

Summary
Borna disease virus (BDV) is a non‐segmented negative‐stranded RNA virus that maintains a strictly neurotropic and persistent infection in affected end hosts. The primary target cells for BDV infection are brain cells, e.g. neurons and astrocytes. The exact mechanism of how infection is propagated between these cells and especially the role of the viral glycoprotein (GP) for cell–cell transmission, however, are still incompletely understood. Here, we use different cell culture systems, including rat primary astrocytes and mixed cultures of rat brain cells, to show that BDV primarily spreads through cell–cell contacts. We employ a highly stable and efficient peptidomimetic inhibitor to inhibit the furin‐mediated processing of GP and demonstrate that cleaved and fusion‐active GP is strictly necessary for the cell‐to‐cell spread of BDV. Together, our quantitative observations clarify the role of Borna disease virus‐glycoprotein for viral dissemination and highlight the regulation of GP expression as a potential mechanism to limit viral spread and maintain persistence. These findings furthermore indicate that targeting host cell proteases might be a promising approach to inhibit viral GP activation and spread of infection.
Keywords: fusion‐mediated virus infection, cleavage of virus glycoprotein, furin inhibitor
Introduction
The classical mammalian Borna disease virus (BDV) naturally infects horses and sheep (Herden et al., 2013) presumably through open nerve endings in the nasal and pharyngeal mucosa and is subsequently transmitted to the central nervous system (CNS). After initial infection, BDV spreads from the olfactory nerve into the brain, including the nucleus caudatus, hippocampus and the cerebral cortex and maintains a strictly neurotropic and persistent infection in immune competent end hosts (Herden et al., 2013). The resulting Borna disease (BD) is a severe and fatal neurological disorder characterized by virus‐induced immunopathological reactions that lead to non‐purulent encephalomyelitis (Herden et al., 2013). Currently, curative therapy is not possible, and vaccination is hampered by the underlying immunopathogenesis. Viral transmission is thought to occur via secretion and excretions of infected animals in case of disseminated virus distribution, e.g. natural infection of clinically healthy reservoir animals (Bourg et al., 2013). The average seroprevalence of BDV‐specific antibodies in clinically healthy horses in central Europe reaches ~20% in endemic regions, whereas it is considerably higher in BDV‐infected flocks, although only very few individual animals develop clinically manifested BD. In addition to horses and sheep, BDV can also infect several other animal species, including rabbits, mice, rats, guinea pigs, dogs, chicken, cows and non‐human primates (reviewed in Kinnunen et al., (2013) and Herden et al., (2013)). Furthermore, avian Bornaviruses have been identified as aetiologic agents of proventricular dilatation disease, which occurs mainly in psittacine birds (Staeheli et al., 2010; Payne et al., 2011; Piepenbring et al., 2012). Bornavirus sequences with a homology of about 60% to the classical BDV have recently been detected in breeders of variegated squirrels suffering from fatal encephalitis and in a contact animal (Hoffmann et al., 2015). Moreover, distantly related Bornavirus sequences have been identified a number of other animals (Kuhn et al., 2015), and BDV‐like gene elements are present in the genome of several mammalian species (Horie et al., 2010; Fujino et al., 2014). These findings demonstrate that Bornaviruses are much more diverse, and the spectrum of infected animals is much wider than anticipated. Although antibodies interacting with BDV were isolated from blood of human patients with psychiatric disorders (Rott et al., 1985), no reliable link between a psychiatric disease and the classical mammalian BDV infection has been demonstrated (Arias et al., 2012; Hornig et al., 2012).
The virions of BDV are spherical, enveloped particles with diameters between 70 and 130 nm (Kohno et al., 1999), which contain a single‐stranded RNA genome of negative polarity (Briese et al., 1992). The genomic RNA has a length of ~8.9 kb and comprises at least six partially overlapping open reading frames (Briese et al., 1994; Cubitt et al., 1994b), which encode the nucleoprotein N (BDV‐N), X‐protein, phosphoprotein P, matrix protein M, glycoprotein GP and the viral polymerase L (Herden et al., 2013). Because of the organization of its genome, BDV has been classified into the order Mononegavirales (Briese et al., 1994; Cubitt et al., 1994b), where it constitutes the prototypic member of the family Bornaviridae, numerous new members of which have been identified in recent years. Unusual among the Mononegavirales, the genome of BDV is transcribed in the nucleus (Briese et al., 1992; Cubitt and de la Torre, 1994a), and gene expression is regulated by differential splicing, transcriptional read‐through and limited nuclear export of viral mRNAs (Briese et al., 1994; Cubitt et al., 1994c; Schneider et al., 1994; Werner‐Keiss et al., 2008).
The replication cycle of BDV begins with receptor binding and fusion with the host endosomal membrane, mediated by the N‐terminal and C‐terminal cleavage products of the glycoprotein GP. GP is initially encoded as a ~57 kDa precursor protein pre‐GP, which is co‐translationally inserted into the endoplasmic reticulum (ER) membrane as a type I membrane protein. Cleavage of the signal peptide from pre‐GP yields the immature GP, whose molecular weight is shifted to ~94 kDa by extensive N‐glycosylation (Schneider et al., 1997). Early during the transport along the secretory pathway from the ER to the plasma membrane, GP is further processed by the host cell protease furin (Gonzalez‐Dunia et al., 1998; Richt et al., 1998; Eickmann et al., 2005). Furin is an endoprotease that preferentially cleaves its substrates at the C‐terminus of the consensus sequence Arg‐X‐(Arg/Lys)‐Arg or at the less common sequence Arg‐X‐X‐Arg. Its physiological role is the activation of numerous cellular proproteins, e.g. matrix metalloproteinase I, nerve cell growth factor, von Willebrand factor or insulin receptor (Seidah and Prat, 2002; Thomas, 2002). In addition, furin is exploited by a number of pathogens, including bacteria and many enveloped viruses, which utilize furin‐mediated cleavage to activate proteins such as bacterial toxins, viral membrane proteins or fusogenic glycoproteins (Thomas, 2002). In the case of BDV, furin cleaves the GP into the distal, amino‐terminal subunit GP‐N (51 kDa) and the carboxy‐terminal, membrane‐anchored subunit of GP (GP‐C) (43 kDa) (Richt et al., 1998; Kiermayer et al., 2002).
Cleavage of the BDV glycoprotein (BDV‐GP) is a prerequisite for the incorporation of the glycoprotein‐spikes into virus particles, which is thought to ensure virion infectivity (Eickmann et al., 2005). GP‐N is exposed on the trimeric glycoprotein‐spike and binds to a still unknown host cell receptor, while GP‐C possesses all the characteristics of a viral fusion protein required for host cell infection (Gonzalez‐Dunia et al., 1998; Perez et al., 2001). While it is generally accepted that the incorporation of cleaved BDV‐GP into virus particles is required for initial infection of cells, studies have called into question the role of BDV‐GP for the subsequent cell‐to‐cell spread, which is presumed to occur by passage of non‐enveloped viral ribonucleoprotein (vRNP) complexes in the CNS (Cubitt and de la Torre, 1994a). In contrast, other studies showed that virus spread is dependent on expression of GP and can be inhibited by neutralizing antibodies directed against GP, demonstrating the necessity of the glycoprotein for BDV dissemination (Bajramovic et al., 2003).
The purpose of this study was to re‐evaluate and clarify if and how BDV spreads from cell to cell and how BDV‐GP contributes to this process. We analysed virus dissemination in two cell lines differing in their capacity to release viral particles and used cultures of astrocytes as well as mixed brain cell cultures as natural host cell models, in order to determine general mechanisms as well as possible differences between cell culture systems. Furthermore, we show that a highly stable and efficient peptidomimetic furin inhibitor 4‐(guanidinomethyl) phenylacetyl‐Arg‐Val‐Arg‐4‐amidinobenzylamide (MI‐0701) (Becker et al., 2012; Hardes et al., 2015; Lu et al., 2015) is an excellent tool to block cleavage and fusion mediated by the glycoprotein of BDV. Using this inhibitor, we demonstrate a strict correlation between BDV‐GP cleavage and successful spread of BDV. Moreover, our quantitative data show the dominant role of activated, fusogenic BDV‐GP for virus spread, indicating that interference with viral glycoprotein activation might be a feasible option to limit or inhibit spread of viruses employing cellular enzymes such as furin.
