

Summary
Indomethacin, a cyclooxygenase‐1 and ‐2 inhibitor widely used in the clinic for its potent anti‐inflammatory/analgesic properties, possesses antiviral activity against several viral pathogens; however, the mechanism of antiviral action remains elusive. We have recently shown that indomethacin activates the double‐stranded RNA (dsRNA)‐dependent protein kinase R (PKR) in human colon cancer cells. Because of the important role of PKR in the cellular defence response against viral infection, herein we investigated the effect of indomethacin on PKR activity during infection with the prototype rhabdovirus vesicular stomatitis virus. Indomethacin was found to activate PKR in an interferon‐ and dsRNA‐independent manner, causing rapid (< 5 min) phosphorylation of eukaryotic initiation factor‐2 α‐subunit (eIF2α). These events resulted in shutting off viral protein translation and blocking viral replication (IC 50 = 2 μM) while protecting host cells from virus‐induced damage. Indomethacin did not affect eIF2α kinases PKR‐like endoplasmic reticulum‐resident protein kinase (PERK) and general control non‐derepressible‐2 (GCN2) kinase, and was unable to trigger eIF2α phosphorylation in the presence of PKR inhibitor 2‐aminopurine. In addition, small‐interfering RNA‐mediated PKR gene silencing dampened the antiviral effect in indomethacin‐treated cells. The results identify PKR as a critical target for the antiviral activity of indomethacin and indicate that eIF2α phosphorylation could be a key element in the broad spectrum antiviral activity of the drug.
Introduction
The double‐stranded RNA (dsRNA)‐dependent protein kinase PKR is an interferon‐induced serine threonine protein kinase that plays an essential role in innate immunity response to viral infection, acting as a sensor of virus replication (García et al., 2006; Dabo and Meurs, 2012). PKR is encoded from a single gene located on human chromosome 2p21‐22, and contains a C‐terminal kinase domain (KD) shared by three other mammalian kinases: the PKR‐like endoplasmic reticulum‐resident protein kinase (PERK or PEK), the heme‐regulated inhibitor kinase (HRI) and the homologue of the Saccharomyces cerevisiae general control non‐derepressible‐2 (GCN2) kinase, all of which phosphorylate the α‐subunit of the eukaryotic initiation factor 2 (eIF2) in response to stress signals (Schneider and Mohr, 2003; Baltzis et al., 2004; Holcik and Sonenberg, 2005; Taylor et al., 2005). The multiprotein complex eIF2 is responsible for recruiting the initiator Met‐tRNAMet to the 40S ribosomal subunit in a guanosine triphosphate (GTP)‐dependent manner (Clemens, 2004; Gebauer and Hentze, 2004; Holcik and Sonenberg, 2005). Phosphorylation of the eIF2 α‐subunit (eIF2α) at residue Ser51 functions as a dominant inhibitor of guanine exchange factor eIF2B and prevents the recycling of GTP on eIF2 that is required for ongoing translation, thus leading to general inhibition of protein synthesis (Clemens, 2004; Taylor et al., 2005).
In addition to the KD shared by the other eIF2α kinases, PKR also contains an N‐terminal dsRNA‐binding domain (dsRBD) consisting of two dsRNA‐binding motifs of 70 amino acids each, connected by a short 20‐aa linker that regulates its activity. In uninfected non‐stressed cells, PKR is found in a monomeric latent state due to the auto‐inhibitory effect of its dsRBDs, which normally occlude the KD (García et al., 2006). Upon binding to dsRNA produced as a virus replication intermediate, PKR undergoes homo‐dimerization and autophosphorylation on Thr446 and Thr451, leading to substrate docking, eIF2α phosphorylation and block of protein synthesis in virally infected cells (Taylor et al., 2005; Dabo and Meurs, 2012). dsRNA‐induced PKR‐mediated eIF2α phosphorylation represents a primary antiviral response in mammalian cells (Schneider and Mohr, 2003; García et al., 2006); in fact production of dsRNA is an obligate part of the genomic replication cycle for RNA viruses as RNA serves as a template for both viral transcription and replication, and dsRNA is often produced by opposing transcription units also during DNA virus replication (Schneider and Mohr, 2003).
In addition to its role in translation, PKR participates in several signalling pathways controlling transcription, including the IκB kinase (IKK)/nuclear factor‐κB pathway (Santoro et al., 2003; Donzé et al., 2004; Dabo and Meurs, 2012), and is implicated in the control of cell growth, differentiation and apoptosis generally with tumour suppression function (Donzé et al., 2004; García et al., 2006).
The prototype rhabdovirus vesicular stomatitis virus (VSV) is a negative‐sense, single‐stranded RNA virus that is widely studied as a model of viral protein translational control (Lyles et al., 2013). Following viral penetration and uncoating, VSV primary mRNAs are synthesized in the host cell cytoplasm by the virus‐associated RNA‐dependent RNA polymerase (RdRp). When viral proteins begin to accumulate, progeny viral genomes are replicated and used for secondary transcription. mRNA from both primary and secondary transcription are similar in structure to host messengers, containing a cap at the 5′‐end and a 3′‐poly(A) tail similar in length to host mRNAs (Connor and Lyles, 2002; Lyles et al., 2013). During VSV replication host translation is rapidly inhibited, an event that is mainly attributed to virus‐induced modifications of host eukaryotic initiation factor‐4F (eIF4F) that enhance translational efficiency of viral mRNA while dampening synthesis of host proteins, including antiviral defence enzymes (Connor and Lyles, 2002; 2005). Previous studies had suggested that the non‐steroidal anti‐inflammatory drug indomethacin (INDO) inhibits VSV replication, possibly affecting transcription of viral messages (Mukherjee and Simpson, 1987).
INDO, a traditional cyclooxygenase‐1 and ‐2 inhibitor, is widely used in the clinic for its potent anti‐inflammatory and analgesic properties (Vane and Botting, 1997). In addition to the anti‐inflammatory/analgesic action, indomethacin has been known for several decades to also possess antiviral properties. Since the initial discovery in the 1960s (Inglot, 1969), in vitro and in vivo studies have confirmed the antiviral activity of indomethacin against several human viral pathogens, including herpes virus (Zhu et al., 2002), rotavirus (Rossen et al., 2004), hepatitis B (Bahrami et al., 2005), human immunodeficiency virus (HIV) (Ramirez et al., 1986; Bourinbaiar and Lee‐Huang, 1995) and the SARS‐CoV coronavirus, agent of the severe acute respiratory syndrome (Amici et al., 2006). The experimental and clinical evidence accumulated suggests a novel therapeutic use of this classical anti‐inflammatory drug during viral infection; however, the lack of understanding of the mechanism of antiviral action is a major drawback for INDO treatment of viral diseases. Indomethacin ability to inhibit the replication of several unrelated DNA and RNA viruses points out to a host‐mediated antiviral mechanism, rather than an effect on virus‐specific enzymes; however, this mechanism remains largely unknown.
We have previously observed that INDO causes a dys‐regulation of protein synthesis in coronavirus‐infected cells (Amici et al., 2006). Herein we investigated the molecular mechanism responsible for the antiviral activity of INDO during VSV infection. We demonstrate that INDO acts at the translational level by rapidly inducing the phosphorylation of the α‐subunit of eIF2. Phosphorylation of eIF2α occurs at concentrations of INDO in the Cmax range detected in patients treated with medium–high doses of the drug (Benet, 1996). Confirming our previous observation in cancer cells (Brunelli et al., 2012), eIF2α phosphorylation was found to be mediated by selective activation of PKR. Interestingly, siPKR silencing dampened the ability of INDO to inhibit VSV protein synthesis, identifying PKR as a key element in the antiviral activity of this drug.
Results
Indomethacin blocks viral protein expression and protects host cells from VSV‐induced damage
To investigate the effect of indomethacin on VSV replication, epithelial monkey MA104 cells were mock infected or infected with VSV [10 plaque‐forming units (PFU) per cell] and treated with different concentrations of INDO after the 1 h adsorption period. Virus yields were determined by TCID50 infectivity assay at 8 h post‐infection (p.i). The effect of INDO on cell viability was determined by MTT assay in mock‐infected cells 24 h after treatment. INDO showed a remarkable antiviral activity against VSV, reducing virus yield dose dependently with an IC50 of 2 μM and LD50 above 800 μM (Fig. 1A). Under similar conditions, INDO was equally active in different types of host cells, including hamster BHK‐21 cells, and human HeLa and HEK293 cells (http://onlinelibrary.wiley.com/doi/10.1111/cmi.12446/suppinfo).
