

Abstract
A convenient approach to antimalarial drug discovery is the use of the organic scaffold of a known antimalarial drug and an organometallic moiety to alter its unwanted properties and/or to optimize its initial effects. This minireview focuses mainly on the discovery of ferroquine, which has emerged from a collaborative French discovery project, and efforts to understand its mechanism of action and resistance.
Keywords: bioorganometallics, drug candidates, ferroquine, malaria, mechanism of action, resistance
Introduction
Malaria is a tropical disease and is common in Africa, south east Asia, and south America. There are 500 million clinical cases of malaria each year. In 2006, an estimated 1.5 to 2.7 million deaths were the result of malaria and most of the deaths occurred in children under five years old. Malaria is caused by blood parasites of the Plasmodium species. P. falciparum is the most widespread and dangerous Plasmodium because it can lead to host death. The burden of malaria is currently increasing because of drug and insecticide resistance and social and environmental changes.
By far the most important factor is the development of resistance by P. falciparum to cheap and effective drugs like chloroquine (CQ).1 In fact, all the quinoline‐based compounds marketed encounter chemoresistance problems.2 Currently, the World Health Organization (WHO) recommends artemisinin (ART) combination therapies (ACTs) for the treatment of malaria. Indeed, combination therapies are preferred to monotherapies as they prevent the development of resistant parasites. These ART therapies associate fast‐acting ART‐derived drugs with other antimalarials with longer half‐lives such as mefloquine (MF). Nevertheless, continuing research is needed to develop new antimalarial drugs which do not induce resistance.
A convenient approach to (antimalarial) drug discovery is based on the modification of those drugs encountering resistance problems. Alternately, the attachment of an organometallic complex to an organic scaffold was already attempted 20 years ago.3 The idea was to use the organic scaffold of the drug for its primary properties (that is, permeability, uptake, transport, binding to a target) and the organometallic moiety to alter its unwanted properties (that is, resistance) and/or to optimize its initial effects.4
Ferrocene (Fc) with its sandwich structure was rapidly identified as the metallocene of choice. Indeed, Fc is a small, rigid, lipophilic molecule which can penetrate cellular membranes. Fc is stable in aqueous, aerobic media, and allows the accessibility to a large variety of derivatives. In addition, the electrochemical behavior of Fc makes it very attractive for biological applications and especially for drug design.5 Among the numerous ferrocene bioconjugates reported,5 one of the famous examples is the structural variation of the anticancer drug tamoxifen to give ferrocifen.6, 7 Another one is ferrocerone, which was developed to treat iron deficiency anemias, and used to be marketed in Russia.8
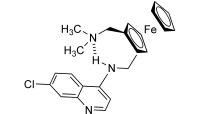
This minireview focuses mainly on the discovery of ferroquine (FQ, SR97193, Figure 1), a new antimalarial, including efforts to understand its mechanism(s) of action and resistance (Table 1).
Figure 1.
Chemical structure of ferroquine, the new antimalarial. The intramolecular H‐bond is indicated with dashed lines.
Table 1.
The publisher did not receive permission from the copyright owner to include this object in this version of this product. Please refer either to the publisher's own online version of this product or the printed product where one exists.
Innovative metallodrug design
Ferrocene conjugates with chloroquine
CQ 1 (Figure 2) is a 4‐aminoquinoline drug long used in the treatment or prevention of malaria. Although its mechanism of action is only partially understood, its therapeutic effectiveness has been attributed to its ability to preferentially concentrate in the food vacuole of the parasite and to inhibit the formation of malarial pigment (or hemozoin).9–12
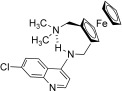
Figure 2.
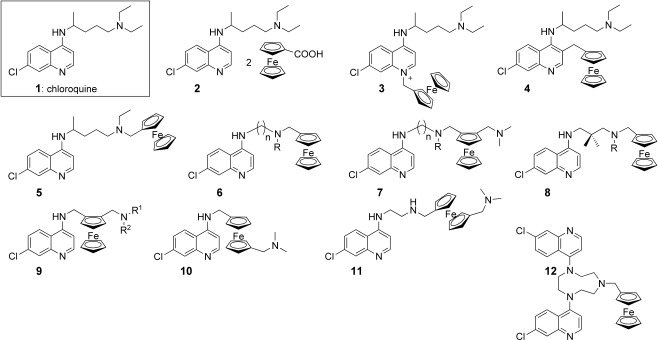
The template, chloroquine 1, and the ferrocene conjugates 2–12. R is an alkyl or a ferrocenylmethyl group, n varying from 2 to 6.
Resistance to CQ was first reported from Columbia and Thailand in the early 1960s, and is now worldwide. CQ‐resistant parasites expel CQ much more rapidly from red blood cells than CQ‐sensitive parasites, and many observations indicated that a P. falciparum transmembrane protein (PfCRT) was involved in this efflux.13–17 Mutations of PfCRT have been described in all CQ‐resistant P. falciparum isolates.
