

Abstract
Influenza A virus (IAV) has evolved multiple mechanisms to compromise type I interferon (IFN) responses. The antiviral function of IFN is mainly exerted by activating the JAK/STAT signalling and subsequently inducing IFN‐stimulated gene (ISG) production. However, the mechanism by which IAV combat the type I IFN signalling pathway is not fully elucidated. In this study, we explored the roles of human microRNAs modulated by IAV infection in type I IFN responses. We demonstrated that microRNA‐30 (miR‐30) family members were downregulated by IAV infection. Our data showed that the forced expression of miR‐30 family members inhibited IAV proliferation, while miR‐30 family member inhibitors promoted IAV proliferation. Mechanistically, we found that miR‐30 family members targeted and reduced SOCS1 and SOCS3 expression, and thus relieved their inhibiting effects on IFN/JAK/STAT signalling pathway. In addition, miR‐30 family members inhibited the expression of NEDD4, a negative regulator of IFITM3, which is important for host defence against influenza viruses. Our findings suggest that IAV utilises a novel strategy to restrain host type I IFN‐mediated antiviral immune responses by decreasing the expression of miR‐30 family members, and add a new way to understand the mechanism of immune escape caused by influenza viruses.
Keywords: influenza virus, microRNA‐30, NEDD4, SOCS1, SOCS3
1. INTRODUCTION
The type I interferon (IFN) system constitutes the first line of host defence against viral invasions (Borden et al., 2007). The antiviral effects of type I IFN, which includes IFN‐α and IFN‐β, are mainly mediated by a great diversity of IFN‐stimulating genes (ISGs). The binding of IFN‐α and IFN‐β to their receptors represents the first step in this signalling transduction, followed by activation of the JAK/STAT cascade, a well‐established major pathway that transmits IFN signals (Murray, 2007). Ligand binding then activates the JAK family, which subsequently phosphorylates STAT1 and STAT2 at Y701 and Y690 by JAK1 and Tyk2, respectively (Levy & Darnell, 2002; Uze, Schreiber, Piehler, & Pellegrini, 2007). These phosphorylated STATs dimerise and form a heterotrimer together with IFF9, called IFN‐stimulated gene (ISG) factor 3, which then initiates the gene transcription of ISGs, such as OAS2, MX2, CXCL10, IFIT3, and IFITM3 (Tang et al., 2007). The JAK/STAT cascade is tightly regulated by several key endogenous cellular mechanisms. Among them, several molecules, such as SOCS1 and SOCS3, can restrict type I IFN signalling by target JAK/STAT cascade response (Alexander, 2002; Giordanetto & Kroemer, 2003; Larsen & Ropke, 2002). ISGs can also be negatively regulated. For example, IFITM3, a cell‐intrinsic factor, which is reported to limit SARS coronavirus, HIV, Ebola virus, and influenza virus, can be degraded through ubiquitination by NEDD4, an E3 ubiquitin ligase (Chesarino, McMichael, & Yount, 2015).
Similar to many other viruses, influenza viruses have acquired multiple strategies to cripple cellular antiviral responses. For example, the NS1 protein of influenza virus has been demonstrated as the main antagonistic factor of host IFN‐β production via inhibition of the RIG‐I‐MAVS signalling pathway (Krug, 2015; Pachler & Vlasak, 2011; Rajsbaum et al., 2012), and the PB1‐F2 protein was identified to block IFN‐β induction by targeting MAVS (Varga, Grant, Manicassamy, & Palese, 2012; Varga et al., 2011). Influenza virus can also modulate the antiviral response at the JAK/STAT pathway level. The HA protein was shown to antagonise the IFN response by inducing the degradation of type I IFN receptor 1 (IFNAR1) (Xia et al., 2015). The NS1 protein was demonstrated to negatively regulate JAK/STAT signalling by elevating SOCS1 and SOCS3 expression (Jia et al., 2010). In addition, influenza virus can restrict JAK/STAT pathway activation via the NF‐κB‐dependent induction of SOCS3 expression (Pauli et al., 2008).
MicroRNAs (miRNAs) are a class of noncoding small RNA molecules, which can regulate gene expressions by binding to the 3′‐untranslated region (3′UTR) of targeted mRNA to induce degradation or suppress translation (Ambros, 2004). Growing evidence has shown that miRNAs are involved in regulating development, apoptosis, host immunity, and viral infection (Bartel, 2004; Cullen, 2013; Wienholds & Plasterk, 2005). MiRNAs can regulate viral infection by modulating the antiviral immune response at multiple levels. MiR‐146 was shown to inhibit type I IFN production by regulating expressions of IRAK1, IRAK2, and TRAF6 (Hou et al., 2009), and miR‐3570 was found to modulate MAVS expression following rhabdovirus infection (Xu, Chu, Cui, & Bi, 2018). Many studies have demonstrated the significant effect of miRNAs on modulating influenza virus infection. For examples, miR‐33a was suggest to disrupt influenza A virus (IAV) replication by targeting APCN1 (Hu et al., 2016); miR‐34a contributed to influenza virus‐mediated apoptosis by binding to BAX (Fan & Wang, 2016); and miR‐let‐7c targeted and inhibited M1 expression of the H1N1 influenza virus (Ma et al., 2012). Although several studies linked miRNAs to type I IFN production mediated by RIG‐I in influenza virus infection (miR‐144, miR‐485, and miR‐136) (Ingle et al., 2015; Rosenberger et al., 2017; Zhao et al., 2015), the underlying regulation of the type I IFN‐mediated antiviral response by miRNAs during influenza virus infection remains unclear.
In this study, we found that microRNA‐30 (miR‐30) family members were downregulated upon IAV infection. Further analysis demonstrated that miR‐30 overexpression suppressed SOCS1, SOCS3, and NEDD4 and thus positively regulated the type I IFN signalling pathway and IFITM3 expression, thereby inhibiting influenza virus infection. For the first time, we identified miR‐30 as a positive regulator of the antiviral response, and our data revealed that influenza virus could disrupt the host antiviral immune response by downregulating miR‐30 expression, which may be a new mechanism of influenza virus inhibition of the host antiviral immune response.
2. RESULTS
2.1. Influenza virus infection downregulates miR‐30 family member expression in A549 cells
Many studies have investigated microRNAs expression upon IAV infection. Previous studies suggested that miR‐30 family members (miR‐30) expression could be modulated by IAV challenge (Y. Li et al., 2010; Y. Li et al., 2011; Tambyah et al., 2013). We hypothesised that miR‐30 may exert some important effects on influenza virus infection. Human miR‐30 family contains five members (miR‐30a–e), which are broadly conserved. We first assessed miR‐30 expression in A549 cells infected with H5N1 influenza virus in a time‐course assay. Sequence analysis indicated that only one or two nucleotides differed among miR‐30a, miR‐30d, and miR‐30e, which have large differences in their 3′ terminals compared with miR‐30b and miR‐30c, while miR‐30b and miR‐30c have significant differences in their 3′ terminal sequences (Figure S1). Therefore, each member of miR‐30 family could be analysed using three specific primers: miR‐30a (which cross‐reacts with miR‐30d and miR‐30e), miR‐30b, and miR‐30c. Our results showed that miR‐30a/d/e, miR‐30b, and miR‐30c were remarkably decreased in A549 cells in a time‐dependent manner following H5N1 infection (Figure 1a–c). However, their expression was not substantially changed at 36 hr post‐infection (hpi) when A549 cells were treated with heat‐inactivated viruses (Figure 1d). MiR‐30 expression was also decreased in H1N1 and H9N2 subtype influenza virus‐infected A549 cells compared with uninfected cells (Figure S2). Poly(I:C), a double‐stranded RNA analogue that can activate type I IFN response, was used as a stimulus to investigate miR‐30 expression. Although poly(I:C) significantly increased IFN‐β and MX1 expression (Figure 1e), it did not alter miR‐30 expression (Figure 1f). Additionally, VSV, another RNA virus, did not downregulate miR‐30 expression (Figure 1g,h). These results strongly indicate that miR‐30 expression can be downregulated in A549 cells upon IAV infection, and this change is dependent on influenza virus replication.
Figure 1.
