

Summary
HIV‐1 matrix protein p17 activates a variety of cell responses which play a critical role in viral replication and infection. Its activity depends on the expression of p17 receptors (p17R) on the surface of target cells. Whether p17 also plays a role in stimulating human monocytes, a major HIV‐1 reservoir, is not known. Here we show that human monocytes constitutively express p17Rs and that p17 selectively triggers these cells to produce MCP‐1. The effect of p17 on MCP‐1 expression was observed at the transcriptional level and was primarily dependent on the activation of the transcription factor AP‐1. p17 increased the binding activity of AP‐1 complexes in a time‐ and dose‐dependent manner. Deletion of the AP‐1 binding sites in the MCP‐1 promoter resulted in the lack of p17‐induced MCP‐1 transcription. In particular, the P3 binding site located between −69 and −63 position seems to be essential to MCP‐1 mRNA induction in p17‐treated monocytes. An ever increasing amount of evidences shows a tight link between biologically dysregulated monocytes, AP‐1 activation, MCP‐1 release and HIV‐1 pathogenesis. Overall our results suggest that p17 may play a critical role in the monocyte‐mediated inflammatory processes, which are suspected to be major precipitating events in AIDS‐defining diseases.
Introduction
Blood monocytes are major in vivo cell targets of HIV‐1 infection. They are known to be less susceptible to viral cytopathic effects and more resistant to HIV‐1‐induced apoptosis than CD4+ T lymphocytes (Herbein et al., 2002; Kedzierska et al., 2003). Due to their ability to survive HIV‐1 infection, and because of a chronic low‐level viral infection, monocytes serve as a major virus reservoir (Ho et al., 1994). Activation of monocytes by infectious and/or inflammatory stimuli increases plasma viral load (Donovan et al., 1996; Goletti et al., 1996) combined with increased numbers of HIV‐1‐positive cells. In addition, activated monocytes can traffic and invade tissues and organs, homing to sites of infection and inflammation (Fischer‐Smith et al., 2001). Productively infected monocytes may therefore ultimately serve as ‘Trojan horses’ for viral dissemination to different organs. Furthermore, a monocyte‐dependent mechanism of viral amplification at local sites of initial infection appears to be a prerequisite for efficient dissemination of infection (Hirsch et al., 1998). The analysis of infected monocytes reveals evidence of signalling cascades leading to upregulation of specific inflammation‐related molecules, which include MCP‐1 (Wahl et al., 2003). MCP‐1 plays an important role in sustaining both inflammation and HIV‐1 replication (Fantuzzi et al., 2003) and is also involved in mass recruitment of such HIV‐1 targets as dendritic cells, monocytes, macrophages and T lymphocytes, into localized sites of infection (Cinque et al., 1998; Fantuzzi et al., 2000; Alfano and Poli, 2001). The pro‐HIV‐1 role of MCP‐1 has been confirmed by the recent finding that HIV‐1 infection and disease progression are influenced by a mutant MCP‐1 allele linked to increased monocyte infiltration of tissues and MCP‐1 levels (Gonzalez et al., 2002). Considerable attention should therefore be given to the mechanisms triggered by HIV‐1 to induce enhanced MCP‐1 production by monocytes.
The HIV‐1 matrix protein p17 is a structural protein that plays an important part in the life cycle of the retrovirus (Fiorentini et al., 2006). It is released by infected cells (Fiorentini et al., 2006), and has recently been shown to accumulates in lymph nodes in the absence of any detectable HIV‐1 replication (Popovic et al., 2005). HIV‐1 p17, as a recombinant protein, exerts a cytokine‐like activity after binding to a specific cellular receptor (p17R) (De Francesco et al., 2002). It is able to modulate the activation and proliferation status of T and NK cells (De Francesco et al., 1998; Vitale et al., 2003) and to enhance HIV‐1 replication (De Francesco et al., 1998). Moreover, it increases the release of such pro‐inflammatory molecules as TNF‐α and IFN‐γ, while counteracting the anti‐inflammatory activity of IL‐4 (De Francesco et al., 2002). It is likely that, through the release of pro‐inflammatory molecules, p17 may induce the activation of monocytes and modulate their functions. However, the potential direct effect of p17 on monocytes has not yet been investigated.
In this study, we show that human monocytes constitutively express p17Rs on their surface and that p17 is capable of triggering monocytes to selectively produce MCP‐1. Furthermore, we show that the p17‐mediated induction of MCP‐1 production is dependent on the activation of the cellular transcriptional factor AP‐1.
Results
Human primary monocytes constitutively express p17Rs on their surface
Biotin‐conjugated p17 was allowed to react with viable peripheral blood mononuclear cells (PBMCs) obtained from healthy donors. As shown in Fig. 1A (left), cell staining with monoclonal antibody (mAb) to CD14 revealed that almost all monocytes constitutively expressed p17Rs on their surface. The presence of p17Rs on human monocytes was confirmed on cells purified by magnetic sorting using anti‐CD14‐coated paramagnetic beads as a specific reagent (Fig. 1A, right). The specificity of the binding was confirmed by displacement of p17/p17R interaction by the anti‐p17 mAb MBS‐3 (data not shown). The stability of the p17R+ phenotype was then evaluated in purified monocyte preparations. Almost 100% of monocytes showed a p17R+ phenotype after 72 h of culture, with a mean fluorescence intensity (MFI) similar to or even higher than that observed on cells soon after their purification (Fig. 1A, right). At the same time, we observed a consistent increase in the MFI of p17Rs expressed on cells stimulated for 16 h with IFN‐γ or LPS. As shown in Fig. 1B (top), IFN‐γ increased the expression of p17Rs on purified monocytes already at a dose as low as 0.2 ng ml−1. A peak of p17R expression was reached at an IFN‐γ dose of 25 ng ml−1. LPS was less efficient than IFN‐γ in upregulating p17Rs expression. A peak in p17R expression was observed at an LPS dose of 10 ng ml−1. A kinetic analysis of p17R expression on IFN‐γ‐ or LPS‐activated monocytes (i.e. 1, 4, 8, 16, 24 and 48 h) showed that p17R expression was enhanced on monocytes within 8–24 h (Fig. 1B, bottom). The slow surface induction of p17Rs is therefore indicative of the de novo synthesis of p17Rs following monocyte activation.
Figure 1.
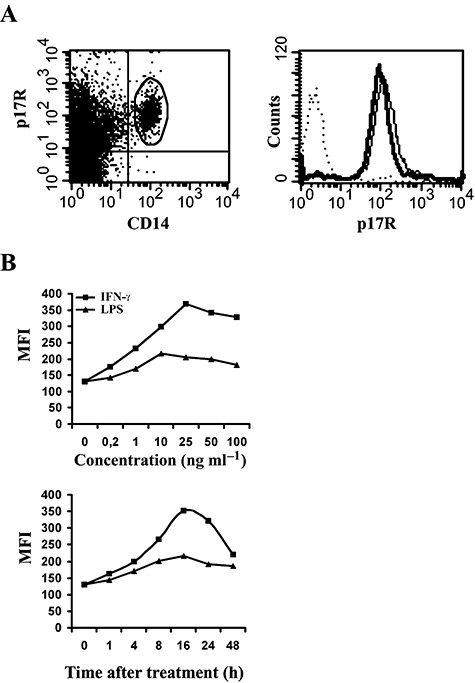
Expression of p17Rs on human monocytes. Biotin‐conjugated p17 was allowed to react with PBMCs or with purified monocytes. Detection of p17 on the cell surface was performed using APC‐conjugated streptavidin as a specific reagent. A. Left. Cells were also stained with FITC‐conjugated anti‐CD14 mAb. Data are displayed as bivariate dot plot and show that approximately all monocytes express p17Rs. Right. The expression of p17Rs on purified monocytes is displayed as hystograms. Histograms: bold, p17R+CD14+ cells within freshly purified monocytes; dotted: CD14+ cells within purified monocytes stained with an unrelated biotinilated protein; solid, p17R+ cells within purified monocytes after 72 h of culture in complete medium. B. Expression of p17Rs on purified activated monocytes. Cells were cultured in the presence of different doses of IFN‐γ or LPS and assayed for p17R expression after 16 h of stimulation (top). The kinetics of p17R upregulation on cells stimulated up to 48 h with optimal doses of IFN‐γ (25 ng ml−1) or LPS (10 ng ml−1) is shown in the bottom panel. These results are representative of three independent experiments.
