

Abstract
The hepatitis E virus (HEV) genome is a single‐stranded, positive‐sense RNA that encodes three proteins including the ORF1 replicase. Mechanisms of HEV replication in host cells are unclear, and only a few cellular factors involved in this step have been identified so far. Here, we used brefeldin A (BFA) that blocks the activity of the cellular Arf guanine nucleotide exchange factors GBF1, BIG1, and BIG2, which play a major role in reshuffling of cellular membranes. We showed that BFA inhibits HEV replication in a dose‐dependent manner. The use of siRNA and Golgicide A identified GBF1 as a host factor critically involved in HEV replication. Experiments using cells expressing a mutation in the catalytic domain of GBF1 and overexpression of wild type GBF1 or a BFA‐resistant GBF1 mutant rescuing HEV replication in BFA‐treated cells, confirmed that GBF1 is the only BFA‐sensitive factor required for HEV replication. We demonstrated that GBF1 is likely required for the activity of HEV replication complexes. However, GBF1 does not colocalise with the ORF1 protein, and its subcellular distribution is unmodified upon infection or overexpression of viral proteins, indicating that GBF1 is likely not recruited to replication sites. Together, our results suggest that HEV replication involves GBF1‐regulated mechanisms.
Abbreviations
- aa
amino acid
- BFA
brefeldin A
- DMEM
Dulbecco's modified Eagle's medium
- ER
endoplasmic reticulum
- ERGIC
ER‐Golgi intermediate compartment
- FCS
foetal calf serum
- FLuc
Firefly luciferase
- GBF1
guanine nucleotide‐exchange factor Golgi brefeldin A resistance Factor 1
- GCA
Golgicide A
- GEF
guanine nucleotide exchange factor
- GLuc
Gaussia luciferase
- gt
genotype
- HCV
hepatitis C virus
- HEV
hepatitis E virus
- MAb
monoclonal antibody
- ORFs
open reading frames
- p.e.
post‐electroporation
- PBS
phosphate buffered saline
- WB
western‐blotting
1. INTRODUCTION
Hepatitis E virus (HEV) is increasingly recognised as the major cause of acute hepatitis worldwide. This virus is annually responsible for 20 million infections with 3.4 million symptomatic cases and 70,000 deaths mainly occurring in less developed regions of the world (Debing, Moradpour, Neyts, & Gouttenoire, 2016). Although infection by HEV is usually self‐resolving, severe forms or chronic infections have been described, mainly in immunocompromised patients. A high rate of mortality has also been reported among pregnant women. In addition, HEV infection has been associated with a broad range of extrahepatic manifestations, including renal injury and a variety of neurological disorders (Kamar, Marion, Abravanel, Izopet, & Dalton, 2016). Four genotypes (gt) are pathogenic in humans. gt1 and gt2 exclusively infect humans, whereas gt3 and gt4 are zoonotic and mainly infect mammals with occasional transmission to humans (Doceul, Bagdassarian, Demange, & Pavio, 2016). In industrialised countries, the most common genotype causing HEV infection is gt3. Importantly, due to the evolution toward chronicity in immunocompromised infected patients, HEV transmission through blood transfusion, resistance of some infected patients to ribavirin and complications in patients with preexisting liver disease, HEV infection is now considered as an emerging problem in industrialised countries (Sayed, Vercouter, Abdelwahab, Vercauteren, & Meuleman, 2015).
HEV has been classified as the sole member of the Orthohepevirus genus within the Hepeviridae family (Smith et al., 2014). It is a quasi‐enveloped virus containing a linear, single‐stranded, positive‐sense RNA genome that encodes three open reading frames (ORFs), namely, ORF1, ORF2, and ORF3 (Tam et al., 1991). ORF1 is the largest gene that encodes a non‐structural polyprotein (ORF1 protein) that contains several functional domains essential for viral replication (Koonin et al., 1992). These functional domains include the methyltransferase (Met), papain‐like cysteine protease, RNA helicase (Hel), and RNA‐dependent RNA polymerase (reviewed in Debing et al., 2016). To date, there is no clear evidence of ORF1 protein processing by protease and antibodies that robustly recognise ORF1 protein are not available. ORF2 encodes the ORF2 viral capsid protein, which is involved in particle assembly, binding to host cells and eliciting neutralising antibodies. Very recently, we demonstrated that during its lifecycle, HEV produces three forms of the ORF2 capsid protein: ORF2i (infectious/intracellular ORF2), ORF2g (glycosylated ORF2), and ORF2c (cleaved ORF2). The ORF2i protein is associated with infectious particles, whereas ORF2g and ORF2c proteins are massively produced glycoproteins that are not associated with infectious particles and are the major antigens present in HEV‐infected patient sera (Montpellier et al., 2017). ORF3 encodes a small multifunctional phosphoprotein that is involved in virion morphogenesis and egress (reviewed in Holla, Ahmad, Ahmad, & Jameel, 2013).
Due to difficulties in efficiently propagating HEV in cell culture, numerous pathways and processes of the HEV lifecycle remain to be elucidated. Notably, mechanisms leading to HEV replication are particularly poorly understood. However, it has been shown that the ORF1 protein might be membrane‐associated and localised in the endoplasmic reticulum‐Golgi intermediate compartment (ERGIC), suggesting that HEV replication might occur within the early secretory pathway (Perttilä, Spuul, & Ahola, 2013). Plus‐strand RNA virus replication occurs in close association with host cell membranes. In infected cells, cellular and viral factors cooperatively generate particular structures resembling organelles that are named viral replication factories. This compartmentalisation allows for coordination of the different steps of the replication cycle, highly efficient RNA replication, and protects the viral genome from cell defence mechanisms. It has to be noted that for a number of viruses, the viral budding site is located near the replication factories, indicating a spatial coordination of replication and viral assembly steps. To induce these massive membrane rearrangements, viruses use cellular factors active on membranes and exploit the cellular pathways involved in membrane homeostasis (reviewed in Paul, 2013). In particular, some viruses divert components from the cellular secretory pathway. Indeed, it has been shown in our laboratory that the guanine nucleotide‐exchange factor Golgi brefeldin A (BFA) resistance Factor 1 (GBF1) is necessary for hepatitis C virus (HCV) replication (Farhat et al., 2016; Goueslain et al., 2010). GBF1 has also been identified as a cellular factor essential for the replication of a number of other viruses such as picornaviruses and coronaviruses (Belov, Feng, Nikovics, Jackson, & Ehrenfeld, 2008; Lanke et al., 2009; van der Linden, van der Schaar, Lanke, Neyts, & van Kuppeveld, 2010; Verheije et al., 2008).
To investigate HEV replication mechanisms, we used BFA, a fungal metabolite inhibiting the activation of Arf proteins, small G‐proteins regulating the cellular secretory pathway. The inactive, cytoplasmic GDP‐bound form of Arf proteins, upon nucleotide exchange to GTP, undergoes conformational changes that allow Arf‐GTP proteins to bind membranes. The active GTP‐bound form of Arf proteins is essential for the formation of secretory vesicles, actin remodelling and phospholipid metabolism by recruiting to membranes effectors that mediate these processes. BFA blocks Arf activation by inhibiting a subset of guanine nucleotide exchange factors (GEFs) that regulate the conversion of Arf‐GDP into Arf‐GTP. In human cells, BFA inhibits the function of three of the 15 known Arf GEFs: GBF1, BIG1, and BIG2, by stabilising normally transient complexes formed between the GEF and Arf‐GDP. Here, we demonstrate that BFA inhibits HEV replication and identified GBF1 as the BFA‐sensitive GEF required for HEV replication.