Combination of the CQ structure, for which the 4‐aminoquinoline moiety is known to target the parasite, with ferrocene has led to the design of FQ, the first organometallic antimalarial (see below).18–20 During this research program, more than 50 ferrocene analogues were synthesized and screened. The group of Professor Chibale in Cape Town (South Africa) also assisted us in this research. Nevertheless, so far none of these compounds prove to be better than FQ.
The envisaged structures were designed to respect the main properties of CQ such as its localization in the food vacuole of the parasite or its propensity to inhibit hemozoin formation. Besides, a less conventional design was also investigated as we could not exclude that the modification of the parent drug might lead to novel properties and alternative mechanisms of action.
First, it was tempting to simply associate CQ and the ferrocenecarboxylic acid by a weak salt‐bridge interaction as in compound 2.21 Indeed, the ferrocene moiety may independently potentiate the activity of CQ by enhancing oxidative stress. Nevertheless, this hypothesis was proven wrong. In vitro tests revealed that salt 2 was even less active than CQ diphosphate, suggesting an antagonist effect between both parts.
Formation of the quaternary ammonium salt 3 by direct condensation of the ferrocenylmethyl (Fem) moiety on the endocyclic nitrogen of the CQ core abolished the activity of the parent molecule on both CQ resistant and sensitive P. falciparum strains.22 The charged species 3 should not be able to cross the membrane. A low in vitro activity was also observed with compound 4 where the quinoline cycle is substituted at the C3 position by Fem.22 The bulky ferrocenyl group should sterically hinder the stacking interaction between the quinoline ring and heme.
Particular attention was devoted to studying the impact of the introduction of the ferrocenyl moiety into the lateral side chain of CQ. Indeed, the length of the side chain and the distance between the two exocyclic nitrogen atoms may both affect resistance against 4‐aminoquinolines by P. falciparum.23, 24 4‐aminoquinolines with shorter (two or three carbon atoms) or longer side chains (10 or 12 carbon atoms) than CQ are more active against CQ‐resistant P. falciparum. It has been suggested that these molecules had an N–N spacing which is less suited for binding with the putative CQ transporter, and are therefore less efficiently extruded from the food vacuole.
A series of CQ analogues 5–8 characterized by the presence of the ferrocenyl group attached to the terminal nitrogen atom of the CQ lateral chain were synthesized and tested.25, 26 Whereas most analogues were found to be more active than CQ, they offered (relatively) disappointing activities compared to FQ. Compounds such as 6 showed an activity which decreased rapidly with the level of CQ‐resistance among the P. falciparum clones tested. Clearly, a cross resistance could be postulated to emerge very quickly. There is no significant correlation between either the liphophilic character or the in vitro inhibition of hemozoin formation and the IC50 values among CQ analogues 6.26 Bis‐ferrocenyl conjugates (compounds such as 6 with R=Fem) led to erratic activities and it was impossible to measure precise IC50 values, because of both stability and solubility problems in the culture medium.26
A decrease of the efficacy was observed between the linear and branched propylamino chain derivatives 8. Introduction of methyl groups in the side chain was not favorable to the antimalarial activity.26 To the contrary, the presence of the ferrocene moiety within the lateral chain (FQ analogues, 9) is the main condition to retain a strong antimalarial activity on CQ‐resistant P. falciparum. All FQ analogues exhibited an antimalarial activity much stronger than CQ itself on CQ‐resistant strains, except when a second ferrocenyl group was introduced on the terminal nitrogen atom. Here again the efficacy of compounds was markedly attenuated.26
As 1,2‐unsymmetrically substituted ferrocenes are chiral molecules, an effort was made to design achiral version of these derivatives. The easiest solution was to move the second substituent to the other cyclopentadienyl cycle. These achiral 1,1′‐substituted ferrocene analogues 10–11 exhibited lower activity against CQ resistant strains than against the CQ sensitive strains.27 Nervertheless, no in vivo data were available for the comparison of the substitution patterns: 1,2 versus 1,1′. A similar trend was also observed for the bis‐quinoline derivatives 12.28
Ferrocene conjugates with other antimalarials
Artemisinin
Ferrocene‐derived artemesinins 15–20 did not show better antimalarial activity than that of the parent compound(s). The analogues 15 (obtained as two separable (α and β) C9 stereoisomers) and 16 (racemate) were prepared from artemisitene 14 (Figure 3).29 These derivatives exhibited IC50 values on P. falciparum tenfold lower than artemisitene, but similar to that of artemisinin.29 Other derivatives (β stereoisomer 17, racemate 18, racemate 19, and α stereoisomer 20) were synthesized merging ferrocene and artemisinin via an ester bond (like in the artesunate structure) or via an ether bond (like in the artemether skeleton). These produced no increase in antimalarial activity compared to dihydroartemisinin itself.30 It was concluded that incorporation of the ferrocene moiety into an artemesinin skeleton did not improve its activity.