Baseline turnover of p17R molecules
It is not yet known how p17R surface expression is regulated. Therefore, we examined the baseline turnover of p17Rs in purified monocytes. Cells were cultured in the presence or absence of the protein synthesis inhibitor cycloheximide and the kinetics of p17R expression was examined over a 24 h time‐course (i.e. 6, 12 and 24 h). Another cell surface receptor, namely HLA‐DR, was included for comparison because of its high stability (Lanzavecchia et al., 1992) and known recycling kinetics in allophycocyanin (APC) (Tulp et al., 1994). As shown in Fig. 2A, cells treated with cycloheximide showed a downregulation of p17R surface expression to the background levels after 24 h of culture. In comparison with the HLA‐DR molecule, whose expression on the surface of monocytes was not affected, as expected, by cycloheximide, p17R clearly showed an accelerated turnover which suggests that a de novo synthesis is required to maintain its normal expression level.
Figure 2.
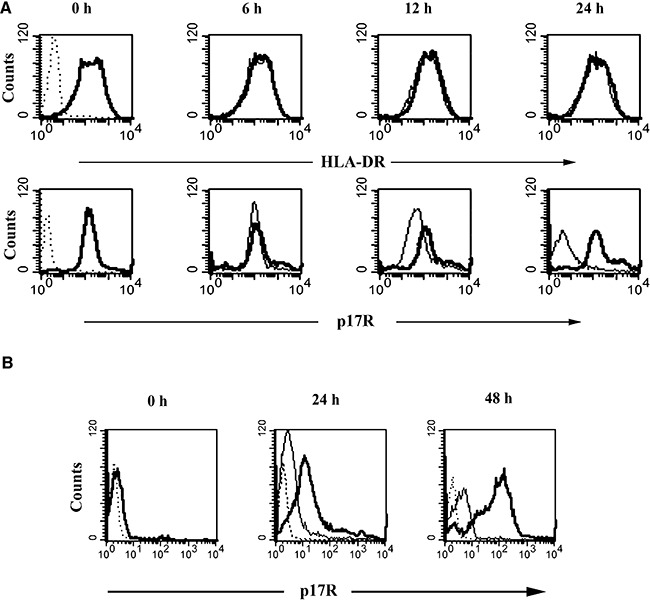
Turnover of p17Rs on the monocyte surface. A. Purified monocytes were stained for p17R and HLA‐DR expression, as described in Experimental procedures. At the beginning of culture (0 h) almost 100% of cells were p17R+ and HLA‐DR+ (bold lines). Controls (dotted lines) represent cells stained with isotype IgG (versus HLA‐DR) or with an unrelated biotinylated protein (versus p17R). The cells were then cultured, for 6, 12 and 24 h in the presence (solid lines) or in the absence (bold lines) of cycloheximide. B. Treatment of monocytes with trypsin for 15 min (0 h) reduced p17R expression (bold line) to almost background levels (dotted line). The cells were then allowed to recover in complete medium in the presence (solid line) or absence (bold line) of cycloheximide. Monocytes were collected 24 h and 48 h after trypsin treatment for p17R staining. These results are representative of three independent experiments.
Treatment of monocytes with trypsin for 15 min at 37°C reduced p17R expression almost to background levels (Fig. 2B). The direct involvement of trypsin in p17R degradation was confirmed by the lack of activity in trypsin inactivated by soybean trypsin inhibitor. Moreover, the effect of trypsin was not due to cell death, as there was no significant increase in the percentage of cells stained with propidium iodide after trypsin treatment, as assessed by flow cytometry (data not shown).
To assess whether the loss of p17Rs on trypsinized cells was transient, trypsin‐treated monocytes were allowed to recover for different periods of time at 37°C before p17 binding analysis. Restoration of p17R expression started approximately after 24 h of culture, and involved 68 ± 13.2% of all monocytes, whereas a total recovery of p17R expression was reached only after 48 h of culture (Fig. 2B). The slow kinetic suggests that protein synthesis may be required. In order to completely exclude the presence of pre‐formed intracellular p17Rs, cells were cultured soon after trypsin treatment in medium containing or not cycloheximide. Re‐expression of p17Rs was completely blocked by cycloheximide, which shows that it is most probably due to protein synthesis rather than to mobilization of a pre‐formed intracellular pool.
Recombinant p17 induces MCP‐1 expression in monocytes
To characterize the effect of p17 on purified monocytes, we investigated the level of different cytokines and chemokines (i.e. IL‐1α, IL‐1β, IL‐6, IL‐8, IL‐12, TNF‐α, MCP‐1, MIP‐1α, MIP‐1β and RANTES) in the medium of p17‐treated or ‐untreated cells 48 h after the beginning of culture. Untreated monocytes constitutively released IL‐8 in a range of 0.5–2.2 ng per 106 cells and, occasionally, also traces of IL‐6 and TNF‐α. The addition of p17 at the beginning of culture induced, in a dose‐dependent manner, the production of MCP‐1 in the cells' supernatant (Fig. 3A), whereas it did not affect the levels of the other cytokines or chemokines tested (data not shown).
Figure 3.
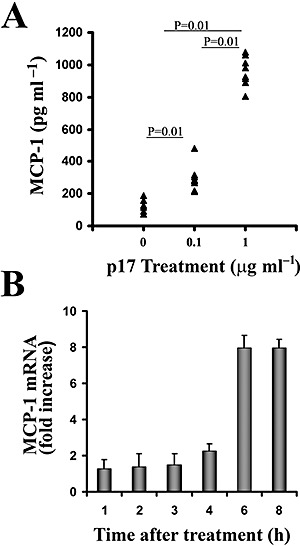
Induction of MCP‐1 production by p17‐treated monocytes. A. Purified monocytes obtained from eight healthy donors were treated or not with p17 at a concentration of 0.1 or 1 μg ml−1. Culture supernatants were collected 48 h after the beginning of culture and analysed for the presence of MCP‐1 by a standard quantitative ELISA. Bars represent the mean value of all samples and P‐values were calculated by the Wilcoxon test. B. Cells treated or not with p17 (1 μg ml−1) were harvested at the specified times, and MCP‐1 and β‐actin levels were analysed by TaqMan rtPCR. MCP‐1 mRNA levels, normalized to β‐actin levels in the same samples, are expressed as fold induction of the levels detected in p17‐untreated cells at the same time points. Bars represent the mean ± SD of triplicate samples.
To assess whether the increase in MCP‐1 protein levels induced by p17 treatment was paralleled by an increase in relative mRNA, we analysed the kinetics of MCP‐1 mRNA appearance by quantitative real‐time PCR TaqMan Assay (rtPCR). Monocytes were treated or not with p17 and total RNA was extracted from cell lysates at 1, 2, 3, 4 and 6 h after the beginning of culture. MCP‐1 messenger levels were quantified according to β‐actin mRNA expression. As shown in Fig. 3B, an enhancement of MCP‐1 mRNA expression (2.4‐fold increase) started 4 h after p17 treatment reaching approximately an eightfold increase 6–8 h after the beginning of treatment.
Activation of AP‐1 transcription factor by p17 in human monocytes
Activation of MCP‐1 gene transcription in different cell systems is known to be under the control of the cellular transcription factors NF‐κB and/or AP‐1 (Shyy et al., 1993). To assess the mechanism(s) whereby p17 induces an increase in MCP‐1 gene expression, we verified whether p17 treatment increases NF‐κB and AP‐1 DNA‐binding activity. Preliminary experiments were run using the TransSignal Array, a protein/DNA array that includes, among others, specific probes for these two transcriptional factors. As shown in Fig. 4A, nuclear extracts from untreated monocytes showed a basal level of AP‐1 DNA‐binding activity that was enhanced after p17 treatment for 1 h at 37°C. Conversely, treatment of monocytes with p17 did not modify NF‐κB DNA‐binding activity which, however, is present at low levels under basal conditions. Data obtained by protein/DNA array analysis were confirmed by electrophoretic mobility shift assay (EMSA). Preliminary experiments using p17 at concentrations ranging from 0.1 to 1 μg ml−1 showed that p17 activated AP‐1 DNA‐binding activity in a dose‐dependent manner with a peak at the 0.4–0.5 μg ml−1 dose (data not shown). Time‐course analysis using 1 μg ml−1 p17 showed that the activation of AP‐1‐binding activity is also time‐dependent and peaks (approximately a threefold increase) at 1 h after the beginning of p17 treatment, after which it declines (Fig. 4B). The specificity of protein–DNA complexes was confirmed by competition experiments using an excess of specific unlabelled oligonucleotide. As AP‐1 proteins bind to DNA as homo‐ or heterodimers of the Jun (c‐Jun, JunD and JunB) and Fos (c‐Fos, Fra1, Fra2 and FosB) proto‐oncogene families, experiments were performed to characterize the proteins involved in p17‐induced AP‐1 protein–DNA complexes. Data obtained by TransAM™ AP‐1 family transcription factor assay showed that c‐Fos, FosB and JunB were the main components of the p17‐inducible AP‐1 complexes (Fig. 4C).