Results
Quantification of BDV release from MDCK and Vero E6 cells
Previous studies have shown that certain cell types, such as HeLa cells, can only be infected with BDV through co‐cultivation with cells already infected with BDV, indicating that the virus spreads via cell–cell contacts (Herzog and Rott, 1980). However, other cell lines including Madin–Darby canine kidney (MDCK) cells, shed low amounts of infectious particles into the supernatants of cell cultures persistently infected with BDV (Stitz et al., 1998). These observations raise the question about the role of cell‐free virus for the dissemination of BDV infection and highlight the need for a comparison of BDV release from different cell types in a more quantitative way. Therefore, we examined the release of BDV from infected MDCK and Vero E6 cells, because both cell lines are similarly susceptible to infection by cell‐free BDV (Herzog and Rott, 1980). We first generated a Vero E6 cell line persistently infected with BDV by incubating non‐infected Vero E6 cells with virus isolated from the supernatant of stably infected MDCK cells. Infection of the cell layer was monitored by immunofluorescence detection of BDV‐N. After 18 cell passages, all cells were infected with BDV (data not shown). We then compared the extent of BDV release from both Vero E6 and MDCK cells. For this, we isolated particles from cell culture supernatants of persistently infected MDCK (MDCK‐BDV) and Vero E6 (Vero E6‐BDV) cells (Fig. 1A) and analysed them by immunoblotting. While the viral nucleoprotein N and the matrix protein M were readily detectable in lysates of both cell lines (Fig. 1A), both proteins could only be detected in the supernatants of BDV‐infected MDCK cells but not in supernatants of BDV‐infected Vero E6 cells (Fig. 1A). This shows that even though BDV infection is propagated in both cell lines, as demonstrated by the similar expression of BDV‐N and BDV‐M, Vero E6 cells infected with BDV do not release detectable amounts of virus into the supernatant.
Figure 1.
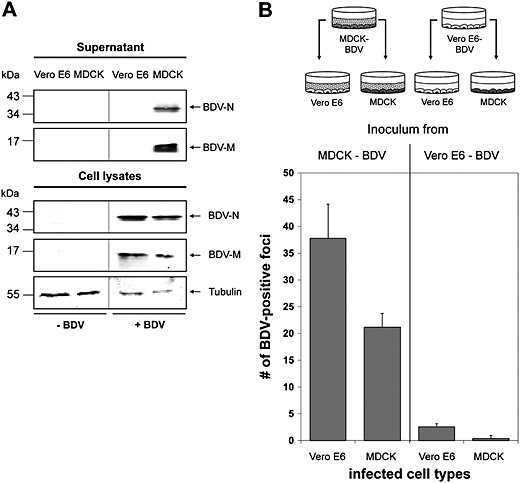
Cell type‐dependent release of free BDV particles. A. Persistently BDV‐infected MDCK and Vero E6 cells were grown to confluency. Uninfected cells served as controls. After further incubation for 4 days, BDV was isolated from the cell culture supernatant and subjected to SDS‐PAGE followed by immunoblotting using BDV‐N‐specific and BDV‐M‐specific antisera. Tubulin present in the cell lysates was used as a loading control and detected using an anti‐tubulin antibody. B. Non‐infected MDCK and non‐infected Vero E6 cells were grown on glass coverslips and incubated for 48 h with pre‐cleared cell culture supernatants from either persistently BDV‐infected MDCK or persistently‐BDV infected Vero E6 cells as depicted. After further incubation for 48 h, each cell culture was fixed, incubated with a BDV‐N specific antibody and stained using an HRP‐coupled secondary antibody and TrueBlue as substrate. BDV‐infected cell foci were counted in three randomly chosen areas.
Next, we determined whether supernatants of MDCK‐BDV and Vero E6‐BDV cells contain infectious virus by parallel infection of Vero E6 and MDCK cells as shown in Fig. 1B. After 4 days of incubation, the infected cell cultures were immunostained for BDV‐N, and immunopositive foci were counted. While the supernatant of MDCK‐BDV cells was infectious for non‐infected MDCK and non‐infected Vero E6 cells (Fig. 1B, left panel in diagram), both cell lines show an extremely small number of immunopositive foci after infection with the supernatant of Vero E6‐BDV cells (Fig. 1B, right panel). Taken together, these observations show that MDCK cells infected with BDV release much more infectious virus particles than BDV‐infected Vero E6 cells.
Kinetics of BDV spread in MDCK and Vero E6 cells
Because MDCK and Vero E6 cells showed significant differences in viral release, we next investigated how BDV infection spreads in these two cell lines. To this end, a small number of persistently BDV‐infected MDCK or Vero E6 cells were mixed with a 400‐fold excess of homologous non‐infected cells. The mixed cultures were grown to confluence in Petri dishes containing coverslips and incubated up to 7 days. Every 24 h, one coverslip was removed from each cell culture, and virus spread was analysed by immunostaining against BDV‐N (Fig. 2A and B). For both cell lines, we observed that infected cells appeared in clusters (foci) that grew in size with prolonged incubation. Furthermore, after longer incubation time, we only detected a very small number of secondary clusters, which contained a considerable lower number of immunostained cells (Fig. 2A and B). Because only single infected cells were present in both cell cultures at 24 h post inoculation, the larger foci specifically originated from primary infected cells, indicating that in both MDCK and Vero E6 cells BDV spreads primarily through contact sites between single, infected cells and neighbouring uninfected cells. In addition, for both cell lines, we observed only very minor differences in the number of infected cells per focus after 168 h of incubation, indicating that the extent of virus spread within the MDCK and Vero E6 cell cultures is comparable (Fig. 2C).
Figure 2.
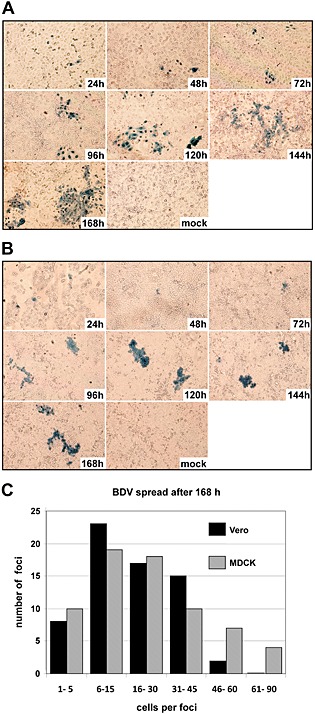
Kinetics of BDV spread in MDCK and Vero E6 cell cultures. Non‐infected MDCK cells (A) and Vero E6 cells (B) were mixed with homologous, infected cells at a ratio of 1 infected to 400 uninfected cells and were grown on glass coverslips. At the indicated time points, individual coverslips were removed; the cells were fixed, permeabilized, incubated with a BDV‐N‐specific antibody and stained using an HRP‐coupled secondary antibody and TrueBlue substrate to visualize BDV‐infected cells. The non‐infected control cells (mock) were analysed at 168 h after seeding on coverslips. (C) Quantification of virus spread. MDCK and Vero E6 cells were infected as described for (A) and (B). After 168 h, the number of immunostained cells per focus was counted and foci were categorized depending on the number of BDV‐positive cells per focus.
Reduction of GP cleavage in cells treated with the furin inhibitor MI‐0701
The observation that spread of BDV infection predominantly occurs via cell–cell contacts (Fig. 2) rather than by cell‐free virus (Fig. 1) raises the question about the impact of BDV‐GP for this spread. The importance of cleaved and fusion‐competent BDV‐GP for virus dissemination through cell–cell contacts is still controversial and has not yet been fully clarified (Bajramovic et al., 2003; Clemente and de la Torre, 2007) We therefore re‐investigated this issue by using MI‐0701, a physiologically stable, very efficient furin inhibitor (Fig. 3A) (Becker et al., 2012) to block the proteolytic processing of BDV‐GP.
Figure 3.
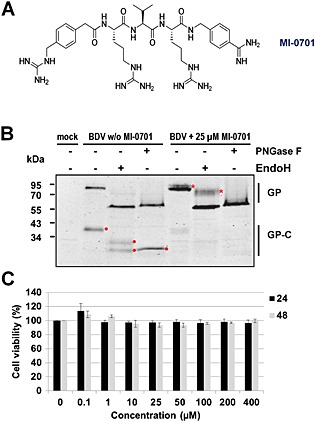
Inhibition of BDV‐GP cleavage by the furin inhibitor MI‐0701.
A. Structure of 4‐(guanidinomethyl)‐phenylacetyl‐Arg‐Val‐Arg‐4‐amidino‐benzylamide, denoted as inhibitor MI‐0701 (Becker et al., 2012).
B. Persistently BDV‐infected MDCK cells were mixed with excess of non‐infected MDCK cells (1:10) and grown in the presence of 25 μM of MI‐0701 or without MI‐0701 as control. After 72 h, the cells were lysed and subjected to lectin precipitation. Aliquots of the cells were either treated or not treated with Endo H or PNGase F respectively. All samples were subjected to SDS‐PAGE and immunoblotting using a BDV‐GPC‐specific rabbit serum that recognizes GP (94 kDa), the cleaved forms of GP‐C (43 kDa) and the respective deglycosylated forms GPdeclyc (57 kDa) and GP‐Cdeglyc (23 kDa) (Richt et al., 1998). The glycosylation variants of non‐cleaved GP are indicated by asterisks, and the positions of GP‐C forms are indicated by dots. Non‐infected MDCK cells treated without inhibitor served as negative control.
(C) Cell viability test: MDCK cells were incubated with culture medium containing variable concentrations of MI‐0701 or left untreated as control. After 24 and 48 h, the cell viability was measured using the MTT assay. The viability of cells not treated with MI‐0701 was set to 100%.