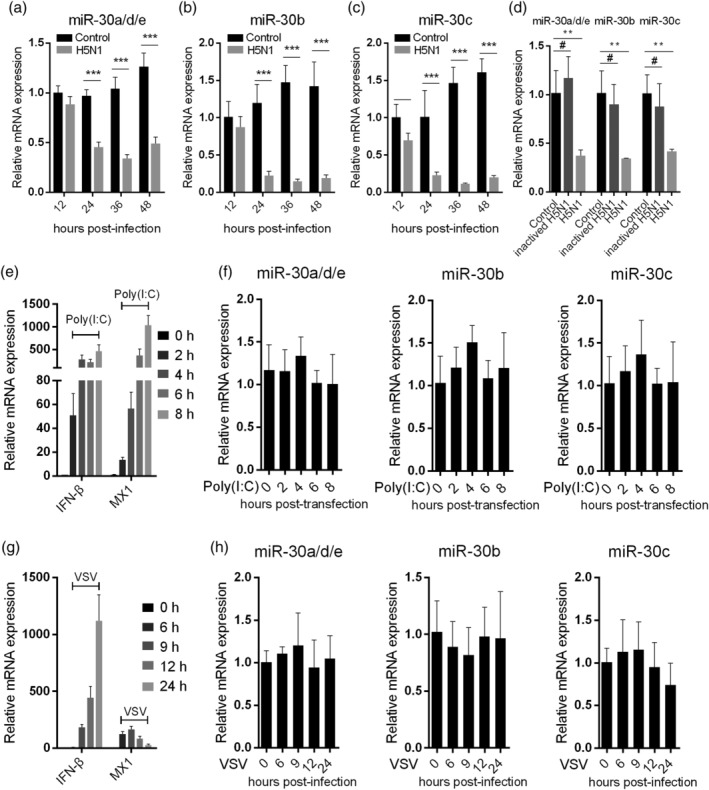
Influenza virus infection downregulates miR‐30 expression. A549 cells were infected with 0.1 MOI of H5N1 influenza virus, and the expression levels of miR‐30a/d/e (a), miR‐30b (b), and miR‐30c (c) were detected at 12, 24, 36, and 48 hpi by qRT‐PCR. (d) A549 cells were infected with heat‐inactivated H5N1, and after 36 hr, miR‐30 expression was detected by qRT‐PCR. A549 cells were transfected with 200 ng poly(I:C), IFN‐β and MX1 (e), and miR‐30 (f) were tested at the indicated time points post‐transfection by qRT‐PCR. A549 cells were infected by VSV with an MOI of 0.1, IFN‐β and MX1 (g), and miR‐30 (h) were detected at the indicated time points post‐infection by qRT‐PCR. The values are shown as the mean and SD and are representative of three independent experiments. The data in (a–c) were analysed using two‐way ANOVA; data in (d) were analysed using Student's t test. The values are shown as the mean and SD and are representative of three independent experiments. #: non‐significant, ***p < .001; **p < .01; *p < .05
2.2. MiR‐30 suppresses influenza virus proliferation
To investigate the biological effect of miR‐30 on viral infection in host cells, we inspected the role of miR‐30a/b/c in H5N1 influenza virus replication in A549 cells. As shown in Figure 2a,b, overexpression of miR‐30a/b/c decreased the influenza virus NP mRNA and protein levels compared with those of the miRNA NC group. Viral titre from the infected A549 cells was also determined. Consistent with the results of the NP mRNA analysis, miR‐30a/b/c mimic transfection significantly suppressed influenza virus proliferation (Figure 2c). MiR‐30a, b, or c mimics transfection also significantly inhibited influenza virus proliferation in primary human alveolar epithelia cells (HAECs) (Figure S3). To further show the effect of miR‐30 on influenza virus infection, we assessed viral infection in the presence of the miR‐30a/b/c inhibitors at different times. The data indicated that miR‐30a/b/c inhibitors significantly promoted viral NP expression (Figure 2d,e) and viral titres in the supernatant (Figure 2f). Given the high sequence similarity of miR‐30a, miR‐30d, and miR‐30e, we did not determine the effect of miR‐30d and miR‐30e on influenza virus infection, but we hypothesised that they have the same role as miR‐30a/b/c in influenza virus infection. These data suggested that miR‐30 could suppress influenza virus replication, while miR‐30 inhibition facilitated influenza virus infection.
Figure 2.
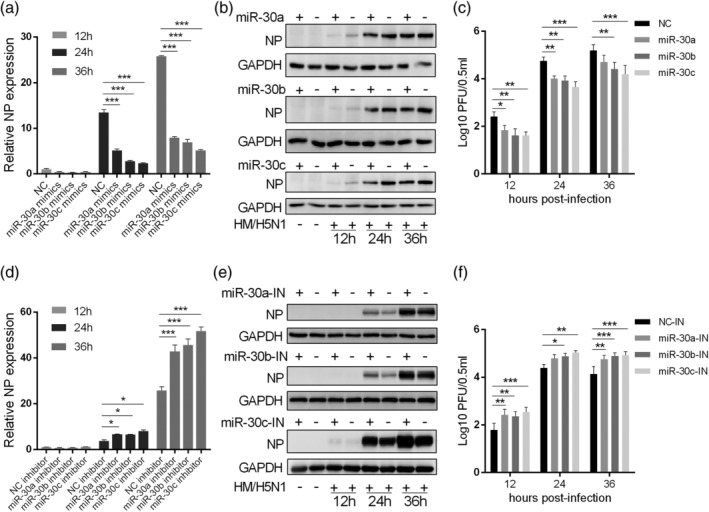
MiR‐30 suppresses influenza virus replication. A549 cells were transfected with 80 nm miR‐30a/b/c mimics (a–c) or 100 nm inhibitors (d–f); 24 hr later, cells were infected with 0.2 MOI of H5N1 influenza virus (HM/H5N1). At the indicated time post‐infection, the RNA, total protein, and the supernatant were collected for NP mRNA (a and d), NP protein (b and e), and viral titre (c and f) detection by qRT‐PCR with GAPDH as housekeeping gene, western blot, and plaque assay analyses, respectively. NC, negative control; IN, inhibitor. The values are shown as the mean and SD and are representative of three independent experiments. Data were analysed using two‐way ANOVA; ***p < .001; **p < .01; *p < .05
2.3. MiR‐30 does not target putative influenza virus genomic RNA
Host miRNAs can affect RNA virus replication either by modulating host gene expression or by directly targeting viral RNA for degradation. To determine whether miR‐30 inhibits influenza virus replication by directly targeting viral RNA, we performed computational analysis of the potential binding sites in viral RNA for miR‐30. Analysis using RNA22 V2 miRNA detection software identified a potential binding site of miR‐30b and miR‐30c at position 952–975 of the M gene sequence from HM/H5N1 in this study (Figure 3a). The prediction was then experimentally tested by a luciferase reporter assay.
Figure 3.
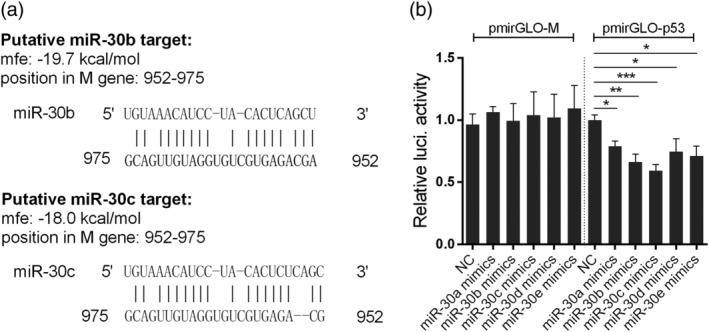
Influenza virus genomic RNA is not the target of miR‐30. (a) Predicted interactions between miR‐30b/c and the influenza virus M gene using RNA22 V2. (b) Effects of miR‐30a/b/c/d/e mimics on the expression of the firefly luciferase gene from the pmirGLO reporter constructs containing the putative miR‐30b/c binding sites from the M gene (pmirGLO‐M) and 3′UTR of human p53 (pmirGLO‐p53). First, 293T cells were co‐transfected with reporter constructs together with miR‐30a/b/c/d/e mimics or NC. After 24 hr, cells were lysed, and luciferase activities were measured. The luciferase activity was normalised to the Renilla luciferase activity, and the data are expressed relative to that of the NC. The values are shown as the mean and SD and are representative of three independent experiments. Data were analysed using Student's t test. ***p < .001; **p < .01; *p < .05
The nucleotide sequence from the M gene of HM/H5N1 was inserted into the pmirGLO vector. Then, 293T cells were transfected with reporter constructs, together with miR‐30a/b/c/d/e or NC mimics, followed by luciferase activity assays at 24 hr post‐transfection. The result showed that miR‐30 mimics transfection did not affect the luciferase expression of reporter constructs that contained the putative target site from the M gene (Figure 3b). As a positive control, miR‐30 mimics transfection significantly reduced the luciferase expression of reporter constructs containing the 3′UTR of p53 compared with NC mimics transfection, which has been reported to be targeted by miR‐30 family (J. Li et al., 2010). Therefore, the inhibitory effect of miR‐30 on IAV proliferation is not due to the directly targeting viral genome RNA, and likely involves the miR‐30‐mediated regulation of host genes.
2.4. SOCS1 and SOCS3 are potential targets of miR‐30
To determine targeted host genes by miR‐30, we conducted a genome‐wide computational prediction. To minimise the number of false‐positive predictions, we performed the analysis using four different prediction algorithms, and only interactions predicted by all the algorithms were chose. The algorithms applied are based on evolutionary conservation of sites (TargetScan and MirTarget2) and seed complementarity (microRNA.org and DIANA‐MicroT). This led us to focus on SOCS1 and SOCS3, which were reported to be involved in the antiviral response (Alexander, 2002; Carow & Rottenberg, 2014; Pothlichet, Chignard, & Si‐Tahar, 2008).
Our analysis identified one putative binding site for miR‐30 in the 3′UTR of human SOCS1 and SOCS3. To examine if miR‐30 can directly interact with these sites and inhibit the expression of the targeted genes, we performed luciferase reporter assays. We first constructed reporter plasmids by cloning the 3′UTRs of the human SOCS1 and SOCS3 genes into the pmirGLO luciferase reporter vector, and mutant SOCS1 and SOCS3 3′UTR luciferase reporter vectors containing the target sequence mutations were generated as controls (Figure 4a). When the reporter vectors were co‐transfected into 293T cells with miR‐30a/b/c mimics, we observed that miR‐30a/b/c mimics substantially decreased the luciferase activities of the SOCS1 3′UTR (Figure 4b) and SOCS3 3′UTR (Figure 4c). In contrast, luciferase activities were not regulated by miR‐30a/b/c mimics when a mutant‐type 3′UTR human SOCS1 or SOCS3 vector was transfected into 293T cells (Figure 4b,c). MiR‐30a/b/c inhibited the luciferase activities of 293T cells in a dose‐dependent manner at 24 hr post‐transfection (Figure 4d,e). However, when the miR‐30a/b/c expression was repressed by miR‐30a/b/c inhibitors (Figure S4), the inhibitory effect was obviously attenuated (Figure 4f,g).
Figure 4.