Figure 1.
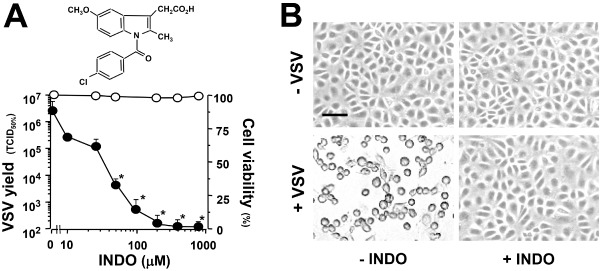
Indomethacin inhibits viral replication and protects host cells from VSV‐induced damage.
A. Structure of indomethacin (upper panel). Antiviral activity of INDO in VSV‐infected MA104 cells (lower panel). Mock‐infected or VSV‐infected cells were treated with different concentrations of INDO after the 1 h adsorption period. VSV yield (8 h p.i.) is expressed in TCID 50% ml−1 (filled circles). Cell viability (24 h) of mock‐infected INDO‐treated cells (empty circles) is expressed as percentage of MTT conversion in untreated cells. Data represent the mean ± standard deviation (SD) of duplicate samples from a representative experiment of three with similar results. *P < 0.01.
B. Mock‐infected (–VSV) or VSV‐infected (+VSV) MA104 cells were treated with 400 μM INDO (+INDO) or control vehicle (−INDO) after the 1 h adsorption period. Indomethacin‐induced cytoprotection at 24 h p.i. is shown. Bar = 100 μm.
VSV infection is generally characterized by a massive cytopathic effect, causing cell shrinkage and loss of adhesion (Fig. 1B). Indomethacin treatment, in addition to inhibiting virus progeny production, was also found to protect MA104 cells from virus‐induced damage. Strikingly, cells treated with 400 μM INDO after viral adsorption showed no sign of infection up to 24 h p.i. (Fig. 1B). This concentration, which was also found not to affect MA104 cell proliferation for at least 24 h (http://onlinelibrary.wiley.com/doi/10.1111/cmi.12446/suppinfo), was used for the following studies, unless differently specified.
Because INDO has been shown to enhance heat shock factor 1 (HSF1) activation under stress conditions and to promote the expression of cytoprotective heat shock proteins (HSP) (Amici et al., 1995; Cotto et al., 1996), which were associated with the establishment of an antiviral state in cells infected with VSV and other viruses (Santoro, 1997; Morimoto and Santoro, 1998), we investigated whether indomethacin could be acting by inducing a cytoprotective heat shock response in host cells. INDO was found not to affect either HSF1 DNA‐binding activity or the accumulation of the 70 kDa HSP (HSP70) in mock‐infected or VSV‐infected MA104 cells (http://onlinelibrary.wiley.com/doi/10.1111/cmi.12446/suppinfo), excluding the involvement of this defence mechanism in drug‐induced cytoprotection.
INDO is a well‐known COX inhibitor (Vane and Botting, 1997). To investigate the effect of different COX‐1 and COX‐2 inhibitors on VSV infection, mock‐infected or VSV‐infected (10 PFU per cell) cells were treated with different concentrations of aspirin, sodium salicylate, the COX‐1 inhibitor SC‐560 or the COX‐2 inhibitor NS‐398 after the 1 h adsorption period. Virus yields were determined by TCID50 infectivity assay at 24 h p.i. In parallel, the effect of the drugs on cell viability was determined by MTT assay in mock‐infected cells 24 h after treatment. The results, shown in http://onlinelibrary.wiley.com/doi/10.1111/cmi.12446/suppinfo, demonstrate that none of the NSAIDs tested was able to mimic the antiviral activity of INDO up to concentrations near the cytotoxic range, suggesting a cyclooxygenase‐independent mechanism.
The fact that a remarkable antiviral activity against VSV could be obtained when INDO treatment was started after virus adsorption indicated that the NSAID was not affecting virus adsorption or entry into the host cell. In addition, pretreatment of MA104 cells with INDO (400 μM) for a 6 h period only modestly affected VSV replication, if the drug was removed before viral infection (http://onlinelibrary.wiley.com/doi/10.1111/cmi.12446/suppinfo). The effect of indomethacin on VSV protein synthesis and localization was then analysed. In a first set of experiments, mock‐infected or VSV‐infected (10 PFU per cell) MA104 cells were treated with INDO after the 1 h adsorption period and, at 6 h p.i., viral proteins were visualized by immunofluorescence microscopy using anti‐VSV antibodies. As shown in Fig. 2, the expression of high levels of cytoplasmic viral proteins at 6 h p.i. was accompanied by a nearly complete disassembly of tubulin filaments in the cytoplasm of infected cells; viral proteins were instead barely detectable in INDO‐treated cells, which were indistinguishable from uninfected controls for size and cytoskeletal arrangement. To investigate the effect of INDO on cellular and viral protein synthesis, mock‐infected and VSV‐infected cells were treated with INDO after virus adsorption and labelled with [35S]‐methionine at 4 h p.i. for the next 4 h. Samples containing equal amounts of proteins or equal amounts of radioactivity were processed for sodium dodecyl sulfate‐polyacrylamide gel electrophoresis (SDS‐PAGE) and autoradiography. As shown in Fig. 3A, INDO treatment strongly inhibited VSV protein expression and prevented the virus‐induced shut‐off of cellular protein synthesis. Next, the effect of INDO on the kinetics of VSV protein synthesis was analysed. Mock‐infected and VSV‐infected cells were treated with 400 μM INDO after virus adsorption and labelled with [35S]‐methionine (1 h pulse) at different times p.i. Indomethacin was found to cause a transient (1–6 h) inhibition of [35S]‐methionine incorporation into proteins in both mock‐infected and VSV‐infected cells (Fig. 3B). Samples containing the same amount of radioactivity were processed for SDS‐PAGE, autoradiography and densitometric analysis. As shown in Fig. 3C, accumulation of VSV proteins was evident starting at 3 h p.i., whereas shut‐off of host cell protein synthesis was observed at later times (4–5 h) p.i. In infected cells, INDO treatment was confirmed to strongly inhibit VSV protein expression and to prevent the virus‐induced shut‐off of cellular protein synthesis up to 6–8 h p.i. (Fig. 3C). It should be pointed out that the synthesis of VSV proteins, in particular the N protein, could be detected starting at 6 h p.i. in INDO‐treated cells.
Figure 2.
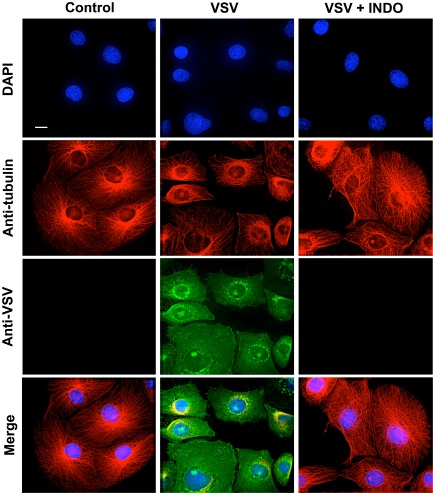
Effect of indomethacin on VSV protein expression and intracellular localization. MA104 cells mock infected or infected with VSV (10 PFU per cell) were treated with 400 μM INDO after the 1 h adsorption period and, at 6 h p.i., viral proteins were visualized by immunofluorescence microscopy using anti‐VSV (green) and anti‐α‐tubulin (red) antibodies. Nuclei are stained with 4′,6‐diamidino‐2‐phenylindole (DAPI) (blue). The overlay of the three fluorochromes is shown (Merge). Images were collected and deconvolved with DeltaVision microscope and software. Bar = 15 μm.
Figure 3.
