

Abstract
The synthesis and antiviral activity of a series of novel polycyclic analogues of the orthopoxvirus egress inhibitor tecovirimat (ST‐246) is presented. Several of these compounds display sub‐micromolar activity against vaccinia virus, and were more potent than cidofovir (CDV). The more active compounds were about 10‐fold more active than CDV, with minimum cytotoxic concentrations above 100 μm. Chemical manipulations of the two carbon–carbon double bonds present in the compounds were carried out to further explore the structure–activity relationships of these new polycyclic imides. Hydrogenation of the two carbon–carbon double bonds decreases antiviral activity, whereas either cyclopropanation or epoxidation of the double bonds fully eliminates the antiviral activity.
Keywords: antiviral agents, drug design, polycycles, vaccinia virus, variola virus
Unorthodox orthopoxvirus inhibitors: A series of novel polycyclic compounds related to the orthopoxvirus egress inhibitor tecovirimat (ST‐246) have been synthesized, several of which display sub‐micromolar activity against vaccinia virus.
Introduction
Poxviruses, double‐stranded DNA viruses that replicate entirely in the cytoplasm, are the largest known animal viruses.1 The most famous members of the poxvirus family are variola virus, the causative agent of smallpox, and vaccinia virus, which shares over 97 % amino acid sequence identity with variola, is used in the variola virus vaccine, and is widely used as a model poxvirus in the laboratory.2
Smallpox is a devastating, highly transmissible, infectious disease with high morbidity and up to 40 % mortality. Following a global, intensive immunization campaign with the vaccinia virus vaccine, the World Health Organization (WHO) declared the worldwide eradication of smallpox in 1980. Three years later, variola virus stocks were either supposedly destroyed or submitted to one of the two WHO‐approved laboratories situated at the US Center for Disease Control and Prevention (CDC) in Atlanta and at the Russian State Research Center of Virology and Biotechnology in Novosibirsk.3
The successful global immunization campaign resulted in a decreased demand for the development of therapies against variola virus because pharmaceutical companies had little interest in developing drugs for a disease that had already been eradicated. However, owing to recent worldwide political developments, variola is nowadays widely regarded as one of the most significant bioterrorist threats.4 In fact, the CDC has placed variola virus at the top of the high‐threat agents list (category A)5 because the impact of a smallpox pandemic in the human population today would be even more catastrophic than during the last century as a result of the vaccination programs being suspended, mainly because the vaccinia virus vaccines have substantial side effects.6 Moreover, there is also a natural public threat arising from the emergence of zoonotic poxvirus infections such as the monkeypox virus, a virus that produces a disease in man that closely resembles smallpox, though less frequently fatal. Monkeypox exists naturally in Africa and several cases are reported in the US every year.7
The bioterrorist threat, the growing concern about zoonotic infections, and the side effects of vaccines have re‐established the need for efficient safe therapies for poxvirus infections.
Recently, several compounds have been identified that inhibit various steps in poxvirus infection, including DNA synthesis and virion morphogenesis.8 The drugs currently recommended for short‐term prophylaxis, adverse vaccination reactions, and emergency treatment against smallpox are cidofovir (CDV) and tecovirimat (ST‐246; Figure 1).
Figure 1.

Structures of CDV and ST‐246.
CDV is an acyclic phosphonate derivative of cytosine that targets viral DNA polymerases. It has a broad‐spectrum antiviral activity, proving to be effective against many poxviruses and herpesviruses. However, the low oral bioavailability of CDV and potential nephrotoxicity associated with its intravenous administration would be an issue in the case of a bioterrorist attack. Prodrugs of CDV have been prepared that significantly enhance its oral bioavailability.9
Tecovirimat, a new, potent, orally active antipoxviral agent recently disclosed by SIGA laboratories, features a polycyclic hydrocarbon moiety and an N‐benzamido substituent.10 It targets the F13L protein of vaccinia virus, a membrane component required for the formation of extracellular viral particles.11 Tecovirimat was shown to inhibit the growth of multiple orthopoxviruses in cell cultures, including two strains of variola virus. In addition, tecovirimat has demonstrated significant antiviral activity in various animal models of the poxvirus disease, including the complete protection of golden‐mantled ground squirrels from lethal doses of monkeypox virus and protection of nonhuman primates from variola virus in lesional disease models.12 The agent demonstrated favorable safety, tolerability, and pharmacokinetics in a double‐blind, randomized, placebo‐controlled phase I ascending‐dose study in healthy human volunteers. These and other results support the use of tecovirimat to prevent smallpox disease in nonvaccinated individuals, as a postexposure therapeutic for use in nonsymptomatic individuals exposed to variola virus, as a treatment for confirmed smallpox infection, and as an adjuvant to vaccination with the smallpox vaccine.13 Recently, it has received both orphan drug designation and fast‐track status from the US Food and Drug Administration (FDA), supporting development of tecovirimat for the prevention and treatment of smallpox infections.14
SIGA has disclosed structure–activity relationships on ST‐246, mainly regarding the effects of the substitution of the aromatic ring. However, few efforts were made with regard to the replacement of the tricyclononene subunit (Figure 2).15
Figure 2.

Polycyclic scaffolds studied by SIGA.
Results and Discussion
For many years, our group has worked on the synthesis of polycyclic hydrocarbon derivatives, mainly from a synthetically oriented point of view.16 We believed that replacement of the tricyclononane subunit of ST‐246 by other polycyclic hydrocarbon structures could lead to new compounds with potential antipoxviral activity.
Dimethyl pentacyclo[6.4.0.02,10.03,7.04,9]dodeca‐5,11‐diene‐8,9‐dicarboxylate (4) is a functionalized polycyclic compound that is easily available from cyclopentadiene and dimethyl acetylenedicarboxylate following a one‐pot procedure.17 For more than 30 years this compound was the starting material for the synthesis of several theoretically interesting polycyclic compounds such as dodecahedrane, a C16‐hexaquinacene, functionalized bisnoradamantane derivatives, and triquinacene derivatives.18
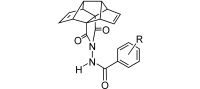
Herein we report the synthesis and potent antiviral activity of several ST‐246 analogues containing a pentacyclo[6.4.0.02,10.03,7.04,9]dodeca‐5,11‐diene moiety. To the best of our knowledge, these are the first biologically active compounds featuring this pentacyclic framework. We also report the synthesis and evaluation of several analogues of ST‐246 containing a tricyclo[3.3.0.03,7]octane (bisnoradamante) skeleton. Note that although ST‐246 and several analogues studied by SIGA had the two carbonyl groups of the imide moiety of the molecule attached to methyne carbon atoms, the compounds herein feature the carbonyl groups attached to quaternary carbon atoms (Figure 3).