Figure 3.
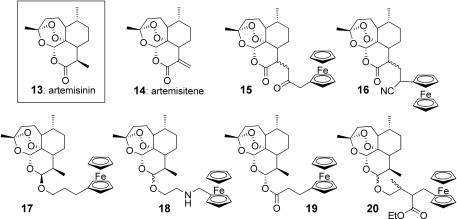
Artemisinin 13, artemisitene 14, and the ferrocene conjugates 15–20.
Mefloquine and quinine
Using a strategy similar to the design of FQ, the quinuclidinyl and the piperidinyl side chains of quinine 21 and mefloquine (MF) 21 were respectively substituted with a ferrocene moiety while maintaining a basic amino group 23, 24 (Figure 4).
Figure 4.
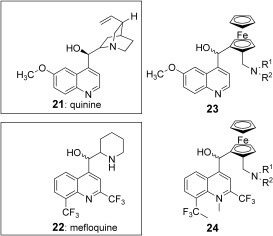
Quinine 21, mefloquine 22 and their respective ferrocene analogues 23 and 24. R1 and R2 are alkyl groups.
In vitro, lower activities than the parent compounds were reported.31 In acidic aqueous solution, these ferrocenyl analogues seemed to be unstable, leading to the formation of the presumably inactive carbeniums.
Atovaquone
Among fourteen ferrocene atovaquone conjugates synthesized and tested both on Toxoplasma gondii and P. falciparum, compounds 26 (Figure 5) with an aliphatic chain of 6, 7, and 8 carbon atoms were active on the atovaquone‐resistant ATO T. gondii clone known to carry a mutation in the ubiquinone binding pocket of the cyt b. As concerns P. falciparum, the products appeared as active on both CQ sensitive and resistant strains, but remained 5–12‐fold less active than atovaquone itself on the same strains.32
Figure 5.
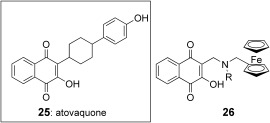
Atovaquone 25 and its respective ferrocene analogues 26. R is an alkyl group.
Miscellaneous
Ferrocenyl sugars were synthesized starting from ellagitanin which has no antimalarial properties per se.33 Some of the derivatives showed antimalarial activities with IC50 values in the micromolar, and, rarely, submicromolar range. Another attempt to inhibit P. falciparum hexose transporter (PfHT) expressed in a heterologous system (xenope oocyte) with 3‐O‐ferrocenyl‐d‐glucose derivatives was performed, but the products failed to show an inhibitory effect upon the transporter.34
Ferrocenyl chalcones were synthesized and, among them, some compounds had an antimalarial activity in the micromolar range.35 Interestingly, the incorporation of the ferrocene moiety in the chalcone template was found to enhance its role in processes that involved free radicals.36 The authors suggested therefore that ferrrocene may participate in redox reaction(s) in relation to their antimalarial activity.36
More recently, an attempt to improve the reversal properties of strychnobrasiline led to the synthesis of a ferrocene–strychnobrasiline conjugate which showed in vitro a synergy with CQ on a resistant clone, but failed to show the same activity in vivo when tested in association with CQ on the P. yoelii N67 clone in a suppressive four‐day test.37
In summary, among all ferrocenic antimalarials synthesized so far, only some 7‐chloro‐4‐aminoquinolines derivatives were found to be promising, usually being active on CQ sensitive and CQ‐resistant clones of P. falciparum. To a lesser extent, some artemisitene derivatives showed interesting properties, but remained at best, equal to artemisinin itself. All other combinations of ferrocene with known antimalarials or other molecules failed to provide a real “lead” for a further development.
Ferroquine
Of all the ferrocenes, FQ was shown to be the most active in vitro and in vivo, and was considered early on as a lead compound (Table 1). Extensive studies were done to test its potential for industrial development.
Formulation
New drug candidates should enter the pharmaceutical development process in a crystalline state. Indeed, molecules in the amorphous state generally exhibit greater chemical instability, enhanced dissolution rates, altered mechanical properties, and greater hygroscopicity. Neutral FQ was selected for drug development, as FQ will become (di)protonated when entering the acidic environment of the stomach. Basic FQ crystallizes in the monoclinic space group P21/n.38 In the solid state, FQ is stabilized by a strong intramolecular hydrogen bond between the anilino nitrogen atom and the tertiary nitrogen atom of the side chain (Figure 1). Nevertheless, this H‐bond is absent in polar solvents (such as water) or when protonated. This flip/flop H‐bond may help transport of FQ through the hydrophobic membranes. Cationic FQ forms stable dimer structures not only in the solid state but also in solution.39 This self‐association process in water is singularly driven by +‐π/+‐π nonbonding interactions.39
Enantiomers
FQ possesses planar chirality due to its 1,2‐unsymmetrically substituted ferrocene moiety (Figure 6). Pure enantiomers (1′R)‐FQ and (1′S)‐FQ were obtained by enzymatic resolution using a biocatalyst.40 Both optical isomers were equally active in vitro on P. falciparum at nanomolar concentrations. In vivo, both enantiomers were slightly less active than the racemic mixture against CQ sensitive and CQ resistant P. vinckei vinckei, suggesting an additive or a synergetic effect between both enantiomers. Moreover, (1′R)‐FQ displayed a better curative effect than (1′S)‐FQ suggesting different pharmacokinetic properties.