To analyse the effect of MI‐0701 on BDV‐GP cleavage, MDCK‐BDV cells were mixed with non‐infected MDCK cells (1:10), co‐cultivated and treated with 25 μM of MI‐0701 for 72 h. Viral glycoproteins were enriched by lectin precipitation from cell lysates and subjected either to Endo H or PNGase F treatment or left untreated before uncleaved GP, and its cleavage product GP‐C were identified by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS‐PAGE) and immunoblotting using a BDV‐GP‐specific antiserum (Fig. 3B). MI‐0701 significantly blocked the proteolytic processing of GP as demonstrated by the reduction of the amount of GP‐C and a corresponding increase of uncleaved GP (Fig. 3B). Uncleaved GP appears as dominant band and a minor band, which represent differently glycosylated GP, as demonstrated by its susceptibility to enzymatic deglycosylation with Endo H and PNGase F respectively (Fig. 3B). Importantly, the cell viability was not affected by treatment of cells at 25 μM or even significantly higher concentrations of MI‐0701 (Fig. 3C) (Becker et al., 2012).
Reduction of BDV‐GP mediated cell‐to‐cell fusion by the furin inhibitor MI‐0701
Correct proteolytic processing by cellular proteases is a prerequisite for the activity of many different fusogenic viral glycoproteins (Klenk and Garten, 1994; Pasquato et al., 2013), including BDV‐GP (Richt et al., 1998). To verify whether the inhibition of BDV‐GP cleavage prevents the fusogenic activity of GP, we analysed the fusion activity of cell surface expressed BDV‐GP in the presence of MI‐0701. For this, we measured syncytia formation when Vero E6 cells persistently infected with BDV were exposed to pH 5.5 for a short time, to simulate the low pH of the endosomal milieu. This treatment triggers fusion of neighbouring BDV‐infected cells (Gonzalez‐Dunia et al., 1998) only when cleaved GP‐N/GP‐C spikes are present. We observed that only infected Vero E6‐BDV cells formed syncytia when exposed to low pH (Fig. 4A). We then analysed syncytia formation between Vero E6‐BDV cells incubated with different concentrations of MI‐0701 and measured polykaryon formation (Fig. 4B and C). Cell‐to‐cell fusion was significantly blocked by the furin inhibitor in a dose‐dependent manner and completely abolished after treatment of cells with 25 μM of MI‐0701. This demonstrates that MI‐0701 efficiently inhibits the fusion activity of BDV‐GP.
Figure 4.
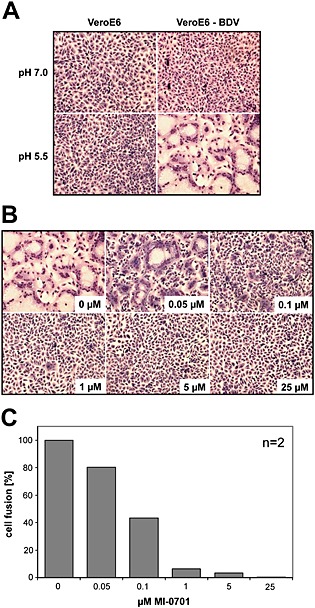
Effect of the inhibition of GP‐cleavage on the fusion activity of surface‐expressed BDV‐GP.
A. Confluent Vero E6 cell cultures either BDV‐infected or non‐infected as control were incubated at a pH of either 7.0 or 5.5 for 5 min, incubated for 90 min in normal culture medium and fixed and stained with Giemsa.
B. BDV‐infected Vero E6 cells were incubated with the indicated concentrations of MI‐0701 and grown to confluence. The cells were then incubated at a pH of 5.5 as described in (A), fixed and stained with Giemsa.
C. Quantification of two independent experiments, conducted as described in (B). For quantification, the number of nuclei within syncytia was divided by the total number of nuclei.
Correlation between cell‐to‐cell spread of BDV and cleavage of GP in MDCK cells
The observation that MI‐0701 blocks the proteolytic processing of GP (Fig. 3) and GP‐mediated fusion (Fig. 4) prompted us to use MI‐0701 to further investigate the role of the viral glycoprotein for the dissemination of BDV in cell culture. For this, we analysed the effect of the furin inhibitor on the spread of BDV in mixed cultures of infected and uninfected cells. To discern cells originally infected with BDV (seed cells) from newly infected cells, we mixed MDCK‐BDV cells with a 100‐fold excess of non‐infected MDCK cells expressing fluorescently labelled sucrose‐isomaltase yellow fluorescent protein (MDCK‐SI‐YFP). The mixed cell cultures were maintained on coverslips for up to 120 h in the presence of MI‐0701 or kept without inhibitor as a control. At different time points, individual samples were fixed, immunostained against BDV‐N and subjected to fluorescence microscopic analysis. Newly infected cells were identified by double fluorescence for yellow fluorescent protein (YFP) and BDV‐N, while seed cells were identified by positive immunostaining of BDV‐N and lack of YFP fluorescence. In the absence of MI‐0701, the first newly infected cells appeared after 48 h in the nearest neighbourhood of seed cells (Fig. 5A), demonstrating BDV transmission from a cell persistently infected with BDV to neighbouring, non‐infected cells. With increased incubation time, we observed a remarkable increase in the number of newly BDV‐infected cells (Fig. 5A: without MI‐0701). In contrast, when the cells were treated with MI‐0701, we observed only a very slow and minor increase in the formation of double fluorescent cell foci over time (Fig. 5B: with 25 μM of MI‐0701). Importantly, newly infected cells were always clustered around seed cells and in contact with each other (Fig. 5A and B). Furthermore, we quantitatively analysed the spread of BDV in MDCK cell cultures in the presence and in the absence of MI‐0701, which demonstrated a significant reduction of viral cell‐to‐cell transmission in the presence of MI‐0701 (Fig. 5C). Taken together, these observations clearly demonstrate not only that the infection with BDV is primarily disseminated by cell‐to‐cell spread but also show that BDV‐GP is highly important for this spread via cell–cell contacts.
Figure 5.
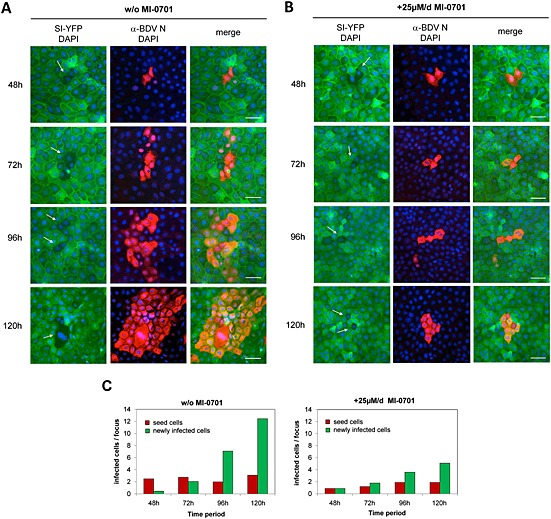
Cell‐to‐cell spread of BDV in MDCK cells in the presence or absence of GP cleavage.
Non‐infected MDCK cells persistently expressing SI‐YFP were mixed with BDV‐infected MDCK cells (seed cells) at a ratio of 100:1 and grown on glass coverslips in the absence (A) and in the presence (B) of 25 μM of MI‐701 respectively. The culture medium was exchanged with fresh medium with or without inhibitor every 24 h. At the indicated time points, individual coverslips were removed and cells were fixed, permeabilized, incubated with a BDV‐N‐specific antibody and stained using a Cy5‐conjugated secondary antibody (red). Nuclei were stained with DAPI (blue). Arrows indicate the position of seed cells of which the nuclei were stained with DAPI. Scale bars, 50 µm. Virus spread inhibition was quantified by using two independent experiments.
C. The MI‐0701 mediated reduction of BDV spread was determined by comparison of the numbers of BDV infected cells, which are double labelled for fluorescence label with Cy5 and YFP (yellow fluorescence protein) at different time points (48, 72, 96 and 120 h).
Correlation between cell‐to‐cell spread of BDV and cleavage of GP in astrocytes
To further evaluate the importance of GP cleavage for the cell‐to‐cell spread of BDV, we next analysed the effect of MI‐0701 on BDV dissemination in primary rat astrocytes. For this, long‐term cultivated primary rat astrocytes, persistently infected with BDV, were mixed with a 100‐fold excess of homologous non‐infected astrocytes and seeded to confluence on coverslips. The mixed cultures were incubated for up to 23 days in the presence or absence of the furin inhibitor. At 2 and 23 days after seeding, individual samples were fixed and analysed for virus spread by immunostaining against BDV‐N (Fig. 6A), and the total number of BDV‐infected cells was determined (Fig. 6B). For astrocytes not treated with MI‐0701, we observed an increase of BDV‐infected cells from 3% on the second day to a total of 38% on day 23 (Fig. 6B). Importantly, cell culture supernatants did not contain infectious virus (data not shown), which indicates that cell–cell contacts are the primary route of BDV dissemination in astrocytes. In contrast to untreated cells, the quantity of infected cells for astrocytes cultured in the presence of MI‐0701 remained nearly constant and increased only very moderately because of cell division (Fig. 6A and B). This demonstrates that the presence of fusion‐active, proteolytically processed BDV‐GP is highly important for the spread of BDV in rat astrocytes through cell–cell contacts and in the absence of cell‐free virus.
Figure 6.