Figure 3.

Polycyclic analogues of ST‐246 reported herein. X=CH2, O.
Synthesis of compounds 3 a–j was accomplished in two steps through condensation of an acyl hydrazide with the known bisnoradamantane anhydride (1),19 followed by thermally induced dehydratation of the carboxylic acids 2 a–j to the N‐benzamido imides 3 a–j (Scheme 1).
Scheme 1.

Synthetic route to N‐benzamido imides 3 a–j: a) NH2NHCOR, diisopropylethylamine, EtOH, reflux, 6 h; b) toluene or xylene, reflux, 24 h.
On the other hand, starting from diester 4,17 hydrolysis and dehydratation led to anhydride 5 in very high yield.20 Reaction of 5 with a series of acyl hydrazides in ethanol led to the corresponding mixtures of acids 6 and benzamido imides 7. Heating of these mixtures in toluene or xylene at reflux for 24 h led to the required imides 7 in good overall yields (Scheme 2).
Scheme 2.

Synthetic route to N‐benzamido imides 7: a) KOH, MeOH, H2O, reflux, 5 h; then HCl; b) acetic anhydride, reflux, 1 h, 85 % yield; c) NH2NHCOR, diisopropylethylamine, EtOH, reflux, 6 h; d) toluene or xylene, reflux, 24 h; e) dimethyldioxirane, acetone, RT; f) H2, Pd/C, 1 atm, EtOH.
To explore the structure–activity relationship (SAR) of these new polycyclic imides further we carried out chemical manipulations of the two carbon–carbon double bonds of 7. Reaction of 7 a, 7 k, and 7 s with an excess of dimethyldioxirane in acetone led to the corresponding diepoxides 8 a, 8 k, and 8 s in high yields (Scheme 2). The stereoselectivity of the epoxidation reaction was unequivocally established for 8 s by X‐ray crystallography (Figure 4).21
Figure 4.

X‐ray diffraction structure of 8 s.
Catalytic hydrogenation of 7 a and 7 d led to 9 a and 9 d, respectively, in nearly quantitative yields. The reduction of the two carbon–carbon double bonds of 7 b was accompanied by hydrogenolysis of the C—Br bond, leading to 9 e in quantitative yield. Finally, we explored the double cyclopropanation of the dienes. Because several attempts to achieve the cyclopropanation of 7 a failed, we carried out the cyclopropanation of anhydride 5 that led to anhydride 10. Reaction of 10 with selected acyl hydrazides, followed by thermally induced dehydratation of the carboxylic acids 11, led to N‐benzamido imides 12 a,k,l,m,o,p in good overall yields (Scheme 3). The stereoselectivity of the cyclopropanation reaction was unequivocally established by X‐ray crystallography (Figure 5).21
Scheme 3.

Synthetic route to N‐benzamido imides 12: a) CH2N2, Pd(OAc)2, Et2O, 98 % yield; b) NH2NHCOR, diisopropylethylamine, EtOH, reflux, 6 h; c) xylene, reflux, 24 h.
Figure 5.

X‐ray diffraction structure of anhydride 10.
The structures of all new compounds were confirmed by elemental analysis and/or accurate mass measurement, as well as IR, 1H NMR, 13C NMR, and mass spectral data. Moreover, the structural features of 3 e, 8 s, and 10 were further confirmed by X‐ray crystallography (Figure 6).21 Interestingly, in 3 e and in 8 s, the plane of the benzamido substituent is essentially orthogonal to the plane of the imide group.
Figure 6.

X‐ray diffraction structure of 3 e.
For the compounds of general structure 3, 7, 8, 9, and 12, CPE (cytopathic effect) reduction assays were performed to determine the antiviral activity against a broad panel of DNA and RNA viruses, that is, herpes simplex virus type 1 and type 2, and vaccinia virus (evaluated in infected human embryonic lung fibroblast (HEL) cells); feline coronavirus and feline herpesvirus (in Crandell–Rees Feline Kidney cells); vesicular stomatitis virus, Coxsackie B4 virus and respiratory syncytium virus (tested in HeLa cells); parainfluenza‐3 virus, reovirus‐1, Sindbis virus, and Punta Toro virus (tested in Vero cells); and influenza virus (in Madin–Darby canine kidney cells).22
In studying the SAR of several analogues of ST‐246, Bailey et al. have previously found that electron‐withdrawing substitution on the meta or para position of the N‐benzamido substituent provided the most potent inhibitors of orthopoxvirus.10, 15 We observed the same trend in our bisnoradamantane derivatives, the p‐nitro (3 c) and 4‐pyridyl (3 f) derivatives being the only active compounds against vaccinia virus (Table 1). For these reasons we only synthesized derivatives of 7, 8, 9, and 12 that have electron‐withdrawing substituents on the benzamide ring.
Table 1.
The publisher did not receive permission from the copyright owner to include this object in this version of this product. Please refer either to the publisher's own online version of this product or the printed product where one exists.
The two carbon–carbon double bonds were shown to be essential for good antiviral activity, as neither the epoxides nor the cyclopropanated derivatives were active (data not shown). The hydrogenation also decreased the potency, with reduced compound 9 a being one order of magnitude less potent than the diene 7 a. Interestingly, within the dienes, although most of the derivatives that were para substituted with one electron‐withdrawing group were active against vaccinia virus in the low micromolar order, introduction of a further electron‐withdrawing group at the meta position led to sub‐micromolar activities. The more active compounds, 7 k, 7 l, and 7 m, showed sub‐micromolar EC50 values that were at least 10 times lower than cidofovir. However, if the electron‐withdrawing substituent was only in the meta position, less potent compounds were found (compare 7 a and 7 r).
Very interestingly, most of the compounds showed no cytotoxicity (as determined by microscopy) at the highest concentration tested in HEL cells (100 μm). None of the compounds displayed considerable activity (defined as an antiviral EC50 value of 20 μm or less) against any of the other viruses tested.
Conclusions
We have synthesized a series of N‐benzamido imides and tested them against vaccinia virus. Although most of the bisnoradamantane derivatives were either inactive or active at high micromolar concentrations, most of the pentacyclo[6.4.0.02,10.03,7.04,9]dodeca‐5,11‐diene derivatives showed low micromolar or sub‐micromolar anti‐vaccinia virus activities. The more active compounds were about 10‐fold more active than cidofovir, with minimum cytotoxic concentrations above 100 μm. Hydrogenation of the two carbon–carbon double bonds decreased the antiviral activity, whereas either the cyclopropanation or the epoxidation of the double bonds fully eliminated the antiviral activity.