Figure 6.
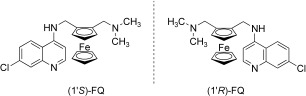
Ferroquine enantiomers.
Metabolism
As illustrated in Figure 7, the metabolic pathway of FQ, based on experiments using animal and human hepatic models, has been proposed.
Figure 7.
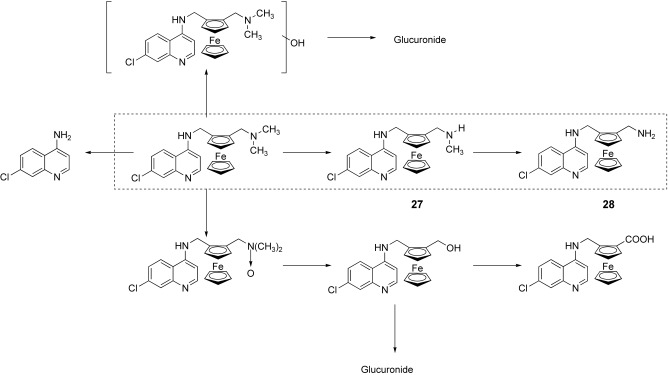
Proposed metabolic pathway of ferroquine in human hepatic models. Main metabolites are in the dashed line box.
FQ is metabolized via a major dealkylation pathway into the mono‐N‐desmethyl FQ 27 and then into di‐N,N‐desmethyl FQ 28.41 Other minor metabolic pathways were also identified. Cytochrome P450 isoforms 2C9, 2C19, and 3A4 and, possibly in some patients, isoform 2D6, are mainly involved in FQ oxidation.
The activity of these two main metabolites was decreased compared to that of FQ; however, the activity of the mono‐N‐desmethyl derivative 27 is significantly higher than that of CQ on both strains, and the di‐N,N‐desmethyl derivative 28 remains more active than CQ on the CQ‐resistant strain.41, 42
As these two metabolites are present in significant concentrations in blood after administration of FQ, they should be involved in the global antimalarial activity of FQ.
Toxicity
FQ responded negatively on the Ames and FETAX (Frog Embryo Teratogenesis Assay Xenopus) tests. FQ also tested negatively in the micronucleus in vitro and in vivo assays conducted under GLP Standards. On the contrary, in the same kind of experiments, CQ was found to be weakly mutagenic and genotoxic.43
Antiviral activity
Although its mode of action is still unknown, CQ has been reported to possess strong antiviral effects on the severe acute respiratory syndrome (SARS) causative agent.44 In this context, FQ was evaluated for its activity against feline and human SARS coronavirus and compared to its parent drug, CQ. Beside its antimalarial activity, FQ was an effective inhibitor of SARS‐CoV replication in Vero cells within the 1–10 μm concentration range. Nevertheless, its low selectivity index of 15 did not allow for pharmaceutical development.45
Specific pharmacology
So far, FQ has been tested on different laboratory P. falciparum strains27, 45, 46 and on seven series of field isolates (total 441) from Gabon, Senegal, and Cambodia.47–51 Figure 8 and Table 2 show the mean IC50 observed from in these different studies.
Figure 8.
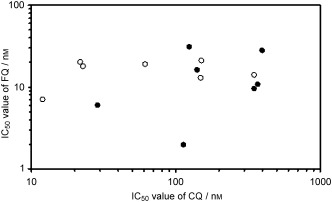
Mean IC50 values of FQ measured on seven different laboratory P. falciparum clones (open circles)27, 45, 46 and on seven sets of field isolates from Gabon, Senegal, and Cambodia (filled circles).47–51
Table 2.
The publisher did not receive permission from the copyright owner to include this object in this version of this product. Please refer either to the publisher's own online version of this product or the printed product where one exists.
No significant correlation appears between CQ and FQ mean IC50 values (Figure 8). Some researchers showed a weak correlation between CQ and FQ responses in different field isolate studies, without significant consequences for clinical applications, but recent investigations demonstrated a probable influence of the initial parasitaemia at the start of the assay, because isolates tested with an identical initial parasitaemia or use of a covariance analysis taking into account the initial parasitaemia did not show a correlated response between FQ and other antimalarials.51
In vivo experiments performed on rodent Plasmodium species showed that whatever the susceptibility of the strain to CQ and the way of administration (Table 3), the curative dose of FQ remained unchanged46 which demonstrated the powerful activity of the drug and its high oral bioavailability, two major qualities for the further development of the drug.