Figure 4.
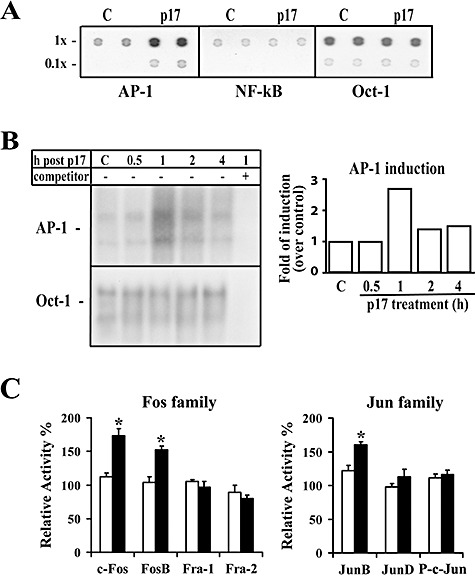
AP‐1 activation in p17‐treated human monocytes. Nuclear protein extracts obtained from monocytes collected after p17 treatment (1 μg ml−1) were analysed for their binding activity to oligonucleotides specific for different transcription factors. A. Representative fluorograms of protein/DNA array analysis of AP‐1, NF‐κB and Oct‐1 DNA‐binding activity in nuclear extracts (10 μg per sample) from human monocytes collected 1 h after p17 treatment. Each transcription factor was evaluated in duplicate at 1× concentration (1×) and at 0.1× concentration (0.1×) of the specific oligonucleotides. B. Representative EMSA autoradiograms using nuclear extracts (10 μg per sample) from human monocytes collected at the specified times after p17 treatment, and AP‐1‐specific radiolabelled oligonucleotides. C: p17‐untreated cells. Protein–DNA complex specificity was confirmed by competition with an excess of unlabelled oligonucleotide probe. DNA‐binding activity to Oct‐1 was used as loading control. The right panel shows a densitometric analysis of changes in the DNA‐binding activity of AP‐1 relative to Oct‐1, expressed as fold of induction over control untreated cells. C. AP‐1 protein family profiling for DNA‐binding activity of an ELISA‐based transcription factor assay kit using nuclear extracts (4 μg per sample) from human monocytes cultured for 1 h in the presence or absence of p17‐ and AP‐1‐specific oligonucleotides immobilized to a 96‐well plate. Levels of Fos or Jun family members in the active AP‐1 complex of p17‐treated or ‐untreated monocytes are expressed as percentage of relative activity where 100% is referred to 0 h values. Statistical analysis was performed by one‐way anova test. *P < 0.05, statistically different compared with 0 h values. Bars represent the mean ± SD of triplicate samples. Representative data from one of three experiments are shown.
Activation of transcription factor AP‐1 by p17 in THP‐1 cells
Due to the large number of primary monocytes required for the analysis of transcription factor DNA‐binding activity in protein nuclear extracts, we further investigated the involvement of AP‐1 transcription factor in p17‐induced MCP‐1 transcriptional events using the monocytic cell line THP‐1, which constitutively expresses p17Rs on its surface (data not shown). Results from EMSA experiments showed that treatment of THP‐1 cells for 4 h at 37°C with p17 (1 μg ml−1) induced AP‐1 activation, with a peak (5.2‐fold increase) at 1 h after treatment, but did not induce any NF‐κB activation (Fig. 5A). EMSA experiments showed that pre‐treatment of exogenous p17 protein with the neutralizing anti‐p17 mAb MBS‐3 blocked AP‐1 activation (data not shown). Further analyses were performed to characterize the specificity and protein composition of AP‐1 complexes induced by p17 treatment in THP‐1 cells. TransAM™ analysis showed a preferential involvement of c‐Fos and JunB in the active AP‐1 complexes (Fig. 5B). Altogether, our results confirm a similar AP‐1 activation pattern by p17 in human primary monocytes and THP‐1 cells, which suggests that this cell line may be used as a model in future studies of the biological activity of p17 in the monocyte compartment. THP‐1 cells were also used as a target to assess the stability of p17 to heat or enzymatic degradation. We therefore treated exogenous p17 with 1% trypsin for 1 h at 37°C or boiled it for 15 min at 100°C. Both treatments completely abolished p17‐induced AP‐1 activity as assessed by EMSA (Fig. 5C), providing evidences that the viral protein is sensitive to protease and heat denaturation.
Figure 5.
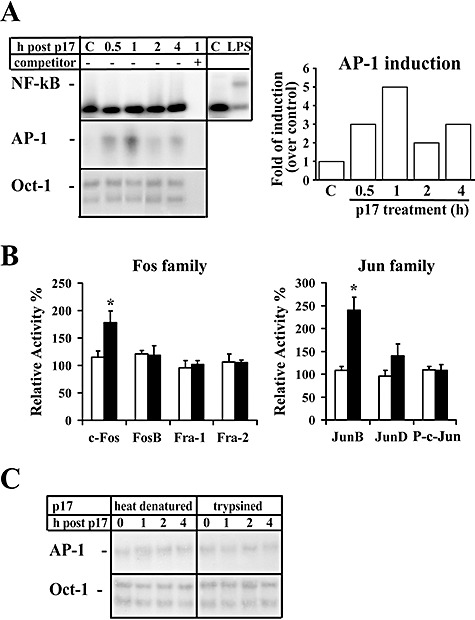
AP‐1 activation in p17‐treated THP‐1 cells. Nuclear protein extracts from THP‐1 cells collected at different times after p17 treatment (1 μg ml−1) were analysed for binding activity to oligonucleotides specific for different transcription factors. A. Representative autoradiograms of EMSA using nuclear extracts (10 μg per sample) from THP‐1 cells at the specified times after p17 treatment and radiolabelled oligonucleotides specific for NF‐κB, AP‐1 or Oct‐1. C: untreated cells; LPS: LPS‐treated cells (1 μg ml−1 LPS for 0.5 h), used as positive control for NF‐κB activation. Protein–DNA complex specificity was confirmed by competition with an excess of unlabelled oligonucleotide probe. The right panel represents a densitometric analysis of changes in DNA‐binding activity of AP‐1 relative to Oct‐1, used as loading control. Data are expressed as fold of induction over control untreated cells. B. AP‐1 family profiling for DNA binding activation from an ELISA‐based kit using nuclear extracts (4 μg per sample) from THP‐1 cells cultured for 1 h in the presence or absence of p17‐ and AP‐1‐specific oligonucleotides immobilized to a 96‐well plate. Levels of Fos or Jun family members in the active AP‐1 complex of p17‐treated or ‐untreated monocytes are expressed as percentage of relative activity where 100% is referred to 0 h values. Statistical analysis was performed by one‐way anova test. *P < 0.05, statistically different compared with 0 h values. Bars represent the mean ± SD of three separate experiments in triplicate. C. Representative EMSA autoradiograms using nuclear extracts from THP‐1 cells collected at the specified times after treatment with boiled or trypsinized p17 and oligonucleotides specific for AP‐1 and for Oct‐1, as loading control.