2. RESULTS
2.1. BFA inhibits HEV replication
In order to investigate the role of the cellular membranes during HEV infection, we used BFA that blocks several membrane trafficking pathways and causes major membrane rearrangements in the host cell (Klausner, Donaldson, & Lippincott‐Schwartz, 1992). The Gaussia luciferase (Gluc)‐encoding subgenomic replicon construct derived from the HEV gt3 Kernow‐C1 p6 strain was modified to produce the subgenomic replicon p6SPGLuc. In this replicon, the first 20 amino acids matching with the signal peptide of Gaussia luciferase were deleted to block secretion of the luciferase. In this context, the amount of intracellular GLuc is proportional to viral RNA synthesis and consequently to HEV replication. In addition, this system allows replication to be monitored independently of protein secretion that is blocked by BFA. Huh‐7.5 cells were electroporated with in vitro‐transcribed p6SPGLuc RNA and luciferase activities were measured at 6, 24, 48, and 72 hr post‐electroporation (p.e.). For each time point, values are presented as fold increase compared to luciferase activities measured at 6 hr p.e. As shown in Figure 1a, the level of p6SPGLuc steadily increased over time to reach a fold increase of 490 times at 72 hr p.e., indicating that the p6SPGLuc replicon efficiently replicates in Huh‐7.5 cells and can be used as a tool to study the HEV replication step. HEV‐p6SPGLuc‐electroporated Huh‐7.5 cells were then treated for 16 hr with different concentrations of BFA (Figure 1b). In parallel, transfected cells were treated with sofosbuvir, an inhibitor of HCV polymerase, which has been recently described as a HEV replication inhibitor (Dao Thi et al., 2016; Figure 1c). Luciferase activities were measured at 24, 48, and 72 hr p.e. For each BFA concentration, values are presented as a percentage of replication compared to non‐treated cells (dimethyl sulfoxide [DMSO]). As expected, treatment of p6SPGLuc‐electroporated Huh‐7.5 cells with sofosbuvir led to a dose‐dependent decrease of HEV replication with a 50% inhibitory concentration (IC50) of 10.6 μM. Interestingly, treatment of electroporated Huh‐7.5 cells with BFA also led to a dose‐dependent decrease of HEV replication with an IC50 of 0.02 μg/ml (Figure 1b), indicating that BFA is an inhibitor of gt3 HEV replication. Next, to verify that the decrease of HEV replication was not due to a toxic effect of the BFA treatment, we performed a 3‐(4,5‐dimethylthiazol‐2‐yl)‐5‐ (3‐carboxymethoxyphenyl)‐2‐(4‐sulfophenyl)‐2H–tetrazolium viability assay on Huh7.5 cells treated for 16 hr with BFA. As shown in Figure 1d, although a weak toxicity was observed at 1 μg/ml, the concentration of 0.1 μg/ml for which HEV replication was reduced by 1 log had no significant toxic effect, indicating that the inhibitory effect of BFA on HEV replication was not due to cell toxicity. In order to confirm the inhibitory effect of BFA on HEV replication in another cell line, we next analysed BFA activity on HEV replication in PLC3 cells, a PLC‐PRF‐5 derived cell clone that highly replicates HEV genome (Montpellier et al., 2017) (Figure 1a, PLC3/p6SPGLuc). As shown in Figure 1e, BFA efficiently inhibited p6SPGLuc replication in transfected PLC3 cells, indicating that the inhibitory effect of BFA on HEV replication is not cell line dependent. We also analysed the antiviral activity of BFA in Huh‐7.5 cells transfected with the gt1 Sar55 Firefly luciferase (FLuc)‐encoding subgenomic replicon (Sar55FLuc) (Figure 1a and 1f). As for gt3, BFA at 0.1 and 1 μg/ml strongly inhibited gt1 replication, indicating that the BFA inhibitory effect on HEV replication is not genotype dependent. Lastly, we performed experiments with the full‐length infectious p6 clone (Shukla et al., 2012). Huh7.5 cells were electroporated with the full‐length p6 strain RNA, and then, BFA was added for 16 hr. At 96 hr post‐transfection, cells were fixed and analysed by immunofluorescence with an anti‐ORF2 capsid protein antibody. As shown in Figure 1g, treatment with BFA at 0.1 and 1 μg/ml led to significant decrease of ORF2‐positive cells, indicating that BFA inhibits the HEV lifecycle.
Figure 1.
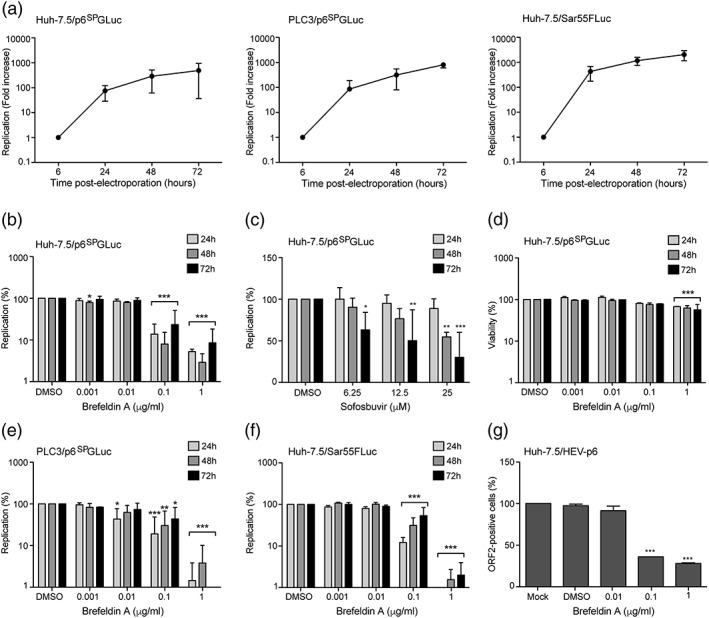
Brefeldin A inhibits hepatitis E virus (HEV) replication. (a) Huh‐7.5 and PLC3 cells were electroporated with in vitro‐transcribed p6SPGLuc RNA or Sar55FLuc RNA. Luciferase activities were measured at 6, 24, 48, and 72 hr post‐electroporation (p.e.). For each time point, values are presented as fold increase compared to luciferase activities measured at 6 hr p.e. (b–d) At 6 hr p.e., HEV‐p6SPGLuc‐electroporated Huh‐7.5 cells were treated for 16 hr with brefeldin A (b and d) or sofosbuvir (c) at indicated concentrations. Luciferase activities (b and c) and viability (d) were quantified at 24, 48, and 72 hr p.e. Values are presented as a percentage of replication compared to cells treated with 0.2% dimethyl sulfoxide (DMSO). PLC3 cells electroporated with p6SPGLuc RNA (e), and Huh‐7.5 cells electroporated with gt1 Sar55FLuc RNA (f) were treated for 16 hr with brefeldin A at indicated concentrations. Luciferase activities were measured at 24, 48, and 72 hr p.e. Values are presented as a percentage of replication compared to cells treated with 0.2% DMSO. (g) Huh‐7.5 cells were electroporated with the full‐length infectious p6 strain RNA and then treated with brefeldin A for 16 hr at indicated concentrations. At 96 hr post‐transfection, cells were fixed and analysed by immunofluorescence with an anti‐ORF2 capsid protein antibody. Values were adjusted to 100% infection for non‐treated cells (mock). Results are presented as mean ± standard deviation of three independent experiments. *, **, and *** mean p‐values below .05, .01, and .001, respectively
Altogether, our results demonstrate that BFA inhibits HEV replication, likely by blocking a cellular factor necessary for this step in the viral lifecycle.
2.2. GBF1 is likely required for HEV replication
BFA inhibits several cellular membrane trafficking pathways, primarily through inhibition of its major cellular targets, members of the Arf GEF family. Among the 15 members of the Arf GEF family in human cells, only three are inhibited by BFA: BIG1, BIG2, and GBF1 (D'Souza‐Schorey & Chavrier, 2006; Gillingham & Munro, 2007). To check which of the three BFA‐sensitive GEFs are required for HEV replication, we analysed the effect of their depletion on HEV replication (Figure 2). However, GBF1 silencing was toxic in our experimental conditions due to the slow kinetics of HEV replication, which requires long depletion times. As an alternative, we used Golgicide A (GCA) that specifically inhibits GBF1 with no effect on BIG1 and BIG2 (Sáenz et al., 2009). Huh7.5 cells were transfected with siRNA pools targeting BIG1, BIG2, BIG1 and BIG2 or a non‐targeting siRNA pool (Figure 2a). Two days post‐transfection, depleted cells were electroporated with the p6SPGLuc replicon, and luciferase activities were measured at 24, 48, and 72 hr p.e., as described previously. As shown in Figure 2b, depletion of BIG1, BIG2 or BIG1 and BIG2 together did not result in any significant decrease of HEV replication levels. In contrast, when p6SPGLuc‐electroporated Huh‐7.5 cells were treated with GCA, which specifically targets GBF1, HEV replication levels were strongly inhibited (Figure 2c) without any significant toxicity (Figure 2d). Altogether, our results suggest that GBF1 is likely the BFA‐ and GCA‐sensitive factor required for HEV replication.
Figure 2.