Table 3.
The publisher did not receive permission from the copyright owner to include this object in this version of this product. Please refer either to the publisher's own online version of this product or the printed product where one exists.
Mechanism of action
The mechanism of action of FQ was studied in comparison to that of CQ. Over the years, the mechanism of CQ has been the subject of a lot of discussions and arguments. Nevertheless there is strong evidence that the action of CQ is correlated with its localization in the food vacuole of the parasite and with its association with hemozoin.53
FQ formed a complex with hematin with a stoichiometry of 1 to 1.54 The free energy of association was estimated to be −7 kcal mol−1, leading to the conclusion that this noncovalent interaction is weak but favorable. It was also noted that these values are similar to those previously reported for CQ. Moreover, in the presence of FQ, hematin is no longer converted into β‐hematin and a dose‐dependent inhibition of β‐hematin formation was obtained. The IC50 of FQ was 0.8 equivalents relative to hematin, whereas the IC50 of CQ was 1.9. This clearly shows that FQ is a strong inhibitor of β‐hematin formation, and even more potent than CQ.54
The molecular electrostatic potential (MEP) surfaces have been computed at the DFT‐B3LYP level of theory for diprotonated FQ and CQ. FQ and CQ show considerable similarity in the quinoline area. As this part of the molecule is thought to interact with hematin by a stacking interaction, a similar mode of interaction between these active drugs (FQ or CQ) and hematin was suggested.54
To get a better understanding of the contribution of the ferrocene moiety to the antimalarial activity of FQ, we have also estimated some of its physicochemical properties. The apparent partition coefficients (log D) of CQ and FQ were measured at vacuolar (5.2) and cytosolic (7.4) pHs. At cytosolic pH, FQ was more than 100‐fold more lipophilic than CQ, whereas the difference in lipophilicity is only slight at vacuolar pH. The pK a values of both drugs allow us to speculate that FQ accumulates at a lower concentration than CQ.54 However, the alternative method based only on the logD values led to a contradictory result. The experimental determination (not straightforward) of the accumulation of FQ inside the food vacuole is now urgently needed to solve this problem.
In conclusion, the activity of FQ may be due to more than one route (Figure 9). Its mechanism of action should be in part similar to that of CQ, based on the inhibition effect on β‐hematin formation, and results in inhibition of hemozoin formation. On another hand, redox activation from the ferrocene to the ferricinium and its implication in radical(s) generation cannot be excluded and is currently under investigation. Moreover, the metallocene altered the shape, volume, lipophilicity, basicity, and electronic profile of the parent molecule and consequently, its pharmacodynamic behavior. The strong activity of FQ on CQ‐resistant clones and isolates of P. falciparum suggests a fundamental difference in interaction with resistance mechanisms of the parasite.
Figure 9.

Proposed structure–activity relationships for ferroquine.
Failure to induce resistance
Induction of resistance to FQ by the 2 % method proposed by Peters52 (derived from the four‐day test) was tested on P. yoelii NS (Figure 10).55 From the original clone PyCQS a clone PyCQR was derived by 17 weeks of CQ pressure at 60 mg kg−1 d−1. CQ ED50 was not modified but IC90 increased (8.23 mg kg−1 d−1).
Figure 10.
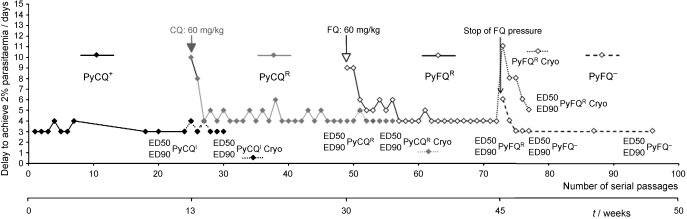
Induction of resistance to FQ tested on P. yoelii NS.55 Delay for the parasite to achieve 2 % blood parasitaemia starting from a 107 parasites infection. PyCQ+: CQ pressure for four days52 followed by three days release. PyCQR: CQ pressure according to the method of 2 % parasitaemia.52 PyFQR: FQ pressure according to the method of 2 % parasitaemia. PyFQ−: release of FQ pressure. Dotted lines, assays done on lines after cryopreservation: only the strain obtained under FQ pressure showed a loss of its resistance to the drug after cryopreservation. ED50 and ED90 indicate the times when resistance to CQ, MF, and FQ were tested on the different strains.