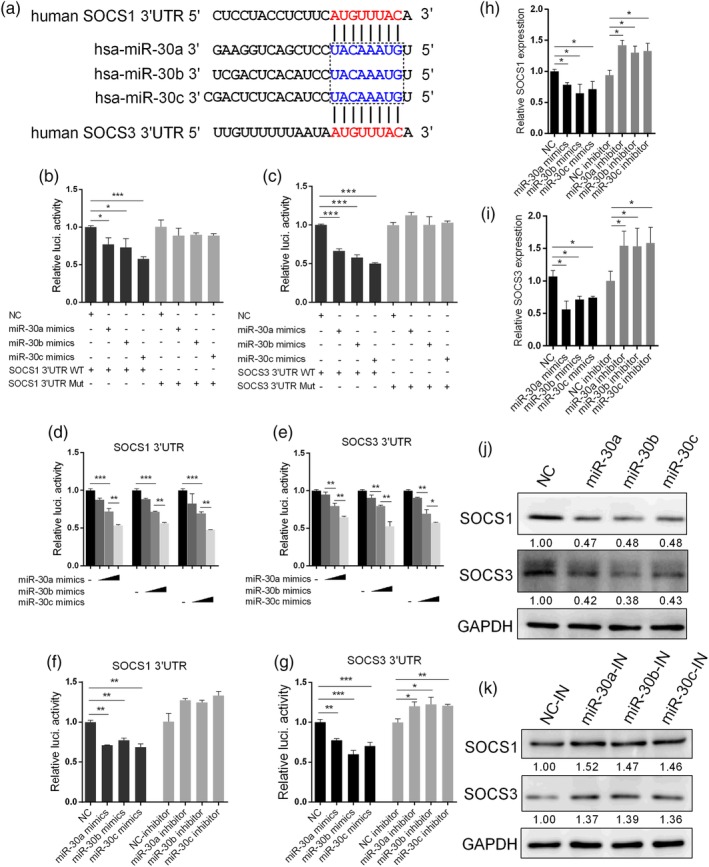
MiR‐30 targets the 3′UTRs of SOCS1 and SOCS3. (a) Predicted target sites of miR‐30a/b/c in the 3′UTRs of SOCS1 and SOCS3. Nucleotides in blue are seed regions of miR‐30a/b/c; nucleotides in red are complementary to the miR‐30a/b/c seed region. (b and c) Effects of miR‐30a/b/c mimics on expression of the firefly luciferase gene from reporter constructs containing the SOCS1 and SOCS3 3′UTRs or SOCS1 and SOCS3 mutated 3′UTRs. First, 293T cells were transfected with NC or miR‐30a/b/c, together with the SOCS1 or SOCS3 3′UTR (3′UTR WT) or 3′UTR mutant (3′UTR Mut), and 24 hr after transfection, cells were harvested to assess luciferase activity. (d and e) MiR‐30a/b/c inhibit the SOCS1 and SOCS3 3′UTRs in a dose‐dependent manner. First, 293T cells were transfected with 30, 60, or 90 nM miR‐30a/b/c mimics, together with SOCS1 or SOCS3 3′UTR, and 24 hr after transfection, cells were lysed for luciferase activity analysis. (f and g) The 293T cells were transfected with 100 nM miR‐30a/b/c inhibitor or NC inhibitor (NC‐inhibitor), together with SOCS1 or SOCS3 3′UTR. Twenty‐four hours later, the cells were lysed for luciferase activity analysis. A549 cells were transfected with 80 nm miR‐30a/b/c mimics or 100 nM miR‐30a/b/c inhibitors, and 36 hr after transfection, the mRNA and protein levels of SOCS1 and SOCS3 were determined by qRT‐PCR (h and i) and western blot analyses (j and k), respectively. The values are shown as the mean and SD and are representative of at least three independent experiments. Data were analysed using Student's t test. ***p < .001; **p < .01; *p < .05
To further determine whether miR‐30a/b/c can target SOCS1 and SOCS3, we examined the expression of endogenous SOCS1 and SOCS3 in A549 cells transfected with mimics or inhibitors of miR‐30a/b/c. As shown in Figure 4, miR‐30a/b/c mimic transfection significantly inhibited SOCS1 (Figure 4h) and SOCS3 mRNA levels (Figure 4i), whereas SOCS1 and SOCS3 expression could be restored by inhibitors. As expected, the SOCS1 and SOCS3 protein levels were also significantly decreased following miR‐30a/b/c overexpression (Figure 4j). MiR‐30a/b/c overexpression also significantly reduced SOCS1 and SOCS3 expression, including mRNA and protein level in HAECs (Figure S5A,B). In contrast, miR‐30a/b/c inhibitors increased the SOCS1 and SOCS3 protein levels (Figure 4k). These data indicated that miR‐30a/b/c functioned similarly in regulating SOCS1 and SOCS3 expression, which may be due to their identical seed region and high sequence similarity (Figure S1). Above all, these results demonstrated that miR‐30 negatively regulates SOCS1 and SOCS3 at the transcriptional level by targeting SOCS1 and SOCS3 3′UTRs.
2.5. MiR‐30 targets the 3′UTR of the NEDD4 transcript
Our in silico analysis also identified three putative seed‐matched sequences (positions 942–949, 960–967, and 1116–1122) for miR‐30 (with miR‐30c as a representative) in the 3′UTR of NEDD4 mRNA (Figure 5a). NEDD4 is an E3 ubiquitin ligase that was reported to promote IAV infection by downregulating the antiviral protein IFITM3 expression (Chesarino et al., 2015). Thus, we investigated whether miR‐30 can target NEDD4. The luciferase reporter assay demonstrated that overexpression of miR‐30a/b/c significantly decreased the luciferase activity of the NEDD4‐WT 3′UTR (Figure 5b), and dose‐dependent inhibitory effects were observed (Figure 5c). In addition, the luciferase activity was slightly inhibited by miR‐30a/b/c inhibitor transfection (Figure 5d). When miR‐30a/b/c were transfected together with the NEDD4 3′UTR containing a mutation in position 960–967 (NEDD4‐Mut2), the inhibitory effect of miR‐30a/b/c was not observed; miR‐30a/b/c mimic transfection also substantially inhibited the luciferase activities of NEDD4‐Mut1 and NEDD4‐Mut3, which contained mutations at 942–949 and 1116–1122 in the predicted binding sites, respectively (Figure 5b). Therefore, these results showed that miR‐30 directly inhibited the NEDD4 3′UTR at position 960–967 of the predicted target site.
Figure 5.
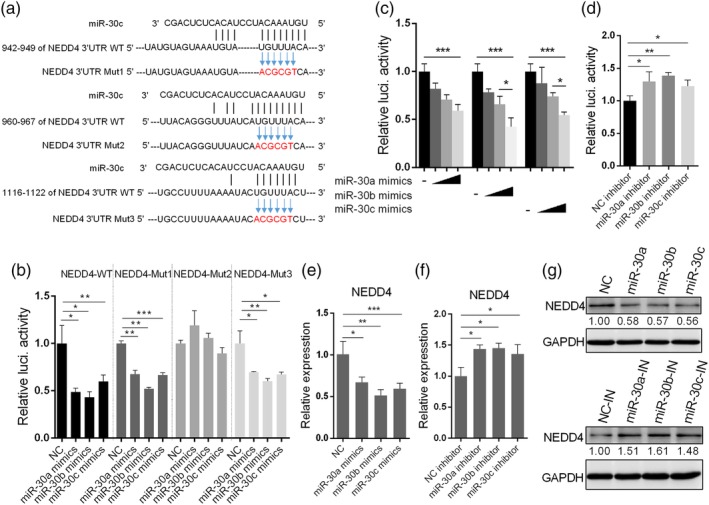
MiR‐30 targets NEDD4. (a) Analysis of NEDD4 3′UTR potential binding sites for miR‐30c; three putative binding sites were predicted (942–949, 960–967, and 1116–1122 in the 3′UTR of NEDD4 mRNA), and nucleotides in red show mutations of the 3′UTR of NEDD4, which is complementary to the seed region of miR‐30c. (b) Effects of miR‐30a/b/c on firefly luciferase activity from reporter constructs containing NEDD3 3′UTR WT or sequences with mutations in three putative positions (NEDD4‐Mut1, Mut2, and Mut3). First, 293T cells were co‐transfected with miR‐30a/b/c mimics and NEDD4 3′UTR or 3′UTR mutants, and 24 hr later, the cells were lysed for luciferase activity analysis. (c) Dose‐dependent inhibitory effects of miR‐30a/b/c on the NEDD4 3′UTR. First, 293T cells were transfected with 30, 60, or 90 nM miR‐30a/b/c, together with the NEDD4 3′UTR. Twenty‐four hours later, luciferase activity was analysed. (d) MiR‐30a/b/c inhibitors increased the NEDD4 3′UTR activity. First, 293T cells were transfected with 100 nM miR‐30a/b/c inhibitors and NEDD4 3′UTR. Twenty‐four hours later, luciferase activity was analysed. A549 cells were transfected with 80 nM miR‐30a/b/c mimics or 100 nM miR‐30a/b/c inhibitors, and 36 hr later, the mRNA and protein levels of NEDD4 were determined by qRT‐PCR (e and f) and western blot analyses (g). The values are shown as the mean and SD and are representative of at least three independent experiments. Data were analysed using Student's t test. ***p < .001; **p < .01; *p < .05
To further demonstrate that miR‐30 targets NEDD4 mRNA, we assayed the NEDD4 mRNA levels in A549 cells following transfection of miR‐30a/b/c mimics or inhibitors. We found that miR‐30a/b/c mimic transfection significantly reduced NEDD4 mRNA (Figure 5e), whereas the inhibitors significantly increased NEDD4 mRNA (Figure 5f). Similarly, the protein level of NEDD4 was reduced by miR‐30a/b/c mimics and was increased by miR‐30a/b/c inhibitors (Figure 5g). Besides, miR‐30a/b/c overexpression also significantly inhibited NEDD4 expression in HAECs (Figure S5C). These results demonstrated that miR‐30a/b/c directly suppressed NEDD4 gene expression at the transcriptional level via targeting the NEDD4 3′UTRs.