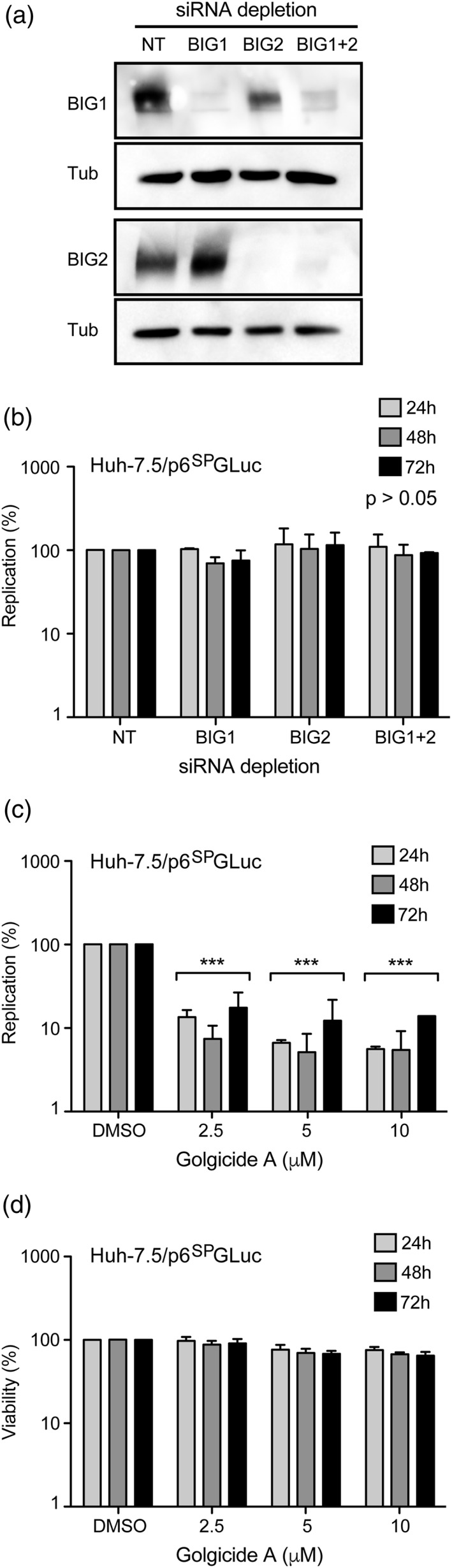
GBF1 is the brefeldin A‐sensitive factor required for hepatitis E virus replication. (a) Huh‐7.5 cells were transfected with siRNA pools targeting BIG1, BIG2, BIG1, and BIG2 or a non‐targeting siRNA pool. Two days post‐transfection, silencing of BIG1, BIG2, or BIG1, and BIG2 was controlled by western blotting with antibodies directed against BIG1 or BIG2. Antibodies directed against tubulin were used to control protein loading. (b) Two days post‐transfection, depleted cells were electroporated with the p6SPGLuc replicon, and luciferase activities were measured at 24, 48, and 72 hr p.e. Values are presented as a percentage of replication compared to cells transfected with non‐targeting siRNA pool. (c and d) hepatitis E virus‐p6SPGLuc‐electroporated Huh‐7.5 cells were treated for 16 hr with Golgicide A at indicated concentrations. Luciferase activities (c) and viability (d) were quantified at 24, 48 and 72 hr p.e. Values are presented as a percentage of replication compared to cells treated with 0.2% of dimethyl sulfoxide. Results in (b), (c), and (d) are presented as mean ± standard deviation of three independent experiments. *** means p‐values below .001
2.3. HEV replication is resistant to BFA in cells expressing a point mutation in GBF1
In a previous study assessing the role of GBF1 in HCV replication, we have isolated BFA‐resistant cell clones derived from the Huh‐7 hepatoma cell line (Farhat et al., 2013). The R2 cell line is resistant to 0.1 μg/ml of BFA and able to support HCV replication in the presence of 100 times more of BFA than the parental Huh‐7 cell line. This resistance is due to a point mutation (M832L) in the sec7 catalytic domain of GBF1, which is known to impair the binding of BFA (Farhat et al., 2013). In order to confirm that BFA inhibition of HEV replication was only related to the effect of the drug on GBF1, we transfected the p6SPGLuc replicon in R2 and parental Huh‐7 cells (Figure 3). BFA was added for 16 hr as in the previous experiments. HEV replication was strongly inhibited by BFA in parental Huh‐7 cells (Figure 3a). In contrast, the replication of p6SPGluc was almost insensitive to BFA in R2 cells regardless of the concentration of BFA (Figure 3b).
Figure 3.
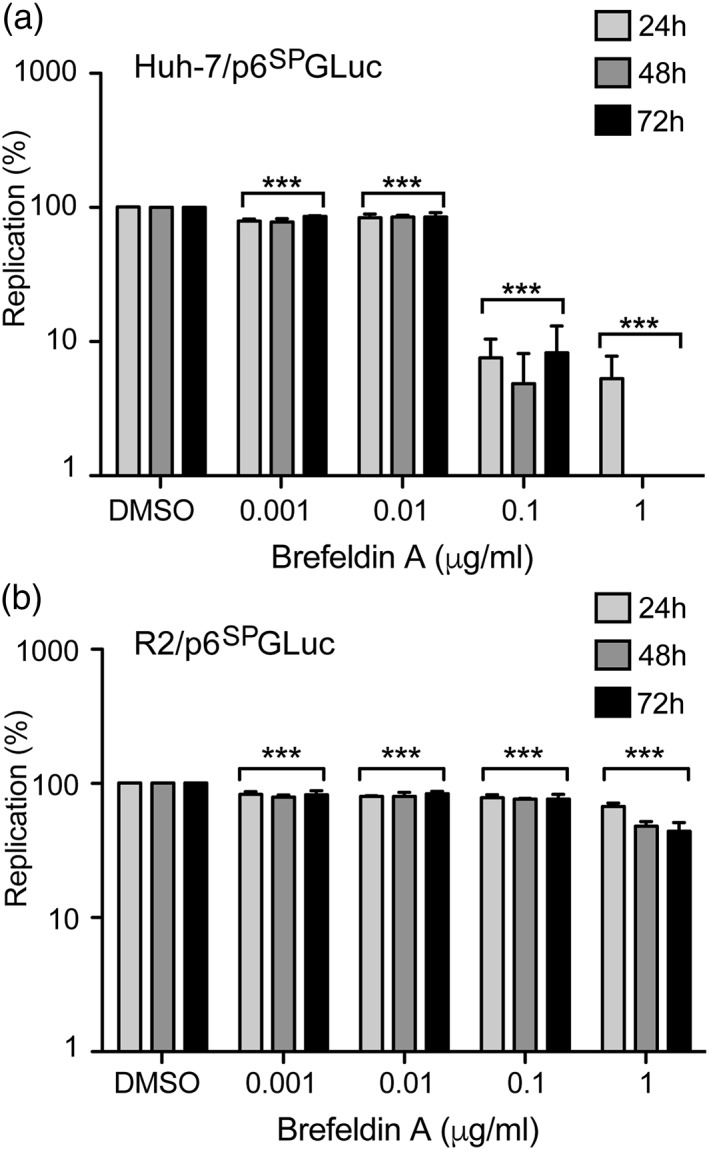
Hepatitis E virus replication in brefeldin A‐resistant cells. Huh‐7 cells (a) and brefeldin A‐resistant R2 cells (b) were electroporated with p6SPGLuc RNA and treated for 16 hr with brefeldin A at indicated concentrations. Luciferase activities were measured at 24, 48, and 72 hr p.e. Values are presented as a percentage of replication compared to cells treated with 0.2% of dimethyl sulfoxide. Results are presented as mean ± standard deviation of three independent experiments. *** means p‐values below .001
Together, these results support the conclusion that GBF1 is the BFA‐sensitive factor that is required for HEV replication, and that the inhibition of HEV replication is not due to a direct effect on the virus.