Experimental Section
Synthesis of 2 a: A mixture of anhydride 1 (103 mg, 0.50 mmol), 4‐trifluoromethylbenzoic acid hydrazide (102 mg, 0.50 mmol), and a drop of diisopropylethylamine in absolute EtOH (2 mL) was heated under reflux for 5 h. Upon cooling to room temperature, H2O (0.2 mL) was added. The precipitate was collected by filtration and washed with cold EtOH (2×2 mL) to give compound 2 a as a white solid (176 mg, 86 % yield); mp: 256–257 °C; 1H NMR (500 MHz, [D6]DMSO): δ=1.15 (s, 6 H, CH3), 1.60 (dd, J=8.0, J′=3.5 Hz, 2 H) and 1.72 (dd, J=8.0, J′=3.5 Hz, 2 H) (2(8)‐Hα and 4(6)‐Hα), 1.80 (d, J=8.0 Hz, 2 H) and 1.87 (d, J=8.0 Hz, 2 H) (2(8)‐Hβ and 4(6)‐Hβ), 7.88 (d, J=8.5 Hz, 2 H, Ar‐3(5)‐H), 8.06 (d, J=8.5 Hz, 2 H, Ar‐2(6)‐H), 9.52 (s, 1 H, NHCO‐C5), 10.53 (s, 1 H, NHCO‐Ar), 11.82 ppm (br s, 1 H, COOH); 13C NMR (100.6 MHz, [D6]DMSO): δ=16.0 (CH3, C3(7)‐CH3), 47.1 (C, C3(7)), 55.7 (CH2) and 56.0 (CH2) (C2(8) and C4(6)), 57.1 (C) and 58.0 (C) (C1 and C5), 123.9 (C, q, J
C−F=272.2 Hz, CF3), 125.4 (CH, q, J
C−F=3.8 Hz, Ar‐C3(5)), 128.4 (CH, Ar‐C2(6)), 131.4 (C, q, J
C−F=31.9 Hz, Ar‐C4), 136.6 (C, Ar‐C1), 164.2 (C, ArCO), 170.9 (C, COOH), 173.6 ppm (C, C5‐CONH); IR (KBr):  =3400–2850 (max. at 3271, 3188, 3086, 3003, 2971, 2891, 2872), 1715, 1678, 1629, 1500, 1479, 1328, 1307, 1168, 1122, 1065, 851, 702, 689 cm−1; MS (EI, 70 eV): m/z (%): 410 (1) [M].+, 393 (2) [M−OH]+, 337 (8), 207 (97) [C11H15O2−CO]+, 189 (22), 173 (100) [CF3−C6H4−CO]+, 161 (53) [C10H13−CO]+, 145 (41), [CF3−C6H4]+, 133 (25), 121 (24); Anal. calcd for C20H21F3N2O4: C 58.53, H 5.16, F 13.89, N 6.83, found: C 58.72, H 5.16, F 13.79, N 6.82.
=3400–2850 (max. at 3271, 3188, 3086, 3003, 2971, 2891, 2872), 1715, 1678, 1629, 1500, 1479, 1328, 1307, 1168, 1122, 1065, 851, 702, 689 cm−1; MS (EI, 70 eV): m/z (%): 410 (1) [M].+, 393 (2) [M−OH]+, 337 (8), 207 (97) [C11H15O2−CO]+, 189 (22), 173 (100) [CF3−C6H4−CO]+, 161 (53) [C10H13−CO]+, 145 (41), [CF3−C6H4]+, 133 (25), 121 (24); Anal. calcd for C20H21F3N2O4: C 58.53, H 5.16, F 13.89, N 6.83, found: C 58.72, H 5.16, F 13.79, N 6.82.
Compounds 2 b–i were prepared in a similar manner to 2 a; full experimental data for these compounds are given in the Supporting Information.
Synthesis of 3 a: A suspension of acid 2 a (155 mg, 0.38 mmol) in xylene (5 mL) was heated under reflux for 24 h in Dean–Stark apparatus. The solution was concentrated in vacuo to give compound 3 a as a white solid (146 mg, 99 % yield). An analytical sample of 3 a was obtained by crystallization from CH2Cl2/pentane; mp: 217–218 °C; 1H NMR (500 MHz, [D6]DMSO): δ=1.22 (s, 6 H, CH3), 1.84–1.89 (complex signal, 4 H, 6(10)‐Hβ and 9(11)‐Hβ), 1.93–1.98 (complex signal, 4 H, 6(10)‐Hα and 9(11)‐Hα), 7.96 (d, J=8.0 Hz, 2 H, Ar‐3(5)‐H), 8.11 (d, J=8.0 Hz, 2 H, Ar‐2(6)‐H), 11.39 ppm (s, 1 H, NH); 13C NMR (100.6 MHz, [D6]DMSO): δ=15.6 (CH3) and 15.7 (CH3) (C7‐CH3 and C8‐CH3), 50.9 (C) and 51.1 (C) (C7 and C8), 53.8 (C, C1(5)), 54.3 (CH2) and 54.5 (CH2) (C6(10) and C9(11)), 123.7 (C, q, J
C−F=272.2 Hz, CF3), 125.8 (CH, q, J
C−F=3.8 Hz, Ar‐C3(5)), 128.6 (CH, Ar‐C2(6)), 132.3 (C, q, J
C−F=32.2 Hz, Ar‐C4), 134.8 (C, Ar‐C1), 163.6 (C, Ar‐CONH), 173.8 ppm (C, C2(4)); IR (KBr):  =3283, 2963, 2901, 2868, 1787, 1726, 1694, 1518, 1496, 1482, 1406, 1381, 1325, 1275, 1169, 1130, 1064, 1015, 855, 816 cm−1; MS (EI, 70 eV): m/z (%): 392 (7) [M].+, 373 (4) [M−F]+, 337 (13), 174 (11), 173 (100) [CF3−C6H4CO]+, 161 (22), 145 (27) [CF3−C6H4]+, 121 (17); Anal. calcd for C20H19F3N2O3: C 61.22, H 4.88, N 7.14, F 14.53, found: C 61.69, H 5.16, N 7.14, F 14.54.
