control of immunity and tolerance by dendritic cell. Pathol Biol (Paris) 51: 59–60. [DOI] [PubMed] [Google Scholar]
- Steinman, R.M. , Pack, M. , and Inaba, K. (1997) Dendritic cell development and maturation. Adv Exp Med Biol 417: 1–6. [DOI] [PubMed] [Google Scholar]
- Tailor, P. , Tamura, T. , and Ozato, K. (2006) IRF family proteins and type I interferon induction in dendritic cells. Cell Res 16: 134–140. [DOI] [PubMed] [Google Scholar]
- Takaoka, A. , and Yanai, H. (2006) Interferon signalling network in innate defence. Cell Microbiol 8: 907–922. [DOI] [PubMed] [Google Scholar]
- Tassaneetrithep, B. , Burgess, T.H. , Granelli‐Piperno, A. , Trumpfheller, C. , Finke, J. , Sun, W. , et al. (2003) DC‐SIGN (CD209) mediates dengue virus infection of human dendritic cells. J Exp Med 197: 823–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teleshova, N. , Kenney, J. , Williams, V. , Van Nest, G. , Marshall, J. , Lifson, J.D. , et al. (2006) CpG‐C ISS‐ODN activation of blood‐derived B cells from healthy and chronic immunodeficiency virus‐infected macaques. J Leukoc Biol 79: 257–267. [DOI] [PubMed] [Google Scholar]
- Thurnher, M. (2007) Lipids in dendritic cell biology: messengers, effectors, and antigens. J Leukoc Biol 81: 154–160. [DOI] [PubMed] [Google Scholar]
- Tough, D.F. (2004) Type I interferon as a link between innate and adaptive immunity through dendritic cell stimulation. Leuk Lymphoma 45: 257–264. [DOI] [PubMed] [Google Scholar]
- Turville, S.G. , Santos, J.J. , Frank, I. , Cameron, P.U. , Wilkinson, J. , Miranda‐Saksena, M. , et al. (2004) Immunodeficiency virus uptake, turnover, and 2‐phase transfer in human dendritic cells. Blood 103: 2170–2179. [DOI] [PubMed] [Google Scholar]
- Vidalain, P.O. , Azocar, O. , Yagita, H. , Rabourdin‐Combe, C. , and Servet‐Delprat, C. (2001) Cytotoxic activity of human dendritic cells is differentially regulated by double‐stranded RNA and CD40 ligand. J Immunol 167: 3765–3772. [DOI] [PubMed] [Google Scholar]
- Wilson, N.S. , and Villadangos, J.A. (2005) Regulation of antigen presentation and cross‐presentation in the dendritic cell network: facts, hypothesis, and immunological implications. Adv Immunol 86: 241–305. [DOI] [PubMed] [Google Scholar]
- De Witte, L. , Abt, M. , Schneider‐Schaulies, S. , Van Kooyk, Y. , and Geijtenbeek, T.B. (2006) Measles virus targets DC‐SIGN to enhance dendritic cell infection. J Virol 80: 3477–3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, K. , Puel, A. , Zhang, S. , Eidenschenk, C. , Ku, C.L. , Casrouge, A. , et al. (2005) Human TLR‐7‐, ‐8‐, and ‐9‐mediated induction of IFN‐alpha/beta and ‐lambda is IRAK‐4 dependent and redundant for protective immunity to viruses. Immunity 23: 465–478. [DOI] [PMC free article] [PubMed] [Google Scholar]