2.4. Expression of wild‐type GBF1 or BFA‐resistant GBF1 mutant rescues HEV replication in BFA‐treated cells
To further confirm that GBF1 is the only host factor sensitive to BFA that is required for HEV replication, we next used a GBF1 complementation assay (Figure 4). Indeed, it has been shown that GBF1 overexpression or expression of the M832L BFA‐resistant GBF1 mutant can rescue HCV replication from BFA inhibition whereas expression of the E794K catalytically inactive GBF1 mutant cannot (Farhat et al., 2016; Goueslain et al., 2010; Jackson & Casanova, 2000; Niu, Pfeifer, Lippincott‐Schwartz, & Jackson, 2005). We therefore transfected Huh‐7.5 cells with plasmids expressing YFP‐fused wildtype GBF1 (GBF1wt), M832L BFA‐resistant GBF1 mutant (GBF1ML) or E794K inactive GBF1 mutant (GBF1EK). A plasmid expressing only the YFP protein was used as a control (YFP). Two days post‐transfection, expression levels of YFP‐fused GBF1 proteins and YFP protein were controlled by western blotting (Figure 4a) and fluorescent microscopy (Figure 4b), and transfected with the full‐length infectious p6 RNA (Figure 4c). Cells were next treated for 16 hr with 75 ng/ml BFA, a concentration that inhibits approximately 60% of the HEV replication. It has to be noted that BFA treatment had no impact on the subcellular localisation of GBF1 proteins (Figure 4b). Four days p.e., cells were fixed and analysed by immunofluorescence with an anti‐ORF2 capsid protein antibody, as described in Figure 1. For each transfection, control DMSO values were set to 100%, and the corresponding percentage was calculated for the BFA treatment condition. As shown in Figure 4c, upon treatment with BFA, the number of ORF2‐positive cells was reduced by approximately 50% in cells transfected with YFP and GBF1EK mutant. In contrast, overexpression of GBF1wt led to an increase of 20% in the number of ORF2‐positive cells, and the expression of the BFA‐resistant mutant M832L restored more than 35% of HEV replication, as compared to DMSO treated cells, indicating a protective effect of functional GBF1 overexpression over BFA‐induced inhibition of HEV replication.
Figure 4.
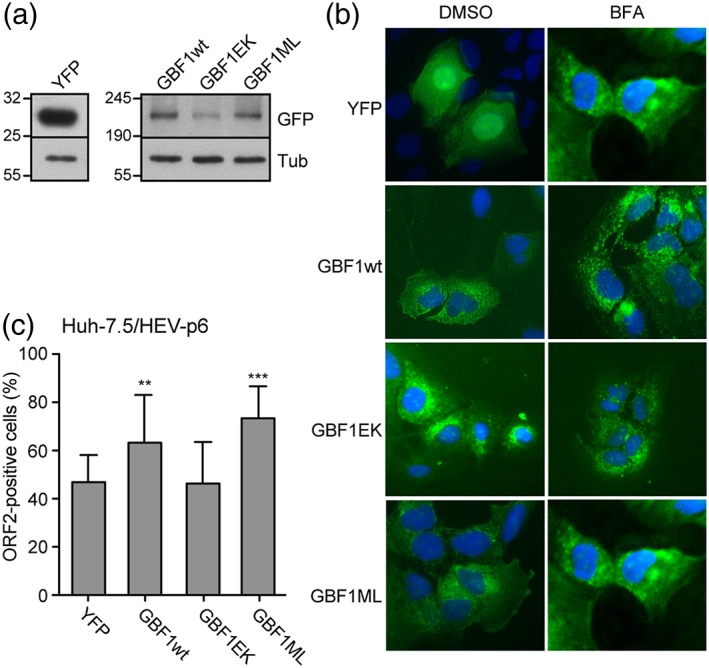
GBF1 complementation assay in cells treated with brefeldin A (BFA). Huh‐7.5 cells were transfected with a plasmid expressing the YFP protein or with plasmids expressing YFP‐fused wildtype GBF1 (GBF1wt), M832L BFA‐resistant GBF1 mutant (GBF1ML), or E794K inactive GBF1 mutant (GBF1EK). Two days post‐transfection, expression levels of YFP‐fused GBF1 proteins, and YFP protein were controlled by western blotting with an anti‐GFP antibody (a) and microscopy (b). Expression of constructs in cells treated with BFA (75 ng/ml) is shown in (b). (c) Two days post‐transfection, cells were electroporated with the full‐length infectious p6 RNA and cultured for 16 hr in the presence of dimethyl sulfoxide (DMSO) or 75 ng/ml BFA. At 96 hr p.e., cells were fixed and analysed by immunofluorescence with an anti‐ORF2 capsid protein antibody. For each construct, the percentage of ORF2‐positive cells in BFA‐treated cells is compared to cells cultured in the absence of BFA. Results are presented as mean ± standard deviation of three independent experiments. ** and *** mean p‐values below .01 and .001, respectively
2.5. GBF1 is likely required for the activity of HEV replication complexes and not for their assembly
Several viruses of the Picornaviridae, Coronaviridae, and Flaviviridae families rely on GBF1 for their replication (Belov et al., 2008; Carpp, Rogers, Moritz, & Aitchison, 2014; Goueslain et al., 2010; Lanke et al., 2009; Liang, Zheng, Bao, & Zhang, 2017; Qin et al., 2014; van der Linden et al., 2010; Verheije et al., 2008; Wang, Du, & Jin, 2014). However, it has been shown that GBF1 is not involved in the formation of poliovirus, mouse hepatitis coronavirus, and HCV replication complexes but rather in their maturation or activity (Belov et al., 2008; Goueslain et al., 2010; Verheije et al., 2008). In order to investigate how GBF1 is involved in HEV replication, we next performed time‐course experiments in which BFA was added for 16 hr at various time points (0, 24, and 48 hr) after electroporation, and replication levels were measured at 24, 48 and 72 hr p.e. (Figure 5). A strong inhibition of HEV replication by BFA was observed whatever the time of addition of the drug, even when BFA was added 48 hr p.e., indicating that BFA is able to inhibit HEV replication in cells in which replication complexes are already formed. Together, these results suggest that GBF1 is required for the activity of HEV replication complexes and not for their assembly.
Figure 5.
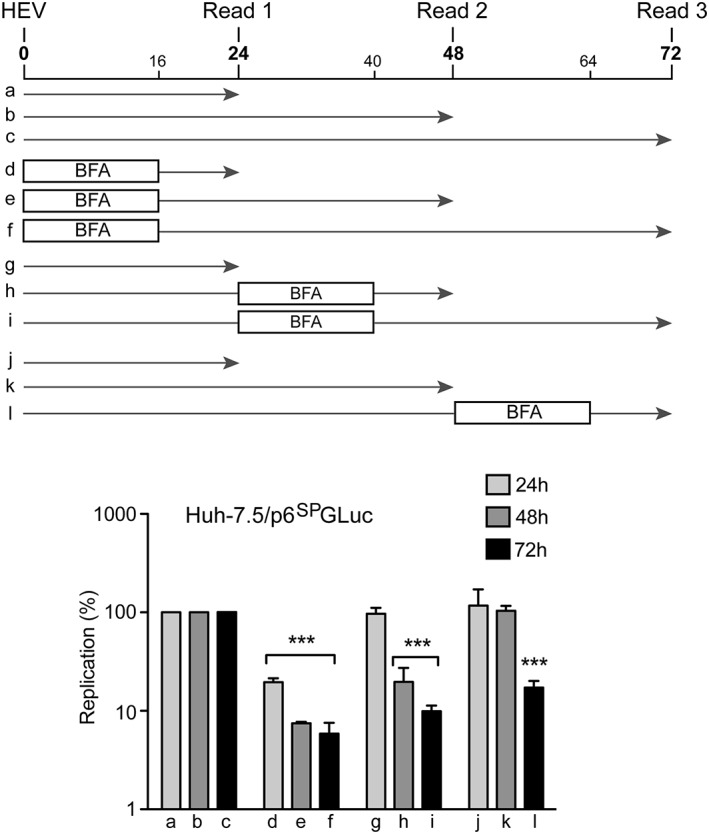
Inhibition of hepatitis E virus replication by brefeldin A (BFA) in time course experiments. Huh‐7.5 cells were electroporated with p6SPGLuc RNA and treated for 16 hr with BFA (0.1μg/ml) at 0 (d–f), 24 (g–i), or 48 hr (j–l) p.e. Luciferase activities were measured at 24, 48, and 72 hr p.e. Values are presented as a percentage of replication compared to cells treated with dimethyl sulfoxide (a–c). Results are presented as mean ± standard deviation of three independent experiments. *** means p‐values below .001
2.6. Subcellular localisation of GBF1 in HEV‐replicating cells and HEV ORF1‐expressing cells
Because we demonstrated that GBF1 is a cellular factor required for HEV replication, we next analysed GBF1 subcellular localisation in HEV‐replicating PLC3 cells using immunofluorescence confocal microscopy (Figure 6). Due to the lack of tools to probe ORF1 protein in HEV‐replicating cells (Lenggenhager et al., 2017), co‐localisation studies of GBF1 with the HEV replicase could not be performed. Therefore, non‐transfected PLC3 cells and cells transfected with the full‐length infectious p6 clone were co‐stained with antibodies directed against GBF1 and the ORF2 capsid protein. As expected, in non‐transfected PLC3 cells, GBF1 staining was observed in Golgi‐like perinuclear structures and in cytoplasmic small dot‐like structures (Figure 6, PLC3). Similar intracellular GBF1 distributions were observed in HEV‐replicating cells (Figure 6, PLC3/HEV‐p6), indicating that the major subcellular localisation of GBF1 is not modified upon HEV replication and therefore is likely not recruited and stably maintained on HEV replication complexes. The same results were obtained in transfected Huh‐7.5 cells (data not shown). As an alternative approach to determining whether GBF1 is recruited to the replication sites, we analysed the subcellular localisation of GBF1 in cells overexpressing the non‐structural ORF1 polyprotein in combination or not with the structural ORF2 and ORF3 proteins (Figure 7). The ORF1 protein was detected with three different antibodies directed against either the Met, Hel, or Pol domain. We observe no difference in the localisation of GBF1 upon expression of the viral proteins. In addition, GBF1 and ORF1 did not co‐stain regardless of the antibody used, strengthening our hypothesis that GBF1 is probably not recruited to HEV replication sites.