=3283, 2963, 2901, 2868, 1787, 1726, 1694, 1518, 1496, 1482, 1406, 1381, 1325, 1275, 1169, 1130, 1064, 1015, 855, 816 cm−1; MS (EI, 70 eV): m/z (%): 392 (7) [M].+, 373 (4) [M−F]+, 337 (13), 174 (11), 173 (100) [CF3−C6H4CO]+, 161 (22), 145 (27) [CF3−C6H4]+, 121 (17); Anal. calcd for C20H19F3N2O3: C 61.22, H 4.88, N 7.14, F 14.53, found: C 61.69, H 5.16, N 7.14, F 14.54.
Compounds 3 b–i were prepared in a similar manner to 3 a; full experimental data for these compounds are given in the Supporting Information.
Synthesis of 3 j: A mixture of acetohydrazide (62 mg, 0.84 mmol), anhydride 1 (155 mg, 0.75 mmol), and a drop of diisopropylethylamine in absolute EtOH (2 mL) was heated under reflux for 6 h. Upon cooling to room temperature, H2O (0.2 mL) was added. The solution (occasionally a suspension) was stored in the refrigerator overnight, and the precipitate was collected by filtration and washed with cold EtOH (2×2 mL). The solid was suspended in xylene (5 mL) and heated under reflux for 24 h in Dean–Stark apparatus. The solution was concentrated in vacuo to give compound 3 j. An analytical sample of 3 j was obtained by crystallization from CH2Cl2; mp: 236–237 °C; 1H NMR (500 MHz, [D6]DMSO): δ=1.19 (s, 6 H, C7(8)‐CH3), 1.74–1.77 (m, 2 H) and 1.82–1.90 (complex signal, 6 H) (6(10)‐Hα, 6(10)‐Hβ, 9(11)‐Hα and 9(11)‐Hβ), 1.97 (s, 3 H CH3CO), 10.40 ppm (s, 1 H, NH); 13C NMR (100.6 MHz, [D6]DMSO): δ=15.6 (CH3) and 15.7 (CH3) (C7‐CH3 and C8‐CH3), 20.3 (CH3, CH3CO), 50.8 (C) and 51.0 (C) (C7 and C8), 53.6 (C, C1(5)), 54.1 (CH2) and 54.5 (CH2) (C6(10) and C9(11)), 167.7 (C, CH3‐CONH), 173.9 ppm (C, C2(4)); IR (KBr):  =3232, 3187, 2972, 2946, 2890, 2868, 1787, 1739, 1669, 1535, 1382, 1314, 1293, 1275, 1208, 1147, 1099, 1012, 984, 861, 819, 744, 620 cm−1; MS (EI, 70 eV): m/z (%): 262 (1) [M].+, 221 (14), 220 (100) [M−C2H2O].+, 192 (63) [M−C2H2O−CO].+, 165 (25), 161 (44), 134 (25), 133 (43), 121 (34), 91 (45), 77 (35); Anal. calcd for C14H18N2O3: C 64.11, H 6.92, N 10.68, found: C 64.05, H 6.91, N 10.44.
=3232, 3187, 2972, 2946, 2890, 2868, 1787, 1739, 1669, 1535, 1382, 1314, 1293, 1275, 1208, 1147, 1099, 1012, 984, 861, 819, 744, 620 cm−1; MS (EI, 70 eV): m/z (%): 262 (1) [M].+, 221 (14), 220 (100) [M−C2H2O].+, 192 (63) [M−C2H2O−CO].+, 165 (25), 161 (44), 134 (25), 133 (43), 121 (34), 91 (45), 77 (35); Anal. calcd for C14H18N2O3: C 64.11, H 6.92, N 10.68, found: C 64.05, H 6.91, N 10.44.
Synthesis of 7 a: This compound was obtained in a similar manner to that described before for compound 3 j. Starting from anhydride 5 (200 mg, 0.88 mmol) and 4‐(trifluoromethyl)benzoic acid hydrazide (180 mg, 0.88 mmol), compound 7 a was isolated as a white solid (285 mg, 78 % yield). An analytical sample of 7 a was obtained by crystallization from CH2Cl2/pentane; mp: 268–269 °C; 1H NMR (500 MHz, [D6]DMSO): δ=2.85 (br s, 1 H) and 2.86 (br s, 1 H) (10‐H and 11‐H), 3.56 (br s, 2 H) and 3.57 (br s, 2 H) (6(9)‐H and 12(15)‐H), 6.06 (s, 2 H) and 6.10 (s, 2 H) (7(8)‐H and 13(14)‐H), 7.90 (d, J=8.0 Hz, 2 H, Ar‐3(5)‐H), 8.06 (d, J=8.0 Hz, 2 H, Ar‐2(6)‐H), 11.31 ppm (s, 1 H, NH); 13C NMR (100.6 MHz, [D6]DMSO): δ=62.1 (CH, C6(9) and C12(15)), 63.7 (CH) and 64.2 (CH) (C10 and C11), 64.9 (C, C1(5)), 123.9 (C, q, J
C−F=272.7 Hz, CF3), 125.9 (CH, q, J
C−F=3.8 Hz, Ar‐C3(5)), 128.8 (CH, Ar‐C2(6)), 131.9 (C, overlapped q, Ar‐C4), 132.2 (CH) and 132.5 (CH) (C7(8) and C13(14)), 134.8 (C, Ar‐C1), 162.8 (C, CONH), 171.9 ppm (C, C2(4)); IR (KBr):  =3312, 3061, 2982, 1722, 1704, 1480, 1407, 1328, 1270, 1244, 1163, 1134, 1117, 1067, 1017, 854, 822, 774, 702 cm−1; MS (EI, 70 eV): m/z (%): 412 (2) [M].+, 393 (4) [M−F]+, 348 (19), 347 (100) [M−C5H5].+, 173 (80) [CF3−C6H4−CO]+, 154 (13), 153 (26), 152 (20), 145 (35) [CF3−C6H4]+; Anal. calcd for C22H15F3N2O3⋅0.5H2O: C 62.71, H 3.83, N 6.65, F 13.53, found: C 62.61, H 3.98, N 6.55, F 13.48.
=3312, 3061, 2982, 1722, 1704, 1480, 1407, 1328, 1270, 1244, 1163, 1134, 1117, 1067, 1017, 854, 822, 774, 702 cm−1; MS (EI, 70 eV): m/z (%): 412 (2) [M].+, 393 (4) [M−F]+, 348 (19), 347 (100) [M−C5H5].+, 173 (80) [CF3−C6H4−CO]+, 154 (13), 153 (26), 152 (20), 145 (35) [CF3−C6H4]+; Anal. calcd for C22H15F3N2O3⋅0.5H2O: C 62.71, H 3.83, N 6.65, F 13.53, found: C 62.61, H 3.98, N 6.55, F 13.48.