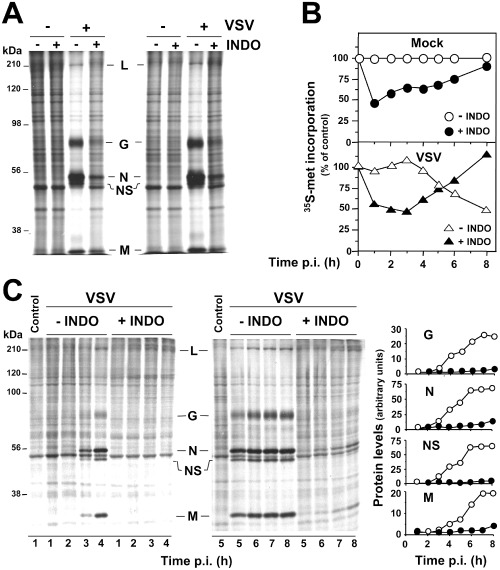
Indomethacin inhibits viral protein synthesis.
A. Mock‐infected (–VSV) or VSV‐infected (+VSV) MA104 cells treated with 400 μM INDO after the 1 h adsorption period were labelled with [35S]‐methionine (4 h pulse) at 4 h p.i. Samples containing equal amounts of proteins (right panel) or equal amounts of radioactivity (left panel) were processed for SDS‐PAGE and autoradiography. VSV proteins L, G, N, NS and M are indicated.
B. Mock‐infected (mock) or VSV‐infected (VSV) MA104 cells treated as in A were labelled with [35S]‐methionine (1 h pulse) at different times p.i. Protein synthesis, determined by [35 S]‐methionine incorporation into proteins of untreated (empty symbols) or INDO‐treated (filled symbols) cells, is expressed as percent of untreated, mock‐infected control.
C. In a parallel experiment, samples containing equal amounts of radioactivity collected at early (1–4 h) and late (5–8 h) times p.i. were processed for SDS‐PAGE and autoradiography (right panel). VSV proteins are indicated as in A. Control cells labelled at 1 and 5 h after mock infection are shown. Levels of viral proteins G, N, NS and M in untreated (empty symbols) or INDO‐treated (filled symbols) samples were determined by densitometric analysis and expressed as arbitrary units (left panel). Data represent results from one experiment representative of three independent experiments with similar results.
The indomethacin‐induced block of viral protein expression is controlled at the translational level via rapid induction of eIF2α phosphorylation
To investigate whether the inhibition of viral protein expression was dependent on transcriptional activation of host antiviral proteins by INDO, cell mRNA transcription was blocked by actinomycin‐D (AMD), an inhibitor of cellular, but nor viral, RNA polymerase (Mukherjee and Simpson, 1987). Mock‐infected and VSV‐infected MA104 cells were treated with INDO in the presence or absence of AMD (5 μg ml−1) after the adsorption period, and then labelled with [35S]‐methionine (2 h pulse) at 6 h p.i. In parallel samples, RNA synthesis was analysed by [3H]‐uridine labelling, and virus yield was determined at 8 h p.i. As shown in Fig. 4 (panels A and B), AMD treatment, which inhibited cellular RNA synthesis by more than 80% ([3H]‐uridine incorporation: uninfected control, 11.60 ± 0.10; uninfected + AMD, 2.08 ± 0.01; VSV‐infected control, 10.50 ± 0.49; VSV‐infected + AMD, 2.19 ± 0.06 c.p.m. × 103/105 cells), did not dampen INDO‐mediated antiviral effects, suggesting that, differently from interferon, host defence protein expression is not required for the antiviral activity.
Figure 4.
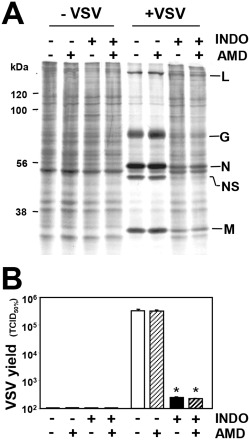
Indomethacin antiviral activity is independent of host protein expression.
A. Mock‐infected (−VSV) or VSV‐infected (+VSV) MA104 cells treated with 400 μM INDO after the 1 h adsorption period in the presence or absence of AMD (5 μg ml−1) were labelled with [35 S]‐methionine (2 h pulse) at 6 h p.i. Samples containing equal amounts of radioactivity were processed for SDS‐PAGE and autoradiography. VSV proteins L, G, N, NS and M are indicated.
B. Virus yield (TCID 50% ml−1) was determined in parallel samples at 8 h p.i. Data represent the mean ± SD of duplicate samples from a representative experiment of two with similar results. *P < 0.01.
Indomethacin was previously suggested to inhibit VSV mRNA transcription (Mukherjee and Simpson, 1987). To establish whether INDO was affecting virus protein expression at transcriptional or translation level, total viral RNA and primary transcript levels were determined by Northern blot analysis in infected cells at 4 h p.i. Viral primary transcript accumulation was obtained by blocking viral protein synthesis (which is essential for viral replication and amplification of viral secondary transcripts) with cycloheximide treatment (50 μg ml−1) started 30 min prior to viral infection. The fact that INDO did not significantly affect VSV primary transcription, whereas it dramatically reduced VSV total RNA levels (Fig. 5A), suggested an effect on viral mRNA translation. A translational block was in fact demonstrated by polysome profile analysis after cell extracts sucrose gradient fractionation. As shown in Fig. 5B, INDO caused a rapid and substantial decrease of heavier polysomes, characteristic of a reduced rate of translation initiation, in both mock‐infected and VSV‐infected cells at 30 min p.i. As indicated earlier, translational inhibition was transient as demonstrated by protein synthesis recovery at later times after treatment (Fig. 3B).
Figure 5.
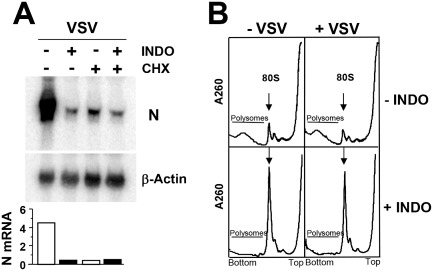
Effect of indomethacin on VSV primary transcription and polysome profiles of VSV‐infected cells.
A. Levels of total and primary VSV N‐mRNA and β‐actin mRNA were determined by Northern blot in cells treated with 400 μM INDO with or without cycloheximide (CHX, 50 μg ml−1) (upper panels). mRNA levels were quantified by MDA, as described in the text; N‐mRNA/β‐actin mRNA ratio for each lane is shown (lower panel).
B. Polysome profiles (5–65% linear sucrose gradient) of mock‐infected (−VSV) or VSV‐infected (+VSV) cells treated with 400 μM INDO (30 min) after virus adsorption. Arrow indicates 80S ribosomes. Data represent results from one experiment representative of two independent experiments with similar results.
Translational control is primarily mediated at the level of initiation (Gebauer and Hentze, 2004; Holcik and Sonenberg, 2005). In particular, phosphorylation of the α‐subunit of translation initiation factor eIF2 is a potent mechanism of translational control (Clemens, 2004; Taylor et al., 2005). The effect of INDO on eIF2α phosphorylation was then investigated in MA104 cells. Mock‐infected and VSV‐infected cells were treated with different concentrations of INDO, and levels of Ser51‐phosphorylated eIF2α (p‐eIF2α) were determined after 30 min. As shown in Fig. 6A, INDO was found to potently induce eIF2α phosphorylation starting at concentrations in the low micromolar range in mock‐infected cells. In VSV‐infected cells, INDO treatment induced eIF2α phosphorylation at concentrations above 10 μM, concomitantly with a substantial decrease in viral yield (see Fig. 1A). eIF2α phosphorylation was a very rapid event starting as soon as 5 min after drug administration and attenuating between 4 and 6 h after treatment (Fig. 6B), concomitantly with the release of protein synthesis inhibition (see Fig. 3B). High cytoplasmic levels of p‐eIF2α were also visualized at 45 min after INDO treatment by immunofluorescence (Fig. 6C).
Figure 6.
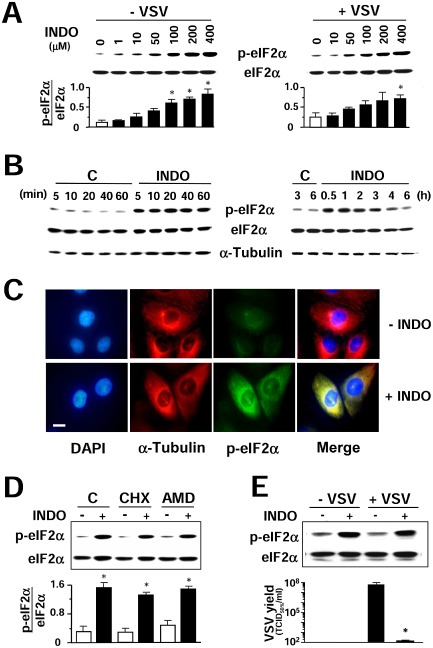
Indomethacin induces eIF2α phosphorylation.