Functional analysis of MCP‐1 gene regulation
The human MCP‐1 gene contains one AP‐1 binding site in the enhancer region (P1) and two AP‐1 binding sites in the promoter region (P2 and P3). To investigate which regions are involved in the transcriptional activation of MCP‐1 by p17, primary monocytes were transfected with different plasmids containing the reporter luciferase gene cloned downstream to the MCP‐1 promoter. MCP‐1 promoter luciferase reporter plasmids used included: pGLM‐PRM containing the proximal promoter region of the MCP‐1 gene, pGLM‐ENH, which contains both the proximal promoter and the distal enhancer regions, pGLM‐PRM‐P2mut mutated into the P2 AP‐1 binding site, and phMCP128, a plasmid containing a MCP‐1 proximal promoter region whose AP‐1 binding sites (P2 and P3) have been deleted (Fig. 6A). Flow cytometric analysis of cells transfected with a control GFP‐encoding plasmid (pmax‐GFP) showed a high transfection efficiency of primary monocyte with over 40% of the viable cells expressing GFP as early as 8 h after pmax‐GFP plasmid transfection (Fig. 6B). Soon after transfection, cells were left untreated or treated with p17 (1 μg ml−1). PMA (20 ng ml−1) or TNF‐α (10 ng ml−1) was used as controls of NF‐κB and AP‐1 activation. PMA and TNF‐α resulted in a significant transcriptional activation (3.9‐ and 2.2‐fold respectively) in cells transfected with the pGLM‐ENH plasmid, in which both the MCP‐1 promoter and enhancer regions are present (not shown). In p17‐treated monocytes the luciferase activity of constructs pGLM‐ENH and pGLM‐PRM was induced approximately twofold (Fig. 6C). The lack of difference between the promoter and the enhancer regions in response to p17, despite the potential AP‐1 binding site in the enhancer region, indicates a critical role for AP‐1 binding sites in the promoter region. Indeed, deletion of both AP‐1 binding sites (phMCP128) resulted in complete loss of p17‐induced activation. To further assess whether p17‐induced MCP‐1 expression was dependent on the P2 or P3 AP‐1 binding site, we mutated the P2 region in the pGLM‐PRM plasmid (pGLM‐PRM‐P2mut) and tested for p17 activation. Treatment with p17 resulted in luciferase expression in both cells transfected with the pGML‐PRM or pGML‐PRM‐P2mut plasmids. This finding suggests that the P3 AP‐1 binding site in the promoter region is necessary for optimal p17 stimulation.
Figure 6.
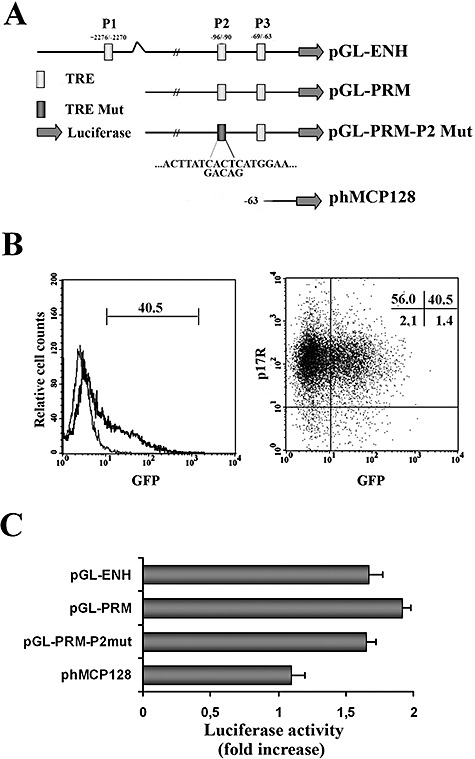
Analysis of p17‐induced MCP‐1 transcriptional activation. A. Schematic representation of the four human MCP‐1 reporter constructs used to perform our experiments. pGL‐ENH contains both the proximal promoter (between −107 and +60) and distal enhancer (between −2742 and −2513) regions of MCP‐1. This construct has three AP‐1 binding sites identified by open boxes: P1 is located in the enhancer region while P2 and P3 are placed in the promoter region. pGL‐PRM contains the proximal promoter region only, whereas pGL‐PRM‐P2 Mut contains the proximal promoter region but has a mutated P2 AP‐1 binding site. pHMCP‐128 is a construct with deletion of both P2 and P3 AP‐1 binding sites. B. The transfection efficiency of purified human monocytes was assessed by flow cytometry using a GFP‐expressing plasmid as a tracer. In the left panel, GFP+ monocytes are displayed in bold in the histogram, as opposed to untransfected cells (thin line). The percentage of GFP+ cells is also reported. Transfection by nucleofection did not impair p17R expression on the surface of monocytes (right). Cells transfected with the pmax‐GFP plasmid were stained with biotin‐conjugated p17 and APC‐conjugated streptavidin to detect p17R expression, and with PI to mark dead cells. Viable monocytes (PI‐) were gated on FSC/PI dot plot (not shown) and an analysis was performed of GFP versus p17R dot plot. Quadrant statistics are shown. C. Purified human monocytes were transiently co‐transfected with the pRL‐TK control vector together with one of the plasmids described in (A). Soon after transfection, cells were treated or not with p17 (1 μg ml−1). Renilla and Firefly luciferase activities were determined 6 h after p17 treatment. Relative activity is expressed as fold stimulation as described in Experimental procedures and represent the mean ± SD of quadruplicate samples from three independent experiments.
Discussion
Monocyte‐mediated inflammatory processes and MCP‐1 production are likely to be major precipitating events in AIDS pathogenesis. An increasing amount of evidence shows a tight link between activated monocytes recruitment, MCP‐1 release and HIV‐1 pathogenesis, especially in AIDS patients suffering from HIV‐1‐associated dementia (HAD). The selective accumulation of MCP‐1 in the cerebrospinal fluid of AIDS patients with HAD, together with the finding that MCP‐1 levels correlate with the degree of dementia (Cinque et al., 1998; Conant et al., 1998; Kelder et al., 1998), substantiates a possible role of MCP‐1 in neuropathogenesis. It is worth noting that the presence of activated monocytes in the brain is a better correlate of HAD than the extent of viral infection (Glass et al., 1995). A chronic inflammatory state of circulating monocytes emerges over time also in HIV‐1 patients receiving highly active anti‐retroviral therapy (HAART). In particular, these elements evolve to a chronic inflammatory state characterized by an increase in chemotactic macrophage markers and a more mature (monocyte/macrophage hybrid) profile (Pulliam et al., 1997; 2004). Considerable interest is focused therefore on the mechanism by which HIV‐1 makes active to induce dysregulation of monocyte activity. Understanding this mechanism will contribute to further identify the linkage between HIV‐1 infection and monocyte activation and, ultimately, to achieve improved control over the replication and spread of the virus, and of the prevention and treatment of immune disorders.
Highly active anti‐retroviral therapy usually leads to prolonged control of HIV‐1 replication. Yet the blocking of HIV‐1 replication is not linked to a complete recovery from immune dysfunction, thus suggesting that mechanisms other than active viral replication are involved in chronic immune perturbation. Structural HIV‐1 proteins have been associated with immune activation and loss of functional competence by different immune cells (Quaranta et al., 2004). Among these, the HIV‐1 matrix protein p17 has been recently identified as a critical determinant in AIDS pathogenesis as it binds to a cellular receptor expressed on immune cells and enhances viral replication and infectivity (De Francesco et al., 1998), through a combination of different effector functions (De Francesco et al., 2002; Vitale et al., 2003). This structural viral protein accumulates and persists in the lymph nodes of patients under HAART (Popovic et al., 2005). Moreover, amounts of the HIV‐1 protein did not noticeably differ in lymph nodes specimens either before initiation of therapy or during treatment where the virus burden significantly decreased. This finding strongly suggests that p17 persists in patients under HAART for a long time in the absence of detectable virus replication making conceivable that their chronic activity on immune cells can lead to immune dysfunction. Furthermore, we have recently shown that p17 is released by infected cells (Fiorentini et al., 2006) and can be detected in sera obtained from HIV‐1‐infected patients using a newly developed p17 capture immunoassay (S. Fiorentini et al., in preparation). The pro‐inflammatory role for p17 is suggested by its ability to induce TNF‐α and IFN‐γ expression on T cells and to counteract the anti‐inflammatory activity of IL‐4 (De Francesco et al., 2002). In the present study we have described the ability of the HIV‐1 matrix protein p17 to perturb the biological activity of primary monocytes, by selectively inducing increased MCP‐1 production and release. This process is mediated by the expression of p17Rs on the monocyte surface.
The biological activity of p17 is exerted after binding to a specific receptor that is absent on resting T cells but expressed on activated T cells. Accordingly, p17 activity is observed only after pre‐activation of T cells with mitogens, which per se exert a biological effect. The in vitro biological activity of p17 has therefore been described so far as a synergistic effect that cannot be completely separated from that triggered by the mitogen itself. This study shows that circulating monocytes constitutively express p17Rs on their surface and for the first time furthers our understanding of signalling triggered by p17/p17R interaction.