2.6. MiR‐30 promotes activation of the JAK/STAT signalling pathway
SOCS1 and SOCS3 were reported to block the JAK/STAT signal pathway (Alexander & Hilton, 2004; Croker, Kiu, & Nicholson, 2008); thus, we first examined the activation of JAK/STAT in response to type I IFN following miR‐30 overexpression. Among miR‐30a/b/c, miR‐30c was selected as a representative to further investigate its function. Exogenous addition of rIFN‐β activated STAT1 in A549 cells, as shown by the enhanced p‐STAT1 levels (Figure 6a). However, p‐STAT1 was elevated by miR‐30c mimic transfection compared with NC transfection (Figure 6a). Similarly, we observed increased p‐STAT1 levels after stimulation by poly(I:C) in miR‐30c‐overexpressing A549 cells compared with NC cells (Figure 6b). We inspected the effect of the p‐STAT1 level upon IAV infection in miR‐30c‐overexpressing A549 cells, and the data showed that miR‐30c also promoted STAT1 activation (Figure 6c).
Figure 6.
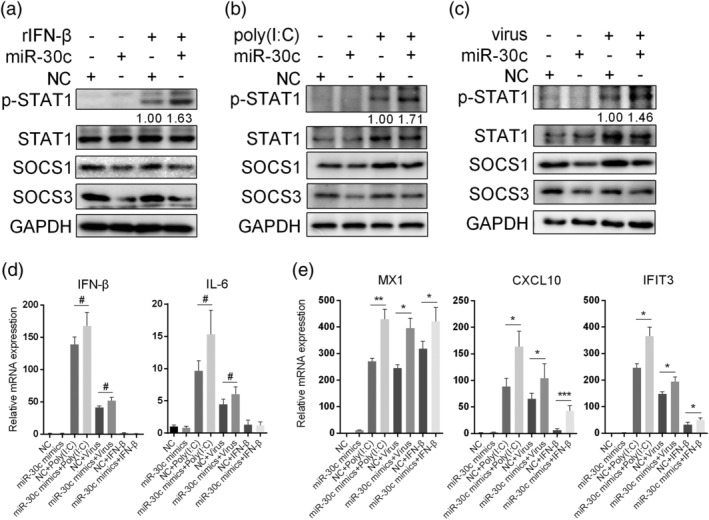
MiR‐30c promotes JAK–STAT signalling pathway activation. A549 cells were transfected with 80 nM NC or miR‐30c mimics, and 36 hr later, cells were treated with 500 U/ml human rIFN‐β for 30 min (a), transfected with 200 ng poly(I:C) for 4 hr (b), or infected with 5 MOI of H5N1 influenza virus for 4 hr (c). Cells were lysed, and cell extracts were analysed by western blot analysis. A549 cells were transfected with miR‐30c mimics; 36 hr later, cells were treated with 100 U/ml rIFN‐β for 3 hr, transfected with 100 ng/ml poly(I:C) for 5 hr, or infected with 3 MOI of H5N1 influenza virus for 8 hr. RNA was extracted for mRNA analyses of IFN‐β, IL‐6 (d), MX1, IFIT3, and CXCL10 (e) by qRT‐PCR. Data are shown as the mean and SD and as one representative of three independent experiments. Data were analysed using Student's t test. #: non‐significant; ***p < .001; **p < .01; *p < .05
The transcription of ISGs is initiated upon JAK/STAT pathway activation. Therefore, we investigated whether miR‐30c affected the expression of ISGs. We first examined IFN‐β and IL‐6 mRNA production in A549 cells activated by poly(I:C) and influenza virus, and then we observed no remarkable difference in IFN‐β and IL‐6 expression when miR‐30c was overexpressed (Figure 6d). However, the induction of ISGs, such as MX1, IFIT3, and CXCL10, was increased in miR‐30c‐overexpressing A549 cells treated by poly(I:C) transfection, influenza virus infection, and rIFN‐β (Figure 6e). These results indicate that miR‐30c increases cellular sensitivity to type I IFN by triggering the type I IFN‐mediated signalling pathway but does not significantly affect IFN‐β production.
2.7. MiR‐30 is positively related to the antiviral protein IFITM3
As NEDD4 was reported to decrease IFITM3 levels by ubiquitinating IFITM3, we hypothesised that miR‐30 could modulate baseline IFITM3 levels. We first determined the effect of NEDD4 on turnover of IFITM3. As shown by the data, NEDD4 overexpression significantly decreased the IFITM3 levels (Figure 7a). When miR‐30c was overexpressed, increased IFITM3 and decreased NEDD4 protein levels were observed (Figure 7b); when A549 cells were transfected with miR‐30 inhibitors, IFITM3 levels were decreased, accompanied by an increase in NEDD4 (Figure 7c). These results suggested that miR‐30c was positively correlated with the antiviral protein IFITM3.
Figure 7.
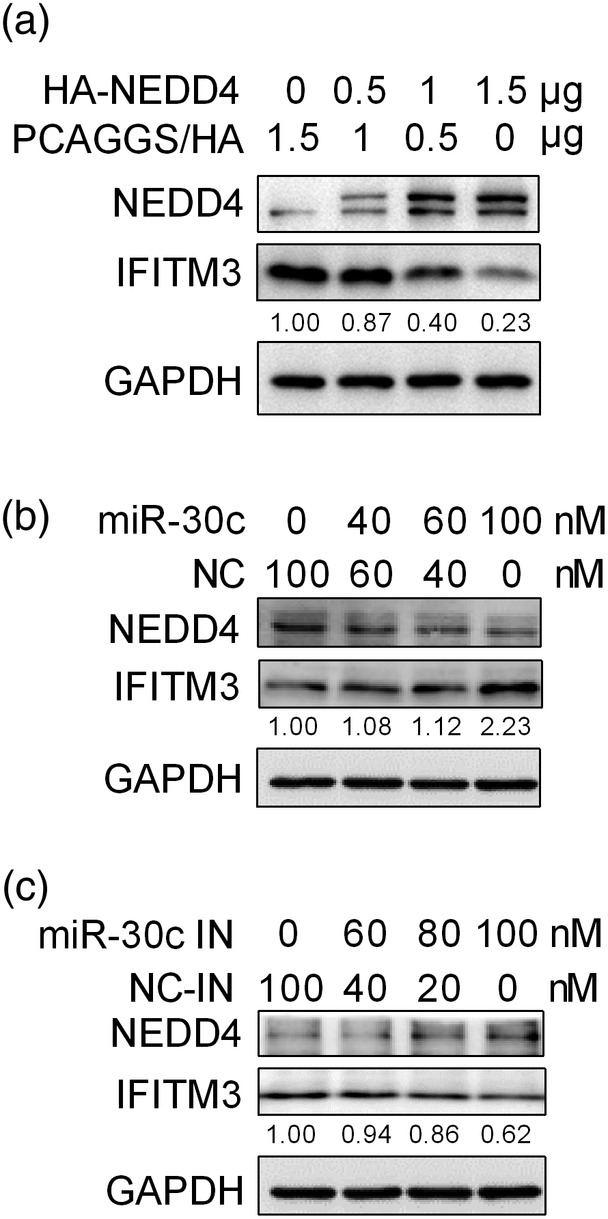
MiR‐30c is positively correlated with IFITM3 protein levels. (a) A549 cells were transfected with control vector (pCAGGS/HA) or 0.5, 1, and 1.5 μg PCAGGS/HA‐NEDD4, and 24 hr after transfection, cells were lysed for IFITM3 detection by western blot analysis. A549 cells were transfected with 0, 40, 60, and 100 nM miR‐30c mimics or negative control mimics (NC) (b) or 0, 60, 80, and 100 nM miR‐30c inhibitors (miR‐30c IN) or control inhibitor (NC‐IN) (c), and 24 hr later, the cells were lysed for IFITM3 and NEDD4 detection by western blot analysis. Data are shown as the mean and SD and as one representative of three independent experiments
2.8. Antiviral function of miR‐30 is mainly through targeting SOCS1, SOCS3, and NEDD4
To elucidate the roles of SOCS1, SOCS3, and NEDD4 in the antiviral function of miR‐30, we carried out experiments of SOCS1, SOCS3, and NEDD4 RNA interference knockdown and overexpression. We confirmed that overexpression of SOCS1, SOCS3, and NEDD4 effectively promoted IAV replication in A549 cells, as shown by the NP protein level and viral titre (Figure 8a–c), whereas, depletion of SOCS1, SOCS3, and NEDD4 by siRNAs significantly inhibited IAV replication in A549 cells (Figure 8d,e), which resembled the effect of miR‐30c overexpression.
Figure 8.