A. Immunoblot using antiphosphoSer‐51‐eIF2α (p‐eIF2α) or eIF2α panspecific antibodies performed on lysates from mock‐infected (−VSV) or VSV‐infected (+VSV) cells treated with different concentrations of INDO (30 min) (upper panel). Graph shows the p‐eIF2α/eIF2α ratio for each sample (lower panel). Values represent the mean ± SD from three experiments. *P < 0.05 versus untreated control.
B. Kinetics of eIF2α phosphorylation following 400 μM INDO treatment was determined as described in A. α‐Tubulin levels are shown as loading control.
C. Immunofluorescence analysis of MA104 cells treated with 400 μM INDO (+INDO) or control diluent (−INDO) for 45 min, and labelled with antiphosphoSer‐51‐eIF2α (p‐eIF2α) (green) and anti‐α‐tubulin (red) antibodies. Nuclei are stained with DAPI (blue). Bar = 15 μm. The overlay of the three fluorochromes is shown (merge). Microscopy was performed on a Leica DM‐IL microscope equipped with UV excitation filters, and the images were captured with a Leica DC‐300 camera using Leica Image‐Manager software.
D. MA104 cells were pretreated for 30 min with control vehicle (C), 50 μg ml−1 cycloheximide (CHX), which inhibited [35S]‐methionine incorporation into proteins by more than 98%, or 5 μg ml−1 AMD, which inhibited [3 H]‐uridine incorporation into acid‐insoluble material by more than 80%, and then treated with INDO (+) or vehicle (−) for 30 min. Lysates were analysed by Western blot using antiphosphoSer‐51‐eIF2α (p‐eIF2α) or eIF2α panspecific antibodies (top panel). Graph shows the p‐eIF2α/eIF2α ratio for each sample (bottom panel). Values represent the mean ± SD from two experiments. *P < 0.05 versus untreated control.
E. Mock‐infected or VSV‐infected BHK‐21 cells were treated with 400 μM INDO after the adsorption period and, at 2 h p.i., were analysed by Western blot as in D (top panel). In parallel, virus yields were determined by TCID 50 infectivity assay at 8 h p.i. (bottom panel). Data represent the mean ± SD of duplicate samples from a representative experiment of two with similar results. *P < 0.05.
Indomethacin‐induced eIF2α phosphorylation did not require protein expression as shown by high levels of p‐eIF2α in cells where RNA transcription or translation was blocked by AMD and cycloheximide treatment respectively (Fig. 6D). Finally, phosphorylation of eIF2α following INDO treatment is not limited to MA104 cells as similar results were obtained in uninfected or VSV‐infected BHK‐21 cells (Fig. 6E) and in HeLa cells (Brunelli et al., 2012).
PKR is responsible for indomethacin‐induced eIF2α phosphorylation
As indicated in the Introduction section, four distinct eukaryotic eIF2α kinases have been identified to date: the dsRNA‐activated protein kinase PKR, PERK, HRI and GCN2 kinase (Schneider and Mohr, 2003; Baltzis et al., 2004; Holcik and Sonenberg, 2005; Taylor et al., 2005). We have recently shown that INDO treatment selectively activates PKR in a cyclooxygenase‐independent manner in human colon cancer cells (Brunelli et al., 2012). To confirm that PKR is the kinase responsible for INDO‐induced eIF2α phosphorylation also in MA104 cells, GCN2, PERK, HRI and PKR kinase activities were analysed in MA104 whole‐cell lysates by in vitro kinase assay using recombinant eIF2α as a substrate after immunoprecipitation with specific antibodies. HRI was not detectable in MA104 cells. INDO did not affect PERK and GCN2 activity, whereas it strongly induced PKR activity (Fig. 7A). Indomethacin‐induced PKR activation was also evidenced in MA104 cells by increased PKR auto‐phosphorylation (Fig. 7B), starting rapidly after drug administration and attenuating between 4 and 6 h after treatment, concomitantly with attenuation of eIF2α‐phosphorylation (Fig. 6B). As shown in Fig. 7C, treatment with the PKR inhibitor 2‐aminopurine resulted in preventing eIF2α phosphorylation in INDO‐treated cells, confirming that PKR is responsible for INDO‐induced eIF2α phosphorylation in MA104 cells. Interestingly, treatment with 2‐aminopurine also caused the attenuation of INDO antiviral activity in VSV‐infected cells (Fig. 7D).
Figure 7.
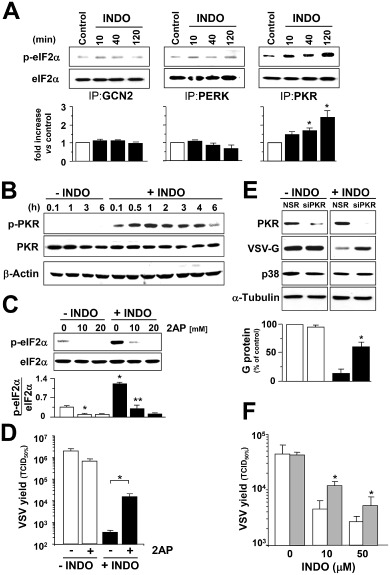
Indomethacin selectively induces PKR activity.
A. At different times after INDO treatment (400 μM) MA104 cell lysates were immunoprecipitated using antibodies against eIF2α kinases GCN2, PERK or PKR and analysed by in vitro kinase assay (upper panels). Plotted values indicate fold increase of the p‐eIF2α/eIF2α ratio in INDO‐treated cells versus control (lower panels). Values represent the mean ± SD from two experiments. *P < 0.05 versus untreated control.
B. Kinetics of PKR phosphorylation was analysed by Western blot using antiphospho‐PKR (p‐PKR) or anti‐PKR (PKR) antibodies in lysates from MA104 cells treated with 400 μM INDO; β‐actin levels are shown as loading control.
C. Immunoblot using antiphosphoSer‐51‐eIF2α (p‐eIF2α) or eIF2α panspecific antibodies performed on lysates from MA104 cells treated for 30 min with INDO and/or different concentrations of 2‐aminopurine (2AP) (upper panel). Graph shows the p‐eIF2α/eIF2α ratio for each sample (lower panel). Values represent the mean ± SD from three independent experiments. *P < 0.05 versus untreated control. **P < 0.05 versus INDO‐treated control.
D. VSV‐infected MA104 cells were treated with 400 μM INDO after the 1 h adsorption period in the presence or absence of 2 mM 2AP. Virus yield (TCID 50% ml−1) was determined at 8 h p.i. Data represent the mean ± SD of duplicate samples from a representative experiment of two with similar results. *P < 0.01.
E. HeLa cells transiently transfected with PKR‐siRNA (siPRK) or non‐specific siRNA (NSR) were infected with VSV and treated with 25 μM INDO or diluent control after the 1 h adsorption period. VSV‐G protein levels at 8 h p.i. were detected by Western blot using polyclonal anti‐VSV‐G antibodies (1:2000 dilution); endogenous PKR and p38 MAPK levels were detected on the same filter by immunoblot (top panels); α‐tubulin levels are shown as loading control. Plotted values indicate the levels of G protein expressed as percent of levels in untreated mock‐transfected control (bottom panel). Values represent the mean ± SD from two independent experiments. *P < 0.05 versus INDO‐treated mock‐transfected cells.
F. HeLa cells transfected with PKR‐siRNA (dashed bars) or non‐specific siRNA (empty bars) were infected with VSV and treated with different concentrations of INDO after the 1 h adsorption period. Virus yields were determined by TCID 50 infectivity assay at 8 h p.i. Data represent the mean ± SD of duplicate samples from a representative experiment of two with similar results. *P < 0.05.