Figure 6.
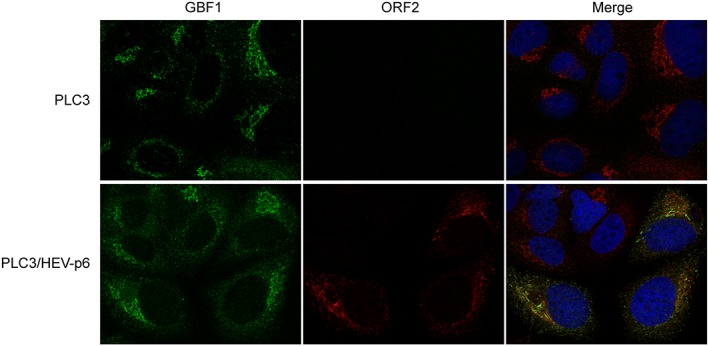
Intracellular distribution of GBF1 in hepatitis E virus (HEV)‐replicating and non‐replicating cells. PLC3 cells were electroporated with water or with the full‐length infectious p6 strain RNA. At 3 days p.e., cells were fixed, permeabilized, and processed for double‐label immunofluorescence for GBF1 (red) and ORF2 (green). Nuclei are in blue. Representative confocal images are shown together with the merge image
Figure 7.
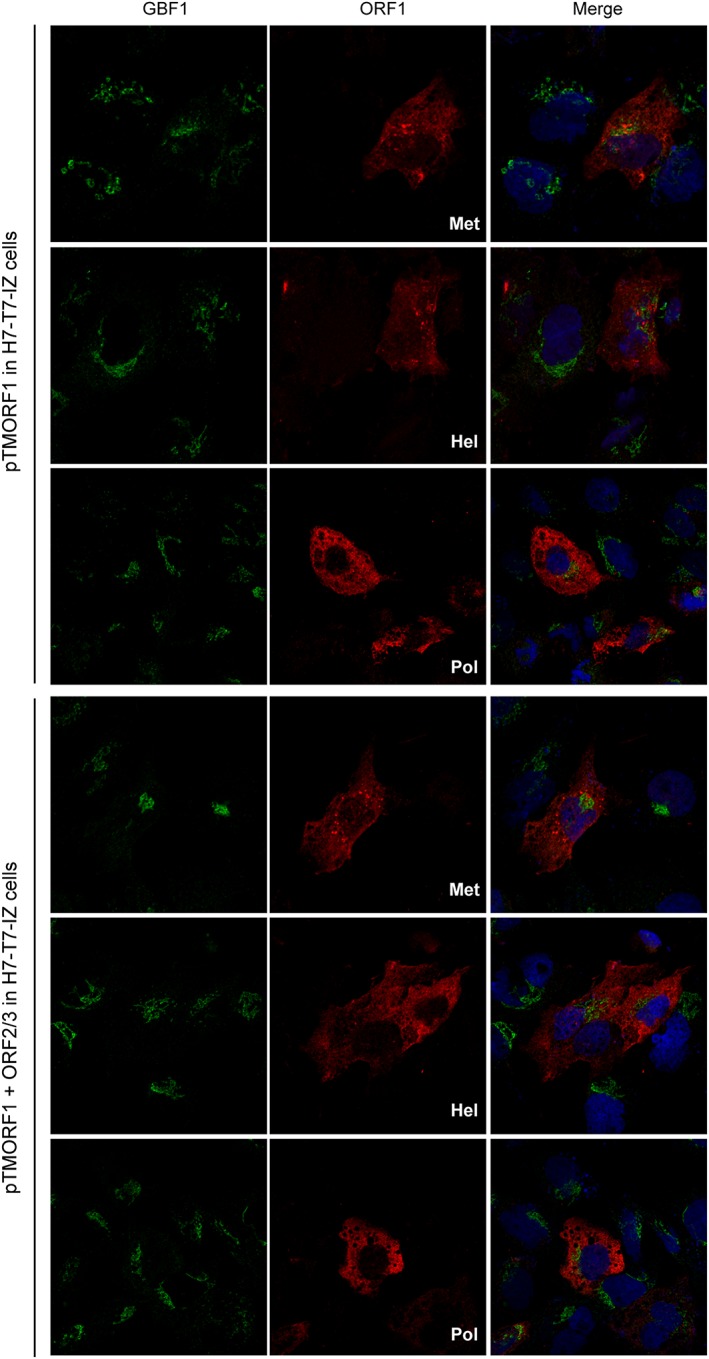
Intracellular distribution of GBF1 in hepatitis E virus ORF1‐expressing cells. H7T7IZ cells were transfected with pTM‐ORF1 or in combination with pTM‐ORF2/3. Twenty four hours post‐transfection, cells were fixed, permeabilized, and processed for double‐label immunofluorescence for GBF1 (green) and ORF1 methyltransferase , helicase, or polymerase domain (red). Nuclei are in blue. Representative confocal images are shown together with the merge image
3. DISCUSSION
Due to difficulties to amplify HEV in cell culture and the absence of tools to analyse HEV non‐structural proteins, mechanisms leading to HEV replication are particularly poorly understood. The site of RNA replication within the host cell has not been identified yet. However, the use of vector systems showed that the non‐structural ORF1 polyprotein might be membrane‐associated and localised in the ERGIC, suggesting that HEV replication might occur within the early secretory pathway (Perttilä et al., 2013). In our study, we show that activity of HEV replication complexes strongly depends on GBF1, a GEF regulating the activity of Arf small G‐proteins, which in turn are key regulators of the cellular secretory pathway. We demonstrate that BFA and GCA, a specific inhibitor of GBF1, inhibit HEV replication and that GBF1 is the only BFA‐sensitive cellular factor required for HEV replication.
GBF1 orchestrates retrograde Golgi‐to‐ER transport by activating Arf proteins that regulate COPI‐coated vesicles transport (D'Souza‐Schorey & Chavrier, 2006). GBF1 also participates in Golgi morphogenesis and lipid droplet metabolism (Jackson & Bouvet, 2014). In addition, GBF1 is hijacked by several positive‐strand RNA viruses including Picornaviridae, Coronaviridae, and Flaviviridae members for their replication (Belov et al., 2008; Carpp et al., 2014; Goueslain et al., 2010; Lanke et al., 2009; Liang et al., 2017; Qin et al., 2014; van der Linden et al., 2010; Verheije et al., 2008; Wang et al., 2014). For instance, GBF1 was shown to interact with poliovirus and coxsackievirus B3 (CVB3) non‐structural protein 3A (Wessels et al., 2006; Wessels et al., 2007). GBF1 overexpression rescues enterovirus replication in cells treated with BFA, whereas its silencing strongly inhibits viral replication (Belov et al., 2008; Lanke et al., 2009). The precise role of GBF1 in enterovirus replication is not clear. It has been proposed that Arf‐activating function of GBF1 would be necessary for enterovirus replication to recruit other cellular factors supporting replication such as the phosphatidylinositol kinase PI4KIII (Hsu et al., 2010). However, more recent data suggest that enterovirus replication requires the N‐terminal region of the GBF1 protein but not its Arf‐GEF activity (Belov, Kovtunovych, Jackson, & Ehrenfeld, 2010; Viktorova, Nchoutmboube, Ford‐Siltz, & Belov, 2015). In contrast, GBF1‐mediated Arf1 activation is crucial for mouse hepatitis coronavirus RNA replication (Verheije et al., 2008). GBF1 is also a host factor required for HCV replication (Goueslain et al., 2010). In contrast to enteroviruses, no interaction between GBF1 and viral proteins has been reported so far. In addition, it has been shown recently that the role of GBF1 in HCV replication is mediated by its Arf‐GEF activity (Farhat et al., 2016). Interestingly, Arf4 and Arf5 were shown to be essential for mediating GBF1 function in HCV replication, yet depletion of these two Arf proteins did not inhibit the secretory pathway, instead affecting lipid metabolism (Farhat et al., 2016). In our study, we demonstrate that GBF1 has essential functions in HEV replication. Further experiments using Arf protein expression knockdown, and mutants of the catalytic Sec7 domain of GBF1 (Farhat et al., 2016), are now necessary to define the importance of Arf‐GEF activity of GBF1 in HEV replication.
