

Abstract
Introduction
Asthma is driven by an inflammatory response against normally harmless environmental inorganic and organic compounds in the respiratory tract. Immune responses to airborne pathogens such as viruses and bacteria may reduce the allergic responses but are also known to trigger asthma attacks and eventually lead to severe disease condition.
Objective
To investigate the role of respiratory pathogens concerning the induction or protection against acute or chronic asthma manifestations.
Methods
We included 131 articles for the final review according to their relevance with the subject.
Results
There is apparently contradictory interaction of respiratory germs in the airways of asthmatics which may be protective on one angle but deleterious on the other.
Conclusion
The relationship between inflammation and remodeling and the pathogenic role of viral and bacterial infection in the airways of asthmatic patients is still highly debatable and incompletely understood.
Keywords: asthma, asthma control, bacteria, infection, virus
Introduction
The definition of asthma is based on the functional disparities and the underlying inflammation:
Asthma is a chronic inflammatory disorder of the airways in which many cells and cellular elements play a role. The chronic inflammation is associated with airway hyperresponsiveness that leads to recurrent episodes of wheezing, breathlessness, chest tightness and coughing, particularly at night or in the early morning. These episodes are usually associated with widespread, but variable, airflow obstruction with the lung that is often reversible either spontaneously or with treatment 1. The asthma nomenclature as well as the definition of disease severity and management goals has changed substantially in recent years. It has moved away from a system predominantly based on lung function and symptoms towards one based on therapeutic methods and doses required to achieve disease control.
Airway inflammation is the central driver of the chronic intermittent nature of asthma symptoms eventually ending in severe asthma attacks 2, 3. The inflammation affects all airways from the upper respiratory tract up to the small airways although its physiological effects are believed to be most pronounced in medium‐sized bronchi. If not prevented or therapeutically inhibited, inflammatory processes may lead to a deterioration of the disease. Asthma attacks are potentially life threatening. Acute worsening has many grounds like insufficient treatment, exercise, allergic triggers, cold air but also infectious microorganisms.
This review provides an overview of research published on the cellular basis of this disease, assesses the role of virus and bacteria in acute and chronic worsening.
Inflammation of the airways
Asthma is based on an aberrant immune response to non‐pathogenic stimuli in the airways leading to a chronic inflammatory response relevant to the pathogenesis of the disease. The inflammation affects all compartments of the airways including the upper respiratory tract and the nose. The major physiological effect comprises the medium‐sized bronchi and the small airways 4. Although the spectrum of clinical symptoms varies widely and typically occur episodically, the underlying chronic inflammation is always present even in the absence of continuous allergen exposure or in a period of relatively minor symptoms. The type of cells and the cellular components involved in the inflammatory processes appear to be comparable regardless of the asthma phenotype, the type of allergy or non‐immunologic triggers like exercise or smoke or age. The complex interaction between the multi‐cellular inflammatory infiltrate and parenchymal pulmonary cells is organised by a complex network of interacting bioactive mediators 5, 6, 7.
Mediators
Numerous cytokines, antibodies and growth factors stimulate or inhibit the immune response, and there is now increasing evidence that the deregulation of endogenous immune regulation processes are at least in part responsible for the manifestation and aggravation of this disease 8. Over 100 mediators have been identified to be involved, among those are:
Chemokines which recruit inflammatory cells like eosinophils or Th2 cells from the blood vessels into the airways. Examples are eotaxin, thymus and activation‐regulated chemokines (TARC) or macrophage‐derived chemokines (MDC) 9.
Cysteinyl leukotrienes which are potent bronchoconstrictors and having proinflammtory potency. They derive from mast cells and eosinophils 10.
Cytokines orchestrate the inflammatory response. Interleukins (IL) like IL‐1ß or tumour necrosis factor alpha (TNF‐α) amplify the inflammation, whereas granulocyte macrophage – colony stimulation factor (GM‐CSF) prolongs eosinophil survival. IL‐5 mediates eosinophil differentiation and survival, and IL‐4 is important for Th2 cell differentiation. IL‐13 is required for IgE (immunoglobulin E) formation 11.
Histamine is released from mast cells, and it has various pro‐inflammatory functions leading, among others, to vasodilatation or to bronchoconstriction 12.
Nitric oxide (NO) functions as a vasodilator. Allergic inflammation in the airways activates the inducible NO synthase in bronchoepithelial cells causing higher NO levels e.g. in asthmatic patients 13, 14, 15.
Prostaglandin D2 is a potent bronchoconstrictor released from mast cells which has chemotactic properties on Th2 cells 16.
Structural changes of the airways
Chronic inflammation is often accompanied by remodeling of the tissues. Remodeling can be seen as an adaption to injury and mechanical demands by changing geometry, structure and properties within the airway wall. Patients with poorly controlled asthma develop progressive persistent airflow limitation with longer disease durations and insufficient or no anti‐inflammatory therapy providing evidence that airway remodeling plays a crucial role in the impairment of lung function 17, 18, 19, 20. It occurs in a wide range of tissues and organs and can be observed in almost all tissues susceptible to repeated chronic injury. In the asthmatic inflammation, it leads to structural changes of the airway which includes changes of the epithelium, subepithelial fibrosis, increased airway smooth muscle mass with hyperplasic and hypertrophic composition, decreased distance between the smooth muscles and the epithelium, thickening of the reticular basement membrane, dysregulated extracellular matrix, mucous gland and goblet cell hyperplasia, vascular changes and oedema 21, 22, 23, 24. Some of these changes are related to the severity of the disease and may result in relatively irreversible narrowing of the airways or are related to airway hyperresponsiveness. The thickness of the reticular basement membrane has been correlated to airflow obstruction and increased airway hyperresponsiveness 25, 26, 27.