To investigate whether PKR‐eIF2α signalling is responsible for the VSV translational block induced by INDO, PKR was down‐regulated by small‐interfering RNA (si)RNA‐mediated silencing. Because MA104 cells were found to be poorly transfectable, HeLa cells were utilized for transfection experiments. HeLa cells were transiently transfected with PKR siRNA or non‐specific siRNA and, after 48 h, were mock infected or infected with VSV, and treated with 25 μM INDO after the 1 h adsorption period. At 8 h p.i. whole‐cell extracts were analysed for endogenous PKR levels and VSV‐G protein levels by immunoblot analysis. p38 MAPK and tubulin levels were determined as control. As shown in Fig. 7E, PKR levels were greatly reduced in PKR siRNA transfected cells, whereas p38 MAPK levels were not altered. As expected, treatment with INDO resulted in a nearly complete inhibition of VSV‐G protein synthesis; however, in PKR‐silenced cells, the ability of the drug to inhibit VSV protein synthesis was greatly impaired, and G protein levels were approximately four times higher than in mock‐silenced cells.
In a parallel experiment, HeLa cells transfected with PKR siRNA or non‐specific siRNA were infected with VSV and treated with different concentrations of INDO after the 1 h adsorption period, and virus yield was determined at 8 h p.i. As shown in Fig. 7F, VSV titres were significantly higher in PKR‐silenced cells as compared with control, confirming that PKR participates in the antiviral activity of INDO. However, PKR silencing only partially restored virus replication in treated cells. This could be the consequence of residual PKR activity due to partial silencing of the kinase; alternatively, other mechanisms may contribute to the antiviral activity of the drug.
It should be pointed out that, as previously reported (Baltzis et al., 2004; Neznanov et al., 2008), treatment with the PERK inducer thapsigargin (1 μM) or, to a minor extent, the GCN2 kinase inducer MG132 (5 μM) was effective in reducing VSV‐G protein levels in MA104 cells (http://onlinelibrary.wiley.com/doi/10.1111/cmi.12446/suppinfo), reinforcing the role of eIF2α phosphorylation in the inhibition of VSV protein synthesis.
Finally, the antiviral activity of INDO was investigated in mouse embryonic fibroblasts (MEF) wild type or knockout for GCN2 or PERK. As previously reported (Baltzis et al., 2004; Berlanga et al., 2006; Krishnamoorthy et al., 2008), both GCN2−/− and PERK−/− cells were found to be more susceptible than GCN2+/+ and PERK+/+ cells to VSV‐induced cytopathic effects and cell death that was approximately 50% higher in knockout than wild‐type cells as determined by MTT assay at 8 h after infection with VSV (10 PFU per cell). As shown in http://onlinelibrary.wiley.com/doi/10.1111/cmi.12446/suppinfo, INDO was equally effective in inhibiting VSV replication in PERK+/+ and PERK−/− cells, as well as in GCN2+/+ and GCN2−/− cells, further emphasizing an important role of PKR in the antiviral activity of INDO.
Discussion
As indicated in the Introduction section, PKR plays a critical role in the antiviral defence mechanism of the host, acting as a sensor of virus replication (Gil et al., 2000; García et al., 2006) and, upon activation, leading to eIF2α phosphorylation and block of protein synthesis in virally infected cells (Taylor et al., 2005; Dabo and Meurs, 2012). We have recently shown that the anti‐inflammatory drug indomethacin is able to activate PKR and cause rapid phosphorylation of eIF2α in human colon carcinoma cells (Brunelli et al., 2012). Because of the important role of PKR in the cellular defence response against viral infection, we have now investigated the effect of indomethacin on PKR activity and eIF2α phosphorylation in virally infected cells.
Using the rhabdovirus VSV as a model in MA104 cells, we demonstrate that indomethacin activates PKR causing a rapid and transient phosphorylation of eIF2α with similar kinetics in uninfected and VSV‐infected cells. As previously reported in cancer cells (Brunelli et al., 2012), indomethacin‐induced eIF2α phosphorylation is mediated by PKR as it is prevented by the PKR inhibitor 2‐aminopurine; in addition, INDO selectively activates PKR, whereas it does not affect the activity of the eIF2α kinases PERK and GCN2 in these cells. In uninfected MA104 cells, PKR‐mediated eIF2α‐phosphorylation resulted in a transient inhibition of protein synthesis; on the other hand, in VSV‐infected cells, eIF2α phosphorylation at early times during infection resulted in shutting off viral protein translation, causing a dramatic inhibition of viral replication and infectious viral particle production. Treatment with 2‐aminopurine and PKR silencing both resulted in the attenuation of INDO antiviral activity in VSV‐infected cells. These results are consistent with previous reports showing that PKR plays an essential role against VSV infection in vitro and in vivo (Stojdl et al., 2000; Durbin et al., 2002), and that inactivation of the eIF2 complex early after infection blocks VSV protein synthesis (Connor and Lyles, 2005). On the other hand, Krishnamoorthy et al. (2008) reported that no significant difference in VSV replication was detected in MEFs containing the serine 51 to alanine mutation of eIF2α as compared with wild‐type cells, indicating that eIF2α phosphorylation does not affect VSV replication under normal, non‐stimulated conditions. Nevertheless, the KD of eIF2α kinases was shown to be both necessary and sufficient to inhibit VSV protein expression and virus replication in human fibrosarcoma HT1080 cells (Krishnamoorthy et al., 2008). It should be pointed out that, as previously reported (Connor and Lyles, 2005), the kinetics of eIF2α phosphorylation may be important for the control of VSV replication, suggesting that inhibition of VSV protein synthesis is achieved if the eIF2 complex is inactivated early after infection. In fact, in our model, pretreatment of host cells with 400 μM INDO for a 6 h period before viral infection, a time at which eIF2α phosphorylation is restored to the basal level, only modestly affected VSV replication as compared to treatment with the drug started after the 1 h adsorption period.
Interestingly, differently from interferon, phosphorylation of eIF2α following INDO treatment does not require host defence protein expression; in fact treatment with AMD, an inhibitor of cellular, but not viral, RNA transcription, did not affect INDO‐mediated eIF2α phosphorylation and antiviral effects.
In addition to the inhibition of infectious viral particle production, it should be emphasized that indomethacin treatment also protected the host cell from virus‐induced damage. Strikingly, cells treated with INDO after viral adsorption showed no sign of infection (cell shrinkage and loss of adhesion) up to 24 h p.i. In particular, INDO treatment prevented the virus‐induced massive disassembly of the host microtubular network. This effect was not associated with the previously described ability of INDO to promote a cytoprotective heat shock response by enhancing HSF1 activity and HSP expression under stress conditions (Amici et al., 1995; Cotto et al., 1996). In fact, we could not detect HSF1 activation or increase in accumulation of the major HSP70 protein in either uninfected or VSV‐infected cells in the presence of the drug.
As anticipated in the Introduction section, a large amount of literature has described in vitro and in vivo antiviral activity of indomethacin against several highly pathogenic human viruses, including herpes virus, rotavirus, hepatitis B, HIV and the SARS‐CoV coronavirus (Ramirez et al., 1986; Bourinbaiar and Lee‐Huang, 1995; Zhu et al., 2002; Rossen et al., 2004; Bahrami et al., 2005; Amici et al., 2006). Despite an extensive amount of studies, the mechanism for this wide spectrum activity has remained elusive for several decades. It should be pointed out that indomethacin antiviral activity was previously attributed to cyclooxygenase inhibition and prostaglandin‐E production during herpes virus (Zhu et al., 2002; Schröer and Shenk, 2008) and rotavirus (Rossen et al., 2004) infection. In addition, the antiviral activity of non‐selective as well as selective COX‐1 and COX‐2 inhibitors has been reported also in vivo against several viruses, including VSV (Chen et al., 2000; 2002; Symensma et al., 2003). In our experimental model, however, INDO activity was not mimicked by the NSAID aspirin and sodium salicylate, nor by the selective COX‐1 inhibitor SC‐560 and COX‐2 inhibitor NS‐398 up to concentrations near the cytotoxic range, suggesting a cyclooxygenase‐independent mechanism. The results described herein demonstrate that indomethacin, instead, triggers a cellular antiviral defence mechanism by rapidly and effectively activating PKR in an interferon‐ and dsRNA‐independent manner. INDO‐triggered PKR signalling effectively blocks viral replication by preventing viral protein translation. The fact that high levels of phosphorylated eIF2α were associated with INDO antiviral activity also during infection with the parainfluenza Sendai virus and the SA‐11 rotavirus (Santoro et al., unpublished results) suggests that activation of the PKR/eIF2α signalling cascade may be implicated in the antiviral effect of INDO against other types of RNA viruses.