Many positive‐strand RNA viruses manipulate the internal membranes of host cells to establish their replication complexes, frequently on the cytosolic leaflet of remodelled membranes. This compartmentalisation allows coordination of the different steps of the replication cycle, resulting in highly efficient RNA replication as well as protection of the viral genome from cell defense mechanisms. These remodelled membranes are characterised by two different types of membrane structures: the invaginated vesicle or spherule type induced for instance by Dengue virus and the double membrane vesicle induced for instance by poliovirus and HCV (reviewed in Paul, 2013). To induce and maintain such membrane rearrangements, viruses usurp cellular factors that are active on membranes. For instance, enteroviruses recruit Arf GEFs to rearrange Golgi and ERGIC membranes (Belov et al., 2006) and ER‐resident reticulon proteins (Tang et al., 2007), whereas alphaviruses subvert amphiphysins localised at the plasma membrane for membrane remodelling and viral RNA replication (Neuvonen et al., 2011). As mentioned previously, GBF1 is essential for the replication of enteroviruses, coronaviruses, and flaviviruses, all of which are viruses inducing remodelling of intracellular membranes. In the present study, we demonstrated that GBF1 is a cellular factor required for the activity of HEV replication complexes. Based on the fact that viruses using GBF1 for their replication induce membrane rearrangements, we can therefore speculate that HEV replication might depend on such membrane reshuffling. Further studies using electron microscopy of cells highly replicating the HEV genome are thus required to test this hypothesis.
In order to determine whether GBF1 is recruited by the ORF1 protein at replication complexes, as observed for poliovirus and CVB3 non‐structural protein 3A (Wessels et al., 2006; Wessels et al., 2007), we analysed the subcellular distribution of GBF1. However, although the full‐length protein or domains of the non‐structural ORF1 protein can be detected by antibodies in cells transfected with vector systems (Lenggenhager et al., 2017; Perttilä et al., 2013), these antibodies fail to detect ORF1 protein in HEV‐replicating cells (data not shown; Lenggenhager et al., 2017). This lack of tools directed against ORF1 prevents us from analysing whether GBF1 is recruited to replication complexes in HEV‐replicating cells. As an indirect approach, we first analysed the subcellular distribution of GBF1 in HEV replicating and non‐replicating cells and found no evidence of a change in its intracellular localisation. As a second alternative approach, we studied the subcellular distribution of GBF1 in cells overexpressing the ORF1 viral replicase and found that GBF1 intracellular pattern was unaffected by viral protein expression. In addition, we did not find any evidence of GBF1‐ORF1 co‐localisation. These results suggest that, as for its involvement in HCV replication, GBF1 might have an indirect role in HEV replication by activating effectors involved in HEV replication. However, further experiments of co‐staining of ORF1 protein and GBF1 in HEV‐replicating cells are needed to elucidate the involvement of GBF1 in HEV replication. The development of tools allowing the probing of ORF1 protein in HEV‐replicating cells is thus essential to characterise the mechanisms of HEV replication and the involvement of GBF1.
In conclusion, our results highlight a functional connection between HEV RNA replication and the early secretory pathway of the host cell. Identifying more precisely the function of GBF1 in HEV replication and a potential effect of HEV replication on intracellular membranes should also provide new insights into the understanding of cellular mechanisms underlying HEV RNA replication.
4. EXPERIMENTAL PROCEDURES
4.1. Chemicals and reagents
Dulbecco's modified Eagle's medium (DMEM), phosphate buffered saline (PBS), foetal calf serum (FCS), and 4′,6‐diamidino‐2‐phenylindole were purchased from Life Technologies. Mowiol 4‐88, and Golgicide A were from Calbiochem. Protease inhibitors cocktail (complete) was from Roche. Sofosbuvir was purchased from Selleckchem (Houston, USA). Other chemicals were from Sigma.
4.2. Cell culture
Huh‐7 (Nakabayashi, Taketa, Miyano, Yamane, & Sato, 1982), Huh‐7.5 (Blight, Mckeating, & Rice, 2002), R2 (Farhat et al., 2013), and PLC3 (Montpellier et al., 2017) cells were grown in DMEM supplemented with 2 mM glutamax‐I and 10% inactivated FCS (DMEM/FCS) at 37 °C/5% CO2.
The Huh‐7‐derived H7T7IZ cells stably expressing the T7 RNA polymerase (Romero‐Brey et al., 2012; kindly provided by Volker Lohmann and Ralf Bartenschlager, University of Heidelberg, Germany) were used for the transfection of the T7 promoter‐driven expression vectors, pTM‐ORF1, and pTM‐ORF2/3 plasmids, allowing the expression of ORF1 and ORF2/3, respectively (Lenggenhager et al., 2017).
4.3. Antibodies
Rabbit anti‐HEV ORF1 polyclonal antibodies against the Met, the Hel, and the polymerase domain were kindly provided by Tero Ahola (University of Helsinki, Finland; Perttilä et al., 2013). Mouse anti‐HEV ORF2 MAb (1E6/IgG2b, antibody registry #AB‐827236) was from Millipore. Mouse anti‐GFP mAb was from Roche. Rabbit anti‐BIG1 and BIG2 antibodies were from Bethyl Laboratories. Mouse anti‐GBF1 (IgG1, antibody registry #AB‐399487) was from BD Biosciences. Mouse anti‐β tubulin was from Sigma. Secondary antibodies were from Jackson ImmunoResearch.
4.4. Viability assay
Sub‐confluent cell cultures grown in 96‐well plates were incubated with BFA, GCA, or DMSO for 16 hr, or incubated all the time with Sofosbuvir, and kept in culture for 24, 48, or 72 hr. A 3‐(4,5‐dimethylthiazol‐2‐yl)‐5‐ (3‐carboxymethoxyphenyl)‐2‐(4‐sulfophenyl)‐2H–tetrazolium based viability assay (CellTiter 96 aqueous non‐radioactive cell proliferation assay from Promega) was conducted as recommended by the manufacturer.
4.5. Plasmids and transfection
Plasmids expressing the cell culture adapted gt3 Kernow C‐1 strain (HEV‐p6, GenBank accession number JQ679013), or the replicon expressing the Gaussia luciferase gene (p6GLuc) were provided by S. U. Emerson (Shukla et al., 2012). The p6GLuc replicon construct was used to generate a new replicon construct (p6SPGLuc) in which the 20 first amino acids matching with the signal peptide of Gaussia luciferase were deleted to block luciferase secretion. Fusion polymerase chain reaction were done with external primers (5′‐GCGGGGTCATGCATGGTATT‐3′, 5′‐ACCCATACGTAGCCTGATCG‐3′) and internal primers (5′‐GATCACCATGAAGCCCACCGAGAACAACGA‐3′, 5′‐TGGGCTTCATGGTGATCCCATGGGCGATGC‐3′). The gt1 Sar55 strain replicon expressing the Firefly luciferase has been described previously (Pudupakam et al., 2009) (kindly provided by X.J. Meng, Virginia Polytechnic Institute and State University, Blacksburg, VA). Capped RNA transcripts were generated with the mMESSAGE mMACHINE® kit (Ambion) and delivered into cells by electroporation using a Gene Pulser Xcell™ apparatus (Bio‐Rad). Plasmids expressing yellow fluorescent protein (YFP), YFP‐tagged GBF1, YFP‐tagged GBF1 E794K, or YFP‐tagged GBF1 M832L have been described previously (Goueslain et al., 2010; Niu et al., 2005).