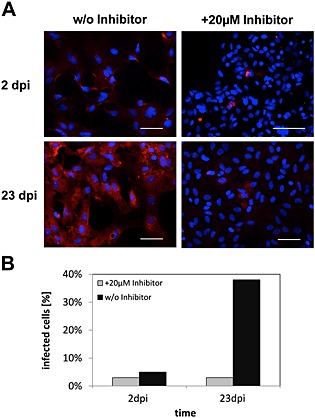
Cell‐to‐cell spread of BDV in primary rat astrocytes in the presence or absence of GP cleavage.
A. Non‐infected primary cortical rat astrocytes were mixed with BDV‐infected primary cortical rat astrocytes at a ratio of 100:1 and co‐cultivated for 23 days in the presence or absence of 20 μM of MI‐0701. At the indicated time points, individual samples were fixed, permeabilized, incubated with an antibody specific for BDV‐N and stained using a Cy5‐conjugated secondary antibody (red). Nuclei were visualized by staining with DAPI. Scale bars, 50 µm.
B. Quantification of total number of infected astrocytes.
Effect of GP cleavage for the spread of BDV in mixed cultures of brain cells
Next, we investigated the importance of cleaved, fusion‐active GP using mixed cultures of brain cells containing primarily astrocytic glia cells and neurons (Ott et al., 2010) as model system for the BDV spread in the brain. To this end, cells were isolated from rat cortical tissue, infected with BDV and maintained in the presence or absence of 10 μM of MI‐0701. To analyse viral spread, individual samples were fixed at 8 and 14 days post infection (dpi) and immunostained against BDV‐N, and the number of infected cells was determined by counting a total of 200 astrocytes and 200 neurons per sample. For cells not treated with MI‐0701, we observed a remarkable increase in BDV‐positive astrocytes over time (Fig. 7A, top panel). At 8 dpi, already 50% of astrocytes were infected, with a further increase to 70% BDV‐positive cells at 14 dpi (Fig. 7B). In contrast, treatment with 10 μM of MI‐0701 resulted in a significantly decreased spread of infection (Fig. 7A, bottom panel) with only 12% of BDV‐positive astrocytes at 8 dpi and a very minor increase to 16% infected cells at 14 dpi (Fig. 7B). Importantly, infection seems to occur via cell‐to‐cell contacts because infected astrocytes were present as single cells early during infection and arranged in small clusters that grew in size over time in the case of non‐treated cells, whereas these clusters did not appear in samples treated with MI‐0701 (Fig. 7A).
Figure 7.
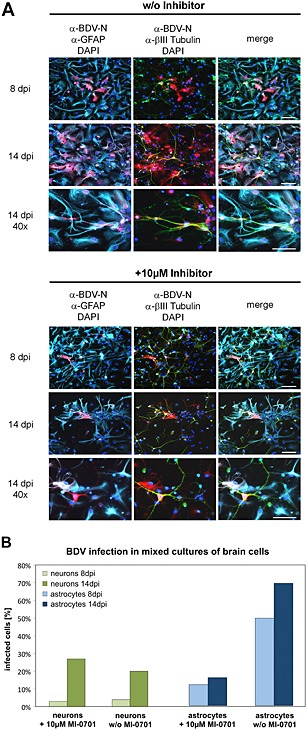
Influence of GP cleavage on the cell‐to‐cell spread of BDV in mixed cultures of brain cells.
A. Cultures of primary rat cortical astrocytes and neurons were infected with BDV and cultured for 14 days in the presence or absence of 10 μM of MI‐0701. At 8 and 14 dpi, individual samples were fixed and permeabilized. BDV was detected using a BDV‐N‐specific antibody and a Texas Red‐conjugated secondary antibody (red). Astrocytes were stained using an α‐GFAP antiserum and a Cy5‐conjugated secondary antibody (cyan). Neurons were detected using an α‐βIII Tubulin antiserum and an FITC‐conjugated secondary antibody (green). Nuclei were stained with DAPI. Scale bars, 50 µm.
B. Quantification of BDV‐infected astrocytes and neurons for an experiment conducted as described in (A).
In contrast to astrocytes, treatment with MI‐0701 seems to have no inhibiting effect on the spread of infection in neurons. We observed an increase in the number of infected neurons for both treated and untreated samples with 3% (with MI‐0701) vs 4% (non‐treated) infected neurons at 8 dpi and a slightly higher increase in the number of infected cells (27%) treated with MI‐0701 versus 20% (non‐treated) BDV‐positive neurons at 14 dpi (Fig. 7B).
In summary, these results show that BDV spreads predominantly via cell‐to‐cell contacts in mixed cultures of brain cells and that the inhibition of BDV‐GP cleavage very efficiently blocks BDV transmission between astrocytes at the applied concentration of MI‐0701, thereby highlighting the importance of BDV‐GP for the cell‐to‐cell spread among these cells.
Discussion
The classical mammalian BDV is a neurotropic virus that spreads in the CNS mainly through astrocytes and neurons (Rott and Becht, 1995). However, the mechanism and the viral components necessary for BDV dissemination between these cells are still unclear (Bajramovic et al., 2003; Clemente and de la Torre, 2007). In this study, we provide evidence that BDV primarily transmits through cell–cell contacts and show that the viral glycoprotein is a key determinant for spread of infection.
Our analysis of BDV spread in different cell lines demonstrates that even though Vero E6 and MDCK cells propagate BDV infection, they differ in their ability to release infectious virus (Fig. 1A). It is notable that Vero E6 cells seem to be more susceptible to BDV infection (Fig. 1B) and form a slightly larger number of smaller infected foci (Fig. 2C), whereas MDCK cells, which release more infectious viral particles form slightly more larger clusters of infected cells (Fig. 2C). One can speculate that the virus uptake, receptor density and/or the viral replication machinery are more efficient in Vero E6 cells, but the mechanism for release of free virus particles is more efficient in MDCK, which leads to the formation of slightly larger foci. Similar observations for the strong difference in the ability to release infectious virus have been made for neurons and astrocytes (Carbone et al., 1993), and together with the lack of detectable, cell‐free virus in the CNS (Gosztonyi et al., Gosztonyi and Ludwig, 1995), this has led to the hypothesis that BDV infection is propagated directly from cell to cell (Herden et al., 2013). Our quantitative analyses show that infection is predominantly transmitted through direct cell–cell contacts despite the cell‐type‐specific differences in viral release (Figs 2, 5 and 6). This indicates that cell‐free virus plays only a rather minor role during BDV infection and further supports the concept that cell‐to‐cell spread is the primary route of BDV dissemination, as schematically outlined in Fig. 8B. Other, alternative routes of BDV spread (discussed in the following) are also shown in Fig. 8.
Figure 8.
Models of potential pathways for the intercellular spread of BDV.
A. Budding and release of cell‐free virus, which infects long distant target cells, (B) nascent virus budding and simultaneous attachment to nearest neighbouring cells, followed by rapid endocytosis into the target cell, (C) GP‐independent intercellular transmission of free viral RNPs using nanotubes.
Our experiments and the previous application of a less specific furin inhibitor on BDV growth indicated that furin and/or some closely related proprotein convertases (members of the eukaryotic subtilase family) are necessary for the cleavage of BDV‐GP (Richt et al., 1998). This raises the question whether other relevant proteases other than furin‐like PCs may play a role for the activation of BDV‐GP, for which there are no indications so far. The only known protease that cleaves BDV‐GP in cell culture assays is exogenously added trypsin. Other trypsin‐like proteases have not been shown to substitute furin. Trypsin‐like proteases, such as type II transmembrane serine proteases including HAT (human airway trypsin‐like protease), TMPRSS2 (transmembrane protease, serine 2), TMPRSS4, TMPRSS13 and matriptase are blocked by various inhibitors quite different from those that are specific for furin and furin‐like proteases (Böttcher‐Friebertshäuser et al., 2013; Garten et al., 2015). It therefore seems highly unlikely that trypsin‐like proteases are suitable as activating proteases for BDV‐GP.
The expression level of BDV‐GP in infected astrocytes is very low during natural infection (Werner‐Keiss et al., 2008), and the fact that GP cleavage is essential for BDV propagation in these cells indicates that even very minor amounts of GP might be sufficient to mediate BDV dissemination through cell–cell contacts. This could be further substantiated by the observation that BDV‐GP can be detected in infected astrocytes only after lectin precipitation (data not shown). Surprisingly, we observed no effect of MI‐0701 on the spread of BDV infection between neurons in the mixed culture system (Fig. 7). A potential explanation for this discrepancy to earlier observations (Bajramovic et al., 2003) is the relatively low concentration of MI‐0701 in our experiments, which was sufficient to inhibit spread in astrocytes (Fig. 7) but might not be sufficient to completely inhibit GP cleavage in neurons. This might be due to the higher amount of BDV‐GP translation in neurons, as shown in the in vivo infection of rats (Werner‐Keiss et al., 2008) or due to differences in inhibitor uptake between astrocytes and neurons. Conducting experiments at higher concentrations of MI‐0701, however, was not successful, because cell viability worsened at concentrations above 10 μM (data now shown), possibly caused by inhibition of the furin‐mediated activation of proβ–nerve growth factor or other essential factors (Lee et al., 2001; Thomas, 2002).