The finding that INDO‐mediated inhibition of viral replication is suppressed in PKR‐silenced cells, but not in GCN2−/− and PERK−/− cells, identifies PKR as a central player in the antiviral activity of the drug. However, the fact that PKR silencing only partially restored virus replication in INDO‐treated cells indicates that other mechanisms may contribute to the antiviral activity of the drug. Moreover, because in addition to its role in translation, PKR participates in several signalling pathways controlling transcription (Santoro et al., 2003; Donzé et al., 2004; García et al., 2006; Dabo and Meurs, 2012), it cannot be excluded that other PKR‐mediated mechanisms may be involved in the control of VSV replication after INDO treatment.
The mechanism by which INDO triggers PKR activity remains to be established. We have previously shown that INDO‐mediated PKR induction is COX independent, and that, intriguingly, indomethacin is able to stimulate PKR activity in the absence of the natural dsRNA inducer, and does not require the presence of interferon (Brunelli et al., 2012). This has now been confirmed in a different model. In addition, it was previously shown that indomethacin was able to induce PKR activity in vitro at levels comparable with the PKR inducer poly(I):poly(C), suggesting a direct effect of the drug in the kinase activation (Brunelli et al., 2012).
Altogether, the results suggest that PKR‐mediated eIF2α phosphorylation may be a key element in the broad spectrum antiviral activity of indomethacin.
Experimental procedures
Cell culture, transfection and treatments
Epithelial monkey kidney MA104 cells, baby hamster kidney BHK‐21 cells, human cervical adenocarcinoma (HeLa) and embryo kidney (HEK293) cells, and GCN2−/−, PERK−/− and wild‐type MEFs (Scheuner et al., 2006), kindly supplied by Dr. Randal Kaufman, were cultured in either MEM199 (MA104), MEM (BHK‐21) or Dulbecco's modified Eagle's medium (HeLa, HEK293, GCN2+/+, GCN2−/−, PERK+/+ and PERK−/−) medium (Invitrogen) supplemented with 10% fetal calf serum, 2 mM glutamine and antibiotics. Cell numbers were evaluated by standard procedures, and cell viability was determined by trypan blue exclusion assay and by 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyl‐tetrazolium bromide (MTT) to MTT formazan conversion assay (Amici et al., 2006). Indomethacin, aspirin, COX‐1 inhibitor SC‐560 and COX‐2 inhibitor NS‐398 (Sigma‐Aldrich) dissolved in ethanol, sodium salicylate, 2‐aminopurine, thapsigargin and MG132 (Sigma‐Aldrich) dissolved in dimethyl sulfoxide (DMSO), or equal amounts of control vehicle were added immediately after the 1 h adsorption period and maintained during the experiment. Ethanol or DMSO vehicles had no effect on VSV replication, eIF2α phosphorylation and PKR activity at all concentrations tested. For PKR silencing, HeLa cells were transiently transfected with 21–23 bp siRNAs heterogeneous mixture (25 nM, New England BioLabs), using TransPass‐R2 Transfection Reagent (New England BioLabs) according to the manufacturer's instructions.
Virus infection and titration
Confluent monolayers of MA104, BHK‐21, HeLa or HEK293 cells and GCN2−/−, PERK−/− and wild‐type MEFs were infected with VSV, Indiana serotype (Orsay, 10 PFU per cell) for 1 h at 37°C. After the adsorption period, the viral inoculum was removed and cell monolayers were washed three times with phosphate‐buffered saline (PBS) and incubated at 37°C in conditioned medium. Microscopical examination of virus‐induced cytopathic effect was performed at 24 h p.i. using a Leica DM‐IL (Leica Microsystems GmbH) inverted microscope and images were captured with a Leica DC‐300 camera using Leica Image‐Manager software. Virus yields were determined by TCID50 (50% tissue culture infective dose) infectivity assay, as described previously (Rossi et al., 1996). IC50 (inhibitory concentration 50%) and LD50 (lethal dose 50%) were calculated using Prism 5.0 software (GraphPad Software Inc.).
Metabolic labelling and polysomal profile analysis
Cells were labelled with L‐[35S]‐methionine (10 μCi/105 cells, 1 h pulse, unless differently specified). After lysis in L‐buffer (20 mM Tris‐Cl pH 7.4, 0.1 M NaCl, 5 mM MgCl2, 1% NP40, 0.5% SDS), samples containing the same amount of radioactivity were separated by SDS‐PAGE (3% stacking gel, 10% resolving gel) and processed for autoradiography and densitometric analysis (Rossi et al., 2000).
For analysis of polysomal profiles, MA104 cells were lysed with PL buffer [10 mM NaCl, 10 mM MgCl2, 10 mM Tris‐HCl pH 7.5, 1% Triton X‐100, 1% sodium deoxycholate, 0.2 U μl−1 RNase inhibitor (Promega Corporation), 1 mM dithiothreitol (DTT)]. After incubation on ice for 1 min, extracts were centrifuged for 1 min in a cold centrifuge and supernatants were loaded onto a 5–65% linear sucrose gradient containing 30 mM Tris‐HCl (pH 7.5), 100 mM NaCl and 10 mM MgCl2 and centrifuged in a Beckman SW 41 rotor (Beckman Coulter, Inc.) for 3 h at 37 000 rpm. Profiles were obtained monitoring the absorbance at 260 nm.
For RNA synthesis analysis, cells were labelled with [3H]‐uridine (10 μCi/2 × 105 cells), 5 h pulse starting at 3 h p.i., and the radioactivity incorporated into acid‐insoluble material was determined as previously described (Amici et al., 2006).
Antibodies and Western blot analysis
Cells were lysed with cold high‐salt extraction (HSB) buffer (Rossi et al., 2000), containing 2 mM DTT, 1 mM phenylmethanesulphonylfluoride, 1 mM orthovanadate, 20 mM β‐glycerophosphate and 1 mM PNPP (Sigma‐Aldrich) and a protease inhibitor cocktail (Complete Mini, Roche). Whole‐cell extracts (20 μg) were separated by SDS‐PAGE, blotted to nitrocellulose and filters were incubated with the following antibodies: rabbit anti‐phosphoSer‐51‐eIF2α (p‐eIF2α) from Calbiochem (Merck KGaA), anti‐eIF2α (FL‐315), anti‐phospho‐PKR (Thr451), anti‐HRI (H‐165), anti‐PERK (H‐300) from Santa Cruz Biotechnology, Inc., anti‐GCN2 and anti‐p38 MAP kinase from Cell Signaling Technology, Inc., or with monoclonal β‐actin (C2), anti‐PKR (B‐10) (Santa Cruz Biotechnology), anti‐HSP70 (Stressgen) or α‐tubulin (Sigma‐Aldrich) antibodies. Polyclonal anti‐VSV‐G antibodies were a kind gift from E. Rodriguez‐Boulan (Cornell University New York). After decoration with peroxidase‐labelled anti‐mouse or anti‐rabbit IgG (ECL, Amersham Biosciences), quantitative evaluation of proteins was determined by Typhoon 8600 imager (GE Healthcare) with the use of ImageQuant software (Brunelli et al., 2012).
Immunoprecipitation and eIF2α kinase assay
PKR kinase assay was described previously (Brunelli et al., 2012). Briefly, protein extracts (150 μg) were subjected to immunoprecipitation using anti‐PKR, ‐PERK, ‐GCN2 or ‐HRI antibodies in the presence of 15 μl of protein A‐Sepharose at 4°C for 12 h. After extensive washing in HSB buffer, immune complexes were resuspended in 25 μl kinase buffer [20 mM Tris‐HCl, pH 7.6, 2.5 mM MgCl2, 2.5 mM Mg(OAc)2, 50 mM ATP] containing 1 mM DTT, and assayed for their ability to phosphorylate recombinant eIF2α (1 μg; Cell Sciences) for 30 min at 30°C. Incubations were terminated by the addition of SDS sample buffer. Phosphorylated eIF2α was detected by Western blot analysis using rabbit anti‐phosphoSer51‐eIF2α antibodies (Calbiochem). Total eIF2α or kinase levels were determined by immunoblot in the same samples for loading control.