Like compound 7 a, derivatives 7 b–d, f, k–u were prepared in a similar manner to 3 j; full experimental data for these compounds are given in the Supporting Information.
Synthesis of 8 a: An excess of a solution of dimethyldioxirane (63 mg, 3.8 mmol) in acetone (1.5 mL) was added to solid compound 7 a (70 mg, 0.17 mmol) and the solution was stirred overnight at room temperature. The solvent was evaporated in vacuo to give compound 8 a as a white solid (75 mg, 99 % yield). An analytical sample of 8 a was obtained by crystallization from acetone. Mp:>290 °C (dec.); 1H NMR (500 MHz, [D6]DMSO): δ=2.14 (br s, 2 H, 11‐H and 12‐H), 3.31 (br s, 4 H, 6(10)‐H and 13(17)‐H), 3.36 (br s, 2 H) and 3.43 (br s, 2 H) (7(9)‐H and 14(16)‐H), 7.95 (d, J=8.3 Hz, 2 H, Ar‐3(5)‐H), 8.11 (d, J=8.3 Hz, 2 H, Ar‐2(6)‐H), 11.62 ppm (s, 1 H, NH); 13C NMR (100.6 MHz, [D6]DMSO): δ=38.4 (CH) and 39.0 (CH) (C11 and C12), 47.3 (CH) and 47.7 (CH) (C7(9) and C14(16)), 54.8 (CH) and 55.1 (CH) (C6(10) and C13(17)), 59.2 (C, C1(5)), 123.7 (C, q, J
C−F=272.8 Hz, CF3), 125.9 (CH, q, J
C−F=3.8 Hz, Ar‐C3(5)), 128.8 (CH, Ar‐C2(6)), 132.5 (C, q, J
C−F=31.8 Hz, Ar‐C4), 134.3 (C, Ar‐C1), 163.4 (C, CONH), 171.0 ppm (C, C2(4)); IR (KBr):  =3248, 3047, 3018, 2985, 1792, 1741, 1703, 1521, 1497, 1407, 1398, 1326, 1305, 1268, 1247, 1164, 1133, 1115, 1100, 1065, 852, 845, 823, 695 cm−1; MS (EI, 70 eV): m/z (%): 444 (4) [M].+, 393 (4) [M−F]+, 173 (100) [CF3−C6H4−CO]+, 145 (29) [CF3−C6H4]+, 81 (20); HRMS (ESI+): m/z [M+H]+ calcd for C22H15F3N2O5+H: 445.1006, found: 445.1007; Anal. calcd for C22H15F3N2O5⋅0.25acetone: C 59.55, H 3.62, N 6.10, F 12.42, found: C 59.15, H 3.67, N 5.88, F 12.15.
=3248, 3047, 3018, 2985, 1792, 1741, 1703, 1521, 1497, 1407, 1398, 1326, 1305, 1268, 1247, 1164, 1133, 1115, 1100, 1065, 852, 845, 823, 695 cm−1; MS (EI, 70 eV): m/z (%): 444 (4) [M].+, 393 (4) [M−F]+, 173 (100) [CF3−C6H4−CO]+, 145 (29) [CF3−C6H4]+, 81 (20); HRMS (ESI+): m/z [M+H]+ calcd for C22H15F3N2O5+H: 445.1006, found: 445.1007; Anal. calcd for C22H15F3N2O5⋅0.25acetone: C 59.55, H 3.62, N 6.10, F 12.42, found: C 59.15, H 3.67, N 5.88, F 12.15.
Compounds 8 k and 8 s were prepared in a similar manner to 8 a; full experimental data for these compounds are given in the Supporting Information.
Synthesis of 9 a: A mixture of benzamide 7 a (142 mg, 0.34 mmol) and Pd/C (5 %, 8 mg) in EtOH (20 mL) was hydrogenated at 1 atm for 18 h. The suspension was filtered and the filtrate was concentrated to dryness in vacuo to give compound 9 a (137 mg, 96 % yield). An analytical sample of 9 a was obtained by crystallization from MeOH; mp: 254–255 °C; 1H NMR (500 MHz, [D6]DMSO): δ=1.40–1.44 (m, 2 H, 13(14)‐Hendo), 1.59–1.67 (complex signal, 6 H, 7(8)‐Hexo, 7(8)‐Hendo and 13(14)‐Hexo), 2.60 (br s, 2 H, 10‐H and 11‐H), 2.72 (br s, 4 H, 6(9)‐H and 12(15)‐H), 7.94 (d, J=8.0 Hz, 2 H, Ar‐3(5)‐H), 8.13 (d, J=8.0 Hz, 2 H, Ar‐2(6)‐H), 11.47 ppm (br s, 1 H, NH); 13C NMR (100.6 MHz, [D6]DMSO): δ=22.5 (CH2) and 22.7 (CH2) (C7(8) and C13(14)), 50.7 (CH) and 51.0 (CH) (C10 and C11), 55.0 (CH) and 55.4 (CH) (C6(9) and C12(15)), 59.8 (C, C1(5)), 123.6 (C, q, J
C−F=272.3 Hz, CF3), 126.0 (CH, q, J
CF=3.8 Hz, Ar‐C3(5)), 128.8 (CH, Ar‐C2(6)), 132.4 (C, q, J
C−F=32.2 Hz, Ar‐C4), 135.0 (C, Ar‐C1), 163.6 (C, CONH), 173.6 ppm (C, C2(4)); IR (KBr):  =3325, 2959, 2875, 1781, 1720, 1700, 1521, 1476, 1329, 1287, 1270, 1163, 1143, 1120, 1093, 1068, 849, 695 cm−1; MS (EI, 70 eV): m/z (%): 416 (30) [M].+, 173 (100) [CF3−C6H4−CO]+, 145 (19) [CF3−C6H4]+; Anal. calcd for C22H19F3N2O3⋅0.25H2O: C 62.78, H 4.67, N 6.66, F 13.54, found: C 62.85, H 4.88, N 6.61, F 13.64.
=3325, 2959, 2875, 1781, 1720, 1700, 1521, 1476, 1329, 1287, 1270, 1163, 1143, 1120, 1093, 1068, 849, 695 cm−1; MS (EI, 70 eV): m/z (%): 416 (30) [M].+, 173 (100) [CF3−C6H4−CO]+, 145 (19) [CF3−C6H4]+; Anal. calcd for C22H19F3N2O3⋅0.25H2O: C 62.78, H 4.67, N 6.66, F 13.54, found: C 62.85, H 4.88, N 6.61, F 13.64.