The route via free infectious virus particles (illustrated in Fig. 8A) plays an important role for long distance virus spread to different tissues within an organism and for spread among other hosts between the same or other species. However, many viruses use different pathways without the need of free infectious virus diffusing through the extracellular environment. This mode of viral spread can be advantageous for persistent infections, such as BDV infection, in order to evade the defense by the host. Such mechanisms of virus transmission to neighbouring cells were recently demonstrated with influenza virus and parainfluenza virus 5 (PIV5) (Roberts et al., 2015). The observation that fusion‐active BDV‐GP is essential for the dissemination via cell–cell contacts raises the question of how exactly GP mediates spread in the absence of cell‐free virus. It has previously been hypothesized that BDV is transmitted as non‐enveloped vRNPs (Gosztonyi et al., 1993; Cubitt and de la Torre, 1994a; Clemente and de la Torre, 2007) (illustrated in Fig. 8C), and a recent study showed the trafficking of viral components through contact sites between cells (Charlier et al., 2013). This cell‐to‐cell transfer of the core infectious viral machinery (RNP complex and polymerase) could occur through intercellular connections (tunnelling nanotubes) (Gerdes and Carvalho, 2008; Sherer and Mothes, 2008) in the absence of a fusion‐mediating viral glycoprotein (Fig. 8C), or through GP‐induced, local fusion events that would permit vRNPs to directly travel from infected cells to uninfected cells. Such a glycoprotein‐mediated transmission via microfusion has been suggested for the spread of measles and canine distemper virus in neurons (Young and Rall, 2009; Wyss‐Fluehmann et al., 2010). This mode of cell‐to‐cell spread not only facilitates rapid viral dissemination but may also promote immune evasion and influence disease outcome. Alternatively, BDV could be transmitted through short‐lived viral particles, which bud at close contact sites between cells and immediately bind and enter adjacent cells in a GP‐dependent manner (Fig. 8B), or are transported either in vesicles or as surface‐attached virus on the surface of tunnelling nanotubes, which enter the cell through GP‐dependent fusion. Such a mode of transport would be compatible with the absence of cell‐free BDV in the brain and has been observed for other neurotropic viruses, e.g. during the transsynaptic passage of rabies virus (Etessami et al., 2000). Interestingly, in BDV‐infected rat brains, uninfected neurons adjacent to BDV‐infected ones harbour virus‐positive dot‐like structures at the outer plasma membrane of their perikaryon, an observation that could substantiate former hypotheses. The transmission pathways described earlier either depend on a fusion‐mediating viral glycoprotein (Fig. 8A and B) or need no viral glycoprotein (Fig. 8C). However, a lack of detectable cleaved glycoprotein per se does not necessarily exclude glycoprotein‐mediated virus transmission as was shown recently for coronaviruses, where the cleavage of the glycoprotein S occurs shortly before the fusion of viral and lysosomal membranes (Burkard et al., 2014). Further ultrastructural studies of the events at contact sites between infected and uninfected cells would be helpful to clarify the detailed mechanism of how GP mediates cell–cell transmission of BDV; however, the extremely low BDV production during infection might render such experiments nearly impossible and beyond the scope of this study.
During experimental infection in rats, GP‐expression is very tightly regulated and restricted to neurons (Richt et al., 1998; Werner‐Keiss et al., 2008). While the GP expression level in these cells is relatively high early during infection, it significantly decreases during the later, chronic stages, in contrast to the expression level of BDV‐N, which stays high even during the chronic phase (Richt et al., 1998; Werner‐Keiss et al., 2008). The observation that GP is essential for cell‐to‐cell spread, the main route of BDV dissemination in neurons (Bajramovic et al., 2003) and astrocytes (this study) suggests that the regulation of GP expression not only circumvents the antiviral immune response (Kiermayer et al., 2002; Eickmann et al., 2005) but might also be a key mechanism to regulate persistence and restrict viral spread through the brain.
In summary, our study emphasizes the role of the viral glycoprotein for the dissemination of BDV and shows that GP mediates an effective virus spread in BDV permissive cell cultures and also in natural host cells. The importance of the glycoprotein for the spread of BDV and many other neurotropic viruses indicates that new antiviral strategies could target cellular enzymes that cleave these viral glycoproteins, thereby interfering with a basic mechanism necessary for viral spread. We found that using a stable and potent peptidomimetic furin inhibitors such as MI‐0701 might be a suitable approach to target glycoprotein activation and suppress viral dissemination and possibly also the persistence of these viruses. The cell‐to‐cell transmission of BDV mediated by cleaved BDV‐GP proved to be the dominant pathway of viral spread besides the minor transmission of free virus passing long distances. However, an intercellular transmission of viral RNPs in distinct cell types without the involvement of BDV‐GP cannot be entirely excluded.
Experimental procedures
Cell culture and infection
MDCK and Vero E6 cells
Vero E6 (African green monkey kidney) cells were grown and maintained in Dulbecco's modified Eagle medium (DMEM, Life Technologies, Carlsbad, CA, USA). MDCK cells were grown and maintained in modified Eagle medium. All growth media were supplemented with 100 U ml−1 of penicillin, 100 U ml−1 of streptomycin and 10% of foetal calf serum (all from Life Technologies, Carlsbad, CA, USA), and all cells were grown at 37°C under 5% of CO2. MDCK cells stably expressing YFP – tagged sucrase‐isomaltase (MDCK SI‐YFP) – were described previously (Jacob and Naim, 2001). Cultivation of MDCK cells persistently infected with Giessen strain of BDV‐He80 was described before (Richt et al., 1991). For generation of a persistently BDV‐infected Vero E6 cell line, Vero E6 cells were grown to 40% confluency and incubated with pre‐cleared (centrifuged at 4500 × g for 10 min at 4°C) virus‐containing supernatant of MDCK cells persistently infected with BDV. After 48 h, the cells were washed with fresh medium and further incubated. The medium was exchanged twice per week, and the cells were cultured over 9 weeks until all cells of the monolayer were infected with BDV. The percentage of BDV‐infected cells was determined by immunofluorescence labelling with a primary antibody that detects BDV‐N.
Rat cortical astrocytes
Rat cortical astrocytes were prepared as described previously (McCarthy and de Vellis, 1980; Ahlemeyer et al., 2013). Briefly, newborn Lewis rats (P1‐2) were killed by decapitation (approved by the Government Commission of Animal Care V54‐19C20/15C‐GI 18/4). Neocortical regions from both hemispheres of all animals were excised after total removal of the meninges and incubated in 0.25% of trypsin in DMEM for tissue digestion for 15 min at 37°C. After incubation, the tissue was passed through a metal sieve (Tissue grinder Kit, Sigma‐Aldrich, St Louis, MO, USA). The flow‐through was collected and centrifuged, and the resulting cell pellet was resuspended in low glucose DMEM (1 g l−1) (Life Technologies) supplemented with 10% of foetal bovine serum ‘Gold’ (PAA), 100 U ml−1 of penicillin, 100 U ml−1 of streptomycin (all from Life Technologies, Carlsbad, CA, USA) and 0.1% of gentamycin (Sigma‐Aldrich, St Louis, MO, USA) and seeded directly into culture flasks. The cells were then incubated at 37°C under 5% of CO2. The next day, the cells were washed once with ice‐cold phosphate‐buffered saline (PBS) to remove cell debris and non‐adherent cells and further incubated while exchanging the culture medium every 3–4 days. Shortly before reaching confluence (7–10 days in vitro), the cells were passaged for the first time followed by another passage after 3 weeks. For infection with BDV, 4 h after the PBS wash at day 1, the cells were incubated with virus‐containing brain homogenates of adult Lewis rats (infected with BDV strain H24). After 1 h, the cells were washed with fresh medium and further maintained as described for non‐infected astrocytes. For the generation of persistently BDV‐infected primary rat astrocytes, the cells were maintained for a total time of 10 months. Infection with BDV was monitored by immunofluorescence labelling using a primary antibody that detects BDV‐N. To test BDV release into the supernatant, MDCK cells were inoculated with supernatant from infected astrocytes, incubated for 4 days and immunostained using a primary antibody that detects BDV‐N.