Trypsin‐sensitive membrane proteins are shown to be involved in p17 binding to human monocytes, while the re‐expression of p17R on monocytes after trypsin treatment required several hours and was impaired by cycloheximide. This suggests that de novo protein synthesis is needed to maintain p17R expression on monocytes rather than p17Rs mobilization from intracellular stores. Interestingly, p17R expression – in terms of mean fluorescence intensity – was strongly up‐modulated on the surface of these cells upon activation of monocytes with IFN‐γ or LPS. The finding that p17R expression is increased by two known monocyte stimulatory molecules warrants the definition of this molecule as a new marker attesting for the monocyte activation status. It is not yet clear, however, why p17R expression is specifically sustained on monocytes but not on T cells.
Several signalling pathways have been hypothesized for the transcriptional regulation of MCP‐1. In the human MCP‐1 gene, TRE and κB enhancer elements exist in the 5′‐flanking region of MCP‐1 gene, thus suggesting a role for AP‐1 and NF‐κB (Shyy et al., 1993). We therefore decided to use the primary human monocyte model to assess p17 signal transduction pathways involved in MCP‐1 gene expression. EMSA revealed that p17 does not cause significant changes to the DNA‐binding activity of NF‐κB, whereas it increases AP‐1 binding in a time‐ and dose‐dependent manner. Induction of AP‐1 in both primary human monocytes and THP‐1 cells was associated with increased levels of c‐Fos and JunB proteins in the active AP‐1 complexes. These results indicate that the p17‐induced AP‐1 complexes responsible for the MCP‐1 transcriptional activity are likely to be c‐Fos/JunB heterodimers. Deletion of AP‐1 binding sites in the enhancer region of human MCP‐1 (P1) or mutation of the P2 AP‐1 binding site in the MCP‐1 proximal promoter region had no effect on the transcriptional activity in response to p17. Instead, deletion of both AP‐1 binding sites (P2 and P3) in the promoter region resulted in reduced p17‐induced MCP‐1 gene transcription levels, close to basal transcription values. Such evidence suggests that the upregulation of MCP‐1 transcription in human monocytes by p17 may depend on AP‐1 activation and binding to the P3 site in the MCP‐1 proximal promoter region. However, a direct evidence for the involvement of the P3 AP‐1 binding site in p17‐triggered MCP‐1 transcription is still lacking, as all our efforts to obtain a MCP‐1 promoter–luciferase construct containing a mutated P3 AP‐1 binding site were unsuccessful.
The finding suggesting that in primary human monocytes AP‐1 could be a major regulator of MCP‐1 transcription, independently of NF‐κB activation, is in line with previous studies (Hanazawa et al., 1993; Wang et al., 1999; Lee et al., 2001; Cho et al., 2002). Moreover, Cho et al. (2002) and Lim and Garzino‐Demo (1999) have clearly shown that the P3 AP‐1 binding site abolished inducible MCP‐1 promoter activity in human endothelial cells and human astrocytoma cells (U‐87) respectively. Taken together, these results suggest that, at least in certain cells, AP‐1 activation can circumvent the NF‐κB pathway by means of different signal transduction pathways leading to the induction of the MCP‐1 gene. Interestingly, our experiments show a peak of AP‐1 activation at 1 h after p17 stimulation and persists, at lower levels, up to 4 h. At the same time, MCP‐1 transcripts increase at 4 h after p17 treatment reaching a plateau at 6 h of stimulation. The kinetics of AP‐1 activation does not apparently well correlate with induction of MCP‐1 gene. This finding may reflect the capability of p17 to trigger an unusual pathway that induce AP‐1 binding to the MCP‐1 promoter leading to the production of MCP‐1 transcripts but also to their stabilization, ultimately allowing their accumulation. Indeed, it is well known that regulation of MCP‐1 transcription is due to both gene transcription activation and mRNA post‐transcriptional stabilization. Different studies have demonstrated that, upon different exogenous stimuli, MCP‐1 mRNA half‐life increases to 3 h while anti‐inflammatory molecules may reduce it to 15 min (Poon et al., 1999; Dhawan et al., 2007). On the other hand, we cannot completely rule out the possibility that p17 may activate other transcription factors that cooperate with AP‐1 to determine the prolonged activation of MCP‐1 transcription.
It is important to note that several viruses have been reported to modulate AP‐1 activity (Sadowska et al., 2003; Kwon et al., 2004; Xie et al., 2005; Holloway and Coulson, 2006). In some cases, the viral protein responsible for AP‐1 activation has been identified, e.g. the hepatitis B virus X protein (Tanaka et al., 2006), the severe acute respiratory syndrome (SARS) coronavirus nucleocapsid protein (He et al., 2003), the papilloma virus E2 protein (Behren et al., 2005), the Epstein–Barr virus latent protein 1 and the parvovirus non‐structural protein (NS) 1 (Kieser et al., 1997). The common exploitation of the AP‐1 pathway points to the wider role of this signalling event and of its downstream gene targets in viral infections. In the case of HIV‐1, AP‐1 activation is a redundant phenomenon, being performed by such viral proteins as tat (Kumar et al., 1998), nef (Biggs et al., 1999), gp120 (Gibellini et al., 1999), vpr (Varin et al., 2005) and, as shown here, by the matrix protein p17. In the case of HIV‐1, AP‐1 activation may not only enhance viral replication via trans‐activation of the HIV‐1‐LTR (Rabbi et al., 1997) but, as demonstrated above for p17, it may also modulate the expression of cellular genes. These effects could not only promote HIV‐1 infection but also lead to functional defects and pathogenetic events, as demonstrated for MCP‐1 and its contribution to the pathogenesis of AIDS dementia. Our results suggest that it is necessary to block the biological activity of the HIV‐1 proteins (including p17) continuously released by infected cells despite the successful blocking of viral replication obtained by HAART. At the same time, the knowledge that p17 induces monocytes to produce MCP‐1 by activating the transcriptional factor AP‐1 should be followed up by new anti‐HIV‐1 therapeutic strategies capable of neutralizing the functions of molecules induced by the HIV‐1 proteins which are known contributors to virulence and pathogenesis.
Experimental procedures
Reagents
Purified, endotoxin‐free recombinant HIV‐1 matrix protein p17 was produced as described previously (De Francesco et al., 2002). An aliquot of the purified p17 preparation was biotinylated by using AH‐NHS‐Biotin (BIOSPA, Milan Italy) according to the manufacturer's instructions. Recombinant human IFN‐γ and TNF‐α were obtained from R&D System (Minneapolis, MN, USA). LPS from Escherichia coli, phorbol 12‐myristate 13‐acetate (PMA) and cycloheximide were supplied by Sigma‐Aldrich (Milan, Italy). MBS‐3, an anti‐p17 mAb that recognizes the p17 epitope responsible for p17R binding and neutralizes the p17/p17R interaction (De Francesco et al., 2002), was produced and purified in our laboratory.
Cells
Peripheral blood mononuclear cells (PBMCs) were isolated by density gradient centrifugation from the blood of healthy HIV‐1‐seronegative donors, who gave informed consent to this research under the Helsinki Declaration. Cells were then plated onto tissue culture dishes in RPMI‐1640 supplemented with 10% (v/v) fetal calf serum (FCS) (Invitrogen, Milan, Italy), 100 U ml−1 penicillin, 100 μg ml−1 streptomycin, 2 mM l‐glutamine (complete medium). Monocytes were purified from PBMCs using a CD14 MicroBeads kit (Miltenyi Biotec, Bologna, Italy) and AutoMacs (Miltenyi Biotec), according to the manufacturer's instructions. The purity of monocytes was assessed after overnight incubation at 37°C in complete medium by CD14 staining and flow cytometry, and found to be greater than 95%. The THP‐1 human monocytic cell line was a gift from Dr Maura Ferrari (Istituto Zooprofilattico Sperimentale, Brescia, Italy). Cells were grown and maintained in culture in complete medium.
Flow cytometry
Staining of cells for p17R expression was performed as already described previously (De Francesco et al., 2002). Briefly, the cells were incubated for 30 min on ice with biotin‐conjugated recombinant p17 at a concentration of 400 ng ml−1. After washing with PBS containing 1% FCS, they were incubated for 30 min on ice with APC‐conjugated streptavidin (BD Biosciences, Milan, Italy). For experiments performed on PBMCs, cells were stained for 30 min on ice with FITC‐conjugated CD14 mAb to identify p17R+ monocytes. All data obtained were analysed with CellQuest software (BD Biosciences). In some experiments cells were also counterstained with PE‐conjugated anti‐HLA‐DR mAb (BD Biosciences).
