

Abstract
Dengue is a systemic viral infection that is transmitted to humans by Aedes mosquitoes. No vaccines or specific therapeutics are currently available for dengue. Lycorine, which is a natural plant alkaloid, has been shown to possess antiviral activities against flaviviruses. In this study, a series of novel lycorine derivatives were synthesized and assayed for their inhibition of dengue virus (DENV) in cell cultures. Among the lycorine analogues, 1‐acetyllycorine exhibited the most potent anti‐DENV activity (EC50=0.4 μm) with a reduced cytotoxicity (CC50>300 μm), which resulted in a selectivity index (CC50/EC50) of more than 750. The ketones 1‐acetyl‐2‐oxolycorine (EC50=1.8 μm) and 2‐oxolycorine (EC50=0.5 μm) also exhibited excellent antiviral activities with low cytotoxicity. Structure–activity relationships for the lycorine derivatives against DENV are discussed. A three‐dimensional quantitative structure–activity relationship model was established by using a comparative molecular‐field analysis protocol in order to rationalize the experimental results. Further modifications of the hydroxy group at the C1 position with retention of a ketone at the C2 position could potentially lead to inhibitors with improved overall properties.
Keywords: alkaloids, antiviral agents, dengue virus, lycorine, structure–activity relationships
Alkaloids that bite back: Dengue is a systemic viral infection that is transmitted to humans by Aedes mosquitoes. Currently, vaccines or specific therapeutics are not available. Lycorine, which is a natural alkaloid, has reportedly demonstrated biological activity against dengue virus (DENV). A series of lycorine derivatives and their anti‐DENV activities are reported herein.
Introduction
Dengue fever is currently a global health threat. Dengue transmission is ubiquitous throughout the tropics, with the highest risk zones in the Americas and Asia. It is estimated that there are 390 million dengue infections annually.1 The pathogen of dengue fever is the dengue virus (DENV), which is a single‐stranded RNA virus that belongs to the Flavivirus genus. DENV is transmitted by Aedes Aegypti. There are four serotypes of DENV (DENV1, DENV2, DENV3, and DENV4). All of these serotypes can cause mild dengue fever and, in some cases, can lead to life‐threatening complications such as dengue hemorrhagic fever (DHF) and dengue shock syndrome (DSS).2 DHF and DSS represent the more severe forms of the disease after DENV infection, and they are commonly reported in children. Other symptoms of DENV infection include fever, low platelet count, hemorrhagic bleeding, and vascular permeability.3 Currently, an effective antiviral therapy or a vaccine for DENV infection is not available. The need to simultaneously immunize and induce long‐lasting protection against all four serotypes poses a particular challenge in the development of a vaccine.4 Therefore, there is a continuing and increasing need to develop effective antiviral agents against DENV.
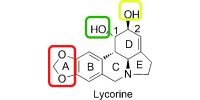
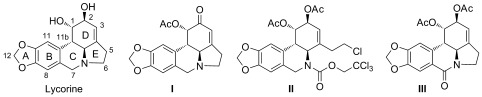
Lycorine (Figure 1), which is one of the major alkaloids that have been isolated from the Amaryllidaceae family of plants, possesses diverse biological properties, including anticancer,5 antiplasmodial,6 antitrypanosomal,7 anti‐inflammatory,8 analgesic,9 and emetic10 properties. Lycorine has also been reported to demonstrate broad‐spectrum inhibitory activities against several viruses, such as poliovirus,11 severe acute respiratory syndrome‐associated coronavirus (SARS‐CoV),12 herpes simplex virus (type 1),13 vaccinia virus,14 bunyaviruses, Punta Toro virus, and Rift Valley fever virus.15 Lycorine has been previously shown to potently inhibit flaviviruses in an in vitro model.16 Lycorine reduced viral titers of West Nile virus (WNV), DENV, and yellow fever virus (YFV) by 102‐ to 104‐fold at a concentration of 1.2 μm. Lycorine primarily exerted its activity through the suppression of viral RNA replication. Mutagenesis studies revealed a single amino acid substitution (Val9Met) in the 2K peptide (which spans the endoplasmic reticulum membrane between the NS4A and NS4B proteins). However, whether the 2K peptide was the direct target of lycorine remains to be determined. Additionally, some synthesized lycorine derivatives have been investigated for their inhibitory activities against WNV. A preliminary structure–activity relationship study indicated that modification of the hydroxy groups at the C1 and C2 positions reduced cytotoxicity, whereas oxidation of the 2‐hydroxy group (Figure 1, I) slightly increased the potency. Compounds with an opened E ring (Figure 1, II) and those that were oxidized at the C7 position (Figure 1, III) demonstrated lower activities.16 To discover novel antiviral agents against DENV, a series of lycorine derivatives were synthesized and assayed to study the structure–activity relationships with respect to their anti‐DENV activity.
Figure 1.
The structure of lycorine and select derivatives.
Results and Discussion
Chemistry
Lycorine (1) is commercially available as a hydrochloride salt. Due to its poor solubility in most organic solvents, derivatives containing A‐ring modifications were synthesized from 1,2‐diacetyllycorine (2), which was formed in high yield by acetylation of both hydroxy groups of 1 with acetic anhydride in the presence of pyridine (Scheme 1).16 The 1,3‐dioxolane ring (A ring) was opened by using boron tribromide, and this was followed by acetylation with acetic anhydride to produce 3. After all of the acetyl groups were removed with HCl/methanol treatment, 4 was treated with different ketones to afford a series of derivatives, 5–9, containing various functionalized A rings. The nitro group of 8 was further reduced to an amino group by using hydrazine monohydrate to produce 9 (Scheme 1).
Scheme 1.
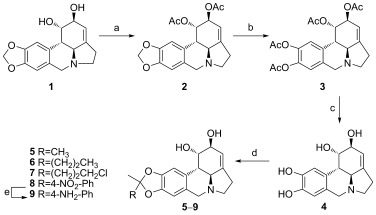
Synthesis of lycorine derivatives with modifications on the A ring. Reagents and conditions: a) Ac2O, Py, 50 °C, 12 h; b) 1. BBr3, CH2Cl2, 0 °C, 1.5 h; 2. Ac2O, Py, RT, o/n; c) HCl, MeOH, 50 °C, 6 h; d) RCOCH3, p‐TsOH, MeOH, 70 °C, 20 h; e) NH2NH2⋅H2O, Pd/C, MeOH, 70 °C, 30 min.
Several C2 derivatives were synthesized to investigate the influence of modifications of the 2‐hydroxy group (Scheme 2). Selective deacetylation at the C2 position of 2 with HCl in methanol afforded 1‐acetyllycorine 10.16 The esters 11–13 and 16–19 were prepared through the reactions of 10 with different acyl chlorides in the presence of pyridine. Additionally, the treatment of 10 with glutaric anhydride in dichloromethane produced derivative 14. With diphenylphosphoryl azide (DPPA) as the condensation agent, Boc‐glycine reacted with 10 to produce 15. The Mitsunobu reaction was applied to 10 in order to furnish 20, which was subsequently converted into 21 through deprotection with hydrazine monohydrate. The inversion of the configuration of 20 was confirmed by using NOSEY analysis, which provided the correlation peaks between H11b/H1, H1/H2, and H2/H11b (Figure 2 a). The 2‐hydroxy group in 1‐acetyllycorine (10) was easily oxidized under Swern conditions to afford the desired ketone 22 16 in good yield. Moreover, 2‐oxolycorine (23)17 was obtained by heating 22 with concentrated HCl in methanol.
Scheme 2.
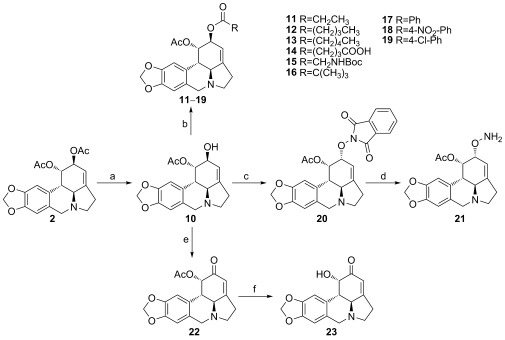
Synthesis of lycorine derivatives with modifications at the 2‐hydroxy group. Reagents and conditions: a) HCl, CH3OH, 55 °C, 1 h; b) RCOCl or (RCO)2O, Py, CH2Cl2, 0 °C; for 15: DPPA, NaHCO3, Boc‐glycine, DMF, 0 °C, o/n; c) N‐hydroxyphthalimide, PPh3, diethylazodicarboxylate, THF, RT, 26 h; d) NH2NH2⋅H2O, CH2Cl2, RT, 1 h; e) DMSO, (COCl)2, Et3N, CH2Cl2, −78 °C, 2 h; f) HCl, CH3OH, 50 °C, 2 h.
Figure 2.
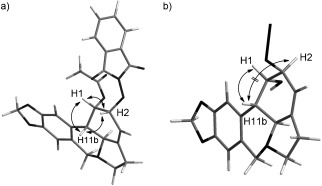
NOESY analysis of a) 20 and b) 25.
Derivatives with various modifications at the C2 position were formed through the introduction of an epoxide or allylic alcohol at the C2 position (Scheme 3). An epoxide ring can be introduced by a two‐step reaction from 10.18 These products possess a trans configuration because the epoxide was below the D ring. Azide 25 and ether 27 were obtained through the opening of the epoxide with the corresponding nucleophilic reagents. The nucleophilic attack of the epoxide occurred at the least hindered side with high regioselectivity to afford 25 and 27, similar to a previous report.19 Based on the bioisosterism, we intended to convert the hydroxy group at the C2 position into an amino group. 2‐Aminolycorine (26) was obtained through reduction of the azide group of 25 with lithium aluminum hydride in THF. A triazole ring was introduced into lycorine by applying “click chemistry”20 on 25 to afford derivatives 28 and 29. The 2‐hydroxy group of 1 is more reactive than the 1‐hydroxy group due to its allylic nature; therefore, the 2‐hydroxy group was selectively modified by using different chloride reagents to yield compounds 30, 31,17 32, and 33.21 The configuration of 25 was confirmed by using NOSEY analysis. A correlation peak was found at H11b/H1, but no correlation between H11b/H2 was observed (Figure 2 b).