Mixed cultures of brain cells
Mixed cultures of glia cells, oligodendrocytes and neurons were prepared from rat cerebral cortices according to Ott et al. (2010). Briefly, a 5 day old Lewis rat was decapitated; the brain was removed under sterile conditions and transferred into a 3 cm Petri dish containing cold, oxygenated GBSS (Gey's balanced salt solution, Sigma‐Aldrich, St Louis, MO, USA) supplemented with 5% of d‐glucose (Sigma‐Aldrich, St Louis, MO, USA). The cerebrum was cut by a fine eye scissor in slices of 2 mm, and the cerebral cortex was removed and cut into small pieces. Using a sterile Pasteur pipette, the tissue was transferred into a 3 cm Petri dish containing cold oxygenated HBSS (Hanks balanced salt solution without Ca2+ and Mg2+, Biochrom Ltd, Cambridge, UK) supplemented with 20 mM HEPES (Life Technologies, Carlsbad, CA, USA). The tissue was then treated with 2.4 U of Dispase‐I (Sigma‐Aldrich, St Louis, MO, USA) in HBSS for 40 min at 37°C under oxygenation and washed three times with 1 mM of EDTA (Sigma‐Aldrich, St Louis, MO, USA) in HBSS. After this, tissue was washed three times with Neurobasal medium A (Life Technologies, Carlsbad, CA, USA) containing 2% of B27 (Life Technologies, Carlsbad, CA, USA), 100 U ml−1 of penicillin, 100 U ml−1 of streptomycin (all from Life Technologies) and 2 mM of l‐glutamine (all from Life Technologies, Carlsbad, CA, USA) and dissolved in complete medium to a final concentration of 1.5 × 105 cells ml−1. Three hundred fifty microlitres of this suspension was transferred to flexiPERM Micro 12 culture dishes (Sarstedt) containing coverslips (MAGV, Rabenau‐Londorf, Germany) treated with 0.1 mg ml−1 of poly‐l‐lysine (Biochrom Ltd, Cambridge, UK). On the next day, the cells were washed with fresh medium, and culture medium was replaced every second day. Infection with BDV was conducted on day 2 after preparation of the cells from tissue with virus‐containing brain homogenates of adult Lewis rats (infected with BDV strain H24) and incubated for 1 h at 37°C. After incubation, the cells were washed with fresh medium. The cells were further maintained in complete Neurobasal medium, and the medium was exchanged with fresh medium every second day. At different time points, individual samples were fixed and subjected to immunofluorescence analysis for the detection of BDV‐N.
Indirect immunofluorescence microscopy
Indirect immunofluorescence staining of MDCK cells
The MDCK cells stably expressing SI‐YFP were mixed with BDV‐infected MDCK cells at a ratio of 100:1, seeded onto coverslips to confluent density and maintained in the presence or absence of the furin inhibitor MI‐0701 (4‐(guanidino‐methyl)‐phenylacetyl‐Arg‐Val‐Arg‐4‐amidinobenzylamide). At different time points, individual samples were fixed with 4% of paraformaldehyde (PFA) for 20 min at room temperature and then permeabilized with 0.1% of Triton X‐100 in PBS, followed by two washings with PBS containing 1% of bovine serum albumin (BSA). Then, the cells were incubated for 1 h at room temperature with a primary antibody (diluted in PBS‐BSA), as indicated. Next, the cells were washed with PBS‐BSA and incubated for 1 h at room temperature with an appropriate secondary antibody (diluted in PBS‐BSA). After repeated PBS washings, cells were mounted with Mowiol4‐88 (Calbiochem, Darmstadt, Germany) and analysed using a Zeiss Axiovert 200M microscope. ImageJ (Schneider et al., 2012) was used for image processing.
Indirect immunofluorescence staining of rat astrocytes
Uninfected rat astrocytes, or uninfected rat astrocytes mixed with BDV‐infected rat astrocytes at a ratio of 100:1, were seeded onto coverslips at confluent density and maintained in the presence or absence of the furin inhibitor MI‐0701. At different time points, individual samples were washed with PBS and fixed with 4% of PFA for 25 min at room temperature, followed by permeabilization with 0.25% of Triton‐X 100 in PBS for 20 min at 37°C. After three washings with PBS, the cells were incubated with 0.2% of PBS‐BSA for 1 h at room temperature. Thereafter, the cells were incubated over night at 4°C with a primary antibody (diluted in 1% of PBS‐BSA) as indicated, followed by repeated washings with PBS and incubation with an appropriate secondary antibody (diluted in PBS‐BSA) for 1 h at room temperature. After three washing steps with PBS, the cell nuclei were stained with DAPI (Carl Roth, Karlsruhe, Germany), and the cells were mounted with Entellan in Toluene (Merck & Co., Kenilworth, NJ, USA) and analysed using a Nikon Eclipse 80i microscope. ImageJ (Schneider et al., 2012) was used for image processing.
Indirect immunofluorescence staining of mixed cultures of rat brain cells
Mixed cell cultures of rat brain cells were grown on coverslips in the presence or absence of the furin inhibitor MI‐0701. At different time points, individual samples were washed with warm PBS (37°C) and fixed with 4% of PFA in PBS for 20 min at room temperature. Afterwards, the cells were permeabilized and incubated for 20 min at room temperature using PBS containing 20% of horse serum, 3% of BSA and 0.25% of Triton X‐100 in PBS. The samples were then incubated overnight at 4°C with the indicated primary antibodies. All antibodies were diluted in PBS containing 3% of BSA and 0.25% of Triton X‐100. On the next day, the samples were washed three times with PBS and incubated for 1 h at room temperature with appropriate secondary antibodies, as indicated. After three washing steps with PBS, the cell nuclei were stained with DAPI (Carl Roth), followed by one washing step using PBS and one washing step using ddH2O. The cells were then mounted with Entellan in Toluene (Merck & Co., Kenilworth, NJ, USA) and analysed using a Nikon Eclipse 80i microscope. ImageJ (Schneider et al., 2012) was used for image processing.
Immunostaining of infected and non‐infected cells using TrueBlue peroxidase substrate
Non‐infected or BDV‐infected Vero E6 or MDCK cells were grown on glass coverslips. At different time points, the cells were washed three times with PBS and fixed with 4% of PFA in DMEM for 30 min at 4°C. After three washing steps with PBS, the cells were permeabilized with 0.3% of Triton X‐110 in PBS for 20 min at room temperature. The samples were then incubated with a BDV‐N specific antibody for 2 h at room temperature, washed three times with PBS containing 0.05% of Tween 20 and incubated with a horseradish peroxidase (HRP)‐coupled secondary antibody for 1 h at room temperature. After three washing steps using PBS, the samples were incubated with TrueBlue substrate (KPL, Gaithersburg, MD, USA) for 10 min at room temperature, washed with ddH2O, dried and analysed using a Nikon Eclipse TS100 microscope.
Antibodies
The BDV‐N was detected using the monoclonal mouse antibody Bo‐18 (kindly provided by J. Richt, Kansas State University). A rabbit polyclonal antibody was used to detect BDV‐M (Kraus et al., 2001). For detection of BDV‐GP, a previously described rabbit antiserum was used (Richt et al., 1998). A monoclonal mouse antibody was used to detect tubulin (Sigma‐Aldrich, St Louis, MO, USA). Primary antibodies used for indirect immunofluorescence were Bo‐18 (diluted 1:100), a polyclonal α‐GFAP serum (Synaptic Systems, Gottingen, Germany, diluted 1:500) and a monoclonal ßIII‐Tubulin antibody (Covance, Princeton, NJ, USA, diluted 1:1000). Secondary antibodies for indirect immunofluorescence were labelled with rhodamine (Dianova, Hamburg, Germany), Cy3 (Dako/Dianova), AlexaFluor 488 (Dianova) or AlexaFluor 647 (Dianova). All secondary antibodies for indirect immunofluorescence were used at a 1:100 dilution. The secondary antibody used for immunostaining with TrueBlue substrate was labelled with HRP (Dako). Secondary antibodies used for detection of proteins using an Odyssey Infrared Imaging System (LI‐COR Biosciences, Lincoln, NE, USA) were labelled with IRDye800 (Rockland Immunochemicals Inc., Limerick, PA, USA) or Alexa Fluor 680 (Life Technologies, Carlsbad, CA, USA).
Isolation of BDV from supernatants of infected cells
Serum‐free cell culture medium of confluent, persistently BDV‐infected MDCK or Vero E6 cells was pre‐cleared by centrifugation and subjected to ultracentrifugation at 80 000 × g for 1 h using a SW41 rotor (Beckmann Technologies Inc., Durham, NC, USA). The pelleted virus was resuspended in 100 µl of H2O, boiled in reducing sodium dodecyl sulfate (SDS) sample buffer and analysed by SDS‐PAGE and immunoblotting.
Quantified cell‐to‐cell fusion assay
The MDCK cells persistently infected with BDV were grown on coverslips in presence or absence of the furin inhibitor MI‐0701. The medium was exchanged with fresh medium containing MI‐0701 every 12 h until the cells reached confluency. To induce cell‐to‐cell fusion, the supernatant was removed; the cells were washed twice with PBS and subjected to pH shift by incubation in citrate buffer at pH 5.5 for 5 min at 37°C. Next, the cells were washed with DMEM without foetal calf serum (FCS) and incubated in DMEM containing standard supplements and 2% of FCS with or without MI‐0701 for 90 min at 37°C. After two additional washings with PBS, the cells were fixed for 20 min with 70% pre‐cooled ethanol (−20°C), and the cell nuclei were stained using Giemsa solution (Merck & Co., Kenilworth, NJ, USA). To quantify cell–cell fusion, the number of nuclei present in syncytia (defined as cells containing >2 nuclei) and the total number of nuclei in five independent areas containing ~200 cells were counted using a Nikon Eclipse TS100 microscope. Fusion activity was determined by dividing the number of nuclei in syncytia by the total number of nuclei.