Protease treatment
Purified monocytes (1 × 106) were treated with 1 ml of trypsin solution (Sigma‐Aldrich) for 15 min at 37°C. Protease activity was stopped by the addition of ice‐cold complete medium or soybean trypsin inhibitor (SBTI, Type II‐S; Sigma‐Aldrich). Control cells were treated with buffer only or using a combination of trypsin and SBTI was added simultaneously. The protease‐treated cells were washed twice in RPMI‐1640 and subsequently assayed for p17R expression, as described above. The viability of cells was > 85% as determined by propidium iodide (2.5 μg ml−1) staining and flow cytometric analysis. For experiments measuring the recovery of p17R expression after trypsin treatment, cells were re‐suspended at a concentration of 1 × 106 cells ml−1 in complete medium and incubated for 48 h at 37°C. In some experiments, monocytes were cultured in the presence of cycloheximide (1 μg ml−1) (Sigma‐Aldrich).
Real‐time PCR
MCP‐1, RANTES, MIP‐1α and MIP‐1β mRNA levels were determined by quantitative rtPCR. Total RNA was extracted from p17‐treated or ‐untreated monocytes using the TRIzol® RNA isolation kit (Invitrogen), according to the manufacturer's instructions. After reverse transcription using ImProm reverse transcriptase (Promega, Milan, Italy), the relative amounts of MCP‐1, RANTES, MIP‐1α, MIP‐1β and β‐actin mRNAs were determined by rtPCR. Amplification was carried out in a 25 μl reaction mix containing 12.5 μl of 2× TaqMan universal PCR master mix (Applied Biosystems, Monza, Italy), and 1.25 μl of primer and FAM dye‐labelled probe (Gene Expression Assays, Applied Biosystems). rtPCR assays were performed in triplicate on a 7500 rtPCR System (Applied Biosystems) with the following programme: 40 cycles at 95°C for 20 s and at 60°C for 1 min. Analysis of rtPCR data was performed with the 2−ΔΔCt method using Relative Quantitation Study software (Applied Biosystems). Quantification of each chemokine cDNA was normalized in each reaction according to the internal β‐actin control. The levels of each chemokine mRNA were expressed as fold difference in p17‐treated cells in comparison with untreated cells (calibrator sample).
ELISA
Cytokines and chemokines, namely IL‐1α, IL‐1β, IL‐6, IL‐8, IL‐12, TNF‐α, MCP‐1, MIP‐1α, MIP‐1β and RANTES, were evaluated in the supernatant of monocytes cultured in the presence or absence of p17 – at a concentration ranging from 0.1 to 1 μg ml−1– by Searchlight Chemiluminescent Arrays (Endogen, Rockford, IL). In some experiments, MCP‐1 quantification in the supernatant of p17‐treated or ‐untreated monocytes was performed using a quantitative ELISA kit (Endogen).
Extraction of nuclear proteins
For each treatment condition, nuclear extracts of THP‐1 cells or primary blood monocytes were prepared from 1 or 2 × 107 cells respectively. The cells were washed with ice‐cold PBS and lysed on ice for 10 min in 5× packed cell volume of lysis buffer containing 10 mM Hepes pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 0.1 mM EGTA, 1 mM DTT, 0.2% Nonidet P‐40 and protease inhibitor cocktail (Sigma‐Aldrich). The nuclei were then pelleted by centrifugation at 3000 r.p.m. at 4°C for 10 min and re‐suspended in 3× packed nuclear volume of high‐salt buffer containing 20 mM Hepes pH 7.9, 1 mM EDTA, 1 mM EGTA, 420 mM NaCl, 20% glycerol, 1 mM DTT and a protease inhibitor cocktail. The suspension was rocked for 20 min at 4°C, gently vortexed and subsequently centrifuged at 10 000 r.p.m. for 30 min at 4°C. The supernatant/nuclear extracts were collected and stored in aliquots at −80°C. The protein concentration was determined by using the Bio‐Rad protein assay kit (Bio‐Rad, Hercules, CA).
Protein/DNA array analysis of transcription factor activation
Nuclear extracts (10 μg of protein per sample) prepared as described above were incubated with biotinylated DNA oligonucleotides of 54 selected transcription factor binding element sequences (TransSignal Array; Panomics, Redwood City, CA). After isolation of protein–DNA complexes, the samples were denaturated and retained binding elements hybridized to membranes containing complementary sequences. Hybridized biotinylated oligonucleotides were visualized using horseradish peroxidase (HRP)‐conjugated streptavidin and ECL reagents (Panomics). Each transcription factor was quantified in duplicate at 1× concentration, and in duplicate at 0.1× oligonucleotide concentration under each condition.
Electrophoretic mobility shift assay (EMSA)
Nuclear extracts (10 μg of protein per sample), obtained as described above from monocytes and THP‐1 cells treated or not with p17 (1 μg ml−1), were incubated with 32P‐labelled AP‐1 or NF‐κB DNA probe (Tiberio et al., 2007), and the mobility of DNA–protein complexes (EMSA) was analysed as described previously (Tiberio et al., 2006). In some experiments recombinant p17 was boiled (20 min at 100°C) or treated with 1% trypsin for 15 min at 37°C. Protease activity was stopped by the addition of ice‐cold complete medium.
For competition experiments, a 100‐fold excess of specific unlabelled probe was incubated with nuclear extracts before addition of the radiolabelled probe. For loading controls, the nuclear extracts were analysed also for the DNA‐binding activity of octamer‐1, whose site is present in many housekeeping genes (Tiberio et al., 2007). Autoradiographic signals were quantified by Molecular Dynamics PhosphoImager (MDP) analysis (Typhoon 8600; Molecular Dynamics, Sunnyvale, CA).
Quantification of AP‐1 activation
In order to quantify AP‐1 activation, and to assess the nature of dimeric AP‐1 complexes induced by p17, we used an ELISA‐based kit (TransAM™; Active Motif, Carlsbad, CA) consisting of an oligonucleotide that contains a tissue‐type plasminogen activator (t‐PA)‐responsive element (TRE) immobilized on solid phase. The assay was run according to the manufacturer's instructions. Briefly, nuclear extracts obtained from both monocytes and THP‐1 cells treated or not with p17 for 1 h at 37°C were allowed to interact with TRE‐coated wells (4 μg of nuclear extract per well). Then, AP‐1 dimers contained in nuclear cell extracts were detected using antibodies directed against c‐Fos, FosB, Fra‐1, Fra‐2, phospho c‐Jun, JunB or JunD. Antibody binding to the AP‐1/TRE complex was detected by adding an HRP‐conjugated secondary antibody.
Plasmids
MCP‐1 promoter–luciferase constructs containing the proximal promoter (between −107 and +60) (pGLM‐PRM) and distal enhancer (between −2742 to −2513) (pGLM‐ENH) regions of MCP‐1 (kindly provided by Dr T. Yoshimura, Laboratory of Immunobiology, National Cancer Institute, Frederick, MD) have already been described (Ueda et al., 1997). Site‐directed mutation was introduced in the AP‐1 binding P2 site of the promoter region with a quick‐change site‐directed mutagenesis kit (Stratagene, La Jolla, CA). Briefly, 15 ng of pGL‐PRM and 125 ng of oligonucleotides: (5′‐CAG CCC ACT TAT GAC AGA TGG AAG ATC CCT CC‐3′/5′‐GGA GGG ATC TTC CAT CTG TCA TAA GTG GGC TG‐3′) were incubated with 2.5 U of Pfu DNA polymerase and cycled 12 times under the following conditions: denaturation at 95°C for 30 s, annealing at 55°C for 1 min and extension at 68°C for 6 min. The reaction mix was cooled on ice for 2 min and digested with 10 U of DpnI at 37°C for 1 h; 2 μl of the reaction mix was transformed into E. coli XL1‐Blue supercompetent cells. Plasmids were isolated from colonies grown overnight at 37°C on ampicillin agar plates, and sequencing was carried out to identify those containing the target mutated sequence of interest (pGLM‐PRM‐P2mut). The plasmid phMCP128 containing deletion of both P2 and P3 AP‐1 binding sites (kindly provided by Dr A. Garzino‐Demo, Institute of Human Virology, University of Maryland, Baltimore, MD) has been previously described (Lim and Garzino‐Demo, 1999).