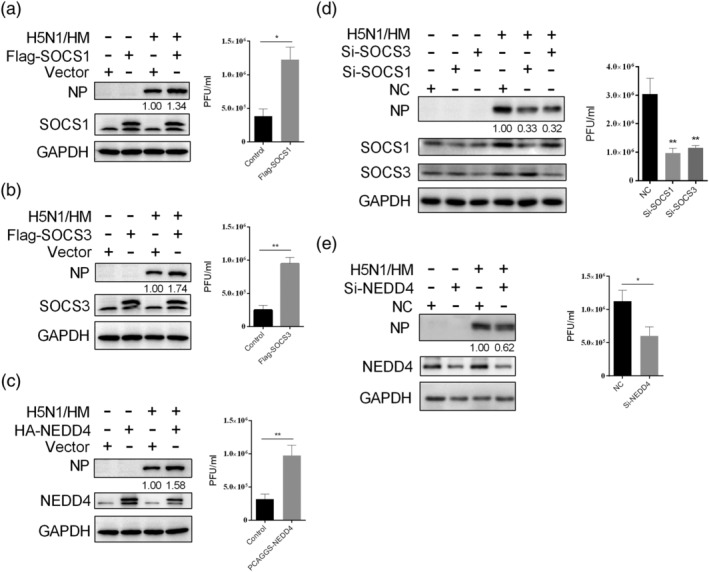
SOCS1, SOCS3, and NEDD4 facilitate influenza virus replication. A549 cells were transfected with SOCS1 (a), SOCS3 (b), or NEDD4 (c) overexpression vector, and 24 hr after transfection, the cells were infected with 0.2 MOI of H5N1 influenza virus (HM/H5N1). After 36 hr, the supernatant was collected, and the cells were lysed for viral titre and NP protein determination by plaque and western blot analyses, respectively. A549 cells were transfected with 60 nM SOCS1 or SOCS3 siRNAs (d) or NEDD4 siRNAs (e); 24 hr later, A549 cells were infected with 0.2 MOI of HM/H5N1, and after 36 hr, the supernatant was collected, and the cells were lysed for viral titre and NP protein determination. The western blot results are shown as one representative of three independent experiments, and viral titres data are expressed as the mean and SD of three independent experiments. Data were analysed using Student's t test. **p < .01; *p < .05
When SOCS1 and SOCS3 were knocked down at the same time, the IAV replication was also obviously inhibited compared with control; when the miR‐30c was overexpressed in SOCS1‐ and SOCS3‐depleted cells, IAV titre was further significantly reduced compared with control (Figure 9a). This could be due to that miR‐30c overexpression could reduce NEDD4 expression. However, when SOCS1, SOCS3, and NEDD4 were all knocked down at the same time, miR‐30c overexpression could not further suppress IAV replication (Figure 9b). The results showed that SOCS1, SOCS3, and NEDD4 were beneficial for influenza virus replication, and antiviral function of miR‐30 is mainly through targeting SOCS1, SOCS3, and NEDD4.
Figure 9.
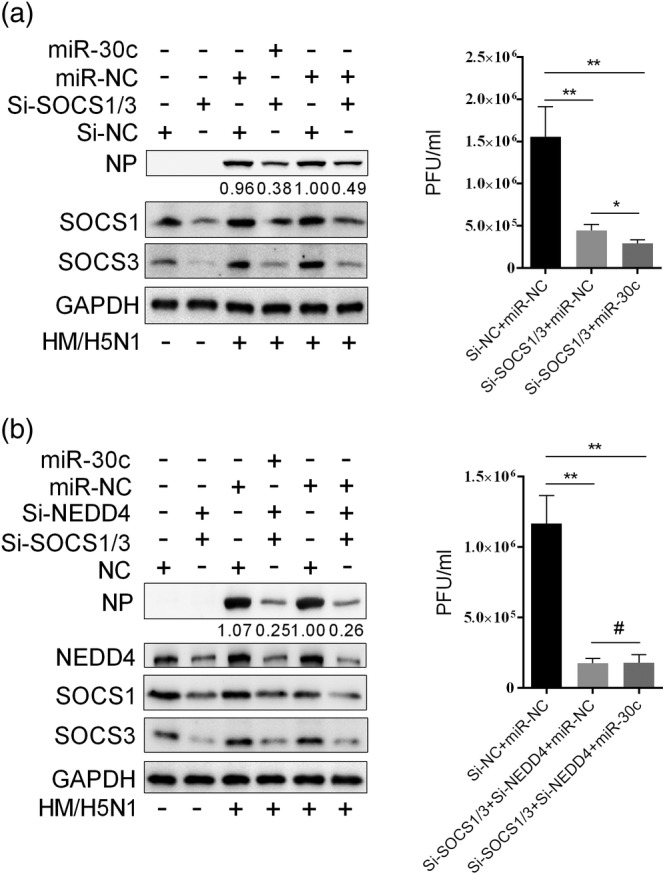
Antiviral function of miR‐30c is mainly dependent on SOCS1, SOCS3, and NEDD4 expression. A549 cells were transfected with 50 nm siRNA of SOCS1 and 50 nm siRNA of SOCS3 at the same time (a) or transfected with 50 nm siRNA of SOCS1, 50 nm siRNA of SOCS3, and 50 nm siRNA of NEDD4 at the same time (b). After 12 hr, 80 nm miR‐30c mimics or miRNA control (miR‐NC) was transfected; 24 hr later, A549 cells were infected with 0.2 MOI of HM/H5N1, and after 30 hr, the supernatant were collected for viral titres determination, and cells were lysed for NP test by western blot. Data are shown as the mean and SD and as one representative of three independent experiments. Data were analysed using Student's t test. #: non‐significant; **p < .01
3. DISCUSSION
In addition to cellular gene expression regulation, miRNAs have been demonstrated to be critical in virus infection, either by targeting viral gene transcripts or by regulating host antiviral response. Thus, the finding that viruses, including influenza virus, modulate the expression of specific cellular miRNAs is not surprising. The miR‐30 family was shown to participate in development, apoptosis, and autophagy (Agrawal, Tran, & Wessely, 2009; J. Li et al., 2010; Pan et al., 2013). Previously, several groups reported that the expression of miR‐30 family members was reduced by influenza virus invasion (Y. Li et al., 2010; Y. Li et al., 2011; Tambyah et al., 2013). However, the exact function of miR‐30 in influenza virus infection has not been determined. Herein, we showed that miR‐30 family members were downregulated by IAV infection. The regulation of miR‐30 family members by IAV infection appears to be independent of subtype, as the H1N1 and H9N2 subtypes also altered the miR‐30 levels (Figure S2). Interestingly, miR‐30 was not affected by VSV infection and poly(I:C) stimulation in A549 cells. Previous studies reported that miR‐30a and miR‐30d were reduced upon West Nile virus infection in HEK293 cells, while miR‐30c was increased by porcine reproductive and respiratory syndrome virus (PRRSV) in porcine alveolar macrophages (Zhang et al., 2016). It is likely that miR‐30 family members can be differentially modulated by various stimulants. Notably, because heat‐inactivated virus failed to alter the levels of miR‐30 family members, and individual overexpression of a virus‐coding protein did not influence miR‐30 expression (data not shown), we speculated that the downregulation of miR‐30 is dependent on influenza virus replication. However, our attempt to discover the mechanism by which influenza virus infection decreases miR‐30 levels failed. We hypothesised that influenza virus infection may affect the transcription of miR‐30 by acting on promoters or through oxidative stress induced by virus infection because influenza virus infection was shown to lead to oxidative stress, which was suggested to reduce the expression of miR‐30 family members in a previous study (J. Li et al., 2010). This mechanism requires further investigation.
We further demonstrated that miR‐30 family members were inversely correlated with influenza virus proliferation. MiR‐30 was reported to participate in various biological processes by targeting different host genes, such as p53, ATG5, BECIN1, and Xlim1/Lhx1 (Agrawal et al., 2009; J. Li et al., 2010; Yu et al., 2012). In this report, we demonstrated that miR‐30 directly targets SOCS1, SOCS3, and NEDD4, which are negative regulators of the antiviral response pathway. Interestingly, a previous study reported that miR‐30 can target JAK1 to positively modulate the IFN response in porcine Marc‐145 and PAM cells (Zhang et al., 2016). However, in our experiments, miR‐30 did not target JAK1 in A549 cells, as shown by dual‐luciferase reporter assays and real‐time polymerase chain reaction (RT‐PCR; data not shown). This discrepancy could be due to the different physiological conditions of the different cell types used in our experiment. It seems that the main targets for one specific miRNA often variable under different physical or pathological conditions or in different cell types to play diverse functions.
Influenza virus has devised multiple strategies to attenuate the type I IFN‐mediated antiviral response. Viral coding proteins (NS1, PB2, and PB1‐F2) were reported to disrupt the signalling pathways of type I IFN synthesis. In addition to the blockade of IFN production, influenza virus can also inhibit JAK/STAT pathway activation to prevent ISG expression. The influenza virus NS1 protein can inhibit JAK/STAT signalling pathway activation by increasing SOCS1 and SOCS3 expression; in addition, influenza viral proteins antagonise the functions of ISG, such as PB2, which was reported to inhibit mouse Mx1 function (Stranden, Staeheli, & Pavlovic, 1993). However, whether miRNAs participate in influenza virus‐mediated inhibition of type I IFN‐mediated antiviral responses is not very clear. In this study, we described a new strategy used by influenza virus to antagonise the type I IFN signalling pathway by decreasing host conserved miRNA expression (miR‐30 family members). MiR‐30 was shown to be a positive regulator of the antiviral response through inhibiting SOCS1, SOCS3, and NEDD4 expression in this study. However, it is hard to verify whether miR‐30 is more or less important in viral pathogenesis than other reported mechanisms. Nevertheless, influenza virus may promote its own survival by decreasing miR‐30 via cooperation with its coding proteins to interfere with the type I IFN‐mediated antiviral response. The antiviral immune response is regulated in an accurate and sophisticated manner by multiple regulators, both negative and positive. The mechanisms of influenza virus‐mediated immune evasion, through viral proteins or host molecules, are also complicated. Other miRNAs are likely involved in modulating influenza virus infection and virus‐mediated immune evasion. Because one miRNA may act as a counterbalance to another, the overall function of these miRNAs in influenza virus infection may not be apparent. In addition to the miRNAs that showed substantial changes following influenza virus infection, miRNAs that showed little alteration may also function in viral infection via the cumulative impact of all or most of them, even if a single miRNA has little effect. Although we showed that miR‐30 is used by influenza virus to counter the host antiviral immune response, we believe that further studies on miRNAs that participate in influenza virus infection are needed.