Lectin precipitation and immunochemical assay for BDV‐GP
Non‐infected or a 1:10 mixture of persistently BDV‐infected or uninfected MDCK cells grown in the presence or absence of MI‐0701 were incubated with 0.05% of trypsin/ EDTA (Life Technologies, Carlsbad, CA, USA) for 30 min at 37°C. Subsequently, the cell suspensions were seeded in culture dishes (3 cm diameter) and grown in presence of the furin inhibitor MI‐0701 at the indicated concentrations. Every 24 h, the medium was replaced by fresh medium containing MI‐0701. After 72 h, the cells were harvested and resuspended in freshly prepared GDK1‐buffer consisting of 50 mM of Tris‐HCl, 150 mM of NaCl, 1 mM of CaCl2, 1 mM of MnCl2, 0.5% of Triton X‐100 and complete protease inhibitor cocktail (Roche Holding AG, Basel, Switzerland) modified according to Kiermayer et al., (2002). After ultrasonication of the cells (90 s at 40 Watt), the cell lysates were subjected to centrifugation at 21 000 × g for 30 min. The soluble supernatants were then incubated with 50 µl of pre‐washed concanavalin A sepharose beads (GE Healthcare, Little Chalfont, UK) for 16 h at 4°C. After the incubation, the beads were washed three times with GDK1‐buffer and either further deglycosylated using peptide‐N‐glycosidase F and Endo H.
Cell viability assay
The viability of cells incubated with the furin inhibitor MI‐0701 was analysed using the MTT 3‐[4,5‐dimethylthiazol‐2‐yl]‐2,5‐diphenyltetrazoliumbromide assay (Mosmann, 1983). Briefly, MDCK cells were grown in 96 well plates to 100% of confluency and incubated in complete medium lacking FCS in the presence of indicated concentrations of MI‐0701 or in its absence. After 24 and 48 h respectively, the cells were washed twice with PBS and then incubated with 100 µl of freshly prepared MTT solution (0.5 mg ml−1 in PBS) for 2 h at 37°C. Subsequently, the MTT solution was removed, and the cells were incubated with 50 µl dimethyl sulfoxide for 1 h at room temperature. The cell viability of control cells not treated with inhibitor and of cells treated with inhibitor cells was determined by measuring the adsorption at a wavelength of 570 nm using a Microplate Spectrophotometer (BioTek, Winooski, VT, USA).
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgements
We thank Ralph Jacob, Institute for Cytobiology und Cytopathology (Philipps University Marburg) for kindly providing us with MDCK SI‐YFP cells and Jürgen A. Richt (Manhattan, Kansas State University, Kansas) for kindly providing the BDV antibodies. We also thank Tanja Labitzke for confirmatory experiments. The work was supported by the Deutsche Forschungsgemeinschaft‐SFB 593 TPB2 to W. G.
Lennartz, F. , Bayer, K. , Czerwonka, N. , Lu, Y. , Kehr, K. , Hirz, M. , Steinmetzer, T. , Garten, W. , and Herden, C. (2016) Surface glycoprotein of Borna disease virus mediates virus spread from cell to cell. Cellular Microbiology, 18: 340–354. doi: 10.1111/cmi.12515.
References
- Ahlemeyer, B. , Kehr, K. , Richter, E. , Hirz, M. , Baumgart‐Vogt, E. , and Herden, C. (2013) Phenotype, differentiation, and function differ in rat and mouse neocortical astrocytes cultured under the same conditions. J Neurosci Methods 212: 156–164. [DOI] [PubMed] [Google Scholar]
- Arias, I. , Sorlozano, A. , Villegas, E. , de Dios Luna, J. , McKenney, K. , Cervilla, J. , et al. (2012) Infectious agents associated with schizophrenia: a meta‐analysis. Schizophr Res 136: 128–136. [DOI] [PubMed] [Google Scholar]
- Bajramovic, J.J. , Münter, S. , Syan, S. , Nehrbass, U. , Brahic, M. , and Gonzalez‐Dunia, D. (2003) Borna disease virus glycoprotein is required for viral dissemination in neurons. J Virol 77: 12222–12231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker, G.L. , Lu, Y. , Hardes, K. , Strehlow, B. , Levesque, C. , Lindberg, I. , et al. (2012) Highly potent inhibitors of proprotein convertase furin as potential drugs for treatment of infectious diseases. J Biol Chem 287: 21992–22003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Böttcher‐Friebertshäuser, E. , Klenk, H.D. , and Garten, W. (2013) Activation of influenza viruses by proteases from host cells and bacteria in the human airway epithelium. Pathog Dis 69: 87–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourg, M. , Herzog, S. , Encarnacao, J.A. , Nobach, D. , Lange‐Herbst, H. , Eickmann, M. , and Herden, C. (2013) Bicolored white‐toothed shrews as reservoir for Borna disease virus, Bavaria, Germany. Emerg Infect Dis 19: 2064–2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briese, T. , de la Torre, J.C. , Lewis, A. , Ludwig, H. , and Lipkin, W.I. (1992) Borna disease virus, a negative‐strand RNA virus, transcribes in the nucleus of infected cells. Proc Natl Acad Sci U S A 89: 11486–11489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briese, T. , Schneemann, A. , Lewis, A.J. , Park, Y.S. , Kim, S. , Ludwig, H. , and Lipkin, W.I. (1994) Genomic organization of Borna disease virus. Proc Natl Acad Sci U S A 91: 4362–4366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkard, C. , Verheije, M.H. , Wicht, O. , van Kasteren, S.I. , van Kuppeveld, F.J. , Haagmans, B.L. , et al. (2014) Coronavirus cell entry occurs through the endo‐/lysosomal pathway in a proteolysis‐dependent manner. PLoS Pathog 10: e1004502, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbone, K.M. , Rubin, S.A. , Sierra‐Honigmann, A.M. , and Lederman, H.M. (1993) Characterization of a glial cell line persistently infected with Borna disease virus (BDV): influence of neurotrophic factors on BDV protein and RNA expression. J Virol 67: 1453–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlier, C.M. , Wu, Y.J. , Allart, S. , Malnou, C.E. , Schwemmle, M. , and Gonzalez‐Dunia, D. (2013) Analysis of Borna disease virus trafficking in live infected cells by using a virus encoding a tetracysteine‐tagged p protein. J Virol 87: 12339–12348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemente, R. , and de la Torre, J.C. (2007) Cell‐to‐cell spread of Borna disease virus proceeds in the absence of the virus primary receptor and furin‐mediated processing of the virus surface glycoprotein. J Virol 81: 5968–5977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cubitt, B. , and de la Torre, J.C. (1994a) Borna disease virus (BDV), a nonsegmented RNA virus, replicates in the nuclei of infected cells where infectious BDV ribonucleoproteins are present. J Virol 68: 1371–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cubitt, B. , Oldstone, C. , and de la Torre, J.C. (1994b) Sequence and genome organization of Borna disease virus. J Virol 68: 1382–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cubitt, B. , Oldstone, C. , Valcarcel, J. , and Carlos de la Torre, J. (1994c) RNA splicing contributes to the generation of mature mRNAs of Borna disease virus, a non‐segmented negative strand RNA virus. Virus Res 34: 69–79. [DOI] [PubMed] [Google Scholar]
- Eickmann, M. , Kiermayer, S. , Kraus, I. , Gössl, M. , Richt, J.A. , and Garten, W. (2005) Maturation of Borna disease virus glycoprotein. FEBS Lett 579: 4751–4756. [DOI] [PubMed] [Google Scholar]
- Etessami, R. , Conzelmann, K.K. , Fadai‐Ghotbi, B. , Natelson, B. , Tsiang, H. , and Ceccaldi, P.E. (2000) Spread and pathogenic characteristics of a G‐deficient rabies virus recombinant: an in vitro and in vivo study. J Gen Virol 81: 2147–2153. [DOI] [PubMed] [Google Scholar]
- Fujino, K. , Horie, M. , Honda, T. , Merriman, D.K. , and Tomonaga, K. (2014) Inhibition of Borna disease virus replication by an endogenous Bornavirus‐like element in the ground squirrel genome. Proc Natl Acad Sci U S A 111: 13175–13180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garten, W. , Braden, C. , Arendt, A. , Peitsch, C. , Baron, J. , Lu, Y. , et al. (2015) Influenza virus activating host proteases: identification, localization and inhibitors as potential therapeutics. Eur J Cell Biol 94: 375–383. [DOI] [PubMed] [Google Scholar]
- Gerdes, H.H. , and Carvalho, R.N. (2008) Intercellular transfer mediated by tunneling nanotubes. Curr Opin Cell Biol 20: 470–475. [DOI] [PubMed] [Google Scholar]
- Gonzalez‐Dunia, D. , Cubitt, B. , and de la Torre, J.C. (1998) Mechanism of Borna disease virus entry into cells. J Virol 72: 783–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosztonyi, G. , and Ludwig, H. (1995) Borna disease – neuropathology and pathogenesis. Curr Top Microbiol Immunol 190: 39–73. [PubMed] [Google Scholar]
- Gosztonyi, G. , Dietzschold, B. , Kao, M. , Rupprecht, C.E. , Ludwig, H. , and Koprowski, H. (1993) Rabies and Borna disease. A comparative pathogenetic study of two neurovirulent agents. Lab Invest 68: 285–295. [PubMed] [Google Scholar]