Under continuous CQ pressure, the ED90 remained stable between 8 and 16 mg kg−1 d−1. The resistance was reversed by verapamil. PyCQR sensitivity remained unchanged towards MF and FQ. From the PyCQR line, a PyFQR line was derived under a 23‐week FQ pressure (60 mg kg−1 d−1). The PyFQR line presented a multiresistant phenotype (ED90 >90 mg kg−1 d−1 for CQ, MF, and FQ) and was only partially reversed by verapamil (for CQ). The resistance to FQ and MF was not fixed genetically, because it disappeared in 4–5 serial passages when the FQ pressure was removed. It was also lost during cryoconservation. Growth of the PyFQR line was very slow in mouse, and only reticulocytes were selectively invaded. PCR amplification of DNA fragments did not show the presence in pymdr1 gene of mutations 86, 1034, 1042, and 1246 already associated with resistance phenotypes to various antimalarials in Plasmodium falciparum ortholog gene pfmdr1 and, in the pycrt gene, of mutation K76T involved in CQ resistance in Plasmodium falciparum. 13–17
These results show that if FQ resistance can be obtained in a rodent malaria parasite, 1) the fit cost of the resistance is extremely high, and the growth is so limited and so slow that sometimes the mouse succeeds in clearing its parasites without treatment, 2) the putative mechanism involved in resistance is probably different from that defined by P. falciparum CQ resistance.
The risk of resistance of human Plasmodium species to FQ might be questioned on the basis of results obtained on rodent strains. Studies on field isolates from Cambodia41, 56 showed that the susceptibility of P. falciparum susceptibility to FQ was not related to pfcrt gene phenotype or to the level of expression of the protein.56 Exposing 1010–1011 P. falciparum W2 strains to a continuous 2‐month pressure of 100 nm FQ did not yield a viable resistant clone. A transcient growth was observed, but parasites were unable to be maintained in culture, even in the absence of FQ.56 Complementary experiments carried out under similar experimental conditions in the presence of 50 nm of FQ (a concentration slightly above the in vitro IC90 for W2 strain) failed to select a viable resistant clone. Only a transcient growth was observed, as in the previous experiments (unpublished data).
These observations showed that the biological fit cost of FQ resistance is very high for P. falciparum, and that in the absence of continuous drug pressure, potentially resistant parasites will be easily concurrenced by nonresistant parasites.
Conclusion and Outlook
Ferroquine is a unique metallocene drug candidate which has emerged from a collaborative French discovery project, and it is the most advanced of malaria drug candidates being developed by Sanofi‐Aventis. Ferroquine is extremely active against both CQ‐sensitive and CQ‐resistant P. falciparum. Phase I clinical trials are now completed. As recommended by the WHO, phase II clinical trials will begin with the examination of efficacy of artemisinin‐based combination therapy (ACT) between artesunate and FQ in malaria patients.57
Acknowledgements
C.B. thanks all the organizers of the Joint Meeting “Medicinal Chemistry in Parasitology: New Avenues in Drug Discovery” in Modena in February 2007. The authors also thank C. Roux for proofreading the manuscript.
References
- 1.World Health Organization. The World Health Report 2006, http://www.who.int/en.
- 2. Reed Z. H., Friede M., Kieny M. P., Curr. Mol. Med. 2006, 6, 231–245. [DOI] [PubMed] [Google Scholar]
- 3. Dombrowski K. E., Baldwin W., Sheats J. E., J. Organomet. Chem. 1986, 302, 281–306. [Google Scholar]
- 4. Top S., Tang J., Vessières A., Carrez D., Provot C., Jaouen G., Chem. Commun. 1996, 955–956. [Google Scholar]
- 5.For a full‐length review on bioorganometallic chemistry of ferrocene, see: van Staveren D. R., Metzler‐Nolte N., Chem. Rev. 2004, 104, 5931–5986. [DOI] [PubMed] [Google Scholar]
- 6. Top S., Vessieres A., Cabestaing C., Laios I., Leclercq G., Provot C., Jaouen G., J. Organomet. Chem. 2001, 637–639, 500–506. [Google Scholar]
- 7. Top S., Vessieres A., Leclercq G., Quivy J., Tang J., Vaissermann J., Huche M., Jaouen G., Chem. Eur. J. 2003, 9, 5223–5236. [DOI] [PubMed] [Google Scholar]
- 8. Nesmeyanov A. N., Bogomolova L. G., Viltcheskaya V., Palitsyne N., Andrianova I., Belozerova O., US Patent 119 356, 1971.