Scheme 3.
Synthesis of lycorine derivatives with modifications at the 2‐hydroxy group. Reagents and conditions: a) 1. POCl3, NaCl, HCl, 30 °C, 3 h; 2. CH3ONa, MeOH, 0 °C, 20 min; b) NaN3, MeOH, 80 °C, 20 min; c) LiAlH4, THF, 0 °C, 1 h; d) CH3ONa, MeOH, RT, 30 h; e) CuSO4, l‐ascorbic acid, THF/H2O (1:1), RT, o/n; f) hexanoyl chloride, Py, 0 °C, 40 min; g) for 31: Ac2O, Py, 50 °C, 1 h; for 32: hexanoyl chloride, CH2Cl2, Py, RT, 1.5 h; for 33: tert‐butyldimethylsilyl chloride, 4‐dimethylaminopyridine, Py, RT, 6 h.
The 2‐hydroxy group is more reactive than the 1‐hydroxy group, so the hydroxy group at the C2 position must be converted into other functional groups or be first protected to investigate the influence of C1‐position modifications. The oxidized product containing a free 1‐hydroxy group, 23, was selected as the starting material for reaction with diverse acyl chloride reagents to produce derivatives 34–41 (Scheme 4).
Scheme 4.
Synthesis of lycorine derivatives with modifications at the C1 position. Reagents and conditions: a) RCOCl, Py, 0 °C.
Anti‐DENV activity and structure–activity relationship (SAR) studies
Previous studies have shown that lycorine potently inhibits DENV, WNV, and YFV in cell cultures. The goal of this study was to investigate the influence of A‐ring modifications of lycorine and the substitution of the 1‐ and/or 2‐hydroxy groups on the activities against DENV. The anti‐DENV activities of these lycorine derivatives were evaluated by using an in vitro cell‐based flavivirus immunodetection (CFI) assay,16 and the results were expressed as EC50 values for antiviral activity and CC50 values for cytotoxicity (Tables 1–1, 2, 3).
Table 1.
The publisher did not receive permission from the copyright owner to include this object in this version of this product. Please refer either to the publisher's own online version of this product or the printed product where one exists.
Table 2.
The publisher did not receive permission from the copyright owner to include this object in this version of this product. Please refer either to the publisher's own online version of this product or the printed product where one exists.
Table 3.
The publisher did not receive permission from the copyright owner to include this object in this version of this product. Please refer either to the publisher's own online version of this product or the printed product where one exists.
Estévez‐Braun et al. reported that the dissection of the A ring decreased the antiplasmodial activity of the lycorine derivatives.6 Similarly, the two derivatives with an opened A‐ring, 3 and 4, demonstrated a dramatic loss of anti‐DENV activity. Various ketones were used to rebuild the 1,3‐dioxolane ring with different substituents at the methylene group, which yielded 5–9. However, none of the derivatives exhibited antiviral activities when evaluated up to a concentration of 300 μm (Table S1 in the Supporting Information). These results indicated the requirement of the 1,3‐dioxolane ring for potency and the intolerance of substitutions at the methylene group of the 1,3‐dioxolane ring.
The biological analysis revealed that the potency of 10 (EC50=0.4 μm) is two times that of lycorine (EC50=0.8 μm), with significantly decreased cytotoxicity (CC50>300 μm), which resulted in an SI value greater than 750. Therefore, an SAR study was initiated with a focus on C2 modification with retention of the 1‐acetyl ester. Hence, 10 was selected as a chemical starting point, and modifications at the 2‐hydroxy group were generated to study their effect on the anti‐DENV activity (Table 1).
The comparison of the esterification products 2 and 11–19 with 10 indicated that esterification of the 2‐hydroxy group generally resulted in a reduction in the antiviral activity. The potencies of the esters 2 and 11–13 decreased with an increasing length of the carbon chain. Interestingly, if a carboxylic acid (in 14; EC50=21.0 μm) or carbamate (in 15; EC50=48.2 μm) was introduced at the terminus of the aliphatic ester chain, improved activities were achieved in comparison to 12 (EC50=120.1 μm), which does not contain this modification. An ester containing a bulky aliphatic group (tert‐butyl group in 16) demonstrated less potency than the acetyl ester 11. Additionally, the derivatives containing aromatic substitution groups at the C2 position, 17–19, exhibited less potency.
The chirality at the C2 position was changed from S to R by using the Mitsunobu reaction on 10 to produce analogues 20 and 21. The decreased activities of 20 and 21 suggest that the stereochemistry may influence the activity of lycorine derivatives against DENV (Table 1). However, the impact of the modification of the hydroxy group cannot be excluded. Therefore, the influence of the chirality at the C2 position remains to be addressed in a future study.
Oxidation of the 2‐hydroxy group into a carbonyl group was reported to slightly increase anti‐WNV activity, with significantly reduced cytotoxicity.16 In our study, the C2 oxidation derivative 22 exhibited slightly lower potency (EC50=1.8 μm) than 1, but the cytotoxicity was dramatically reduced (CC50>300 μm), with an SI value greater than 166.
Efforts to explore modifications of the 2‐hydroxy while retaining the 1‐acetyl ester led to the discovery of ketone 22, which exhibited low micromolar inhibition against DENV with a significant reduction in the cytotoxicity. Further removal of the acetyl ester at the C1 position led to 23 (EC50=0.5 μm, CC50=56.6 μm, SI=113.2), which exhibited slightly higher anti‐DENV potency and less cytotoxicity than lycorine (EC50=0.8 μm, CC50=25.1 μm, SI=31.4; Table 2). The hydroxy group at the C1 position was retained, and substitution with relatively broad structural features at the C2 position was investigated. The hydroxy group at the C2 position was converted into various functional groups, including azide, amino, ether, ester, amide, triazole, siloxane, and carboxyl groups. Generally, the functional‐group transformations of the 2‐hydroxy group resulted in a loss of activity (Table 2). The ester, ether, and siloxane derivatives resulted in better activities than amino, azide, amide, and triazole derivatives among this series. Based on bioisosterism, the hydroxy group at the C2 position could potentially be converted into an amino group.22 However, amino derivative 26 was much less active than lycorine, as were the azide and amide derivatives 25 and 30. The anti‐DENV activity of triazole derivatives 28 and 29 also dramatically decreased. Overall, the results suggested that the 2‐hydroxy group plays an important role in the anti‐DENV activity.
Ketone 23 exhibited relatively improved antiviral properties, so the impact of modifying the C1 substituent while retaining the ketone at the C2 position was investigated. The acylation of the 1‐hydroxy group yielded esters 34–41. Evaluation of the biological properties indicated that linear aliphatic chain substitutions at the C1 position (in 22 and 34–37) resulted in improved antiviral activities relative to those with bulky (in 38) and aromatic substitutions (in 39–41; Table 3). Among all of the linear aliphatic esters, inhibitors with shorter carbon chains were less cytotoxic, although their inhibitory activities against DENV were similar.
QSAR study
To achieve a quantitative understanding of the relationship between the DENV inhibitory activity and the molecular structure of the synthesized lycorine derivatives, a three‐dimensional quantitative structure–activity relationship (3D‐QSAR) study was performed by using a comparative molecular‐field analysis (CoMFA) protocol implemented in the Tripos Sybyl‐X 2.0 software.
Herein, we discussed the influence of modifications at the C1 and/or C2 position. A total of 31 compounds (1, 2, 10–19, 22, 23, and 25–41) were selected for the QSAR study. Derivatives 20, 21, and 24 were excluded from QSAR analysis because these compounds contain significant conformational changes, and their number is insufficient to yield a meaningful analysis. Ester 10 was chosen as the template because of its high inhibitory activity. The sample partial‐least‐squares (SAMPLS) analysis method was used to linearly correlate the CoMFA fields to the inhibitory activity values. The cross‐validation analysis was performed by using the leave‐one‐out (LOO) method, in which one compound was removed from the dataset, and its activity was predicted by using the model derived from the remainder of the dataset. The cross‐validated conventional correlation coefficient (r 2) that resulted in an optimum number of components (ONC) and lowest standard error of prediction was considered for further analysis.
The PLS results of the CoMFA model are summarized in Table 4. The experimental versus predicted pEC50 values are summarized in Table S2 in the Supporting Information and are graphically depicted in Figure 3. The cross‐validation coefficient (q 2), which indicates the predictive capacity, and the conventional correlation coefficient (r 2), which indicates the self‐consistence, are two pivotal parameters to evaluate the quality of the PLS statistics. A q 2 value over 0.3 is considered significant for the case of significant correlation in more than 95 % of the trials.23 For the CoMFA model used in this study, the q 2 and r 2 values were 0.575 and 0.998, respectively. The PLS results indicated that the CoMFA model demonstrated a good predictive ability.
Table 4.
The publisher did not receive permission from the copyright owner to include this object in this version of this product. Please refer either to the publisher's own online version of this product or the printed product where one exists.
Figure 3.
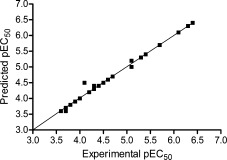
Plot of the experimental versus predicted activities based on the CoMFA model.