In summary, we demonstrated that influenza virus infection significantly downregulated the expression of miR‐30 family members, which modulated SOCS1, SOCS3, and NEDD4 to impair the host antiviral response and promote influenza virus replication. We present a new strategy used by influenza virus to counter the type I IFN‐mediated antiviral immune response by engaging miRNAs, which may help to elucidate the mechanism of immune escape by influenza virus.
4. EXPERIMENTAL PROCEDURES
4.1. Viruses and cells
Influenza virus strains A/duck/Hubei/hangmei01/2006 (H5N1) (HM/H5N1), A/Puerto Rico/8/34 (H1N1) (PR8/H1N1), and A/duck/Hubei/W1/2004 (H9N2) (W1/H9N2) were propagated in the allantoic cavities of 9‐ to 11‐day‐old fertile SPF chicken eggs. Viral titres were determined by plaque assays in Madin‐Darby canine kidney (MDCK) cells. Experiments with the H5N1 virus were conducted in an Animal Biosafety Level 3 laboratory (BSL‐3), Huazhong Agricultural University, and complied with the institutional biosafety manual. Lung carcinoma cells (A549) were cultured in F12 medium (HyClone, Logan, UT), and human embryonic kidney cells (293T) were cultured in RPMI 1640 medium (HyClone); MDCK and primary human alveolar epithelial cells (HAECs) were maintained in Dulbecco's modified Eagle's medium (DMEM) (HyClone). All media were supplemented with 10% heat‐inactivated foetal bovine serum (FBS) (HyClone). All cells were cultured in a 37°C humidified incubator with 5% CO2.
4.2. Reagents and antibodies
Lipofectamine 2000 was obtained from Invitrogen (Carlsbad, CA). Lipo8000 was purchased from Beyotime (Shanghai, China). The double‐stranded RNA mimic poly(I:C) was obtained from Sigma–Aldrich (Saint Louis, MO). SYBR Green I Master Mix was purchased from Roche (Penzberg, Germany). Human recombinant IFN‐β (rIFN‐β) was purchased from Sino Biological (Beijing, China). The scrambled negative control RNA (NC), miR‐30a/b/c/d/e mimics and inhibitors, SOCS1‐specific short interfering RNA (siRNA), SOCS3‐specific siRNA, and NEDD4‐specific siRNA oligonucleotides are listed in Table S1 and were synthesised by GenePharma (Suzhou, China).
Rabbit anti‐STAT1, anti‐STAT1‐Y701, and anti‐SOCS3 were obtained from Cell Signalling Technology (Beverly, MA). Rabbit anti‐SOCS1, anti‐NEDD4, and anti‐IFITM3 were obtained from ABclonal Biotechnology (Cambridge, MA). Rabbit anti‐NP and mouse monoclonal anti‐GAPDH were obtained from GeneTex (San Antonio, TX) and California Bioscience (Coachella, CA), respectively.
4.3. Plasmids
The 3′UTRs of human SOCS1, SOCS3, and NEDD4 were cloned from the genomic DNA of A549 cells and inserted into the pmirGLO Dual‐Luciferase miRNA Target Expression Vector (Promega). The mutant 3′UTRs of human SOCS1, SOCS3, and NEDD4 pmirGLO (SOCS1 3′UTR Mut, SOCS3 3′UTR Mut, and NEDD4 3′UTR Mut, respectively) were constructed by PCR using specific primers. Human SOCS1 and SOCS3 were cloned from the cDNA of A549 cells into pCMV3‐N‐Flag, and human NEDD4 was inserted into pCAGGS‐HA. All primers for cloning are shown in Table S2.
4.4. Quantitative real‐time PCR assay
Lipofectamine 2000 and Lipo8000 were applied to transfect small RNAs and DNA constructs, respectively. For qRT‐PCR determination of mRNA or miRNA, the total RNA of cells subjected to different treatments were extracted by Trizol, and a total of 1 μg RNA was reversely transcribed, as described previously (Lin et al., 2015). The miRNAs and mRNA levels were normalised to that of U6 and GAPDH, respectively. The relative expressions of miRNAs and mRNAs were determined via SYBR Green‐based qRT‐PCR under an ABI ViiA 7 PCR system. The qRT‐PCR primers used here are shown in Table 1.
Table 1.
Primers for qRT‐PCR
| Primers | Sequence (5′→3′) |
|---|---|
| miR‐30a‐d‐e‐RT | GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACCTTCCA |
| miR‐30b‐RT | GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACAGCTGA |
| miR‐30c‐RT | GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACGCTGAG |
| U6‐RT | GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACAAAATA |
| miR‐30a‐d‐e‐F | GCTGTAAACATCCTCGACTGGAAG |
| miR‐30b‐F | CGCTGTAAACATCCTACACTCAGCT |
| miR‐30c‐F | CGCTGTAAACATCCTACACTCTCAGC |
| Universal miR‐R | CAGTGCAGGGTCCGAGGT |
| U6‐F | TCGTATCCAGTGCGAATACCTCGGAC |
| SOCS1‐F | TTGGAGGGAGCGGATGGGTGTAG |
| SOCS1‐R | AGAGGTAGGAGGTGCGAGTTCAGGTC |
| SOCS3‐F | CCAGCATAGGAAAGCCACATAC |
| SOCS3‐R | GCCAATACTTACTGGGCTGACA |
| NEDD4‐F | TCCAATGATCTAGGGCCTTTACC |
| NEDD4‐R | TCCAACCGAGGATCTTCCCAT |
| IFN‐β‐F | GCTTGGATTCCTACAAAGAAGCA |
| IFN‐β‐R | ATAGATGGTCAATGCGGCGTC |
| IL‐6‐F | AGGAGACTTGCCTGGTGAAA |
| IL‐6‐R | CAGGGGTGGTTATTGCATCT |
| CXCL10‐F | TGGCATTCAAGGAGTACCTCTC |
| CXCL10‐R | CTTGATGGCCTTCGATTCTG |
| MX1‐F | GTTTCCGAAGTGGACATCGCA |
| MX1‐R | CTGCACAGGTTGTTCTCAGC |
| IFIT3‐F | AGAAAAGGTGACCTAGACAAAGC |
| IFIT3‐R | CCTTGTAGCAGCACCCAATCT |
| GAPDH‐F | GCCAAGGCTGTGGGCAAGG |
| GAPDH‐R | GGAGGAGTGGGTGTCGCTG |
4.5. Prediction of miR‐30 family member targeting sites
The miR‐30 family member targets were predicted and selected using MicroRNA.org (Betel, Wilson, Gabow, Marks, & Sander, 2008), DIANA‐MicroT (Alexiou et al., 2010), TargetScan (Lewis, Burge, & Bartel, 2005), MirTarget2 (Wong & Wang, 2015), and RNA22 V2 (Miranda et al., 2006). MicroRNA and DIANA‐MicroT analyses were based on seed complementarity, with target predict score threshold 0.7; TargetScan predicts targets according to aggregate P ct score of the longest 3′UTR isoform, which ranks based upon the confidence that targeting is evolutionary conserved; MirTarget2 analysis predicts gene targets with more than 60 target prediction score; RNA22 analysis is performed under sensitivity more than 63% and specificity more than 61%.
4.6. Dual‐luciferase reporter assays
To validate the targeted genes by miR‐30, luciferase reporter vectors containing wild‐type SOCS1, SOCS3, and NEDD4 3′UTR or their mutants in seed regions were co‐transfected with control mimics, miR‐30a/b/c mimics, NC inhibitor, or miR‐30a/b/c inhibitors into 293T cells. Twenty‐four hours later, cells were lysed, and supernatants were collected for luciferase activity test through the Dual‐Luciferase test reagent provided by Promega. All obtained luciferase values were normalised against those of the Renilla luciferase control. For each experiment, at least three independent experiments were performed, and each experiment was performed in triplicate.
4.7. Viral infection and titre determination
A549 cells that were transfected with oligonucleotides or plasmids for 24 hr were challenged with influenza viruses at the indicated multiplicity of infection (MOI). After 1 hr of viral adsorption at 37°C, the cells were washed twice with phosphate buffered saline (PBS) and the supernatants were replaced with F12 medium containing 1% FBS. After that, the cells were cultured in a 37°C humidified incubator with 5% CO2. At the indicated time post‐infection, the supernatants were collected, and the viral titres were analysed by plaque assays in MDCK cells.
4.8. Western blotting
Treated cells were first washed twice with cold PBS and then were lysed in RIPA buffer (Sigma) on ice for 15 min and supplemented with protease and phosphatase inhibitors cocktail (APExBIO). The lysate was centrifuged at 12,000 rpm/min for 10 min at 4°C, and supernatants were collected for protein concentration determination by bicinchoninic acid (BCA) assay (Beyotime Biotechnology, China) and equalised with lysis buffer. For western blot analysis, equal amounts of the extracts were separated on 10–12% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS‐PAGE) gels and were subsequently transferred onto pure nitrocellulose membranes (GE). Membranes were then blocked in 1–2% bovine serum albumin (BSA) in Tris Buffered Saline Tween (TBST) buffer for about 1 hr at room temperature, washed once for 5 min, and then incubated with indicated primary antibodies for 2 hr at room temperature. After incubating with HRP‐conjugated anti‐rabbit or anti‐mouse secondary antibodies for 1 hr at room temperature, signals were visualised by ECL reagent (Advansta, San Jose, CA). The densitometry of protein expression was quantified relative to GAPDH by ImageJ software; in the densitometry analysis, control was set as 1.00.