For detection of PKR autophosphorylation, whole‐cell extracts from MA104 cells treated with INDO or control vehicle were analysed by Western blot using anti‐phospho‐PKR antibodies and anti‐PKR antibody as loading control.
RNA extraction and Northern blot
Total RNA from uninfected and virus‐infected cells was isolated using TRIzol (Invitrogen), according to the manufacturer's specification and stored at −20°C. For detection of VSV mRNA, aliquots of total RNA (5 μg) were fractionated on 1% agarose/formaldehyde gels, transferred onto Hybond‐N nylon membranes (Amersham Biosciences – GE Healthcare), and hybridized with [32P]‐labelled pN4 plasmid (Gallione et al., 1981). After being stripped, filters were rehybridized with a plasmid specific for the β‐actin gene 5′ end‐labelled by T4 kinase with [γ‐32P] ATP (Amersham Biosciences), as a loading control (Amici et al., 2006). RNA was quantified by Molecular Dynamics Typhoon‐8600 imager (GE Healthcare) with the use of ImageQuant (Amersham‐GE Healthcare) software [Molecular Dynamics Analysis (MDA)].
Immunofluorescence microscopy
MA104 cells grown on coverslips were fixed with 4% paraformaldehyde and permeabilized with 0.5% Triton X‐100 PBS. After nuclear staining with 4′,6‐diamidino‐2‐phenylindole (Sigma‐Aldrich), cells were incubated with polyclonal anti‐VSV or anti‐phospho‐eIF2α antibodies, and monoclonal anti‐α‐tubulin antibodies. After washing, cells were incubated with fluorescein isothiocyanate‐conjugated anti‐rabbit and rhodamine‐conjugated anti‐mouse antibodies for VSV or phospho‐eIF2α and α‐tubulin respectively. Images were captured and deconvolved with a DeltaVision microscope (Applied‐Precision LLC) using the SoftWoRx‐2.50 software (Applied Precision). The overlay of the three fluorochromes is shown. eIF2α images were captured with a Leica DMLB microscope equipped with UV excitation filters and Leica DC300 camera using Leica Image‐Manager500 software.
Electrophoretic mobility shift assay (EMSA)
Aliquots of total extracts (12 μg) prepared after lysis in high‐salt extraction buffer (Rossi et al., 2000) were incubated with a [32P]‐labelled HSE DNA probe (Rossi et al., 2006) followed by analysis of DNA‐binding activity by EMSA on non‐denaturing 4% polyacrylamide gels (Caselli et al., 2007). Specificity of protein–DNA complexes was verified by competition with unlabeled probes and supershift with polyclonal antibodies specific for HSF1, as described (Rossi et al., 2006). Quantitative evaluation of HSF–HSE complex formation was determined by Typhoon 8600 imager with the use of ImageQuant software.
Statistical analysis
Statistical analysis was performed using the Student's t‐test for unpaired data. In virus titration experiments, data represent the mean ± standard deviation of duplicate samples from a representative experiment of three with similar results. P‐values of < 0.05 were considered significant.
Conflicts of interest
All contributors declare that they have no conflict of interest.
Supporting information
Fig. S1. Indomethacin inhibits VSV replication in different types of cells.
Fig. S2. Effect of INDO on MA104 cell proliferation.
Fig. S3. Indomethacin cytoprotective activity is independent of the host heat shock response.
Fig. S4. Different COX inhibitors do not mimic the antiviral effect of INDO.
Fig. S5. Effect of INDO treatment before and after VSV infection.
Fig. S6. Effect of thapsigargin and MG132 on VSV‐G protein levels.
Fig. S7. Effect of INDO on VSV replication in GCN2−/− and PERK−/− cells.
Acknowledgements
We thank Dr. Enrique Rodriguez‐Boulan (Cornell University Medical School, New York, NY) for the anti‐VSV antibodies, Dr. J.K. Rose (Yale University School of Medicine, New Haven, CT) for the pN4 plasmid and Dr. Randal Kaufman (University of Michigan Medical Center, Ann Arbor, MI) for providing GCN2−/−, PERK−/− and wild‐type MEFs. We also thank Palma Mattioli for assistance with the DeltaVision Imaging system. This work was supported by grants from the Italian Ministry of University and Scientific Research (PRIN project no. 2010PHT9NF‐006).
Amici, C. , La Frazia, S. , Brunelli, C. , Balsamo, M. , Angelini, M. , and Santoro, M. G. (2015), Inhibition of viral protein translation by indomethacin in vesicular stomatitis virus infection: role of eIF2α kinase PKR . Cell Microbiol, 17, 1391–1404. doi: 10.1111/cmi.12446.
References
- Amici, C. , Rossi, A. , and Santoro, M.G. (1995) Aspirin enhances thermotolerance in human erythroleukemic cells: an effect associated with the modulation of the heat shock response. Cancer Res 55: 452–457. [PubMed] [Google Scholar]
- Amici, C. , Di Caro, A. , Ciucci, A. , Chiappa, L. , Castelletti, C. , Martella, V. , et al. (2006) Indomethacin has a potent antiviral activity against SARS coronavirus. Antivir Ther 11: 1021–1030. [PubMed] [Google Scholar]
- Bahrami, H. , Daryani, N.E. , Haghpanah, B. , Moayyeri, A. , Moghadam, K.F. , Mirmomen, S. , and Kamangar, F. (2005) Effects of indomethacin on viral replication markers in asymptomatic carriers of hepatitis B: a randomized, placebo‐controlled trial. Am J Gastroenterol 100: 856–861. [DOI] [PubMed] [Google Scholar]
- Baltzis, D. , Qu, L.K. , Papadopoulou, S. , Blais, J.D. , Bell, J.C. , Sonenberg, N. , and Koromilas, A.E. (2004) Resistance to vesicular stomatitis virus infection requires a functional cross talk between the eukaryotic translation initiation factor 2alpha kinases PERK and PKR. J Virol 78: 12747–12761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benet, L.Z. (1996) Design and optimization of dosage regiments; pharmacokinetic data. In Goodman & Gilman's: The Pharmacological Basis of Therapeutics. Hardman, J.G. , Limbird, L.E. , Molinoff, P.B. , Ruddon, R.W. , and Gilman, A.G. (eds). New York: McGraw‐Hill, pp. 1707–1793. [Google Scholar]
- Berlanga, J.J. , Ventoso, I. , Harding, H.P. , Deng, J. , Ron, D. , Sonenberg, N. , et al. (2006) Antiviral effect of the mammalian translation initiation factor 2α kinase GCN2 against RNA viruses. EMBO J 5: 1730–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourinbaiar, A.S. , and Lee‐Huang, S. (1995) The non‐steroidal anti‐inflammatory drug, indomethacin, as an inhibitor of HIV replication. FEBS Lett 360: 85–88. [DOI] [PubMed] [Google Scholar]
- Brunelli, C. , Amici, C. , Angelini, M. , Fracassi, C. , Belardo, G. , and Santoro, M.G. (2012) The non‐steroidal anti‐inflammatory drug indomethacin activates the eIF2α kinase PKR, causing a translational block in human colorectal cancer cells. Biochem J 443: 379–386. [DOI] [PubMed] [Google Scholar]
- Caselli, E. , Fiorentini, S. , Amici, C. , Di Luca, D. , Caruso, A. , and Santoro, M.G. (2007) Human herpesvirus 8 acute infection of endothelial cells induces monocyte chemoattractant protein 1‐dependent capillary‐like structure formation: role of the IKK/NF‐κB pathway. Blood 109: 2718–2726. [DOI] [PubMed] [Google Scholar]
- Chen, N. , Warner, J.L. , and Reiss, C.S. (2000) NSAID treatment suppresses VSV propagation in mouse CNS. Virology 276: 44–51. [DOI] [PubMed] [Google Scholar]
- Chen, N. , Restivo, A. , and Reiss, C.S. (2002) Selective inhibition of COX‐2 is beneficial to mice infected intranasally with VSV. Prostaglandins Other Lipid Mediat 67: 143–155. [DOI] [PubMed] [Google Scholar]
- Clemens, M.J. (2004) Targets and mechanisms for the regulation of translation in malignant transformation. Oncogene 23: 3180–3188. [DOI] [PubMed] [Google Scholar]