Compounds 9 d and 9 e were prepared in a similar manner to 9 a; full experimental data for these compounds are given in the Supporting Information.
Synthesis of 10: Excess of an ethereal solution of diazomethane (100 mL) was added to a mixture of anhydride 5 (200 mg, 0.88 mmol) and Pd(OAc)2 (5 mg, 0.02 mmol) and the suspension was stirred overnight at room temperature. The mixture was filtered and the filtrate was dried with anhydrous Na2SO4 and concentrated in vacuo to give a yellow solid, which was subjected to silica‐gel column chromatography (hexane/EtOAc mixtures). Upon elution with hexane/EtOAc 95:5, anhydride 10 (220 mg, 98 % yield) was obtained as a white solid. An analytical sample of 10 was obtained by crystallization from a mixture CH2Cl2/pentane; mp: 180 °C (sublimes); 1H NMR (500 MHz, CDCl3): δ=0.21 (dt, J=5.6, J′=7.3 Hz, 2 H, 8(15)‐Hanti), 0.30 (dt, J=5.6 Hz, J′=3.3 Hz, 2 H, 8(15)‐Hsyn), 1.19 (dd, J=7.3 Hz, J′=3.3 Hz, 4 H, 7(9,14,16)‐H), 1.99–2.01 (m, 2 H, 11(12)‐H), 2.92 ppm (dd, J=1.8, J′=0.8 Hz, 4 H, 6(10,13,17)‐H); 13C NMR (100.6 MHz, CDCl3): δ=2.1 (CH2, C8(15)), 9.6 (CH, C7(9,14,16)), 39.7 (CH, C11(12)), 56.6 (CH, C6(10,13,17)), 66.3 (C, C1(5)), 170.7 ppm (C, C2(4); IR (KBr):  =3024, 2965, 1839, 1778, 1265, 1203, 1068, 1030, 911, 849, 816, 755, 679 cm−1; MS (EI, 70 eV): m/z (%): 254 (16) [M].+, 210 (54) [M−CO2].+, 182 (29) [M−C2O3].+, 167 (100), 166 (42), 165 (80), 153 (36), 152 (43), 141 (42), 128 (52), 115 (46), 104 (21), 103 (23), 91 (31) 79 (60); Anal. calcd for C16H14O3: C 75.57, H 5.55, found: C 75.49, H 5.56.
=3024, 2965, 1839, 1778, 1265, 1203, 1068, 1030, 911, 849, 816, 755, 679 cm−1; MS (EI, 70 eV): m/z (%): 254 (16) [M].+, 210 (54) [M−CO2].+, 182 (29) [M−C2O3].+, 167 (100), 166 (42), 165 (80), 153 (36), 152 (43), 141 (42), 128 (52), 115 (46), 104 (21), 103 (23), 91 (31) 79 (60); Anal. calcd for C16H14O3: C 75.57, H 5.55, found: C 75.49, H 5.56.
Synthesis of 12 a: This compound was obtained in a similar manner to that described before for compound 3 j. Starting from anhydride 10 (200 mg, 0.79 mmol) and 4‐(trifluoromethyl)benzoic acid hydrazide (162 mg, 0.79 mmol), compound 12 a was isolated as a white solid (182 mg, 52 % yield). An analytical sample of 12 a was obtained by crystallization from a mixture CH2Cl2/pentane; mp: 258–259 °C; 1H NMR (500 MHz, [D6]DMSO): δ=0.13–0.20 (m, 2 H, 8‐Hanti and 15‐Hanti), 0.31–0.37 (m, 2 H, 8‐Hsyn and 15‐Hsyn), 0.96 (dd, J=7.1, J′=3.0 Hz, 2 H) and 1.04 (dd, J=7.1, J′=3.2 Hz, 2 H) (7(9)‐H and 14(16)‐H), 2.02–2.06 (m, 2 H, 11‐H and 12‐H), 2.86 (br s, 4 H, 6(10)‐H and 13(17)‐H), 7.94 (d, J=8.2 Hz, 2 H, Ar‐3(5)‐H), 8.12 (d, J=8.2 Hz, 2 H, Ar‐2(6)‐H), 11.43 ppm (s, 1 H, NH); 13C NMR (125.7 MHz, [D6]DMSO): δ=1.8 (CH2) and 2.0 (CH2) (C8 and C15), 9.5 (CH) and 9.6 (CH) (C7(9) and C14(16)), 38.3 (CH) and 38.6 (CH) (C11 and C12), 54.4 (CH) and 54.7 (CH) (C6(10) and C13(17)), 62.5 (C, C1(5)), 123.7 (C, q, J
C−F=272.8 Hz, CF3), 125.8 (CH, q, J
C−F=3.9 Hz, Ar‐C3(5)), 128.6 (CH, Ar‐C2(6)), 132.2 (C, q, J
C−F=32.3 Hz, Ar‐C4), 134.7 (C, Ar‐C1), 163.2 (C, CONH), 172.9 ppm (C, C2(4)); IR (KBr):  =3351, 3079, 3012, 2965, 1780, 1718, 1694, 1473, 1332, 1291, 1265, 1167, 1143, 1128, 1116, 1096, 1067, 854, 817 cm−1; MS (EI, 70 eV): m/z (%): 440 (4) [M].
+, 421 (2) [M−F]+, 267 (4) [M−(CF3−C6H4−CO)]+, 173 (100) [CF3−C6H4−CO]+, 145 (22) [CF3−C6H4]+; Anal. calcd for C24H19F3N2O3: C 65.45, H 4.35, N 6.36, F 12.94, found: C 65.55, H 4.59, N 6.20, F 12.76.
=3351, 3079, 3012, 2965, 1780, 1718, 1694, 1473, 1332, 1291, 1265, 1167, 1143, 1128, 1116, 1096, 1067, 854, 817 cm−1; MS (EI, 70 eV): m/z (%): 440 (4) [M].
+, 421 (2) [M−F]+, 267 (4) [M−(CF3−C6H4−CO)]+, 173 (100) [CF3−C6H4−CO]+, 145 (22) [CF3−C6H4]+; Anal. calcd for C24H19F3N2O3: C 65.45, H 4.35, N 6.36, F 12.94, found: C 65.55, H 4.59, N 6.20, F 12.76.
Like compound 12 a, derivatives 12 k–m, o–p were prepared in a similar manner to 3 j; full experimental data for these compounds are given in the Supporting Information.