4.9. Statistical analysis
The results are expressed as the mean ± SD. Data analysis was performed using Student's t test or two‐way analysis of variance (ANOVA). Differences between means were considered significant at p values of <.05.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
AUTHOR CONTRIBUTIONS
M.J. and X.L. designed the study; X.L., S.Y., P.R., and X.S. conducted the experiments; X.L. wrote the manuscript; M.J. and X.L. analysed the data.
Supporting information
Figure S1. Sequence comparison among miR‐30 family members. Nucleotides in red are seed region; black rectangle shows the difference in nucleotides among miR‐30a, d, and e.
Figure S2. H1N1 and H9N2 influenza virus infection decreased miR‐30 expression. A549 cells were infected with 0.5 MOI PR8/H1N1 and W1/H9N2 influenza virus. Expression of miR‐30 was detected at 12, 24, 36, and 48 hpi by qRT‐PCR. The values are shown as the mean and SD. ***p < .001; *p < .05.
Figure S3. MiR‐30a/b/c inhibited influenza virus proliferation in HAECs. HAECs were transfected with 120 nm miR‐30a (A), miR‐30b (B), miR‐30c (C), or negative control (NC). Twenty‐four hours later, 0.1 MOI HM/H5N1 was used to infect HAECs. At 30 hr post‐infection, the supernatant was collected for plaque test, and the cells were lysed for NP determination using western blot. The values are shown as the mean and SD and are representative of three independent experiments. Data was analysed using Student's t test. ***p < .001; **p < .01.
Figure S4. MiR‐30a/b/c inhibitors transfection significantly repressed miR‐30a/b/c expression. 293T cells were transfected with 100 nM miR‐30a/b/c inhibitors or control NC‐inhibitor. Twenty‐four hours later, the cells were harvested for RNA extraction. Then, miR‐30a/b/c expression was determined by qRT‐PCR. Expression of miR‐30a/b/c was normalised to U6. The values are shown as the mean and SD and was analysed using Student's t test. ***p < .001; **p < .01.
Figure S5. MiR‐30c suppresses SOCS1, SOCS3, and NEDD4 expression in HAECs. 120 nm miR‐30c mimics or NC were transfected into HAECs, and 24 hr after transfection, cells were harvested for RNA and protein extract. The mRNA and protein of SOCS1 (A), SOCS3 (B), and NEDD4 (C) were determined by qRT‐PCR and western blot, respectively. The values are shown as the mean and SD and are representative of three independent experiments. Data were analysed using Student's t test. **p < .01; *p < .01.
Table S1. Supporting Information.
ACKNOWLEDGEMENTS
This work was supported by the National Natural Science Foundation of China (No. 31702212), National Key Research and Development Program of China (No. 2016YFD0500205), and China Postdoctoral Science Foundation (No. 2017M612483). The funders had no roles in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Lin X, Yu S, Ren P, Sun X, Jin M. Human microRNA‐30 inhibits influenza virus infection by suppressing the expression of SOCS1, SOCS3, and NEDD4. Cellular Microbiology. 2020;22:e13150 10.1111/cmi.13150
Funding information China Postdoctoral Science Foundation, Grant/Award Number: 2017M612483; National Key Research and Development Program of China, Grant/Award Number: 2016YFD0500205; National Natural Science Foundation of China, Grant/Award Number: 31702212
REFERENCES
- Agrawal, R. , Tran, U. , & Wessely, O. (2009). The miR‐30 miRNA family regulates Xenopus pronephros development and targets the transcription factor Xlim1/Lhx1. Development, 136(23), 3927–3936. 10.1242/dev.037432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, W. S. (2002). Suppressors of cytokine signalling (SOCS) in the immune system. Nature Reviews. Immunology, 2(6), 410–416. 10.1038/nri818 [DOI] [PubMed] [Google Scholar]
- Alexander, W. S. , & Hilton, D. J. (2004). The role of suppressors of cytokine signaling (SOCS) proteins in regulation of the immune response. Annual Review of Immunology, 22, 503–529. 10.1146/annurev.immunol.22.091003.090312 [DOI] [PubMed] [Google Scholar]
- Alexiou, P. , Maragkakis, M. , Papadopoulos, G. L. , Simmosis, V. A. , Zhang, L. , & Hatzigeorgiou, A. G. (2010). The DIANA‐mirExTra web server: From gene expression data to microRNA function. PLoS One, 5(2), e9171 10.1371/journal.pone.0009171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambros, V. (2004). The functions of animal microRNAs. Nature, 431(7006), 350–355. 10.1038/nature02871 [DOI] [PubMed] [Google Scholar]
- Bartel, D. P. (2004). MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell, 116(2), 281–297. [DOI] [PubMed] [Google Scholar]
- Betel, D. , Wilson, M. , Gabow, A. , Marks, D. S. , & Sander, C. (2008). The microRNA.org resource: Targets and expression. Nucleic Acids Research, 36(Database issue), D149–D153. 10.1093/nar/gkm995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borden, E. C. , Sen, G. C. , Uze, G. , Silverman, R. H. , Ransohoff, R. M. , Foster, G. R. , & Stark, G. R. (2007). Interferons at age 50: Past, current and future impact on biomedicine. Nature Reviews. Drug Discovery, 6(12), 975–990. 10.1038/nrd2422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carow, B. , & Rottenberg, M. E. (2014). SOCS3, a major regulator of infection and inflammation. Frontiers in Immunology, 5, 58 10.3389/fimmu.2014.00058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesarino, N. M. , McMichael, T. M. , & Yount, J. S. (2015). E3 ubiquitin ligase NEDD4 promotes influenza virus infection by decreasing levels of the antiviral protein IFITM3. PLoS Pathogens, 11(8), e1005095 10.1371/journal.ppat.1005095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croker, B. A. , Kiu, H. , & Nicholson, S. E. (2008). SOCS regulation of the JAK/STAT signalling pathway. Seminars in Cell & Developmental Biology, 19(4), 414–422. 10.1016/j.semcdb.2008.07.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen, B. R. (2013). How do viruses avoid inhibition by endogenous cellular microRNAs? PLoS Pathogens, 9(11), e1003694 10.1371/journal.ppat.1003694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan, N. , & Wang, J. (2016). MicroRNA 34a contributes to virus‐mediated apoptosis through binding to its target gene Bax in influenza A virus infection. Biomedicine & Pharmacotherapy, 83, 1464–1470. 10.1016/j.biopha.2016.08.049 [DOI] [PubMed] [Google Scholar]
- Giordanetto, F. , & Kroemer, R. T. (2003). A three‐dimensional model of suppressor of cytokine signalling 1 (SOCS‐1). Protein Engineering, 16(2), 115–124. [DOI] [PubMed] [Google Scholar]
- Hou, J. , Wang, P. , Lin, L. , Liu, X. , Ma, F. , An, H. , … Cao, X. (2009). MicroRNA‐146a feedback inhibits RIG‐I‐dependent Type I IFN production in macrophages by targeting TRAF6, IRAK1, and IRAK2. Journal of Immunology, 183(3), 2150–2158. 10.4049/jimmunol.0900707 [DOI] [PubMed] [Google Scholar]
- Hu, Y. , Jiang, L. , Lai, W. , Qin, Y. , Zhang, T. , Wang, S. , & Ye, X. (2016). MicroRNA‐33a disturbs influenza A virus replication by targeting ARCN1 and inhibiting viral ribonucleoprotein activity. The Journal of General Virology, 97(1), 27–38. 10.1099/jgv.0.000311 [DOI] [PubMed] [Google Scholar]
- Ingle, H. , Kumar, S. , Raut, A. A. , Mishra, A. , Kulkarni, D. D. , Kameyama, T. , … Kumar, H. (2015). The microRNA miR‐485 targets host and influenza virus transcripts to regulate antiviral immunity and restrict viral replication. Science Signaling, 8(406), ra126 10.1126/scisignal.aab3183 [DOI] [PubMed] [Google Scholar]
- Jia, D. , Rahbar, R. , Chan, R. W. , Lee, S. M. , Chan, M. C. , Wang, B. X. , … Fish, E. N. (2010). Influenza virus non‐structural protein 1 (NS1) disrupts interferon signaling. PLoS One, 5(11), e13927 10.1371/journal.pone.0013927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krug, R. M. (2015). Functions of the influenza A virus NS1 protein in antiviral defense. Current Opinion in Virology, 12, 1–6. 10.1016/j.coviro.2015.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen, L. , & Ropke, C. (2002). Suppressors of cytokine signalling: SOCS. APMIS, 110(12), 833–844. [DOI] [PubMed] [Google Scholar]
- Levy, D. E. , & Darnell, J. E., Jr. (2002). Stats: Transcriptional control and biological impact. Nature Reviews. Molecular Cell Biology, 3(9), 651–662. 10.1038/nrm909 [DOI] [PubMed] [Google Scholar]
- Lewis, B. P. , Burge, C. B. , & Bartel, D. P. (2005). Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell, 120(1), 15–20. 10.1016/j.cell.2004.12.035 [DOI] [PubMed] [Google Scholar]