- Connor, J.H. , and Lyles, D.S. (2002) Vesicular stomatitis virus infection alters the eIF4F translation initiation complex and causes dephosphorylation of the eIF4E binding protein 4E‐BP1. J Virol 70: 10177–10187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor, J.H. , and Lyles, D.S. (2005) Inhibition of host translation during vesicular stomatitis virus infection. J Biol Chem 280: 13512–13519. [DOI] [PubMed] [Google Scholar]
- Cotto, J.J. , Kline, M. , and Morimoto, R.I. (1996) Activation of heat shock factor 1 DNA binding precedes stress‐induced serine phosphorylation. Evidence for a multistep pathway of regulation. J Biol Chem 271: 3355–3358. [DOI] [PubMed] [Google Scholar]
- Dabo, S. , and Meurs, E.F. (2012) dsRNA‐dependent protein kinase PKR and its role in stress, signaling and HCV infection. Viruses 4: 2598–2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donzé, O. , Deng, J. , Curran, J. , Sladek, R. , Picard, D. , and Sonenberg, N. (2004) The protein kinase PKR: a molecular clock that sequentially activates survival and death programs. EMBO J 23: 564–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durbin, R.K. , Mertz, S.E. , Koromilas, A.E. , and Durbin, J.E. (2002) PKR protection against intranasal vesicular stomatitis virus infection is mouse strain dependent. Viral Immunol 15: 41–51. [DOI] [PubMed] [Google Scholar]
- Gallione, C.J. , Greene, J.R. , Iverson, L.E. , and Rose, J.K. (1981) Nucleotide sequences of the mRNA's encoding the vesicular stomatitis virus N and NS proteins. J Virol 39: 529–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García, M.A. , Gil, J. , Ventoso, I. , Guerra, S. , Domingo, E. , Rivas, C. , and Esteban, M. (2006) Impact of protein kinase PKR in cell biology: from antiviral to antiproliferative action. Microbiol Mol Biol Rev 70: 1032–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebauer, F. , and Hentze, M.W. (2004) Molecular mechanisms of translational control. Nature Rev Mol Cell Biol 5: 827–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil, J. , Alcamí, J. , and Esteban, M. (2000) Activation of NF‐κB by the dsRNA‐dependent protein kinase, PKR involves the IκB kinase complex. Oncogene 19: 1369–1378. [DOI] [PubMed] [Google Scholar]
- Holcik, M. , and Sonenberg, N. (2005) Translational control in stress and apoptosis. Nature Rev Mol Cell Biol 6: 6318–6327. [DOI] [PubMed] [Google Scholar]
- Inglot, A.D. (1969) Comparison of the antiviral activity in vitro of some non‐steroidal anti‐inflammatory drugs. J Gen Virol 4: 203–214. [DOI] [PubMed] [Google Scholar]
- Krishnamoorthy, J. , Mounir, Z. , Raven, J.F. , and Koromilas, A.E. (2008) The eIF2α kinases inhibit vesicular stomatitis virus replication independently of eIF2α phosphorylation. Cell Cycle 7: 2346–2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyles, D.S. , Kuzmin, I.V. , and Rupprecht, C.E. (2013) Rhabdoviridae. In Field's Virology. Knipe, D.M. , and Howley, P. (eds). New York: Raven Press, pp. 885–922. [Google Scholar]
- Morimoto, R.I. , and Santoro, M.G. (1998) Stress‐inducible responses and heat shock proteins: new pharmacologic targets for cytoprotection. Nature Biotechnol 16: 833–838. [DOI] [PubMed] [Google Scholar]
- Mukherjee, P.K. , and Simpson, R.W. (1987) Transcriptionally defective nucleocapsids of vesicular stomatitis virus from cells treated with indomethacin. Virology 156: 25–31. [DOI] [PubMed] [Google Scholar]
- Neznanov, N. , Dragunsky, E.M. , Chumakov, K.M. , Neznanova, L. , Wek, R.C. , Gudkov, A.V. , and Banerjee, A.K. (2008) Different effect of proteasome inhibition on vesicular stomatitis virus and poliovirus replication. PLoS ONE 3: e1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez, J. , Alcami, J. , Arnaiz‐Villena, A. , Regueiro, J.R. , Rioperez, E. , Yague, J. , and Fernandez, F. (1986) Indomethacin in the relief of AIDS symptoms. Lancet 2: 570. [DOI] [PubMed] [Google Scholar]
- Rossen, J.W. , Bouma, J. , Raatgeep, R.H. , Büller, H.A. , and Einerhand, A.W. (2004) Inhibition of cyclooxygenase activity reduces rotavirus infection at a postbinding step. J Virol 78: 9721–9730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi, A. , Elia, G. , and Santoro, M.G. (1996) 2‐Cyclopenten‐1‐one, a new inducer of heat shock protein 70 with antiviral activity. J Biol Chem 271: 32192–32196. [DOI] [PubMed] [Google Scholar]
- Rossi, A. , Kapahi, P. , Natoli, G. , Takahashi, T. , Chen, Y. , Karin, M. , and Santoro, M.G. (2000) Anti‐inflammatory cyclopentenone prostaglandins are direct inhibitors of IκB kinase. Nature 403: 103–118. [DOI] [PubMed] [Google Scholar]
- Rossi, A. , Ciafrè, S. , Balsamo, M. , Pierimarchi, P. , and Santoro, M.G. (2006) Targeting the heat shock factor 1 by RNA interference: a potent tool to enhance hyperthermochemotherapy efficacy in cervical cancer. Cancer Res 66: 7678–7685. [DOI] [PubMed] [Google Scholar]
- Santoro, M.G. (1997) Antiviral activity of cyclopentenone prostanoids. Trends Microbiol 5: 276–281. [DOI] [PubMed] [Google Scholar]
- Santoro, M.G. , Rossi, A. , and Amici, C. (2003) NF‐kappaB and virus infection: who controls whom. EMBO J 22: 2552–2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheuner, D. , Patel, R. , Wang, F. , Lee, K. , Kumar, K. , Wu, J. , et al. (2006) Double‐stranded RNA‐dependent protein kinase phosphorylation of the alpha‐subunit of eukaryotic translation initiation factor‐2 mediates apoptosis. J Biol Chem 281: 21458–21468. [DOI] [PubMed] [Google Scholar]
- Schneider, R.J. , and Mohr, I. (2003) Translation initiation and viral tricks. Trends Biochem Sci 28: 130–136. [DOI] [PubMed] [Google Scholar]
- Schröer, J. , and Shenk, T. (2008) Inhibition of cyclooxygenase activity blocks cell‐to‐cell spread of human cytomegalovirus. Proc Natl Acad Sci USA 105: 19468–19473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stojdl, D.F. , Abraham, N. , Knowles, S. , Marius, R. , Brasey, A. , Lichty, B.D. , et al. (2000) The murine double‐stranded RNA‐dependent protein kinase PKR is required for resistance to vesicular stomatitis virus. J Virol 74: 9580–9585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symensma, T.L. , Martinez‐Guzman, D. , Jia, Q. , Bortz, E. , Wu, T.T. , Rudra‐Ganguly, N. , et al. (2003) COX‐2 induction during murine gammaherpesvirus 68 infection leads to enhancement of viral gene expression. J Virol 77: 12753–12763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor, S.S. , Haste, N.M. , and Ghosh, G. (2005) PKR and eIF2alpha: integration of kinase dimerization, activation, and substrate docking. Cell 122: 823–825. [DOI] [PubMed] [Google Scholar]
- Vane, J.R. , and Botting, R.M. (1997) Mechanism of action of anti‐inflammatory drugs. Adv Exp Med Biol 433: 131–138. [DOI] [PubMed] [Google Scholar]
- Zhu, H. , Cong, J.P. , Yu, D. , Bresnahan, W.A. , and Shenk, T.E. (2002) Inhibition of cyclooxygenase 2 blocks human cytomegalovirus replication. Proc Natl Acad Sci USA 99: 3932–3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Indomethacin inhibits VSV replication in different types of cells.
Fig. S2. Effect of INDO on MA104 cell proliferation.
Fig. S3. Indomethacin cytoprotective activity is independent of the host heat shock response.
Fig. S4. Different COX inhibitors do not mimic the antiviral effect of INDO.
Fig. S5. Effect of INDO treatment before and after VSV infection.
Fig. S6. Effect of thapsigargin and MG132 on VSV‐G protein levels.
Fig. S7. Effect of INDO on VSV replication in GCN2−/− and PERK−/− cells.