As shown in Table 4, the contributions of steric and electrostatic fields were 57.3 % and 42.7 %, respectively, which indicated that the SAR of the C1 and/or C2 position is more influenced by steric effects than by electrostatic effects. The CoMFA contour map of the steric field is presented in Figure 4 a. The green contours indicate favorable regions for a sterically bulky group, whereas the yellow contours indicate less favorable regions for a bulky group. This phenomenon can be observed for compounds 11–13, in that their inhibitory activities decreased according to the length of the aliphatic chain at the C2 position and the CoMFA steric‐field contour maps for these compounds gradually extended into the yellow region. In contrast, the CoMFA steric‐field contour maps for compounds 14 and 15 lie near the green area; therefore, these compounds exhibited relatively high potency. Moreover, the flexibility of the carbon linear chain of 34–37 resulted in contours in the green region near the C1 position, which yielded improved activities relative to those of 39–41, for which the aromatic substitutions were found in the yellow region. Similarly, in the CoMFA contour maps of the electrostatic field (Figure 4 b), the blue/red regions favored positively/negatively charged groups, respectively. For example, 18 and 19 possessed higher potency than 17 because electron‐withdrawing groups (NO2 and Cl) may increase the positive charge of the phenyl ring. This is in contrast to the results for compounds 39–41, because negatively charged groups are favored around the C1 position.
Figure 4.
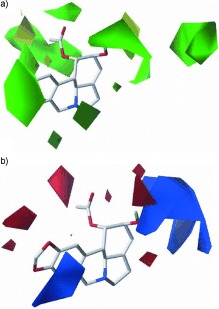
Contour maps of the CoMFA model with compound 10 as the reference molecule. a) Steric contour maps of the CoMFA model. Green indicates regions in which bulky groups increase the activity, whereas yellow indicates regions in which bulky groups decrease the activity. b) Electrostatic contour maps of the CoMFA model. Blue indicates regions in which more positively charged groups increase the activity, whereas red indicates regions in which more negatively charged groups increase the activity.
Conclusions
In summary, a series of lycorine derivatives were synthesized and evaluated against DENV in a cellular assay. Compound 10 (EC50=0.4 μm) was found to be the most potent antiviral agent and demonstrated the best SI value (>750). Derivatives 22 (EC50=1.8 μm) and 23 (EC50=0.5 μm) also exhibited excellent inhibitory activity with low cytotoxicity. The structure–activity relationships of lycorine derivatives against DENV were investigated. Further modifications at the 1‐hydroxy group with retention of the ketone at the C2 position could potentially lead to inhibitors with improved overall properties. A 3D‐QSAR model by using a CoMFA protocol was established to rationalize the experimental results.
Experimental Section
General
All solvents were treated by using standard techniques before use. All reagents were purchased from commercial suppliers and were used without further purification. 1H and 13C NMR spectra were recorded on a Bruker Avance‐400 (400 MHz) NMR spectrometer (1H: 400 MHz; 13C: 100 MHz) in CDCl3, CD3OD, or [D6]DMSO with tetramethylsilane as an internal standard. Chemical shifts are reported in parts per million (δ in ppm), and coupling constants are provided in hertz (Hz). ESI‐MS spectra were recorded on a Shimadzu LCMS‐2020 apparatus. HRMS were recorded on a high‐resolution ESI‐FTICR mass spectrometer (Varian 7.0 T). All tested compounds displayed purities of >95 %. Silica gel TLC (GF254) was used to monitor the reactions, and TLC plates were visualized under UV light at 254 nm and 365 nm. Flash chromatography was performed by using silica gel (200–300 mesh).
CFI assay
A CFI assay was performed as previously described.24 Briefly, A549 cells were infected with DENV‐2 (multiplicity of infection of 0.3) in the presence of twofold serial dilutions of test compounds. After incubation at 37 °C for 48 h, viral antigen production was quantified by immune detection with the 4G2 antibody and goat anti‐mouse immunoglobulin G conjugated with horseradish peroxidase as primary and secondary antibodies, respectively. The concentration of the compounds that decreased viral envelope protein production by 50 % (EC50) was calculated by using nonlinear regression analysis.
QSAR method
In total, 31 compounds were used to generate the model. All structures were constructed with the SYBYL‐X 2.0 sketcher and were assigned Gasteiger–Hückel charges, followed by energy minimization by using the Tripos force field, prior to being stored in a SYBYL database. Database alignment was subsequently performed based on a lycorine scaffold, and compound 10 was used as a template because of its high potency. The biological activity EC50 values (with units of mol L−1) were converted into the negative logarithm pEC50 values, which were then merged into the compound database.
The CoMFA method was applied to perform 3D‐QSAR analysis by using the SYBYL‐X 2.0 software. The CoMFA region was automatically generated. This method generated a single lattice, which overlapped all included compounds, and extended it by at least 4 Å along all axes. Energy cutoff values of 30 kcal mol−1 were set for both steric and electrostatic interactions. After addition of CoMFA fields, PLS was used to generate a linear regression, which correlates descriptors with inhibitory activities. Subsequently, cross‐validation was performed by using LOO methodology to obtain the ONC and the predictive ability. The correlation coefficient q 2 was calculated to be 0.575 for 10 components. Subsequently, non‐cross‐validation was performed for 10 components to obtain the r 2 value.
Chemistry
1,2‐Diacetyllycorine (2): Ac2O (9.5 mL) was added to a solution of lycorine (3.17 g, 11 mmol) in pyridine (8.0 mL), and the solution was stirred at 50 °C for 12 h. Subsequently, MeOH (25 mL) was added, and the mixture was stirred at RT for 3 h. The solvent was removed in vacuo, followed by the addition of CH2Cl2 (50 mL) and water (40 mL). The organic layer was washed with saturated aq NaHCO3 and then brine and was dried over anhydrous Na2SO4. The solution was concentrated, and the crude residue was purified by using silica gel chromatography (CH2Cl2/EtOAc/CH3OH, 10:10:1) to yield 2 as a white solid (3.78 g, 93.3 %). 1H NMR (400 MHz, CDCl3): δ=1.95 (s, 3 H), 2.08 (s, 3 H), 2.40 (m, 1 H), 2.65 (m, 2 H), 2.77 (d, J=10.4 Hz, 1 H), 2.86 (d, J=10.0 Hz, 1 H), 3.37 (m, 1 H), 3.53 (d, J=14.0 Hz, 1 H), 4.16 (d, J=14.0 Hz, 1 H), 5.26 (s, 1 H), 5.53 (s, 1 H), 5.74 (s, 1 H), 5.91 (s, 2 H), 6.57 (s, 1 H), 6.75 ppm (s, 1 H); 13C NMR (100 MHz, CDCl3): δ=21.0, 21.2, 28.7, 40.5, 53.6, 56.9, 61.2, 69.2, 70.9, 101.0, 105.0, 107.3, 113.8, 126.5, 129.4, 146.2, 146.4, 169.8, 170.0 ppm; ESI‐MS: m/z 372 [M+H]+; HRMS: m/z calcd for C20H22NO6 [M+H]+: 372.1442, found: 372.1445.
8,9‐nor‐1,2,8,9‐Tetraacetyllycorine (3): BBr3 (80 mg, 0.32 mmol) was added dropwise to a solution of 2 (60 mg, 0.16 mmol) in anhydrous CH2Cl2 (5 mL). The mixture was stirred at 0 °C for 1.5 h, and MeOH (5 mL) was slowly added to quench the reaction. The solvent was removed in vacuo. The crude residue was dissolved in anhydrous pyridine (2 mL), followed by the addition of Ac2O (2 mL). The mixture was stirred at RT overnight. The solvent was removed in vacuo, followed by the addition of CH2Cl2 (25 mL) and water (25 mL). The organic layer was washed with saturated aq NaHCO3 and then brine and was dried over anhydrous Na2SO4. The solution was concentrated, and the crude residue was purified by using silica gel chromatography (EtOAc/CH3OH, 10:1) to yield 3 as a pale yellow solid (28 mg, 39.5 %). 1H NMR (400 MHz, CDCl3): δ=1.99 (s, 3 H), 2.04 (s, 3 H), 2.27 (s, 6 H), 2.41 (m, 1 H), 2.65 (m, 2 H), 2.86 (d, J=10.0 Hz, 1 H), 3.01 (s, 2 H), 3.38 (m, 1 H), 3.58 (d, J=14.0 Hz, 1 H), 4.22 (d, J=14.8 Hz, 1 H), 5.36 (s, 1 H), 5.78 (s, 1 H), 6.09 (d, J=4.4 Hz, 1 H), 6.93 (s, 1 H), 7.00 ppm (s, 1 H); 13C NMR (100 MHz, CDCl3): δ=20.6, 20.7, 20.8, 20.9, 28.3, 43.6, 53.8, 56.1, 61.1, 65.3, 70.9, 115.1, 119.5, 122.1, 131.9, 134.8, 140.5, 140.7, 144.9, 168.3, 168.4, 170.3, 170.8 ppm; ESI‐MS: m/z 444 [M+H]+; HRMS: m/z calcd for C23H26NO8 [M+H]+: 444.1615, found: 444.1623.