DNA transfection and luciferase assay
Purified monocytes were co‐transfected by nucleofection using the Human Monocyte kit (Amaxa AG, Cologne, Germany) with the individual luciferase (Firefly) test plasmids (2 μg ml−1) and a Renilla luciferase expression plasmid (pRL‐TK as an internal control to signal the total cellular transcription level (Promega). Cells were then plated into a 24‐well plate (106 cells per well) and exposed or not to p17 (1 μg ml−1) in complete medium. After 6 h, they were harvested and luciferase expression was quantified using a Dual‐Luciferase reporter assay system, according to the manufacturer's instructions (Promega). Test values were corrected for the luciferase activity value of the internal control plasmid pRL‐TK. The results for the p17 exposed cells are expressed as fold induction of luciferase activity by the same construct under control conditions, taking the control (no p17 added) value as 1.
Acknowledgements
This work was supported by a grant from the Istituto Superiore di Sanità AIDS Projects (Grant 40G.16) and by Ministero dell'Università e della Ricerca, PRIN projects.
References
- Alfano, M. , and Poli, G. (2001) Cytokine and chemokine based control of HIV infection and replication. Curr Pharm Des 7: 993–1013. [DOI] [PubMed] [Google Scholar]
- Behren, A. , Simon, C. , Schwab, B.M. , Loetzsch, E. , Brodbeck, S. , Huber, E. , et al. (2005) Papillomavirus E2 protein induces expression of the matrix metalloproteinase‐9 via the extracellular signal‐regulated kinase/activator protein‐1 pathway. Cancer Res 65: 11613–11621. [DOI] [PubMed] [Google Scholar]
- Biggs, T.E. , Cooke, S.J. , Barton, C.H. , Harris, M.P. , Saksela, K. , and Mann, D.A. (1999) Induction of activator protein 1 (AP‐1) in macrophages by human immunodeficiency virus type‐1 NEF is a cell‐type‐specific response that requires both hck and MAPK signalling events. J Mol Biol 290: 21–35. [DOI] [PubMed] [Google Scholar]
- Cho, N.‐H. , Seong, S.‐Y. , Huh, M.‐S. , Kim, N.‐H. , Choi, M.‐S. , and Kim, I.‐S. (2002) Induction of the gene encoding macrophage chemoattractant protein 1 by Orientia tsutsugamushi in human endothelial cells involves activation of transcription factor activator protein 1. Infect Immun 70: 4841–4850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cinque, P. , Vago, L. , Mengozzi, M. , Torri, V. , Ceresa, D. , Licenzi, E. , et al. (1998) Elevated cerebrospinal fluid levels of monocyte chemotactic protein‐1 correlate with HIV‐1 encephalitis and local viral replication. AIDS 12: 1327–1332. [DOI] [PubMed] [Google Scholar]
- Conant, K. , Garzino‐Demo, A. , Nath, A. , McArthur, J.C. , Halliday, W. , Power, C. , et al. (1998) Induction of monocyte chemoattractant protein‐1 in HIV‐1 tat‐stimulated astrocytes and elevation in AIDS dementia. Proc Natl Acad Sci USA 95: 3117–3121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Francesco, M.A. , Baronio, M. , Fiorentini, S. , Signorini, C. , Bonfanti, C. , Ponesi, C. , et al. (2002) HIV‐1 matrix protein p17 increases the production of proinflammatory cytokines and couteracts IL‐4 activity by binding to a cellular receptor. Proc Natl Acad Sci USA 99: 9972–9977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Francesco, M.A. , Caruso, A. , Fallacara, F. , Canaris, A.D. , Dima, F. , Poiesi, C. , et al. (1998) HIV p17 enhances lymphocyte proliferation and HIV‐1 replication after binding to a human serum factor. AIDS 12: 245–252. [DOI] [PubMed] [Google Scholar]
- Dhawan, L. , Liu, B. , Blaxall, B.C. , and Taubman, M.B. (2007) A novel role for the glucocorticoid receptor in the regulation of monocyte chemoattractant protein‐1 mRNA stability. J Biol Chem 282: 10146–10152. [DOI] [PubMed] [Google Scholar]
- Donovan, R.M. , Bush, C.E. , Markowitz, N.P. , Baxa, D.M. , and Saravolatz, L.D. (1996) Changes in viral load markers during AIDS‐associated opportunistic diseases in human immunodeficiency virus‐infected persons. J Infect Dis 174: 401–403. [DOI] [PubMed] [Google Scholar]
- Fantuzzi, L. , Conti, L. , Gauzzi, M.C. , Eid, P. , Del Corno, M. , Varano, B. , et al. (2000) Regulation of chemokine/cytokine network during in vitro differentiation and HIV‐1 infection of human monocytes: possible importance in the pathogenesis of AIDS. J Leukoc Biol 68: 391–399. [PubMed] [Google Scholar]
- Fantuzzi, L. , Spadaro, F. , Vallanti, G. , Canini, I. , Ramoni, C. , Licenzi, E. , et al. (2003) Endogenous CCL2 (monocyte chemotactic protein‐1) modulates human immunodeficiency virus type‐1 replication and affects cytoskeleton organization in human monocyte‐derived macrophages. Blood 102: 2334–2337. [DOI] [PubMed] [Google Scholar]
- Fiorentini, S. , Marini, E. , Caracciolo, S. , and Caruso, A. (2006) Functions of the HIV‐1 matrix protein p17. New Microbiol 29: 1–10. [PubMed] [Google Scholar]
- Fischer‐Smith, T. , Croul, S. , Svertiuk, A.E. , Capini, C. , L'Hereux, D. , Regulier, E.G. , et al. (2001) CNS invasion by CD14+/CD16+ peripheral blood‐derived monocytes in HIV dementia: perivascular accumulation and reservoir of HIV infection. J Neurovirol 6: 528–541. [DOI] [PubMed] [Google Scholar]
- Gibellini, D. , Re, M.C. , Bassini, A. , Guidotti, L. , Catani, I. , La Placa, M. , and Zauli, G. (1999) HIV‐1 gp120 induces the activation of both c‐fos and c‐jun immediate‐early genes in HEL megakaryocytic cells. Br J Haematol 104: 81–86. [DOI] [PubMed] [Google Scholar]
- Glass, J.D. , Fedor, H. , Wesselingh, S.L. , and McArthur, J.C. (1995) Immunocytochemical quantitation of human immunodeficiency virus in the brain: correlation with dementia. Ann Neurol 38: 755–762. [DOI] [PubMed] [Google Scholar]
- Goletti, D. , Weissman, D. , Jackson, R.W. , Graham, N.M. , Vlahov, D. , Klein, R.S. , et al. (1996) Effect of Mycobacterium tuberculosis on HIV replication. Role of immune activation. J Immunol 175: 1271–1278. [PubMed] [Google Scholar]
- Gonzalez, E. , Rovin, R.H. , Sen, L. , Cooke, G. , Dhanda, R. , Mummidi, S. , et al. (2002) HIV‐1 infection and AIDS dementia are influenced by a mutant MCP‐1 allele linked to increased monocyte infiltration of tissues and MCP‐1 levels. Proc Natl Acad Sci USA 99: 13795–13800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanazawa, S. , Takeshita, A. , Amano, S. , Semba, T. , Nirazuka, T. , Katoh, H. , and Kitano, S. (1993) Tumor necrosis factor‐α induced expression of monocyte chemoattractant JE via fos and jun genes in clonal osteoblastic MC3T3‐E1 cells. J Biol Chem 268: 9526–9532. [PubMed] [Google Scholar]
- He, R. , Leeson, A. , Andonov, A. , Li, Y. , Bastien, N. , Cao, J. , et al. (2003) Activation of AP‐1 signal transduction pathway by SARS coronavirus nucleocapsid protein. Biochem Biophys Res Commun 311: 870–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbein, G. , Coaquette, A. , Perez‐Bercoff, D. , and Pacino, G. (2002) Macrophage activation and HIV infection: can the Trojan horse turn into a fortress? Curr Mol Med 2: 723–738. [DOI] [PubMed] [Google Scholar]