4.6. Luciferase assays
P24 wells were seeded in with 2 × 106 cells that were electroporated with 2.5 μg of p6SPGLuc or Sar55FLuc RNA. Drugs (BFA, GCA, or Sofosbuvir) were added 2 hr p.e. and kept for 16 h. At 6, 24, 48, and 72 hr p.e., cells were lysed with the buffer provided by the manufacturer (Promega), and Gaussia or Firefly luciferase activities were determined with the corresponding luciferase assay system (Promega) and using a TriStar LB941 luminometer (Berthold). Luciferase activities at 6 hr p.e. were used to determine electroporation efficiencies.
4.7. RNA interference
RNA interference experiments were carried out with pools of four different synthetic double‐stranded siRNAs to the same target (on‐target plus smart pool reagents from Dharmacon). The control used in this study was the on‐target plus non‐targeting siRNA #1 (D‐001810‐01‐20). For siRNA transfection, 3 μl of lipofectamine RNAi MAX (Life Technologies) was added to 0.5 ml of D‐PBS and incubated for 3 min. In a six‐well plate, 2.5 μl of siRNA at 20 μM was spotted in the centre of a well. In case of double siRNA transfection, 1.25 μl of each siRNA was used. Then, the diluted transfection reagent was added to the siRNA, and the mixture was incubated for 30 min at room temperature. At the end of this incubation, 2.5 × 105 freshly trypsinised cells in a volume of 2 ml of culture medium were added to the transfection mix, and the cells were returned to 37 °C. Two days post‐transfection, cells were trypsinised, and p6SPGLuc replicon RNA was electroporated, as described previously.
4.8. GBF1 complementation
Huh‐7.5 cells seeded in six‐well plates were transfected with 0.5 μg of plasmids expressing YFP‐fused wild type GBF1, YFP‐fused mutant GBF1, or YFP protein with the Trans‐IT LT1 reagent following the instructions of the manufacturer (Mirus). Two days post‐transfection, expression of GBF1 proteins was controlled by western blotting and fluorescence microscopy. Cells were then electroporated with the infectious full‐length p6 RNA and cultured for 16 hr in the presence of BFA (75 ng/ml). At 96 hr p.e., cells were fixed with methanol and stained with an anti‐ORF2 antibody (1E6). For each transfection with GBF1 proteins or YFP, ORF2‐positive cells values were adjusted to 100% for cells cultured in the presence of DMSO.
4.9. Immunoblotting
Transfected cells were rinsed three times with cold PBS and lysed at 4 °C for 20 min in a buffer containing 50 mM of TrisCl, 100 mM of NaCl, 2 mM of ethylenediaminetetraacetic acid , 1% of Triton‐X, 0.1% of sodium dodecyl sulfate, pH 7.5, 1 mM of phenylmethylsulfonyl fluoride, and a protease inhibitors cocktail (Complete). Insoluble material was removed by centrifugation at 4 °C. The proteins were resolved by sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred onto nitrocellulose membranes (Hybond‐ECL; Amersham) using a Trans‐Blot apparatus (Bio‐Rad). Proteins of interest were revealed with specific primary antibodies, followed by species‐specific secondary antibodies conjugated to peroxidase. Proteins were visualised using enhanced chemiluminescence (ECL Plus; GE healthcare). The signals were recorded using a LAS 3000 apparatus (Fujifilm).
4.10. Immunofluorescence microscopy
Indirect immunofluorescence labelling was performed as previously described (Rouille et al., 2006). Nuclei were stained with 4′,6‐diamidino‐2‐phenylindole. Cells transfected with the full‐length p6 RNA were stained with a mouse anti‐ORF2 MAb (1E6), and positive cells were counted for each condition.
For confocal microscopy analyses, PLC3 cells transfected with the full‐length p6 RNA were co‐stained with GBF1 and ORF2 antibodies. H7T7IZ cells transfected with pTM‐ORF1 or in combination with pTM‐ORF2/3 were co‐stained with GBF1 and ORF1 Met, Hel, or polymerase domain antibodies.
DISCLOSURES
All authors disclose no conflicts.
ACKNOWLEDGEMENTS
We thank S. Ung for his technical assistance. We are grateful to S. U. Emerson, X. J. Meng, V. Lohmann, R. Bartenschlager, and T. Ahola for providing us with reagents. We thank A. Weber for his contribution. We thank the imaging core facility of the BioImaging Center Lille‐Nord de France for access to the instruments.
Farhat R, Ankavay M, Lebsir N, et al. Identification of GBF1 as a cellular factor required for hepatitis E virus RNA replication. Cellular Microbiology. 2018;20:e12804 10.1111/cmi.12804
REFERENCES
- Belov, G. A. , Altan‐Bonnet, N. , Kovtunovych, G. , Jackson, C. L. , Lippincott‐Schwartz, J. , & Ehrenfeld, E. (2006). Hijacking components of the cellular secretory pathway for replication of poliovirus RNA. Journal of Virology, 81, 558–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belov, G. A. , Feng, Q. , Nikovics, K. , Jackson, C. L. , & Ehrenfeld, E. (2008). A critical role of a cellular membrane traffic protein in poliovirus RNA replication. PLoS Pathogens, 4, e1000216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belov, G. A. , Kovtunovych, G. , Jackson, C. L. , & Ehrenfeld, E. (2010). Poliovirus replication requires the N‐terminus but not the catalytic Sec7 domain of ArfGEF GBF1. Cellular Microbiology, 12, 1463–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blight, K. J. , Mckeating, J. A. , & Rice, C. M. (2002). Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. Journal of Virology, 76, 13001–13014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpp, L. N. , Rogers, R. S. , Moritz, R. L. , & Aitchison, J. D. (2014). Quantitative proteomic analysis of host‐virus interactions reveals a role for Golgi brefeldin A resistance factor 1 (GBF1) in dengue infection. Molecular & Cellular Proteomics, 13, 2836–2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dao Thi, V. L. , Debing, Y. , Wu, X. , Rice, C. M. , Neyts, J. , Moradpour, D. , & Gouttenoire, J. (2016). Sofosbuvir inhibits hepatitis E virus replication in vitro and results in an additive effect when combined with ribavirin. Gastroenterology, 150, 82–85.e4. [DOI] [PubMed] [Google Scholar]
- Debing, Y. , Moradpour, D. , Neyts, J. , & Gouttenoire, J. (2016). Update on hepatitis E virology: Implications for clinical practice. Journal of Hepatology, 65, 200–212. [DOI] [PubMed] [Google Scholar]
- Doceul, V. , Bagdassarian, E. , Demange, A. , & Pavio, N. (2016). Zoonotic hepatitis E virus: Classification, animal reservoirs and transmission routes. Virus, 8, 270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Souza‐Schorey, C. , & Chavrier, P. (2006). ARF proteins: Roles in membrane traffic and beyond. Nature Reviews. Molecular Cell Biology, 7, 347–358. [DOI] [PubMed] [Google Scholar]
- Farhat, R. , Goueslain, L. , Wychowski, C. , Belouzard, S. , Fénéant, L. , Jackson, C. L. , … Rouillé, Y . (2013). Hepatitis C virus replication and Golgi function in brefeldin a‐resistant hepatoma‐derived cells. PLoS One, 8, e74491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farhat, R. , Séron, K. , Ferlin, J. , Fénéant, L. , Belouzard, S. , Goueslain, L. , … Rouillé, Y . (2016). Identification of class II ADP‐ribosylation factors as cellular factors required for hepatitis C virus replication. Cellular Microbiology, 18, 1121–1133. [DOI] [PubMed] [Google Scholar]
- Gillingham, A. K. , & Munro, S. (2007). The small G proteins of the Arf family and their regulators. Annual Review of Cell and Developmental Biology, 23, 579–611. [DOI] [PubMed] [Google Scholar]
- Goueslain, L. , Alsaleh, K. , Horellou, P. , Roingeard, P. , Descamps, V. , Duverlie, G. , … Rouillé, Y . (2010). Identification of GBF1 as a cellular factor required for hepatitis C virus RNA replication. Journal of Virology, 84, 773–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holla, R. , Ahmad, I. , Ahmad, Z. , & Jameel, S. (2013). Molecular virology of hepatitis E virus. Seminars in Liver Disease, 33, 003–014. [DOI] [PubMed] [Google Scholar]
- Hsu, N.‐Y. , Ilnytska, O. , Belov, G. , Santiana, M. , Chen, Y.‐H. , Takvorian, P. M. , … Altan‐Bonnet, N . (2010). Viral reorganization of the secretory pathway generates distinct organelles for RNA replication. Cell, 141, 799–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson, C. L. , & Bouvet, S. (2014). Arfs at a glance. Journal of Cell Science, 127, 4103–4109. [DOI] [PubMed] [Google Scholar]