8,9‐nor‐8,9‐Dihydroxyllycorine (4): Concd HCl (3 mL) was added to a solution of 3 (133 mg, 0.30 mmol) in MeOH (10 mL), and the mixture was stirred at 50 °C for 6 h. The solution was made basic (pH 8) with Et3N, and the solvent was removed in vacuo. The crude residue was purified by using silica gel chromatography (EtOAc/CH3OH, 10:1) to yield 4 as a white solid (78 mg, 94.5 %). 1H NMR (400 MHz, CDCl3): δ=2.77 (m, 2 H), 3.00 (m, 1 H), 3.55 (m, 2 H), 3.89 (m, 1 H), 4.18 (d, J=13.8 Hz, 1 H), 4.27 (d, J=13.9 Hz, 1 H), 4.46 (s, 1 H), 4.72 (d, J=3.8 Hz, 1 H), 5.63 (s, 1 H), 6.72 (s, 1 H), 6.95 ppm (s, 1 H); 13C NMR (100 MHz, CDCl3): δ=29.5, 43.2, 54.1, 55.1, 63.0, 67.6, 71.1, 112.9, 115.4, 122.9, 124.9, 127.3, 136.7, 145.5, 147.0 ppm; ESI‐MS: m/z 276 [M+H]+; HRMS: m/z calcd for C15H18NO4 [M+H]+: 276.1236, found: 276.1233.
General procedure for 5–8: Anhydrous ketone (20 equiv) and p‐TsOH (0.2 equiv) were added to a solution of 4 in MeOH (2 mL), and the mixture was heated at 70 °C for 20 h. The solvent was removed in vacuo, followed by the addition of CH2Cl2 (25 mL) and water (25 mL). The organic layer was dried over anhydrous Na2SO4 and filtered. The filtrate was concentrated, and the crude residue was purified by using silica gel chromatography to yield the product.
12,12‐Dimethyllycorine (5): Following the previously described general procedure, 4 (148 mg, 0.54 mmol) yielded 5 as a pale solid (117 mg, 68.8 %). 1H NMR (400 MHz, CD3OD): δ=1.32 (s, 3 H), 1.44 (s, 3 H), 2.79 (m, 2 H), 3.03 (d, J=11.2 Hz, 1 H), 3.37–3.44 (m, 1 H), 3.58 (m, 1 H), 3.66 (d, J=11.6 Hz, 1 H), 4.13 (d, J=13.6 Hz, 1 H), 4.31 (d, J=13.6 Hz, 1 H), 4.80 (s, 1 H), 5.12 (d, J=5.6 Hz, 1 H), 5.80 (s, 1 H), 6.74 (s, 1 H), 6.97 ppm (s, 1 H); 13C NMR (100 MHz, CD3OD): δ=25.8, 27.5, 30.0, 43.5, 54.7, 55.4, 62.1, 73.1, 75.9, 110.2, 113.3, 115.1, 120.9, 124.7, 127.1, 140.9, 142.5, 146.5 ppm; ESI‐MS: m/z 316 [M+H]+; HRMS: m/z calcd for C18H22NO4 [M+H]+: 316.1549, found: 316.1550.
12‐Methyl‐12‐(4‐nitrobenzoyl)lycorine (8): Following the previously described general procedure, 4 (100 mg, 0.58 mmol) yielded 8 as a pale solid (14 mg, 10.4 %). 1H NMR (400 MHz, [D6]DMSO): δ=1.63 (s, 3 H), 2.08–2.01 (m, 1 H), 2.26–2.14 (m, 1 H), 2.32 (m, 2 H), 2.67 (d, J=10.0 Hz, 1 H), 3.07 (m, 1 H), 3.25 (d, J=14.0 Hz, 1 H), 3.88 (d, J=14.0 Hz, 1 H), 5.08 (s, 1 H), 5.15 (d, J=5.7 Hz, 1 H), 5.40 (s, 1 H), 6.48 (s, 1 H), 6.85 (s, 1 H), 7.59 (d, J=8.8 Hz, 2 H), 8.13 ppm (d, J=8.8 Hz, 2 H); 13C NMR (100 MHz, [D6]DMSO): δ=26.9, 28.0, 43.5, 52.8, 55.5, 60.7, 73.0, 75.7, 107.3, 112.3, 113.9, 115.8, 123.0, 125.0, 126.3, 126.4, 143.5, 143.6, 145.8, 146.9, 150.7 ppm; ESI‐MS: m/z 423 [M+H]+; HRMS: m/z calcd for C23H22N2O6 [M+H]+: 423.1551, found: 423.1548.
12‐Methyl‐12‐(4‐aminobenzoyl)lycorine (9): Pd/C (30 mg) and NH2NH2⋅H2O (10 mg, 0.20 mmol) were added to a solution of 8 (43 mg, 0.10 mmol) in MeOH (5 mL), and the mixture was heated to 70 °C for 30 min. The solution was concentrated in vacuo, and the crude residue was purified by using silica gel chromatography (EtOAc/CH3OH, 5:1) to yield 9 as a pale solid (33 mg, 84.0 %). 1H NMR (400 MHz, [D6]DMSO): δ=1.59 (s, 3 H), 2.19 (m, 1 H), 2.43 (br s, 2 H), 2.53 (d, J=10.4 Hz, 1 H), 2.63 (d, J=10.4 Hz, 1 H), 3.17–3.08 (m, 1 H), 3.28 (d, J=13.9 Hz, 1 H), 3.89 (d, J=13.8 Hz, 1 H), 4.92 (m, 1 H), 5.06 (m, 1 H), 5.50 (m, 1 H), 6.40 (d, J=8.6 Hz, 2 H), 6.45 (s, 1 H), 6.78 (s, 1 H), 7.00 ppm (d, J=8.6 Hz, 2 H); 13C NMR (100 MHz, [D6]DMSO): δ=24.5, 28.2, 44.0, 52.9, 55.4, 60.6, 72.0, 74.5, 107.9, 112.4, 112.7, 113.8, 116.4, 125.3, 126.1, 126.4, 130.1, 143.4, 143.5, 145.5, 148.2 ppm; ESI‐MS: m/z 393 [M+H]+; HRMS: m/z calcd for C28H25N2O4 [M+H]+: 393.1809, found: 383.1811.
1‐Acetyllycorine (10): Concentrated HCl (20 mL) was added to a solution of 2 (0.96 g, 2.58 mmol) in MeOH (100 mL), and the solution was stirred at 55 °C for 1 h. The mixture was basified (pH 8) with saturated aq NaHCO3, followed by the addition of CH2Cl2 (25 mL). The combined organic layers were washed with brine (25 mL), dried over anhydrous Na2SO4, and filtered. The filtrate was concentrated in vacuo, and the crude residue was purified by using silica gel chromatography (EtOAc/CH3OH, 10:1) to yield 10 as a white solid (0.50 g, 58.9 %). 1H NMR (400 MHz, CDCl3): δ=1.94 (s, 3 H), 2.37 (m, 1 H), 2.63 (m, 2 H), 2.74 (d, J=10.4 Hz, 1 H), 2.85 (d, J=10.4 Hz, 1 H), 3.35 (m, 1 H), 3.50 (d, J=14.0 Hz, 1 H), 4.25 (d, J=14.4 Hz, 1 H), 4.18 (s, 1 H), 5.54 (s, 1 H), 5.61 (s, 1 H), 5.92 (s, 2 H), 6.57 (s, 1 H), 6.66 ppm (s, 1 H); 13C NMR (100 MHz, CDCl3): δ=21.1, 28.5, 39.0, 53.7, 56.8, 61.6, 69.2, 72.7, 101.0, 104.9, 107.3, 117.6, 127.0, 129.1, 143.3, 146.2, 146.4, 170.8 ppm; ESI‐MS: m/z 330 [M+H]+; HRMS: m/z calcd for C18H20NO5 [M+H]+: 330.1341, found: 330.1344.
General procedure for 11–13 and 16–19: Acyl chloride or anhydride (1.2 equiv) in anhydrous CH2Cl2 (5 mL) was slowly added to a solution of 10 (1 equiv) in anhydrous pyridine (2 mL) over 15 min at 0 °C. The solution was stirred at 0 °C until TLC analysis indicated that all of the starting material had been consumed. Subsequently, CH2Cl2 (10 mL) and water (10 mL) were added. The organic layer was washed with saturated aq NaHCO3 and brine, dried over anhydrous Na2SO4, and filtered. The filtrate was concentrated in vacuo, and the crude residue was purified by using silica gel chromatography (petroleum ether/EtOAc, 2:1) to yield the products.
1‐Acetyl‐2‐propionyllycorine (11): Following the previously described general procedure, 10 (200 mg, 0.61 mmol) yielded 11 as a white solid (113 mg, 53.4 %). 1H NMR (400 MHz, CDCl3): δ=1.29–0.95 (m, 1 H), 1.96 (d, J=4.9 Hz, 1 H), 2.49–2.29 (m, 1 H), 2.65 (s, 1 H), 2.79 (s, 1 H), 2.87 (s, 1 H), 3.38 (d, J=3.9 Hz, 1 H), 3.54 (d, J=13.7 Hz, 1 H), 4.17 (dd, J=13.9, 4.1 Hz, 1 H), 5.27 (s, 1 H), 5.53 (s, 1 H), 5.74 (s, 1 H), 5.92 (d, J=4.9 Hz, 1 H), 6.58 (d, J=4.6 Hz, 1 H), 6.76 ppm (d, J=4.5 Hz, 1 H); 13C NMR (100 MHz, CDCl3): δ=9.0, 20.9, 27.6, 28.7, 40.5, 53.6, 56.9, 61.2, 69.3, 70.7, 101.0, 105.1, 107.3, 113.9, 126.6, 129.4, 146.0, 146.2, 146.4, 170.0, 173.2 ppm; ESI‐MS: m/z 386 [M+H]+; HRMS: m/z calcd for C21H24NO6 [M+H]+: 386.1604, found: 386.1598.