Supporting information
Detailed facts of importance to specialist readers are published as ”Supporting Information”. Such documents are peer‐reviewed, but not copy‐edited or typeset. They are made available as submitted by the authors.
miscellaneous_information
Acknowledgements
P.C. and S.V. acknowledge financial support from the Ministerio de Educación y Ciencia (Project CTQ2008‐03768). M.D. and E.T. thank the Ministerio de Educación y Ciencia (Beca FPU) and Generalitat de Catalunya (Beca FI), respectively. L.N. acknowledges receipt of a grant from the National Institutes of Health (NIAID/NIH Grant AI 062540‐01) as well as technical assistance from Leentje Persoons.
References
- 1. Moss B. in Fields Virology, 5th ed. (Eds.: D. M. Knipe, P. M. Howley), Lippincott Williams & Wilkins, Philadelphia, 2007, pp. 2905–2946. [Google Scholar]
- 2. Massung R. F., Liu L. I., Qi J., Knight J. C., Yuran T. E., Kerlavage A. R., Parsons J. M., Venter J. C., Esposito J. J., Virology 1994, 201, 215–240. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Bray M., Buller M., Clin. Infect. Dis. 2004, 38, 882–889; [DOI] [PubMed] [Google Scholar]
- 3b. Henderson D. A., Smallpox: The Death of a Disease, Prometheus Books, 2009. [Google Scholar]
- 4.
- 4a. Whitley R. J., Antiviral Res. 2003, 57, 7–12; [DOI] [PubMed] [Google Scholar]
- 4b. Bolken T. C., Hruby D. E., Antiviral Res. 2008, 77, 1–5. In fact, there appears to be precedent for use of the variola virus as a biological weapon during the French and Indian wars (1754–1765), see: 17765333 [Google Scholar]
- 4c. Stearn E. W., Stearn A. E., The Effect of Smallpox on the Destiny of the Amerindian, Bruce Humphries, Boston, 1945; [Google Scholar]
- 4d. Henderson D. A., Inglesby T. V., Bartlett J. G., Ascher M. S., Eitzen E., Jahrling P. B., Hauer J., Layton M., McDade J., Osterholm M. T., O'Toole T., Parker G., Perl T., Russell P. K., Tonat K., JAMA J. Am. Med. Assoc. 1999, 281, 2127–2137. [DOI] [PubMed] [Google Scholar]
- 5.Centers for Disease Control and Prevention, “Bioterrorism Agents/Diseases” to be found under http://www.bt.cdc.gov/agent/agentlist‐category. asp (accessed September 27, 2010).
- 6.
- 6a. Lane J. M., Ruben F. L., Abrutyn E., Millar J. D., JAMA J. Am. Med. Assoc. 1970, 212, 441–444; [PubMed] [Google Scholar]
- 6b. Baggs J., Chen R. T., Damon I. K., Rotz L., Allen C., Fullerton K. E., Casey C., Nordenberg D., Mootrey G., Clin. Infect. Dis. 2005, 40, 1133–1140; [DOI] [PubMed] [Google Scholar]
- 6c. Schwartz B., Lebwohl M., Int. J. Dermatol. 2005, 44, 289–292; [DOI] [PubMed] [Google Scholar]
- 6d. Reif D. M., McKinney B. A., Motsinger A. A., Chanock S. J., Edwards K. M., Rock M. T., Moore J. H., J. E. Crowe Jr., J. Infect. Dis. 2008, 198, 16–22; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6e. Jacobs B. L., Langland J. O., Kibler K. V., Denzler K. L., White S. D., Holechek S. A., Wong S., Huynh T., Baskin C. R., Antiviral Res. 2009, 84, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.
- 7a. Huhn G. D., Bauer A. M., Yorita K., Graham M. B., Sejvar J., Likos A., Damon I. K., Reynolds M. G., Kuehnert M. J., Clin. Infect. Dis. 2005, 41, 1742–1751; [DOI] [PubMed] [Google Scholar]
- 7b. Stittelaar K. J., Neyts J., Naesens L., van Amerongen G., van Lavieren R. F., Holý A., De Clercq E., Niesters H. G. M., Fries E., Maas C., Mulder P. G. H., van der Zeijst B. A. M., Osterhaus A. D. M. E., Nature 2006, 439, 745–748; [DOI] [PubMed] [Google Scholar]
- 7c. Parker S., Nuara A., Buller R. M. L., Schultz D. A., Future Microbiol. 2007, 2, 17–34; [DOI] [PubMed] [Google Scholar]
- 7d. Chastel C., Pathol. Biol. 2009, 57, 175–183; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7e. Bray M., Am. J. Trop. Med. Hyg. 2009, 80, 499–500; [PubMed] [Google Scholar]
- 7f. Stabenow J., Buller R. M., Schriewer J., West C., Sagartz J. E., Parker S., J. Virol. 2010, 84, 3909–3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.
- 8a. Harrison S. C., Alberts B., Ehrenfeld E., Enquist L., Fineberg H., McKnight S. L., Moss B., O'Donnell M., Ploegh H., Schmid S. L., Walter K. P., Theriot J., Proc. Natl. Acad. Sci. USA 2004, 101, 11178–11192; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8b. Roy A., Schneller S. W., Keith K. A., Hartline C. B., Kern E. R., Bioorg. Med. Chem. 2005, 13, 4443–4449; [DOI] [PubMed] [Google Scholar]
- 8c. Clercq E. De in Antiviral Drug Discovery for Emerging Diseases and Bioterrorism Threats (Ed. P. F. Torrence), John Wiley & Sons, 2005, pp. 83–113; [Google Scholar]
- 8d. Kern E. R. in Antiviral Drug Discovery for Emerging Diseases and Bioterrorism Threats (Ed.: P. F. Torrence), John Wiley & Sons, 2005, pp. 331–351; [Google Scholar]
- 8e. El Omari K., Stammers D. K., Expert Opin. Drug Discovery 2007, 2, 1263–1272; [DOI] [PubMed] [Google Scholar]
- 8f. Smee D. F., Antiviral Chem. Chemother. 2008, 19, 115–124; [DOI] [PubMed] [Google Scholar]
- 8g. Altmann S. E., Jones J. C., Schultz‐Cherry S., Brandt C. R., Virology 2009, 388, 248–259; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8h. Kern E. R., Prichard M. N., Quenelle D. C., Keith K. A., Tiwari K. N., Maddry J. A., J. A. Secrist III , Antimicrob. Agents Chemother. 2009, 53, 572–579; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8i. De Clercq E., Viruses 2010, 2, 1322–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.