- 9. D. J. Sullivan Jr., Matile H., Ridley R. G., Goldberg D. E., J. Biol. Chem. 1998, 273, 31103–31107. [DOI] [PubMed] [Google Scholar]
- 10. Ginsburg H., Ward S. A., Bray P. G., Parasitol. Today 1999, 15, 357–360. [DOI] [PubMed] [Google Scholar]
- 11. Pagola S., Stephens P. W., Bohle D. S., Kosar A. D., Madsen S. K., Nature 2000, 404, 307–310. [DOI] [PubMed] [Google Scholar]
- 12. Hempelmann E., Egan T. J., Tr. Parasitol. 2002, 18, 11. [DOI] [PubMed] [Google Scholar]
- 13. Sanchez C. P., Stein W., Lanzer M., Biochemistry 2003, 42, 9383–9394. [DOI] [PubMed] [Google Scholar]
- 14. Fidock D. A., Nomura T., Talley A. K., Cooper R. A., Dzekunov S. M., Ferdig M. T., Ursos L. M., Sidhu A. B., Naude B., Deitsch K. W., Su X. Z., Wootton J. C., Roepe P. D., Wellems T. E., Mol. Cell 2000, 6, 861–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Djimde A., Doumbo O. K., Cortese J. F., Kayentao K., Doumbo S., Diourte Y., Dicko A., Su X. Z., Nomura T., Fidock D. A., Wellems T. E., Plowe C. V., Coulibaly D., N. Engl. J. Med. 2001, 344, 257–263. [DOI] [PubMed] [Google Scholar]
- 16. Howard E. M., Zhang H., Roepe P. D., J. Membr. Biol. 2002, 190, 1–8. [DOI] [PubMed] [Google Scholar]
- 17. TE T. E. Wellems, Science 2002, 298, 124–126.12364789 [Google Scholar]
- 18. Brocard J., Lebibi J., Maciejewski L., French Patent 9505532, 1995.
- 19. Brocard J., Lebibi J., Maciejewski L., International Patent PCT/FR 96/00721, 1996.
- 20. Biot C., Glorian G., Maciejewski L., Brocard J., Domarle O., Blampain G., Millet P., Georges A. J., Abessolo H., Dive D., Lebibi J., J. Med. Chem. 1997, 40, 3715–3718. [DOI] [PubMed] [Google Scholar]
- 21. Domarle O., Blampain G., Agnaniet H., Nzadiyabi T., Lebibi J., Brocard J., Maciejewski L., Biot C., Georges A. J., Millet P., Antimicrob. Agents Chemother. 1998, 42, 540–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.C. Biot, PhD thesis, University of Lille 1, Villeneuve‐d′Ascq, FRANCE, 1998.
- 23. De D., Krogstad F. M., Cogswell F. B., Krogstad D. J., Am. J. Trop. Med. Hyg. 1996, 55, 579–583. [DOI] [PubMed] [Google Scholar]
- 24. De D., Krogstad F. M., Byers L. D., Krogstad D. J., J. Med. Chem. 1998, 41, 4918–4926. [DOI] [PubMed] [Google Scholar]
- 25. Chibale K., Moss J. R., Blackie M., van Schalkwyk D., Smith P. J., Tetrahedron Lett. 2000, 41, 6231–6235. [Google Scholar]
- 26. Biot C., Daher W., Ndiaye C. M., Melnyk P., Pradines B., Chavain N., Pellet A., Fraisse L., Pelinski L., Jarry C., Brocard J., Khalife J., Forfar‐Bares I., Dive D., J. Med. Chem. 2006, 49, 4707–4714. [DOI] [PubMed] [Google Scholar]
- 27. Beagley P., Blackie M. A. L., Chibale K., Clarkson C., Meijboom R., Moss J. R., Smith P. J., Su H., Dalton Trans. 2003, 3046–3051. [Google Scholar]
- 28. Biot C., Dessolin J., Ricard I., Dive D., J. Organomet. Chem. 2004, 689, 4678–4682. [Google Scholar]
- 29. Paitayatat S., Tarnchompoo B., Thebtaranonth Y., Yuthavong Y., J. Med. Chem. 1997, 40, 633–638. [DOI] [PubMed] [Google Scholar]
- 30. Delhaes L., Biot C., Berry L., Maciejewski L. A., Camus D., Brocard J. S., Dive D., Bioorg. Med. Chem. 2000, 8, 2739–2745. [DOI] [PubMed] [Google Scholar]
- 31. Biot C., Delhaes L., Maciejewski L. A., Mortuaire M., Camus D., Dive D., Brocard J. S., Eur. J. Med. Chem. 2000, 35, 707–714. [DOI] [PubMed] [Google Scholar]
- 32. Baramee A., Coppin A., Mortuaire M., Pelinski L., Tomavo S., Brocard J., Bioorg. Med. Chem. 2006, 14, 1294–1302. [DOI] [PubMed] [Google Scholar]
- 33. Itoh I., Shirakami S., Ishida N., Yamashita Y., Yoshida T., Kim H. S., Wataya Y., Bioorg. Med. Chem. Lett. 2000, 10, 1657–1659. [DOI] [PubMed] [Google Scholar]
- 34. Fayolle M., Ionita M., Krishna S., Morina C., Patelb A. P., Bioorg. Med. Chem. Lett. 2006, 16, 1267–1271. [DOI] [PubMed] [Google Scholar]