1‐Acetyl‐2‐phenyllycorine (17): Following the previously described general procedure, 10 (260 mg, 0.79 mmol) yielded 17 as a pale yellow oil (173 mg, 50.6 %). 1H NMR (400 MHz, CDCl3): δ=1.99 (s, 3 H), 2.55 (m, 1 H), 2.71 (s, 2 H), 2.99 (d, J=10.5 Hz, 1 H), 3.11 (d, J=10.5 Hz, 1 H), 3.54–3.37 (m, 1 H), 3.65 (d, J=14.1 Hz, 1 H), 4.21 (d, J=14.1 Hz, 1 H), 5.56 (s, 1 H), 5.67 (s, 1 H), 5.90 (s, 2 H), 6.60 (s, 1 H), 6.78 (s, 1 H), 7.41 (t, J=7.5 Hz, 2 H), 7.55 (t, J=7.3 Hz, 1 H), 8.04 ppm (d, J=7.7 Hz, 2 H); 13C NMR (100 MHz, CDCl3): δ=20.9, 28.8, 40.1, 53.5, 56.4, 61.1, 69.0, 70.9, 101.1, 105.1, 107.4, 114.2, 126.5, 128.0, 128.4, 129.0, 129.8, 129.8, 132.1, 133.2, 145.8, 146.5, 146.7, 165.2, 170.0 ppm; ESI‐MS: m/z 434 [M+H]+; HRMS: m/z calcd for C25H24NO6 [M+H]+: 434.1571, found: 434.1597.
1‐Acetyl‐2‐(glutaric acid)lycorine (14): Glutaric anhydride (114 mg, 1 mmol) was added to a solution of 10 (146 mg, 0.44 mmol) in CH2Cl2 (5 mL), and the mixture was stirred at 40 °C for 10 min. The solvent was removed in vacuo, and the crude residue was purified by using silica gel chromatography (CH2Cl2/CH3OH, 10:1) to yield 14 as a white solid (180 mg, 92.3 %). 1H NMR (400 MHz, CDCl3): δ=1.86 (m, 2 H), 1.92 (s, 3 H), 2.24–2.36 (m, 4 H), 2.68 (m, 2 H), 2.84 (m, 1 H), 2.93 (d, J=10.8 Hz, 1 H), 3.21 (d, J=10.4 Hz, 1 H), 3.36 (m, 1 H), 3.82 (d, J=14.0 Hz, 1 H), 4.07 (d, J=14.0 Hz, 1 H), 5.21 (s, 1 H), 5.54 (s, 1 H), 5.69 (s, 1 H), 5.87 (s, 2 H), 6.58 (s, 1 H), 6.64 ppm (s, 1 H); 13C NMR (100 MHz, CDCl3): δ=20.2, 20.9, 28.6, 33.5, 39.2, 53.3, 55.8, 60.9, 69.0, 70.4, 101.1, 105.1, 107.5, 114.7, 126.4, 128.1, 144.5, 146.6, 146.9, 170.0, 171.8, 176.6 ppm; ESI‐MS: m/z 442 [M−H]−; HRMS: m/z calcd for C23H24NO8 [M−H]−: 442.1507, found: 442.1505.
1‐Acetyl‐2‐(Boc‐Gly)lycorine (15): DPPA (0.55 g, 4 mmol) and NaHCO3 (0.42 g, 5 mmol) were added to a solution of 10 (0.16 g, 0.5 mmol) and Boc‐glycine (0.11 g, 2 mmol) in DMF (3 mL). The mixture was stirred at 0 °C overnight and then filtered. The filtrate was concentrated in vacuo, and the crude residue was purified by using silica gel chromatography (petroleum ether/EtOAc, 1:1) to yield 15 as a white solid (90 mg, 38.0 %). 1H NMR (400 MHz, CDCl3): δ=1.38 (s, 9 H), 2.34 (m, 1 H), 2.58 (m, 2 H), 2.72 (d, J=10.4 Hz, 1 H), 2.79 (d, J=11.2 Hz, 1 H), 3.30 (m, 1 H), 3.46 (d, J=14.0 Hz, 1 H), 3.86 (s, 2 H), 4.08 (d, J=14.0 Hz, 1 H), 5.25 (s, 1 H), 5.44 (s, 1 H), 5.65 (s, 1 H), 5.84 (s, 2 H), 6.50 (s, 1 H), 6.65 ppm (s, 1 H); 13C NMR (100 MHz, CDCl3): δ=20.9, 27.4, 28.3, 28.7, 40.3, 42.5, 53.5, 56.8, 61.2, 69.0, 71.7, 101.0, 105.0, 107.3, 113.3, 126.3, 129.3, 146.4, 146.5, 146.8, 169.3, 169.9 ppm; ESI‐MS: m/z 487 [M+H]+; HRMS: m/z calcd for C25H31N2O8 [M+H]+: 487.2075, found: 487.2078.
1‐Acetyl‐2‐(R)‐N‐hydroxyphthalimidelycorine (20): Triphenylphosphine (1645 mg, 6.28 mmol) and N‐hydroxyphthalimide (1024 mg, 6.28 mmol) were added to a solution of 10 (1034 mg, 3.14 mmol) in anhydrous THF (10 mL), followed by the dropwise addition of diethylazodicarboxylate (1092 mg, 6.28 mmol). The mixture was stirred at RT for 26 h. The solvent was removed in vacuo, followed by the addition of CH2Cl2 (20 mL) and water (20 mL). The organic layer was washed with brine (25 mL), dried over anhydrous Na2SO4, and filtered. The filtrate was concentrated, and the crude residue was purified by using silica gel chromatography (petroleum ether/EtOAc, 2:1) to yield 20 as a pale yellow solid (600 mg, 40.3 %). 1H NMR (400 MHz, CDCl3): δ=2.06 (s, 3 H), 2.44–2.50 (m, 1 H), 2.61–2.76 (m, 2 H), 2.92 (d, J=10.4 Hz, 1 H), 3.08 (d, J=9.6 Hz, 1 H), 3.41 (t, J=7.6 Hz, 1 H), 3.52 (d, J=14.0 Hz, 1 H), 4.11 (d, J=14.0 Hz, 1 H), 5.35 (m, 1 H), 5.67 (d, J=4.4 Hz, 1 H), 5.70 (s, 1 H), 5.92 (d, J=14.8 Hz, 2 H), 6.48 (s, 1 H), 6.85 (s, 1 H), 7.76 (m, 2 H), 7.85 ppm (m, 2 H); 13C NMR (100 MHz, CDCl3): δ=21.2, 28.3, 29.7, 44.2, 53.8, 56.7, 62.3, 64.7, 84.6, 101.0, 106.0, 107.2, 114.3, 122.7, 123.6, 125.5, 128.9, 129.0, 133.4, 134.6, 146.5, 163.9, 170.6 ppm; ESI‐MS: m/z 475 [M+H]+; HRMS: m/z calcd for C26H23N2O7 [M+H]+: 475.1500, found: 475.1503.
1‐Acetyl‐2‐(R)‐hydroxylaminelycorine (21): NH2NH2⋅H2O (25 mg, 0.50 mmol) was added to a solution of 20 (118 mg, 0.25 mmol) in MeOH (2 mL) and CH2Cl2 (2 mL), and the mixture was stirred at RT for 1 h. The solvent was removed in vacuo, and the crude residue was purified by using silica gel chromatography (EtOAc/ CH2Cl2/CH3OH, 10:10:1) to yield 21 as a pale yellow solid (103 mg, 97.6 %). 1H NMR (400 MHz, CD3OD): δ=1.96 (s, 3 H), 2.50–2.65 (m, 3 H), 2.75 (d, J=10.0 Hz, 1 H), 3.13 (d, J=10.0 Hz, 1 H), 3.32 (m, 1 H), 3.56 (d, J=14.0 Hz, 1 H), 4.09 (d, J=14.0 Hz, 1 H), 4.54 (s, 1 H), 4.75 (s, 1 H), 5.44 (s, 1 H), 5.90 (s, 2 H), 6.61 (s, 1 H), 6.88 ppm (s, 1 H); 13C NMR (100 MHz, CD3OD): δ=19.4, 29.1, 45.0, 54.9, 57.6, 62.9, 65.5, 87.7, 102.4, 106.1, 108.2, 116.3, 129.0, 129.9, 146.1, 147.9, 148.3, 172.2 ppm; ESI‐MS: m/z 345 [M+H]+; HRMS: m/z calcd for C18H20N2O5 [M+H]+: 345.1445, found: 345.1448.
1‐Acetyl‐2‐oxolycorine (22): DMSO (0.24 mL, 3.1 mmol) in anhydrous CH2Cl2 (1.5 mL) was added dropwise to a solution of (COCl)2 (0.22 mL, 2.5 mmol) in anhydrous CH2Cl2 (5 mL) at −78 °C. The mixture was stirred at −78 °C for 30 min, followed by the slow addition of 10 (329 mg, 1 mmol) in anhydrous CH2Cl2 (6 mL). The reaction was stirred at −78 °C for an additional 1.5 h, followed by addition of Et3N (0.86 mL, 6.2 mmol). The mixture was stirred for an additional 10 min, and brine (10 mL) was added to quench the reaction. The solvent was removed in vacuo, followed by the addition of CH2Cl2 (25 mL) and water (25 mL). The organic layer was washed with saturated aq NaHCO3 (20 mL) and then brine (20 mL), dried over anhydrous Na2SO4, and filtered. The filtrate was concentrated, and the crude residue was purified by using silica gel chromatography (petroleum ether/EtOAc, 1:1) to yield 22 as a white solid (280 mg, 85.6 %). 1H NMR (400 MHz, CDCl3): δ=1.91 (s, 3 H), 2.48 (m, 1 H), 2.82 (m, 2 H), 3.13 (d, J=9.6 Hz, 1 H), 3.21 (d, J=10.4 Hz, 1 H), 3.41 (m, 1 H), 3.56 (d, J=14.0 Hz, 1 H), 4.12 (d, J=14.0 Hz, 1 H), 5.89–5.85 (m, 2 H), 5.93 (s, 1 H), 5.94 (s, 1 H), 6.52 (s, 1 H), 6.67 ppm (s, 1 H); 13C NMR (100 MHz, CDCl3): δ=20.7, 30.0, 45.5, 53.2, 56.3, 62.3, 69.0, 101.0, 105.3, 107.3, 120.3, 125.2, 128.9, 146.6, 146.7, 169.1, 169.5, 192.9 ppm; ESI‐MS: m/z 328 [M+H]+; HRMS: m/z calcd for C18H18NO5 [M+H]+: 328.1185, found: 328.1185.