- 9a. De Clercq E., Med. Res. Rev. 2008, 28, 929–953; [DOI] [PubMed] [Google Scholar]
- 9b. Hostetler K. Y., Antiviral Res. 2009, 82, A84–A98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bailey T. R., Rippin S. R., Opsitnick E., Burns C. J., Pevear D. C., Collett M. S., Rhodes G., Tohan S., Huggins J. W., Baker R. O., Kern E. R., Keith K. A., Dai D., Yang G., Hruby D., Jordan R., J. Med. Chem. 2007, 50, 1442–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Duraffour S., Vigne S., Vermeire K., Garcel A., Vanstreels E., Daelemans D., Yang G., Jordan R., Hruby D. E., Crance J.‐M., Garin D., Andrei G., Snoeck R., Antiviral Ther. 2008, 13, 977–990. [PubMed] [Google Scholar]
- 12.
- 12a. Duraffour S., Snoeck R., de Vos R., van Den Oord J. J., Crance J.‐M., Garin D., Hruby D. E., Jordan R., De Clercq E., Andrei G., Antiviral Ther. 2007, 12, 1205–1216; [PubMed] [Google Scholar]
- 12b. Smith S. K., Olson V. A., Karem K. L., Jordan R., Hruby D. E., Damon I. K., Antimicrob. Agents Chemother. 2009, 53, 1007–1012; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12c. Jordan R., Goff A., Frimm A., Corrado M. L., Hensley L. E., Byrd C. M., Mucker E., Shamblin J., Bolken T. C., Wlazlowski C., Johnson W., Chapman J., Twenhafel N., Tyavanagimatt S., Amantana A., Chinsangaram J., Hruby D. E., Huggins J., Antimicrob. Agents Chemother. 2009, 53, 1817–1822; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12d. Huggins J., Goff A., Hensley L., Mucker E., Shamblin J., Wlazlowski C., Johnson W., Chapman J., Larsen T., Twenhafel N., Karem K., Damon I. K., Byrd C. M., Bolken T. C., Jordan R., Hruby D. E., Antimicrob. Agents Chemother. 2009, 53, 2620–2625; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12e. Grosenbach D. W., Berhanu A., King D. S., Mosier S., Jones K. F., Jordan R. A., Bolken T. C., Hruby D. E., Proc. Natl. Acad. Sci. USA 2010, 107, 838–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.
- 13a. Kaiser J., Science 2007, 316, 1418–1419; [DOI] [PubMed] [Google Scholar]
- 13b. Sbrana E., Jordan R., Hruby D. E., Mateo R. I., Xiao S.‐Y., Siirin M., Newman P. C., Da Rosa A. P. A. Travassos, Tesh R. B., Am. J. Trop. Med. Hyg. 2007, 76, 768–773; [PubMed] [Google Scholar]
- 13c. Quenelle D. C., Prichard M. N., Keith K. A., Hruby D. E., Jordan R., Painter G. R., Robertson A., Kern E. R., Antimicrob. Agents Chemother. 2007, 51, 4118–4124; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13d. Nalca A., Hatkin J. M., Garza N. L., Nichols D. K., Norris S. W., Hruby D. E., Jordan R., Antiviral Res. 2008, 79, 121–127; [DOI] [PubMed] [Google Scholar]
- 13e. Jordan R., Tien D., Bolken T. C., Jones K. F., Tyavanagimatt S. R., Strasser J., Frimm A., Corrado M. L., Strome P. G., Hruby D. E., Antimicrob. Agents Chemother. 2008, 52, 1721–1727; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13f. Grosenbach D. W., Jordan R., King D. S., Berhanu A., Warren T. K., Kirkwood‐Watts D. L., Tyavanagimatt S., Tan Y., Wilson R. L., Jones K. F., Hruby D. E., Vaccine 2008, 26, 933–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.To be found under http://www.siga.com (accessed September 27, 2010).
- 15.
- 15a. Jordan R., Bailey T. R., Rippin S. R., Dai D. (SIGA Technologies, Inc WO 2008/130348, 2008;
- 15b. Jordan R., Bailey T. R., Rippin S. R. (Viropharma Inc.), WO 2004/112718, 2004.
- 16.See, for example:
- 16a. Camps P., Font‐Bardia M., Pérez F., Solans X., Vázquez S., Angew. Chem. 1995, 107, 1011–1012; [Google Scholar]; Angew. Chem. Int. Ed. Engl. 1995, 34, 912–914; [Google Scholar]
- 16b. Camps P., Fernández J. A., Vázquez S., Font‐Bardia M., Solans X., Angew. Chem. 2003, 115, 4183–4185; [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2003, 42, 4049–4051; [DOI] [PubMed] [Google Scholar]
- 16c. Ayats C., Camps P., Fernández J. A., Vázquez S., Chem. Eur. J. 2007, 13, 1522–1532. [DOI] [PubMed] [Google Scholar]
- 17.
- 17a. Paquette L. A., Wyvratt M. J., J. Am. Chem. Soc. 1974, 96, 4671–4673; [Google Scholar]
- 17b. McNeil D., Vogt B. R., Sudol J. J., Theodoropulos S., Hedaya E., J. Am. Chem. Soc. 1974, 96, 4673–4674; [Google Scholar]
- 17c. Taylor R. J., Pelter M. W., Paquette L. A., Org. Synth. Coll. Vol. VIII 1993, 298–302. [Google Scholar]
- 18.
- 18a. Paquette L. A., Chem. Rev. 1989, 89, 1051–1065; [Google Scholar]
- 18b. Park H., Paquette L. A., J. Org. Chem. 1980, 45, 5378–5379; [Google Scholar]
- 18c. Camps P., Coll O., Pérez F., Vázquez S., Synthesis 1997, 668–672. [Google Scholar]
- 19. Camps P., Lukach A. E., Rossi R. A., J. Org. Chem. 2001, 66, 5366–5373. [DOI] [PubMed] [Google Scholar]
- 20. Camps P., Pujol X., Vázquez S., Org. Lett. 2000, 2, 4225–4228. [DOI] [PubMed] [Google Scholar]
- 21.CCDC 752677 (3 e), 783974 (8 s), and 783975 (10) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/cgi-bin/catreq.cgi.
- 22. Derudas M., Brancale A., Naesens L., Neyts J., Balzarini J., McGuigan C., Bioorg. Med. Chem. 2010, 18, 2748–2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Detailed facts of importance to specialist readers are published as ”Supporting Information”. Such documents are peer‐reviewed, but not copy‐edited or typeset. They are made available as submitted by the authors.
miscellaneous_information