- Jackson, C. L. , & Casanova, J. E. (2000). Turning on ARF: The Sec7 family of guanine‐nucleotide‐exchange factors. Trends in Cell Biology, 10, 60–67. [DOI] [PubMed] [Google Scholar]
- Kamar, N. , Marion, O. , Abravanel, F. , Izopet, J. , & Dalton, H. R. (2016). Extrahepatic manifestations of hepatitis E virus. Liver International, 36, 467–472. [DOI] [PubMed] [Google Scholar]
- Klausner, R. D. , Donaldson, J. G. , & Lippincott‐Schwartz, J. (1992). Brefeldin a: Insights into the control of membrane traffic and organelle structure. The Journal of Cell Biology, 116, 1071–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin, E. V. , Gorbalenya, A. E. , Purdy, M. A. , Rozanov, M. N. , Reyes, G. R. , & Bradley, D. W. (1992). Computer‐assisted assignment of functional domains in the nonstructural polyprotein of hepatitis E virus: Delineation of an additional group of positive‐strand RNA plant and animal viruses. Proceedings of the National Academy of Sciences of the United States of America, 89, 8259–8263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanke, K. H. W. , van der Schaar, H. M. , Belov, G. A. , Feng, Q. , Duijsings, D. , Jackson, C. L. , … van Kuppeveld, F. J. M . (2009). GBF1, a guanine nucleotide exchange factor for Arf, is crucial for coxsackievirus B3 RNA replication. Journal of Virology, 83, 11940–11949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenggenhager, D. , Gouttenoire, J. , Malehmir, M. , Bawohl, M. , Honcharova‐Biletska, H. , Kreutzer, S. , … Weber, A . (2017). Visualization of hepatitis E virus RNA and proteins in the human liver. Journal of Hepatology, 67, 471–479. [DOI] [PubMed] [Google Scholar]
- Liang, W. , Zheng, M. , Bao, C. , & Zhang, Y. (2017). CSFV proliferation is associated with GBF1 and Rab2. Journal of Biosciences, 42, 43–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Linden, L. , van der Schaar, H. M. , Lanke, K. H. W. , Neyts, J. , & van Kuppeveld, F. J. M. (2010). Differential effects of the putative GBF1 inhibitors Golgicide A and AG1478 on enterovirus replication. Journal of Virology, 84, 7535–7542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montpellier, C. , Wychowski, C. , Sayed, I. M. , Meunier, J.‐C. , Saliou, J.‐M. , Ankavay, M. , … Cocquerel, L . (2017). Hepatitis E virus lifecycle and identification of 3 forms of the ORF2 capsid protein. Gastroenterology, 10.1053/j.gastro.2017.09.020. [DOI] [PubMed] [Google Scholar]
- Nakabayashi, H. , Taketa, K. , Miyano, K. , Yamane, T. , & Sato, J. (1982). Growth of human hepatoma cells lines with differentiated functions in chemically defined medium. Cancer Research, 42, 3858–3863. [PubMed] [Google Scholar]
- Neuvonen, M. , Kazlauskas, A. , Martikainen, M. , Hinkkanen, A. , Ahola, T. , & Saksela, K. (2011). SH3 domain‐mediated recruitment of host cell amphiphysins by alphavirus nsP3 promotes viral RNA replication. PLoS Pathogens, 7, e1002383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu, T.‐K. , Pfeifer, A. C. , Lippincott‐Schwartz, J. , & Jackson, C. L. (2005). Dynamics of GBF1, a brefeldin A‐sensitive Arf1 exchange factor at the Golgi. Molecular Biology of the Cell, 16, 1213–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul, D. (2013). Architecture and biogenesis of plus‐strand RNA virus replication factories. WJV, 2, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perttilä, J. , Spuul, P. , & Ahola, T. (2013). Early secretory pathway localization and lack of processing for hepatitis E virus replication protein pORF1. Journal of General Virology, 94, 807–816. [DOI] [PubMed] [Google Scholar]
- Pudupakam, R. S. , Huang, Y. W. , Opriessnig, T. , Halbur, P. G. , Pierson, F. W. , & Meng, X. J. (2009). Deletions of the hypervariable region (HVR) in open reading frame 1 of hepatitis E virus do not abolish virus infectivity: Evidence for attenuation of HVR deletion mutants in vivo. Journal of Virology, 83, 384–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin, Y. , Lin, L. , Chen, Y. , Wu, S. , Si, X. , Wu, H. , … Zhong, Z . (2014). Curcumin inhibits the replication of enterovirus 71 in vitro. Acta Pharmaceutica Sinica B, 4, 284–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero‐Brey, I. , Merz, A. , Chiramel, A. , Lee, J.‐Y. , Chlanda, P. , Haselman, U. , … Bartenschlager, R . (2012). Three‐dimensional architecture and biogenesis of membrane structures associated with hepatitis C virus replication. PLoS Pathogens, 8, e1003056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouille, Y. , Helle, F. , Delgrange, D. , Roingeard, P. , Voisset, C. , Blanchard, E. , … Dubuisson, J . (2006). Subcellular localization of hepatitis C virus structural proteins in a cell culture system that efficiently replicates the virus. Journal of Virology, 80, 2832–2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sáenz, J. B. , Sun, W. J. , Chang, J. W. , Li, J. , Bursulaya, B. , Gray, N. S. , & Haslam, D. B. (2009). Golgicide a reveals essential roles for GBF1 in Golgi assembly and function. Nature Chemical Biology, 5, 157–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayed, I. M. , Vercouter, A.‐S. , Abdelwahab, S. F. , Vercauteren, K. , & Meuleman, P. (2015). Is hepatitis E virus an emerging problem in industrialized countries? Hepatology, 62, 1883–1892. [DOI] [PubMed] [Google Scholar]
- Shukla, P. , Nguyen, H. T. , Faulk, K. , Mather, K. , Torian, U. , Engle, R. E. , & Emerson, S. U. (2012). Adaptation of a genotype 3 hepatitis E virus to efficient growth in cell culture depends on an inserted human gene segment acquired by recombination. Journal of Virology, 86, 5697–5707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, D. B. , Simmonds, P. , members of the International Committee on the Taxonomy of Viruses Hepeviridae Study Group , Jameel, S. , Emerson, S. U. , Harrison, T. J. , … Purdy, M. A . (2014). Consensus proposals for classification of the family Hepeviridae. Journal of General Virology, 95, 2223–2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam, A. W. , Smith, M. M. , Guerra, M. E. , Huang, C. C. , Bradley, D. W. , Fry, K. E. , & Reyes, G. R. (1991). Hepatitis E virus (HEV): Molecular cloning and sequencing of the full‐length viral genome. Virology, 185, 120–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, W. F. , Yang, S. Y. , Wu, B. W. , Jheng, J. R. , Chen, Y. L. , Shih, C. H. , … Horng, J. T . (2007). Reticulon 3 binds the 2C protein of enterovirus 71 and is required for viral replication. Journal of Biological Chemistry, 282, 5888–5898. [DOI] [PubMed] [Google Scholar]
- Verheije, M. H. , Raaben, M. , Mari, M. , Lintelo Te, E. G. , Reggiori, F. , van Kuppeveld, F. J. M. , … de Haan, C. A. M . (2008). Mouse hepatitis coronavirus RNA replication depends on GBF1‐mediated ARF1 activation. PLoS Pathogens, 4, e1000088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viktorova, E. G. , Nchoutmboube, J. , Ford‐Siltz, L. A. , & Belov, G. A. (2015). Cell‐specific establishment of poliovirus resistance to an inhibitor targeting a cellular protein. Journal of Virology, 89, 4372–4386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, J. , Du, J. , & Jin, Q. (2014). Class I ADP‐ribosylation factors are involved in enterovirus 71 replication. PLoS One, 9, e99768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessels, E. , Duijsings, D. , Lanke, K. H. W. , Melchers, W. J. G. , Jackson, C. L. , & van Kuppeveld, F. J. M. (2007). Molecular determinants of the interaction between coxsackievirus protein 3A and guanine nucleotide exchange factor GBF1. Journal of Virology, 81, 5238–5245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessels, E. , Duijsings, D. , Lanke, K. H. W. , van Dooren, S. H. J. , Jackson, C. L. , Melchers, W. J. G. , & van Kuppeveld, F. J. M. (2006). Effects of picornavirus 3A proteins on protein transport and GBF1‐dependent COP‐I recruitment. Journal of Virology, 80, 11852–11860. [DOI] [PMC free article] [PubMed] [Google Scholar]