2‐Oxolycorine (23): Concentrated HCl (2 mL) was added to a solution of 22 (117 mg, 0.36 mmol) in MeOH (10 mL), and the solution was stirred at 50 °C for 2 h. The solvent was removed in vacuo, and the residue was purified by using silica gel chromatography (EtOAc, 100 %) to yield 23 as a gray solid (89 mg, 87.0 %). 1H NMR (400 MHz, CDCl3): δ=2.51 (m, 1 H), 2.80–2.89 (m, 2 H), 3.12 (d, J=9.6 Hz, 1 H), 3.23 (d, J=9.6 Hz, 1 H), 3.45 (m, 1 H), 3.61 (d, J=14.4 Hz, 1 H), 4.16 (d, J=14.4 Hz, 1 H), 4.54 (d, J=3.0 Hz, 1 H), 5.99–5.89 (m, 3 H), 6.59 (s, 1 H), 6.75 ppm (s, 1 H); 13C NMR (100 MHz, CDCl3): δ=30.0, 46.0, 53.4, 56.4, 61.8, 70.3, 101.1, 105.0, 107.5, 119.7, 126.1, 129.2, 146.6, 146.7, 169.2, 197.5 ppm; ESI‐MS: m/z 286 [M+H]+; HRMS: m/z calcd for C16H15NO4 [M+H]+: 286.1079, found: 286.1076.
1,2‐α‐Epoxylycorine (24): 10 (156 mg, 0.47 mmol) was added to a solution of NaCl (5 mg, 0.1 mmol) in POCl3 (2 mL), and the mixture was stirred at 30 °C for 1 h, followed by addition of concentrated HCl solution (0.1 mL). After being left to stir at 30 °C for an additional 2 h, the mixture was poured into ice water, basified (pH 8) with 25 % aq NH3, and extracted with CH2Cl2 (2×25 mL). The organic layer was dried over anhydrous Na2SO4 and filtered. The filtrate was concentrated in vacuo, and the crude residue was purified by using silica gel chromatography (EtOAc/CH3OH, 10:1) to yield a white solid (132 mg, 80.9 %). The product was dissolved in MeOH (20 mL), followed by addition of CH3ONa (0.5 g). The mixture was stirred at 0 °C for 20 min, followed by the addition of CH2Cl2 (15 mL) and water (15 mL). The organic layer was dried over anhydrous Na2SO4 and filtered. The filtrate was concentrated in vacuo, and the crude residue was purified by using silica gel chromatography (EtOAc, 100 %) to yield 24 as a white solid (44 mg, 42.9 %). 1H NMR (400 MHz, CDCl3): δ=2.43 (m, 1 H), 2.61 (m, 1 H), 2.80 (t, J=13.6 Hz, 2 H), 3.21 (m, 1 H), 3.51 (t, J=4.0 Hz, 1 H), 3.58 (d, J=14.0 Hz, 1 H), 3.96 (d, J=4.0 Hz, 1 H), 4.08 (d, J=14.0 Hz, 1 H), 5.76 (d, J=1.6 Hz, 1 H), 5.94 (d, J=4.4 Hz, 2 H), 6.60 (s, 1 H), 7.04 ppm (s, 1 H); 13C NMR (100 MHz, CDCl3): δ=29.3, 40.8, 49.5, 54.0, 54.5, 57.0, 63.1, 101.0, 105.4, 107.5, 112.6, 129.2, 129.5, 146.2, 146.4, 147.9 ppm; ESI‐MS: m/z 270 [M+H]+; HRMS: m/z calcd for C16H16NO3 [M+H]+: 270.1125, found: 270.1130.
2‐Azidolycorine (25): NaN3 (401 mg, 6.2 mmol) was added to a solution of 24 (166 mg, 0.62 mmol) in MeOH (10 mL), and the solution was heated to 80 °C for 20 min. The solvent was removed in vacuo, followed by the addition of CH2Cl2 (20 mL) and water (20 mL). The organic layer was dried over anhydrous Na2SO4 and filtered. The filtrate was concentrated, and the crude residue was purified by using silica gel chromatography (petroleum ether/EtOAc, 2:1) to yield 25 as a yellow solid (150 mg, 77.5 %). 1H NMR (400 MHz, CDCl3): δ=2.37 (m, 1 H), 2.57 (d, J=10.4 Hz, 1 H), 2.63 (m, 2 H), 2.82 (d, J=10.0 Hz, 1 H), 3.28 (m, 1 H), 3.49 (d, J=14.0 Hz, 1 H), 3.91 (s, 1 H), 4.07 (d, J=14.0 Hz, 1 H), 4.34 (s, 1 H), 5.51 (s, 1 H), 5.91 (d, J=3.2 Hz, 2 H), 6.58 (s, 1 H), 6.67 ppm (s, 1 H); 13C NMR (100 MHz, CDCl3): δ=28.7, 41.2, 53.7, 56.9, 60.9, 62.7, 69.3, 101.1, 104.7, 107.8, 112.6, 127.1, 129.5, 145.8, 146.4, 146.8 ppm; ESI‐MS: m/z 313 [M+H]+; HRMS: m/z calcd for C16H17N4O3 [M+H]+: 313.1295, found: 313.1299.
2‐Aminolycorine (26): LiAlH4 (168 mg, 4.4 mmol) in anhydrous THF (3 mL) was added to a solution of 25 (69 mg, 0.22 mmol) in anhydrous THF (2 mL), and the mixture was stirred at 0 °C for 1 h. Water (15 mL) was slowly added to quench the reaction, followed by the addition of CH2Cl2 (20 mL). The organic layer was dried over anhydrous Na2SO4 and filtered. The filtrate was concentrated, and the crude residue was purified by using silica gel chromatography (CH2Cl2/CH3OH, 5:1) to yield 26 as a gray solid (40 mg, 63.5 %). 1H NMR (400 MHz, CD3OD): δ=2.44–2.33 (m, 1 H), 2.68–2.45 (m, 3 H), 2.85 (d, J=10.0 Hz, 1 H), 3.26 (m, 2 H), 3.48 (d, J=14.0 Hz, 1 H), 3.59 (s, 1 H), 4.05 (d, J=14.2 Hz, 1 H), 4.49 (s, 1 H), 5.41 (s, 1 H), 5.83 (s, 2 H), 6.55 (s, 1 H), 6.79 ppm (s, 1 H); 13C NMR (100 MHz, CD3OD): δ=29.6, 41.45, 54.7, 55.9, 57.8, 62.2, 69.9, 102.4, 106.2, 108.4, 115.8, 128.6, 130.3, 146.2, 147.9, 148.3 ppm; ESI‐MS: m/z 287 [M+H]+; HRMS: m/z calcd for C16H19N2O3 [M+H]+: 287.1390, found: 287.1395.
2‐Methoxylycorine (27): 24 (93 mg, 0.27 mmol) was added to a solution of CH3ONa (145 mg, 2.7 mmol) in MeOH (6 mL), and the mixture was stirred at RT for 30 h. The solvent was removed in vacuo, followed by addition of CH2Cl2 (20 mL) and brine (20 mL). The organic layer was dried over anhydrous Na2SO4 and filtered. The filtrate was concentrated in vacuo, and the crude residue was purified by using silica gel chromatography (EtOAc, 100 %) to yield 27 as a gray solid (45 mg, 55.4 %). 1H NMR (400 MHz, CDCl3): δ=2.35 (m, 1 H), 2.60 (m, 2 H), 2.67 (d, J=10.8 Hz, 1 H), 2.69 (d, J=10.0 Hz, 1 H), 3.48 (s, 3 H), 3.49 (d, J=13.6 Hz, 1 H), 3.80 (s, 1 H), 4.11 (d, J=14.0 Hz, 1 H), 4.57 (s, 1 H), 5.58 (s, 1 H), 5.92 (d, J=5.2 Hz, 2 H), 6.59 (s, 1 H), 6.83 ppm (s, 1 H); 13C NMR (100 MHz, CDCl3): δ=20.8, 30.0, 45.4, 53.2, 56.3, 62.3, 67.0, 101.1, 105.4, 107.3, 120.4, 125.2, 128.9, 146.6, 146.7, 169.2, 169.6, 193.0 ppm; ESI‐MS: m/z 302 [M+H]+; HRMS: m/z calcd for C17H20NO4 [M+H]+: 302.1387, found: 302.1380.
General procedure for 28 and 29: Phenylacetylene (0.92 mmol), CuSO4 (15 mg, 0.09 mmol) and l‐ascorbic acid (32 mg, 0.18 mmol) were added to a solution of 25 (143 mg, 0.46 mmol) in THF (3 mL) and water (3 mL), and the mixture was stirred at RT overnight. The solution was basified (pH 8) with saturated aq NaHCO3, followed by addition of CH2Cl2 (15 mL) and water (15 mL). The organic layer was dried over anhydrous Na2SO4 and filtered. The filtrate was concentrated in vacuo, and the crude residue was purified by using silica gel chromatography to yield the product.