- Hardes, K. , Becker, G.L. , Lu, Y. , Dahms, S.O. , Köhler, S. , Beyer, W. , et al. (2015) Novel furin inhibitors with potent anti‐infectious activity. Chem Med Chem 10: 1218–1231. [DOI] [PubMed] [Google Scholar]
- Herden, C. , Briese, T. , Lipkin, I.W. , and Richt, J.A. (2013) Bornaviridae. Philadelphia, Pennsylvania: Wolters Kluwer/Lippincott Williams & Wilkins, pp. 2 volumes, 1124–1150. [Google Scholar]
- Herzog, S. , and Rott, R. (1980) Replication of Borna disease virus in cell cultures. Med Microbiol Immunol 168: 153–158. [DOI] [PubMed] [Google Scholar]
- Hoffmann, B. , Tappe, D. , Höper, D. , Herden, C. , Boldt, A. , Mawrin, C. , et al. (2015) A variegated squirrel Bornavirus associated with fatal human encephalitis. N Engl J Med 373: 154–162. [DOI] [PubMed] [Google Scholar]
- Horie, M. , Honda, T. , Suzuki, Y. , Kobayashi, Y. , Daito, T. , Oshida, T. , et al. (2010) Endogenous non‐retroviral RNA virus elements in mammalian genomes. Nature 463: 84–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornig, M. , Briese, T. , Licinio, J. , Khabbaz, R.F. , Altshuler, L.L. , Potkin, S.G. , et al. (2012) Absence of evidence for Bornavirus infection in schizophrenia, bipolar disorder and major depressive disorder. Mol Psychiatry 17: 486–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob, R. , and Naim, H.Y. (2001) Apical membrane proteins are transported in distinct vesicular carriers. Curr Biol 11: 1444–1450. [DOI] [PubMed] [Google Scholar]
- Kiermayer, S. , Kraus, I. , Richt, J.A. , Garten, W. , and Eickmann, M. (2002) Identification of the amino terminal subunit of the glycoprotein of Borna disease virus. FEBS Lett 531: 255–258. [DOI] [PubMed] [Google Scholar]
- Kinnunen, P.M. , Palva, A. , Vaheri, A. , and Vapalahti, O. (2013) Epidemiology and host spectrum of Borna disease virus infections. J Gen Virol 94: 247–262. [DOI] [PubMed] [Google Scholar]
- Klenk, H.‐D. , and Garten, W. (1994) Activation cleavage of viral spike proteins by host proteases In Cellular Receptors for Animal Viruses. Cold Spring Habor Laboratory Press: New York, USA, pp. 241–280. [Google Scholar]
- Kohno, T. , Goto, T. , Takasaki, T. , Morita, C. , Nakaya, T. , Ikuta, K. , et al. (1999) Fine structure and morphogenesis of Borna disease virus. J Virol 73: 760–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraus, I. , Eickmann, M. , Kiermayer, S. , Scheffczik, H. , Fluess, M. , Richt, J.A. , and Garten, W. (2001) Open reading frame III of Borna disease virus encodes a nonglycosylated matrix protein. J Virol 75: 12098–12104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn, J.H. , Dürrwald, R. , Bào, Y. , Briese, T. , Carbone, K. , Clawson, A.N. , et al. (2015) Taxonomic reorganization of the family Bornaviridae. Arch Virol 160: 621–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, R. , Kermani, P. , Teng, K.K. , and Hempstead, B.L. (2001) Regulation of cell survival by secreted proneurotrophins. Science 294: 1945–1948. [DOI] [PubMed] [Google Scholar]
- Lu, Y. , Hardes, K. , Dahms, S.O. , Böttcher‐Friebertshäuser, E. , Steinmetzer, T. , Than, M.E. , et al. (2015) Peptidomimetic furin inhibitor MI‐701 in combination with oseltamivir and ribavirin efficiently blocks propagation of highly pathogenic avian influenza viruses and delays high level oseltamivir resistance in MDCK cells. Antiviral Res 120: 89–100. [DOI] [PubMed] [Google Scholar]
- McCarthy, K.D. , and de Vellis, J. (1980) Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue. J Cell Biol 85: 890–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosmann, T. (1983) Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 65: 55–63. [DOI] [PubMed] [Google Scholar]
- Ott, D. , Murgott, J. , Rafalzik, S. , Wuchert, F. , Schmalenbeck, B. , Roth, J. , and Gerstberger, R. (2010) Neurons and glial cells of the rat organum vasculosum laminae terminalis directly respond to lipopolysaccharide and pyrogenic cytokines. Brain Res 1363: 93–106. [DOI] [PubMed] [Google Scholar]
- Pasquato, A. , Ramos da Palma, J. , Galan, C. , Seidah, N.G. , and Kunz, S. (2013) Viral envelope glycoprotein processing by proprotein convertases. Antiviral Res 99: 49–60. [DOI] [PubMed] [Google Scholar]
- Payne, S. , Covaleda, L. , Jianhua, G. , Swafford, S. , Baroch, J. , Ferro, P.J. , et al. (2011) Detection and characterization of a distinct Bornavirus lineage from healthy Canada geese (Branta canadensis). J Virol 85: 12053–12056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez, M. , Watanabe, M. , Whitt, M.A. , and de la Torre, J.C. (2001) N‐terminal domain of Borna disease virus G (p56) protein is sufficient for virus receptor recognition and cell entry. J Virol 75: 7078–7085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piepenbring, A.K. , Enderlein, D. , Herzog, S. , Kaleta, E.F. , Heffels‐Redmann, U. , Ressmeyer, S. , et al. (2012) Pathogenesis of avian Bornavirus in experimentally infected cockatiels. Emerg Infect Dis 18: 234–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richt, J.A. , Vande Woude, S. , Zink, M.C. , Clements, J.E. , Herzog, S. , Frese, K. , et al. (1991) Molecular and immunopathological studies of Borna disease virus infection in rats. Behring Inst Mitt 89: 163–176. [PubMed] [Google Scholar]
- Richt, J.A. , Fürbringer, T. , Koch, A. , Pfeuffer, I. , Herden, C. , Bause‐Niedrig, I. , and Garten, W. (1998) Processing of the Borna disease virus glycoprotein gp94 by the subtilisin‐like endoprotease furin. J Virol 72: 4528–4533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts, K.L. , Manicassamy, B. , and Lamb, R.A. (2015) Influenza A virus uses intercellular connections to spread to neighboring cells. J Virol 89: 1537–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rott, R. , and Becht, H. (1995) Natural and experimental Borna disease in animals In Borna Disease. Koprowski H., Lipkin W.I., (eds). Springer Berlin Heidelberg, pp. 17–30. [DOI] [PubMed] [Google Scholar]
- Rott, R. , Herzog, S. , Fleischer, B. , Winokur, A. , Amsterdam, J. , Dyson, W. , and Koprowski, H. (1985) Detection of serum antibodies to Borna disease virus in patients with psychiatric disorders. Science 228: 755–756. [DOI] [PubMed] [Google Scholar]
- Schneider, P.A. , Schneemann, A. , and Lipkin, W.I. (1994) RNA splicing in Borna disease virus, a nonsegmented, negative‐strand RNA virus. J Virol 68: 5007–5012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider, P.A. , Hatalski, C.G. , Lewis, A.J. , and Lipkin, W.I. (1997) Biochemical and functional analysis of the Borna disease virus G protein. J Virol 71: 331–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider, C.A. , Rasband, W.S. , and Eliceiri, K.W. (2012) NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9: 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidah, N.G. , and Prat, A. (2002) Precursor convertases in the secretory pathway, cytosol and extracellular milieu. Essays Biochem 38: 79–94. [DOI] [PubMed] [Google Scholar]
- Sherer, N.M. , and Mothes, W. (2008) Cytonemes and tunneling nanotubules in cell–cell communication and viral pathogenesis. Trends Cell Biol 18: 414–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staeheli, P. , Rinder, M. , and Kaspers, B. (2010) Avian Bornavirus associated with fatal disease in psittacine birds. J Virol 84: 6269–6275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stitz, L. , Nöske, K. , Planz, O. , Furrer, E. , Lipkin, W.I. , and Bilzer, T. (1998) A functional role for neutralizing antibodies in Borna disease: influence on virus tropism outside the central nervous system. J Virol 72: 8884–8892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas, G. (2002) Furin at the cutting edge: from protein traffic to embryogenesis and disease. Nat Rev Mol Cell Biol 3: 753–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner‐Keiss, N. , Garten, W. , Richt, J.A. , Porombka, D. , Algermissen, D. , Herzog, S. , et al. (2008) Restricted expression of Borna disease virus glycoprotein in brains of experimentally infected Lewis rats. Neuropathol Appl Neurobiol 34: 590–602. [DOI] [PubMed] [Google Scholar]
- Wyss‐Fluehmann, G. , Zurbriggen, A. , Vandevelde, M. , and Plattet, P. (2010) Canine distemper virus persistence in demyelinating encephalitis by swift intracellular cell‐to‐cell spread in astrocytes is controlled by the viral attachment protein. Acta Neuropathol 119: 617–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young, V.A. , and Rall, G.F. (2009) Making it to the synapse: measles virus spread in and among neurons. Curr Top Microbiol Immunol 330: 3–30. [DOI] [PMC free article] [PubMed] [Google Scholar]