- 35. Wu X., Wilairat P., Go M. L., Bioorg. Med. Chem. Lett. 2002, 12, 2299–2302. [DOI] [PubMed] [Google Scholar]
- 36. Wu X., Tiekink E. R., Kostetski I., Kocherginsky N., Tan A. L., Khoo S. B., Wilairat P., Go M. L., Eur. J. Pharm. Sci. 2006, 27, 175–187. [DOI] [PubMed] [Google Scholar]
- 37. Razafimahefa D., Pelinski L., Martin M. T., Ramanitrahasimbola D., Rasoanaivo P., Brocard J., Bioorg. Med. Chem. Lett. 2005, 15, 1239–1241. [DOI] [PubMed] [Google Scholar]
- 38. Biot C., Taramelli D., Forfar‐Bares I., Maciejewski L. A., Boyce M., Nowogrocki G., Brocard J. S., Basilico N., Olliaro P., Egan T. J., Mol. Pharm. 2005, 2, 185–193. [DOI] [PubMed] [Google Scholar]
- 39. Buisine E., de Villiers K., Egan T. J., Biot C., J. Am. Chem. Soc. 2006, 128, 12122–12128. [DOI] [PubMed] [Google Scholar]
- 40. Delhaes L., Biot C., Berry L., Delcourt P., Maciejewski L. A., Camus D., Brocard J. S., Dive D., ChemBioChem 2002, 3, 418–423. [DOI] [PubMed] [Google Scholar]
- 41. Daher W., Pelinski L., Klieber S., Sadoun F., Meunier V., Bourrie M., Biot C., Guillou F., Fabre G., Brocard J., Fraisse L., Maffrand J. P., Khalife J., Dive D., Drug Metab. Dispos. 2006, 34, 667–682. [DOI] [PubMed] [Google Scholar]
- 42. Biot C., Delhaes L., N′Diaye C. M., Maciejewski L. A., Camus D., Dive D., Brocard J. S., Bioorg. Med. Chem. 1999, 7, 2843–2847. [DOI] [PubMed] [Google Scholar]
- 43. Chatterjee T., Muhkopadhyay A., Khan K. A., Giri A. K., Mutagenesis 1998, 13, 619–624. [DOI] [PubMed] [Google Scholar]
- 44. Vincent M. J., Bergeron E., Benjannet S., Erickson B. R., Rollin P. E., Ksiazek T. G., Seidah N. G., Nichol S. T., Virol. J. 2005, 2: 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Biot C., Daher W., Chavain N., Fandeur T., Khalife J., Dive D., De Clercq E., J. Med. Chem. 2006, 49, 2845–2849. [DOI] [PubMed] [Google Scholar]
- 46. Delhaes L., Abessolo H., Biot C., Berry L., Delcourt P., Maciejewski L., Brocard J., Camus D., Dive D., Parasitol. Res. 2001, 87, 239–244. [DOI] [PubMed] [Google Scholar]
- 47. Atteke C., Ndong J. M., Aubouy A., Maciejewski L., Brocard J., Lebibi J., Deloron P., J. Antimicrob. Chemother. 2003, 51, 1021–1024. [DOI] [PubMed] [Google Scholar]
- 48. Pradines B., Fusai T., Daries W., Laloge V., Rogier C., Millet P., Panconi E., Kombila M., Parzy D., J. Antimicrob. Chemother. 2001, 48, 179–184. [DOI] [PubMed] [Google Scholar]
- 49. Pradines B., Tall A., Rogier C., Spiegel A., Mosnier J., Marrama L., Fusai T., Millet P., Panconi E., Trape J. F., Parzy D., Trop. Med. Int. Health 2002, 7, 265–270. [DOI] [PubMed] [Google Scholar]
- 50. Chim P., Lim P., Sem R., Nhem S., Maciejewski L., Fandeur T., Ann. Trop. Med. Parasitol. 2004, 98, 419–424. [DOI] [PubMed] [Google Scholar]
- 51. Kreidenweiss A., Kremsner P. G., Dietz K., Mordmüller B., Am. J. Trop. Med. Hyg. 2006, 75, 1178–1181. [PubMed] [Google Scholar]
- 52. Peters W. in Chemotherapy, and Drug Resistance in Malaria, Vol. 1, Liverpool School of Tropical Medicine, Liverpool, 1987, p. 145–273. [Google Scholar]
- 53. D. J. Sullivan Jr., Gluzman I. Y., Russell D. G., Goldberg D. E., Proc. Natl. Acad. Sci. USA 1996, 93, 11865–11870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Biot C., Taramelli D., Forfar‐Bares I., Maciejewski L. A., Boyce M., Nowogrocki G., Brocard J. S., Basilico N., Olliaro P., Egan T. J., Mol. Pharm. 2005, 2, 185–193. [DOI] [PubMed] [Google Scholar]
- 55.L. Delhaes, PhD thesis, University of Lille 2, Lille, FRANCE, 2000.
- 56. Daher W., Biot C., Fandeur T., Jouin H., Pelinski L., Viscogliosi E., Fraisse L., Pradines B., Brocard J., Khalife J., Dive D., Malar. J. 2006, 5, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Fraisse L., D. Ter‐Namissian International Patent PCT/FR2006/000842, 2006.