2‐(4‐Phenyltriazole)lycorine (28): Following the previously described general procedure, 25 (143 mg, 0.46 mmol) yielded 28 as a pale yellow solid (120 mg, 63.0 %). 1H NMR (400 MHz, CDCl3): δ=2.47 (m, 1 H), 2.75 (m, 3 H), 3.03 (d, J=10.4 Hz, 1 H), 3.45 (m, 1 H), 3.57 (d, J=14.4 Hz, 1 H), 4.19 (d, J=14.0 Hz, 1 H), 4.75 (s, 1 H), 5.58 (s, 2 H), 5.79 (d, J=3.2 Hz, 2 H), 6.51 (s, 1 H), 6.62 (s, 1 H), 2.27 (d, J=7.2 Hz, 1 H), 7.34 (t, J=7.6 Hz, 2 H), 7.71 (d, J=7.2 Hz, 2 H), 7.77 ppm (s, 1 H); 13C NMR (100 MHz, CDCl3): δ=14.2, 28.9, 41.1, 53.7, 56.9, 60.7, 64.0, 70.5, 101.0, 105.1, 107.5, 112.2, 119.0, 125.7, 126.7, 128.2, 128.8, 129.4, 130.2, 146.4, 146.5, 147.3, 147.4 ppm; ESI‐MS: m/z 415 [M+H]+; HRMS: m/z calcd for C24H23N4O3 [M+H]+: 415.1765, found: 415.1764.
2‐(4‐Pentanetriazole)lycorine (29): Following the previously described general procedure, 25 (170 mg, 0.55 mmol) yielded 29 as a pale yellow solid (42 mg, 18.7 %). 1H NMR (400 MHz, CDCl3): δ=0.87 (t, J=6.8 Hz, 3 H), 1.31 (m, 4 H), 1.60 (m, 2 H), 2.43–2.50 (m, 1 H), 2.61–2.67 (m, 3 H), 2.77 (m, 2 H), 2.98 (d, J=10.4 Hz, 1 H), 3.45 (m, 1 H), 3.59 (d, J=14.0 Hz, 1 H), 4.19 (d, J=14.0 Hz, 1 H), 4.68 (s, 1 H), 5.52 (s, 1 H), 5.56 (s, 1 H), 5.88 (d, J=4.5 Hz, 2 H), 6.58 (s, 1 H), 6.66 (s, 1 H), 7.29 ppm (s, 1 H); 13C NMR (100 MHz, CDCl3): δ=14.0, 22.4, 25.7, 28.9, 29.0, 29.7, 31.5, 41.2, 53.7, 56.9, 60.7, 63.5, 70.8, 101.0, 104.9, 107.6, 112.2, 119.8, 126.6, 129.7, 146.5, 146.6, 147.0, 148.3 ppm; ESI‐MS: m/z 409 [M+H]+; HRMS: m/z calcd for C23H29N4O3Na [M+Na]+: 431.2054, found: 431.2048.
2‐Amidohexanoyllycorine (30): Hexanoyl chloride (67 mg, 0.50 mmol) in CH2Cl2 (2 mL) was slowly added to a solution of 26 (95 mg, 0.33 mmol) in anhydrous pyridine (2 mL) at 0 °C. The solution was stirred at 0 °C for 40 min. The solvent was removed, followed by addition of CH2Cl2 (15 mL) and water (15 mL). The organic layer was dried over anhydrous Na2SO4 and filtered. The filtrate was concentrated in vacuo, and the crude residue was purified by using silica gel chromatography (EtOAc, 100 %) to yield 30 as a pale yellow oil (69 mg, 54.4 %). 1H NMR (400 MHz, CDCl3): δ=0.86 (t, J=6.8 Hz, 3 H), 1.24–1.29 (m, 4 H), 1.58–1.62 (m, 2 H), 2.15 (m, 2 H), 2.36 (m, 1 H), 2.64 (m, 3 H), 2.91 (d, J=10.0 Hz, 1 H), 3.35 (m, 1 H), 3.52 (d, J=14.0 Hz, 1 H), 4.13 (d, J=14.0 Hz, 1 H), 4.35 (s, 1 H), 4.58 (d, J=6.8 Hz, 1 H), 5.35 (s, 1 H), 5.91 (d, J=7.6 Hz, 2 H), 6.35 (d, J=8.0 Hz, 1 H), 6.57 (s, 1 H), 6.69 ppm (s, 1 H); 13C NMR (100 MHz, CDCl3): δ=13.0, 21.4, 24.5, 27.5, 30.4, 35.7, 40.0, 52.0, 52.6, 55.8, 60.0, 68.4, 99.9, 103.9, 106.5, 114.4, 126.9, 127.9, 142.3, 145.3, 145.7, 171.7 ppm; ESI‐MS: m/z 385 [M+H]+; HRMS: m/z calcd for C22H29N2O4 [M+H]+: 385.2122, found: 385.2118.
2‐Acetyllycorine (31): Anhydrous Ac2O (2 mL) was added to a solution of 1 (150 mg, 0.52 mmol) in pyridine (5 mL), and the mixture was stirred at 50 °C for 1 h. The solvent was removed in vacuo, followed by addition of CH2Cl2 (20 mL) and water (20 mL). The organic layer was washed with saturated aq NaHCO3 and then brine, dried over anhydrous Na2SO4, and filtered. The filtrate was concentrated in vacuo, and the crude residue was purified by using silica gel chromatography (petroleum ether/EtOAc, 1:1) to yield 31 as a white solid (130 mg, 76.0 %). 1H NMR (400 MHz, [D6]DMSO): δ=2.03 (s, 3 H), 2.23 (m, 1 H), 2.43 (m, 1 H), 2.53 (m, 2 H), 2.67 (d, J=10.4 Hz, 1 H), 2.85 (d, J=10.4 Hz, 1 H), 3.21 (m, 1 H), 3.34 (d, J=13.6 Hz, 1 H), 4.04 (d, J=14.4 Hz, 1 H), 4.37 (s, 1 H), 5.18 (s, 1 H), 5.35 (s, 1 H), 5.95 (d, J=7.2 Hz, 2 H), 6.69 (s, 1 H), 6.86 ppm (s, 1 H); 13C NMR (100 MHz, [D6]DMSO): δ=21.0, 28.2, 41.2, 53.1, 56.6, 60.5, 67.0, 73.3, 100.6, 105.3, 107.0, 113.3, 128.4, 129.6, 145.4, 145.7, 146.2, 169.6 ppm; ESI‐MS: m/z 330 [M+H]+; HRMS: m/z calcd for C18H20NO5 [M+H]+: 330.1341, found: 330.1340.
2‐Hexanoyllycorine (32): Hexanoyl chloride (162 mg, 1.2 mmol) in CH2Cl2 (2 mL) was slowly added to a solution of 1 (287 mg, 1 mmol) in anhydrous pyridine (5 mL), and the solution was stirred at RT for 1.5 h, followed by the addition of CH2Cl2 (20 mL) and water (20 mL). The organic layer was washed with saturated aq NaHCO3 and then brine, dried over anhydrous Na2SO4, and filtered. After solvent evaporation, the crude residue was purified by using silica gel chromatography (petroleum ether/EtOAc, 1:1) to yield 32 as a colorless oil (60 mg, 16.0 %). 1H NMR (400 MHz, CDCl3): δ=0.88 (m, 3 H), 1.35–1.27 (m, 4 H), 1.68–1.58 (m, 2 H), 2.31 (t, J=7.6 Hz, 2 H), 2.53 (m, 1 H), 2.66 (m, 2 H), 2.72 (d, J=10.4 Hz, 1 H), 2.98 (d, J=10.4 Hz, 1 H), 3.42–3.30 (m, 1 H), 3.62 (d, J=14.0 Hz, 1 H), 4.12 (d, J=14.0 Hz, 1 H), 4.48 (s, 1 H), 5.31 (s, 1 H), 5.47 (s, 1 H), 5.91 (d, J=6.4 Hz, 2 H), 6.79 (s, 1 H), 6.59 ppm (s, 1 H); 13C NMR (100 MHz, CDCl3): δ=13.9, 22.3, 24.6, 28.8, 31.3, 34.4, 41.0, 53.6, 56.3, 60.4, 68.7, 73.3, 101.0, 104.8, 107.7, 114.2, 127.5, 129.2, 145.2, 146.3, 146.7, 173.5 ppm; ESI‐MS: m/z 386 [M+H]+; HRMS: m/z calcd for C22H28NO5 [M+H]+: 386.1962, found: 386.1965.
2‐tert‐Butyldimethylsilyloxylycorine (33): tert‐Butyldimethylsilyl chloride (453 mg, 3.0 mmol) and 4‐dimethylaminopyridine (366 mg, 3.0 mmol) were added to a solution of 1 (287 mg, 1.0 mmol) in anhydrous pyridine (5 mL), and the mixture was stirred at RT for 6 h. The solvent was removed in vacuo, followed by addition of CH2Cl2 (25 mL) and water (25 mL). The organic layer was washed with saturated aq NaHCO3 and then brine, dried over anhydrous Na2SO4, and filtered. The filtrate was concentrated in vacuo, and the crude residue was purified by using silica gel chromatography (EtOAc, 100 %) to yield 33 as a colorless oil (168 mg, 42.0 %). 1H NMR (400 MHz, CDCl3): δ=0.09 (s, 3 H), 0.12 (s, 3 H), 0.87 (s, 9 H), 2.30 (m, 1 H), 2.57 (m, 2 H), 2.68 (d, J=10.4 Hz, 1 H), 2.77 (d, J=10.4 Hz, 1 H), 3.30 (m, 1 H), 3.45 (d, J=14.0 Hz, 1 H), 4.10 (d, J=14.0 Hz, 1 H), 4.24 (s, 1 H), 4.37 (s, 1 H), 5.39 (s, 1 H), 5.89 (s, 2 H), 6.56 (s, 1 H), 6.79 ppm (s, 1 H); 13C NMR (100 MHz, CDCl3): δ=−4.7, −4.4, 18.1, 25.9, 28.6, 40.8, 53.9, 57.1, 60.9, 72.0, 72.5, 100.8, 104.6, 107.6, 118.3, 128.2, 130.1, 141.8, 146.0, 146.3 ppm; ESI‐MS: m/z 402 [M+H]+; HRMS: m/z calcd for C22H32NO4Si [M+H]+: 402.2095, found: 402.2096.