- Li, J. , Donath, S. , Li, Y. , Qin, D. , Prabhakar, B. S. , & Li, P. (2010). miR‐30 regulates mitochondrial fission through targeting p53 and the dynamin‐related protein‐1 pathway. PLoS Genetics, 6(1), e1000795 10.1371/journal.pgen.1000795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Y. , Chan, E. Y. , Li, J. , Ni, C. , Peng, X. , Rosenzweig, E. , … Katze, M. G. (2010). MicroRNA expression and virulence in pandemic influenza virus‐infected mice. Journal of Virology, 84(6), 3023–3032. 10.1128/JVI.02203-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Y. , Li, J. , Belisle, S. , Baskin, C. R. , Tumpey, T. M. , & Katze, M. G. (2011). Differential microRNA expression and virulence of avian, 1918 reassortant, and reconstructed 1918 influenza A viruses. Virology, 421(2), 105–113. 10.1016/j.virol.2011.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, X. , Huang, C. , Shi, J. , Wang, R. , Sun, X. , Liu, X. , … Jin, M. (2015). Investigation of pathogenesis of H1N1 influenza virus and swine Streptococcus suis serotype 2 co‐infection in pigs by microarray analysis. PLoS One, 10(4), e0124086 10.1371/journal.pone.0124086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, Y. J. , Yang, J. , Fan, X. L. , Zhao, H. B. , Hu, W. , Li, Z. P. , … Wang, S. Q. (2012). Cellular microRNA let‐7c inhibits M1 protein expression of the H1N1 influenza A virus in infected human lung epithelial cells. Journal of Cellular and Molecular Medicine, 16(10), 2539–2546. 10.1111/j.1582-4934.2012.01572.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda, K. C. , Huynh, T. , Tay, Y. , Ang, Y. S. , Tam, W. L. , Thomson, A. M. , … Rigoutsos, I. (2006). A pattern‐based method for the identification of microRNA binding sites and their corresponding heteroduplexes. Cell, 126(6), 1203–1217. 10.1016/j.cell.2006.07.031 [DOI] [PubMed] [Google Scholar]
- Murray, P. J. (2007). The JAK‐STAT signaling pathway: Input and output integration. Journal of Immunology, 178(5), 2623–2629. [DOI] [PubMed] [Google Scholar]
- Pachler, K. , & Vlasak, R. (2011). Influenza C virus NS1 protein counteracts RIG‐I‐mediated IFN signalling. Virology Journal, 8, 48 10.1186/1743-422X-8-48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan, W. , Zhong, Y. , Cheng, C. , Liu, B. , Wang, L. , Li, A. , … Liu, S. (2013). MiR‐30‐regulated autophagy mediates angiotensin II‐induced myocardial hypertrophy. PLoS One, 8(1), e53950 10.1371/journal.pone.0053950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauli, E. K. , Schmolke, M. , Wolff, T. , Viemann, D. , Roth, J. , Bode, J. G. , & Ludwig, S. (2008). Influenza A virus inhibits type I IFN signaling via NF‐kappaB‐dependent induction of SOCS‐3 expression. PLoS Pathogens, 4(11), e1000196 10.1371/journal.ppat.1000196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pothlichet, J. , Chignard, M. , & Si‐Tahar, M. (2008). Cutting edge: Innate immune response triggered by influenza A virus is negatively regulated by SOCS1 and SOCS3 through a RIG‐I/IFNAR1‐dependent pathway. Journal of Immunology, 180(4), 2034–2038. 10.4049/jimmunol.180.4.2034 [DOI] [PubMed] [Google Scholar]
- Rajsbaum, R. , Albrecht, R. A. , Wang, M. K. , Maharaj, N. P. , Versteeg, G. A. , Nistal‐Villan, E. , … Gack, M. U. (2012). Species‐specific inhibition of RIG‐I ubiquitination and IFN induction by the influenza A virus NS1 protein. PLoS Pathogens, 8(11), e1003059 10.1371/journal.ppat.1003059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberger, C. M. , Podyminogin, R. L. , Diercks, A. H. , Treuting, P. M. , Peschon, J. J. , Rodriguez, D. , … Aderem, A. (2017). miR‐144 attenuates the host response to influenza virus by targeting the TRAF6‐IRF7 signaling axis. PLoS Pathogens, 13(4), e1006305 10.1371/journal.ppat.1006305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranden, A. M. , Staeheli, P. , & Pavlovic, J. (1993). Function of the mouse Mx1 protein is inhibited by overexpression of the PB2 protein of influenza virus. Virology, 197(2), 642–651. 10.1006/viro.1993.1639 [DOI] [PubMed] [Google Scholar]
- Tambyah, P. A. , Sepramaniam, S. , Mohamed Ali, J. , Chai, S. C. , Swaminathan, P. , Armugam, A. , & Jeyaseelan, K. (2013). microRNAs in circulation are altered in response to influenza A virus infection in humans. PLoS One, 8(10), e76811 10.1371/journal.pone.0076811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, X. , Gao, J. S. , Guan, Y. J. , McLane, K. E. , Yuan, Z. L. , Ramratnam, B. , & Chin, Y. E. (2007). Acetylation‐dependent signal transduction for type I interferon receptor. Cell, 131(1), 93–105. 10.1016/j.cell.2007.07.034 [DOI] [PubMed] [Google Scholar]
- Uze, G. , Schreiber, G. , Piehler, J. , & Pellegrini, S. (2007). The receptor of the type I interferon family. Current Topics in Microbiology and Immunology, 316, 71–95. [DOI] [PubMed] [Google Scholar]
- Varga, Z. T. , Grant, A. , Manicassamy, B. , & Palese, P. (2012). Influenza virus protein PB1‐F2 inhibits the induction of type I interferon by binding to MAVS and decreasing mitochondrial membrane potential. Journal of Virology, 86(16), 8359–8366. 10.1128/JVI.01122-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varga, Z. T. , Ramos, I. , Hai, R. , Schmolke, M. , Garcia‐Sastre, A. , Fernandez‐Sesma, A. , & Palese, P. (2011). The influenza virus protein PB1‐F2 inhibits the induction of type I interferon at the level of the MAVS adaptor protein. PLoS Pathogens, 7(6), e1002067 10.1371/journal.ppat.1002067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wienholds, E. , & Plasterk, R. H. (2005). MicroRNA function in animal development. FEBS Letters, 579(26), 5911–5922. 10.1016/j.febslet.2005.07.070 [DOI] [PubMed] [Google Scholar]
- Wong, N. , & Wang, X. (2015). miRDB: An online resource for microRNA target prediction and functional annotations. Nucleic Acids Research, 43(Database issue), D146–D152. 10.1093/nar/gku1104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia, C. , Vijayan, M. , Pritzl, C. J. , Fuchs, S. Y. , McDermott, A. B. , & Hahm, B. (2015). Hemagglutinin of influenza A virus antagonizes type I interferon (IFN) responses by inducing degradation of type I IFN receptor 1. Journal of Virology, 90(5), 2403–2417. 10.1128/JVI.02749-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, T. , Chu, Q. , Cui, J. , & Bi, D. (2018). Inducible microRNA‐3570 feedback inhibits the RIG‐I‐dependent innate immune response to rhabdovirus in teleost fish by targeting MAVS/IPS‐1. Journal of Virology, 92(2). 10.1128/JVI.01594-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, Y. , Cao, L. , Yang, L. , Kang, R. , Lotze, M. , & Tang, D. (2012). microRNA 30A promotes autophagy in response to cancer therapy. Autophagy, 8(5), 853–855. 10.4161/auto.20053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Q. , Huang, C. , Yang, Q. , Gao, L. , Liu, H. C. , Tang, J. , & Feng, W. H. (2016). MicroRNA‐30c modulates type I IFN responses to facilitate porcine reproductive and respiratory syndrome virus infection by targeting JAK1. Journal of Immunology, 196(5), 2272–2282. 10.4049/jimmunol.1502006 [DOI] [PubMed] [Google Scholar]
- Zhao, L. , Zhu, J. , Zhou, H. , Zhao, Z. , Zou, Z. , Liu, X. , … Jin, M. (2015). Identification of cellular microRNA‐136 as a dual regulator of RIG‐I‐mediated innate immunity that antagonizes H5N1 IAV replication in A549 cells. Scientific Reports, 5, 14991 10.1038/srep14991 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Sequence comparison among miR‐30 family members. Nucleotides in red are seed region; black rectangle shows the difference in nucleotides among miR‐30a, d, and e.
Figure S2. H1N1 and H9N2 influenza virus infection decreased miR‐30 expression. A549 cells were infected with 0.5 MOI PR8/H1N1 and W1/H9N2 influenza virus. Expression of miR‐30 was detected at 12, 24, 36, and 48 hpi by qRT‐PCR. The values are shown as the mean and SD. ***p < .001; *p < .05.
Figure S3. MiR‐30a/b/c inhibited influenza virus proliferation in HAECs. HAECs were transfected with 120 nm miR‐30a (A), miR‐30b (B), miR‐30c (C), or negative control (NC). Twenty‐four hours later, 0.1 MOI HM/H5N1 was used to infect HAECs. At 30 hr post‐infection, the supernatant was collected for plaque test, and the cells were lysed for NP determination using western blot. The values are shown as the mean and SD and are representative of three independent experiments. Data was analysed using Student's t test. ***p < .001; **p < .01.
Figure S4. MiR‐30a/b/c inhibitors transfection significantly repressed miR‐30a/b/c expression. 293T cells were transfected with 100 nM miR‐30a/b/c inhibitors or control NC‐inhibitor. Twenty‐four hours later, the cells were harvested for RNA extraction. Then, miR‐30a/b/c expression was determined by qRT‐PCR. Expression of miR‐30a/b/c was normalised to U6. The values are shown as the mean and SD and was analysed using Student's t test. ***p < .001; **p < .01.
Figure S5. MiR‐30c suppresses SOCS1, SOCS3, and NEDD4 expression in HAECs. 120 nm miR‐30c mimics or NC were transfected into HAECs, and 24 hr after transfection, cells were harvested for RNA and protein extract. The mRNA and protein of SOCS1 (A), SOCS3 (B), and NEDD4 (C) were determined by qRT‐PCR and western blot, respectively. The values are shown as the mean and SD and are representative of three independent experiments. Data were analysed using Student's t test. **p < .01; *p < .01.
Table S1. Supporting Information.