- Hirsch, V.M. , Sharkey, M.E. , Brown, C.R. , Brichacek, B. , Goldstein, S. , Wakefield, J. , et al. (1998) Vpx is required for dissemination and pathogenesis of SIV(SM) PBj: evidence of macrophage‐dependent viral amplification. Nat Med 4: 1401–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho, W.Z. , Cherukrui, R. , and Douglas, S.D. (1994) The macrophage and HIV‐1. Immunol Ser 60: 569–587. [PubMed] [Google Scholar]
- Holloway, G. , and Coulson, B.S. (2006) Rotavirus activates JNK and p38 signaling pathways in intestinal cells, leading to AP‐1‐driven transcriptional responses and enhanced virus replication. J Virol 80: 10624–10633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedzierska, K. , Crowe, S.M. , Turnville, S. , and Cunningham, A.L. (2003) The influence of cytokines, chemokines and their receptors on HIV‐1 replication in monocytes and macrophages. Rev Med Virol 13: 39–56. [DOI] [PubMed] [Google Scholar]
- Kelder, W. , McArthur, J.C. , Nance‐Sproson, T. , McClernon, D. , and Griffin, D.E. (1998) Beta‐chemokines MCP‐1 and RANTES are selectively increased in cerebrospinal fluid of patients with human immunodeficiency virus‐associated dementia. Ann Neurol 44: 831–835. [DOI] [PubMed] [Google Scholar]
- Kieser, A. , Kilger, E. , Gires, O. , Ueffing, M. , Kolch, W. , and Hammerschmidt, W. (1997) Epstein–Barr virus latent membrane protein‐1 triggers AP‐1 activity via the c‐Jun N‐terminal kinase cascade. EMBO J 16: 6478–6485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, A. , Manna, S.K. , Dhawan, S. , and Aggarwal, B.B. (1998) HIV‐Tat protein activates c‐Jun N‐terminal kinase and activator protein‐1. J Immunol 161: 776–781. [PubMed] [Google Scholar]
- Kwon, D. , Fuller, A.C. , Palma, J.P. , Choi, I.H. , and Kim, B.S. (2004) Induction of chemokines in human astrocytes by picornavirus infection requires activation of both AP‐1 and NF‐kappa B. Glia 45: 287–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanzavecchia, A. , Reid, P.A. , and Watts, C. (1992) Irreversible association of peptides with class II MHC molecules in living cells. Nature 357: 249–252. [DOI] [PubMed] [Google Scholar]
- Lee, S.K. , Kim, B.S. , Yang, W.S. , Kim, S.B. , Park, S.K. , and Park, J.S. (2001) High glucose induces MCP‐1 expression partly via tyrosine kinase‐AP‐1 pathway in peritoneal mesothelial cells. Kidney Int 60: 55–64. [DOI] [PubMed] [Google Scholar]
- Lim, S.P. , and Garzino‐Demo, A. (1999) The human immunodeficiency virus type 1 tat protein up‐regulates the promoter activity of the beta‐chemokine monocyte chemoattractant protein 1 in the human astrocytoma cell line U‐87 MG: role of SP‐1, AP‐1 and NF‐κB consensus sites. J Virol 74: 1632–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon, M. , Liu, B. , and Taubman, M.B. (1999) Identification of a novel dexamethasone‐sensitive RNA‐destabilizing region on rat monocyte chemoattractant protein 1 mRNA. Mol Cell Biol 19: 6471–6478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popovic, M. , Tenner‐Racz, K. , Pelser, C. , Stellbrink, H.J. , Van Lunzen, J. , Lewis, G. , et al. (2005) Persistence of HIV‐1 structural proteins and glycoproteins in lymph nodes of patients under highly active antiretroviral therapy. Proc Natl Acad Sci USA 102: 14807–14812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulliam, L. , Gascon, R. , Stubblebine, M. , McGuire, D. , and McGrath, M.S. (1997) Unique monocyte subset in patients with AIDS dementia. Lancet 349: 692–695. [DOI] [PubMed] [Google Scholar]
- Pulliam, L. , Sun, B. , and Rempel, H. (2004) Invasive chronic inflammatory monocyte phenotype in subjects with high HIV‐1 viral load. J Neuroimmunol 157: 93–98. [DOI] [PubMed] [Google Scholar]
- Quaranta, M.G. , Mattioli, B. , Giordani, L. , and Viora, M. (2004) HIV‐1 Nef equipe dendritic cells to reduce survival and function of CD8+ T cells: a mechanism of immune evasion. FASEB J 18: 1459–1461. [DOI] [PubMed] [Google Scholar]
- Rabbi, M.F. , Saifuddin, M. , Gu, D.S. , Kagnoff, M.F. , and Roebuch, K.A. (1997) U5 region of the human immunodeficiency virus type 1 long terminal repeat contains TRE‐like cAMP‐responsive elements that bind both AP‐1 and CREB/ATF proteins. Virology 233: 235–245. [DOI] [PubMed] [Google Scholar]
- Sadowska, B. , Barrucco, R. , Khalili, K. , and Safak, M. (2003) Regulation of human polyomavirus JC virus gene transcription by AP‐1 in glial cells. J Virol 77: 665–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyy, Y.J. , Ly, Y.S. , and Kolattukudy, P.E. (1993) Activation of MCP‐1 gene expression is mediated through multiple signalling pathways. Biochem Biophys Res Commun 192: 693–699. [DOI] [PubMed] [Google Scholar]
- Tanaka, Y. , Kanai, F. , Ichimura, T. , Tateishi, K. , Asaoka, Y. , Guleng, B. , et al. (2006) The hepatitis B virus X protein enhances AP‐1 activation through interaction with Jab1. Oncogene 25: 633–642. [DOI] [PubMed] [Google Scholar]
- Tiberio, L. , Tiberio, G. , Bardella, L. , Cervi, E. , Cerea, K. , Dreano, M. , et al. (2006) Mechanisms of interleukin‐6 protection against ischemia‐reperfusion injury in rat liver. Cytokine 34: 131–142. [DOI] [PubMed] [Google Scholar]
- Tiberio, G. , Tiberio, L. , Benetti, A. , Cervi, E. , Pandolfo, G. , Dreano, M. , et al. (2007) Interleukin‐6 sustains hepatic regeneration in cirrhotic rat. Hepatogastroenterology 54: 878–883. [PubMed] [Google Scholar]
- Tulp, A. , Verwoerd, D. , Dobberstein, B. , Ploegh, H.L. , and Pieters, J. (1994) Isolation and characterization of the intracellular MHC class II compartment. Nature 369: 120–126. [DOI] [PubMed] [Google Scholar]
- Ueda, A. , Ishigatsubo, Y. , Okubo, T. , Yoshimura, T. (1997) Transcriptional regulation of the human monocyte chemoattractant protein‐1 gene. Cooperation of two NF‐kappa B sites and NF‐kappa B/Rel subunit specificity. J Biol Chem 272: 31092–31099. [DOI] [PubMed] [Google Scholar]
- Varin, A. , Decrion, A.‐Z. , Sabbah, E. , Quivy, V. , Sire, J. , Van Lint, C. , et al. (2005) Synthetic Vpr protein activates activator protein‐1, c‐Jun N‐terminal kinase, and NF‐κB and stimulates HIV‐1 transcription in promonocytic cells and primary macrophages. J Biol Chem 280: 42557–42567. [DOI] [PubMed] [Google Scholar]
- Vitale, M. , Caruso, A.M.A. , De Francesco, M.A. , Rodella, L. , Bozzo, L. , Garrafa, E. , et al. (2003) HIV‐1 matrix protein p17 enhances the proliferative activity of natural killer cells and increases their ability to secrete proinflammatory cytokines. Br J Haematol 120: 337–343. [DOI] [PubMed] [Google Scholar]
- Wahl, S.M. , Greenwell‐Wild, T. , Peng, G. , Ma, G. , Orenstein, J.M. , and Vazquez, N. (2003) Viral and host cofactors facilitate HIV‐1 replication in macrophages. J Leukoc Biol 74: 726–735. [DOI] [PubMed] [Google Scholar]
- Wang, N. , Verna, L. , Hardy, S. , Forsayeth, J. , Zhu, Y. , and Stemerman, M.B. (1999) Adenovirus‐mediated overexpression of c‐Jun and c‐Fos induces intercellular adhesion molecule‐1 and monocyte chemoattractant protein‐1 in human endothelial cells. Arterioscler Thromb Vasc Biol 19: 2078–2084. [DOI] [PubMed] [Google Scholar]
- Xie, J. , Pan, H. , Yoo, S. , and Gao, S.‐J. (2005) Kaposi's sarcoma‐associated herpesvirus induction of AP‐1 and interleukin 6 during primary infection mediated by multiple mitogen‐activated protein kinase pathways. J Virol 79: 15027–15037. [DOI] [PMC free article] [PubMed] [Google Scholar]