General procedure for 34–41: Acyl chloride (1.2 equiv) was added to a solution of 23 (1 equiv) in anhydrous pyridine (5 mL), and the solution was stirred at 0 °C until TLC analysis indicated that all of the starting material had been consumed. This was followed by addition of CH2Cl2 (20 mL) and water (20 mL). The organic layer was washed with saturated aq NaHCO3 and brine, dried over anhydrous Na2SO4, and filtered. The filtrate was concentrated in vacuo, and the crude residue was purified by using silica gel chromatography to yield the products.
1‐Propionyl‐2‐oxolycorine (34): Following the previously described general procedure, 23 (200 mg, 0.70 mmol) yielded 34 as a white solid (183 mg, 76.5 %). 1H NMR (400 MHz, CDCl3): δ=0.93 (t, J=6.7 Hz, 3 H), 2.13 (s, 2 H), 2.46 (d, J=8.3 Hz, 1 H), 2.79 (s, 2 H), 3.11 (d, J=9.2 Hz, 1 H), 3.20 (d, J=8.4 Hz, 1 H), 3.39 (s, 1 H), 3.53 (d, J=13.9 Hz, 1 H), 4.10 (d, J=14.0 Hz, 1 H), 5.85 (s, 2 H), 5.92 (s, 2 H), 6.50 (s, 1 H), 6.64 ppm (s, 1 H); 13C NMR (100 MHz, CDCl3): δ=9.0, 27.4, 30.0, 45.4, 53.2, 56.3, 62.3, 68.8, 101.0, 105.4, 107.3, 120.4, 125.2, 128.7, 146.6, 169.0, 173.0, 193.1 ppm; ESI‐MS: m/z 342 [M+H]+; HRMS: m/z calcd for C19H20NO5 [M+H]+: 342.1341, found: 342.1338.
1‐Benzoyl‐2‐oxolycorine (39): Following the previously described general procedure, 23 (200 mg, 0.70 mmol) yielded 39 as a white solid (158 mg, 58.0 %). 1H NMR (400 MHz, CDCl3): δ=2.60 (q, J=8.4 Hz, 1 H), 2.92 (s, 2 H), 3.34 (d, J=9.8 Hz, 1 H), 3.40 (d, J=10.1 Hz, 1 H), 3.56–3.47 (m, 1 H), 3.64 (d, J=14.0 Hz, 1 H), 4.20 (d, J=14.1 Hz, 1 H), 5.87 (d, J=13.9 Hz, 2 H), 6.05 (s, 1 H), 6.25 (s, 1 H), 6.55 (s, 1 H), 6.83 (s, 1 H), 7.36 (t, J=7.4 Hz, 2 H), 7.50 (t, J=7.4 Hz, 1 H), 7.88 ppm (d, J=7.8 Hz, 2 H); 13C NMR (100 MHz, CDCl3): δ=30.0, 45.7, 53.3, 56.3, 62.6, 69.5, 101.1, 105.4, 107.3, 120.6, 125.0, 128.3, 128.6, 129.4, 129.9, 133.2, 146.7, 165.1, 168.9, 192.8 ppm; ESI‐MS: m/z 390 [M+H]+; HRMS: m/z calcd for C23H20NO5 [M+H]+: 390.1341, found: 390.1330.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
miscellaneous_information
Acknowledgements
The authors are grateful to the Novartis Institute for Tropical Diseases (NITD), Singapore, for financial support. This work was supported by the National Natural Science Foundation of China (grant no. 21202087), the National Basic Research Program of China (973 program, grant no. 2013CB911104), the Fundamental Research Funds for the Central Universities (grant no. 65124002), the Specialized Research Fund for the Doctoral Program of Higher Education from the Ministry of Education of China (grant no. 20120031120049), the Tianjin Science and Technology Program (grant nos. 13JCYBJC24300 and 13JCQNJC13100), the Scientific Research Starting Foundation of Returned Overseas Chinese Scholars from the Ministry of Education of China, and the “111” Project of the Ministry of Education of China (Project No. B06005).
Contributor Information
Dr. Lu‐Qing Shang, Email: shanglq@nankai.edu.cn.
Dr. Pei‐Yong Shi, Email: pei_yong.shi@novartis.com.
Prof. Dr. Zheng Yin, Email: zheng_yin@nankai.edu.cn.
References
- 1. Bhatt S., Gething P. W., Brady O. J., Messina J. P., Farlow A. W., Moyes C. L., Drake J. M., Brownstein J. S., Hoen A. G., Sankoh O., Myers M. F., George D. B., Jaenisch T., Wint G. R. W., Simmons C. P., Scott T. W., Farrar J. J., Hay S. I., Nature 2013, 496, 504–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Schüller A., Yin Z., Chia C. S. B., Doan D. N. P., Kim H.‐K., Shang L. Q., Loh T. P., Hill J., Vasudevan S. G., Antiviral Res. 2011, 92, 96–101. [DOI] [PubMed] [Google Scholar]
- 3. Murrell S., Wu S.‐C., Butler M., Biotechnol. Adv. 2011, 29, 239–247. [DOI] [PubMed] [Google Scholar]
- 4. Dong H., Zhang B., Shi P. Y., Antiviral Res. 2008, 80, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lamoral‐Theys D., Decaestecker C., Mathieu V., Dubois J., Kornienko A., Kiss R., Evidente A., Pottier L., Mini‐Rev. Med. Chem. 2010, 10, 41–50. [DOI] [PubMed] [Google Scholar]
- 6. Cedrón J. C., Gutiérrez D., Flores N., Ravelo Á. G., Estévez‐Braun A., Bioorg. Med. Chem. 2010, 18, 4694–4701. [DOI] [PubMed] [Google Scholar]
- 7. Toriizuka Y., Kinoshita E., Kogure N., Kitajima M., Ishiyama A., Otoguro K., Yamada H., Omura S., Takayama H., Bioorg. Med. Chem. 2008, 16, 10182–10189. [DOI] [PubMed] [Google Scholar]
- 8. Çitoǧlu G., Tanker M., Gümüşel B., Phytother. Res. 1998, 12, 205–206. [Google Scholar]
- 9. Tanker M., Çitoglu G., Gümühel B., Hener B., Int. J. Pharmacogn. 1996, 34, 194–197. [Google Scholar]
- 10. Kretzing S., Abrahama G., Seiwert B., Ungemach F. R., Krügel U., Regenthal R., Toxicon 2011, 57, 117–124. [DOI] [PubMed] [Google Scholar]
- 11. Hwang Y. C., Chu J. J‐H., Yang P. L., Chen W., Yates M. V., Antiviral Res. 2008, 77, 232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Li S. Y., Chen C., Zhang H., Guo H., Wang H., Wang L., Zhang X., Hua S., Yu J., Xiao P., Li R., Tan X., Antiviral Res. 2005, 67, 18–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Renard‐Nozaki J., Kim T., Imakura Y., Kihara M., Kobayashi S., Res. Virol. 1989, 140, 115–128. [DOI] [PubMed] [Google Scholar]
- 14. Zhou M., Deng L., Kashanchi F., Brady J. N., Shatkin A. J., Kumar A., Proc. Natl. Acad. Sci. USA 2003, 100, 12666–12671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gabrielsen B., Monath T. P., Huggins J. W., Kefauver D. F., J. Nat. Prod. 1992, 55, 1569–1581. [DOI] [PubMed] [Google Scholar]
- 16. Zou G., Puig‐Basagoiti F., Zhang B., Qing M., Chen L., Pankiewicz K. W., Felczak K., Yuan Z., Shi P. Y., Virology 2009, 384, 242–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Evidente A., Cicala M. R., Randazzo G., Riccio R., Calabrese G., Liso R., Arrloonit O., Phytochemistry 1983, 22, 2193–2196. [Google Scholar]
- 18. Toda J., Sano T., Tsuda Y., Kaneda M., Iitaka Y., Tetrahedron Lett. 1980, 21, 369–370. [Google Scholar]
- 19. Wildman W. C., Heimer N. E., J. Am. Chem. Soc. 1967, 89, 5265–5269. [DOI] [PubMed] [Google Scholar]
- 20. Rostovtsev V. V., Green L. G., Fokin V. V., Sharpless K. B., Angew. Chem. 2002, 114, 2708–2711; [Google Scholar]; Angew. Chem. Int. Ed. 2002, 41, 2596–2599. [DOI] [PubMed] [Google Scholar]
- 21. McNulty J., Nair J. J., Little J. R. L., Brennan J. D., Bastida J., Bioorg. Med. Chem. Lett. 2010, 20, 5290–5294. [DOI] [PubMed] [Google Scholar]
- 22. Patani G. A., LaVoie E. J., Chem. Rev. 1996, 96, 3147–3176. [DOI] [PubMed] [Google Scholar]
- 23. Clark M., Cramer R. D., Quant. Struct.‐Act. Relat. 1993, 12, 137–145. [Google Scholar]
- 24. Wang Q. Y., Patel S. J., Vangrevelinghe E., Xu H. Y., Rao R., Jaber D., Schul W., Gu F., Heudi O., Ma N. L., Poh M. K., Phong W. Y., Keller T. H., Jacoby E., Vasudevan S. G., Antimicrob. Agents Chemother. 2009, 53, 1823–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
miscellaneous_information