Many cells contribute to tissue injury and repair process. These include inflammatory and structural cells. Th‐2 type cells dominate over Th‐1 type cells promote subepithelial fibrosis and hyperplasia of airway smooth muscle cells 28.
Regulatory T‐cells exhibit cytokine‐dependent suppressive mechanism via IL‐10 and TGF‐ß secretion. They induce via – among others – the transcription of IL‐6, TNF‐α and extracellular matrix proteins (ECM) which leads to subendothelial fibrosis, collagen deposition and increased accumulation of actin‐containing smooth muscle cells 29.
Regulated by proinflammatory chemokines such as RANTES (regulation on activation, normal T cell expressed and secreted), IL‐5 and eotaxin, eosinophils accumulate in the airways of asthmatics. They produce many proinflammatory and remodeling cytokines including IL‐6, IL‐11 or IL‐17 3, 30, 31.
Airway epithelial cells and mesenchymal cells form a trophic unit. Epithelial damage occurs typically as a response to the environmental exposure, results in production of signals that act on the underlying mesenchyme to propagate and amplify inflammatory and remodeling responses in the submucosa. Allergen sensitisation may be the consequence of a defective airway epithelium leading to an activation of dendritic cells towards promoting a Th2 response. Activated epithelial cells produce, via STAT‐6 activation (e.g. by infiltrating T lymphocytes), numerous chemokines including IL‐8, RANTES and eotaxin. Further, they release growth factors such as epidermal growth factor (EGF), platelet‐derived growth factor (PDGF), TGF‐ß and vascular endothelial growth factor (VEGF). All of these advocate airway smooth muscle proliferation, angiogenesis and ECM protein deposition.
The airway smooth muscle cells are the most important effort cells of the whole inflammatory process. Evidence of either hypertrophy, hyperplasia, or both is reported in mild, moderate and severe asthma, and it is correlated with the severity of the disease. Myocytes proliferate in response to growth factors and inflammatory mediators. They can also increase in size and shape and migrate. Airway smooth muscle cells not only respond to inflammatory processes, they themselves participate in the remodeling process through their release of cytokines like TGF‐ß, TNF‐α, IL‐1ß, IL‐8, Interferon‐gamma (IFN‐γ), chemokines like RANTES, eotaxin, MIP‐1α (macrophage inflammatory protein 1alpha) and ECM proteins 2, 32, 33, 34, 35. Many of those cytokines induce the expression of toll‐like receptors (TLRs) on airway smooth muscle cells. Gram‐negative bacteria were suggested to promote airway hyperresponsiveness through TLRs on those cells. Infections in general or viral/bacterial components activate TLRs and thus contribute to remodeling 36, 37, 38.
Fibroblasts differentiate in response to various stimuli into myofibroblasts which secrete, among other inflammatory mediators, ECM proteins 39, 40. In severe asthma, or insufficiently with anti‐inflammatory drug‐treated asthma, subepithelial fibrosis occurs because of increases deposition of ECM proteins, including collagens I, III, V, proteoglycan, tenascin and fibronectin. The deposition of ECM proteins are regulated by myofibroblasts and the imbalance between matrix metalloproteinase (MMP‐9), which degrades collagen 4 and tissue inhibitor of matrix metalloproteinase (TIMP‐1) being both secreted by fibroblasts 41.
Mucus glands are activated in asthma. Increase mucus hypersecretion because of goblet cell proliferation is a common feature in poorly controlled asthma. Predominantly IL‐1ß, IL‐9, IL‐13, TNF‐α and COX‐2 and their associated intracellular signalling pathways have been shown to be involved in the upregulation of mucin synthesis further promoting goblet cell hyperplasia. However, goblet cell hyperplasia is not a general feature in asthma and only found in a subgroup of patients 42.
Environmental factors such as allergens but also virus and bacterial infection lead to the destruction of the epithelial barrier in the airways and thus contributing to remodeling 43. Vice versa inflammatory damage of the airways alleviates infection eventually leading to an asthma exacerbation. This explains the susceptibility of asthmatic airways to respiratory germs and the impact on the health of poorly controlled asthmatic patients 44, 45. Furthermore, inhaled (virus, bacteria) and even ingested germs (e.g. parasites) interact with the immune system which may enhance or restrain the immunogenic response utilising the same inflammatory pathways as described above 46, 47, 48. Infections in early life are linked with subsequent respiratory morbidity, but they were also protective towards the eventual development of allergic diseases and possible asthma as proposed by the hygiene hypothesis 49, 50. Wheezing illnesses including asthma exacerbations are associated with viral or bacterial respiratory infections in patients of all ages 51, 52.
The role of viral infections
Interestingly, viral infection can be both inducers of wheezing but also protectors against the development of allergic disease. Respiratory viral infections are associated with asthma exacerbations which are seen in children as well as in adults. It is not clear if respiratory infections instigate disease progression or intensify severity of the disease, because wheezing episodes due to respiratory infections may diminish with age, but for some individuals they start in early life marking the beginning of a life‐long asthma carrier.
Virus trigger in children
There are many virus known to trigger wheezing events including respiratory syncytial virus (RSV), human rhinovirus (HRV), metapneumovirus, parainfluenza and coronavirus. The relation between viral infections and the prevalence in asthma has been best studied in RSV and HRV 53. Natural and experimental viral infection induces activation of transcription factors like NF‐κB. This initiate gene transcription of cytokine and chemokine production such as IL‐1ß, IL‐6, IL‐8, IL‐11, membrane cofactor protein 1, MIP‐1α and RANTES 28, 54, 55, 56. These mediators promote tissue infiltration by T cells, eosinophils and basophils. When activated they themselves, release mediators such as eosinophil cationic protein, histamine and with the immunological reaction also virus‐specific IgE. The persistence and severity of the inflammation seem to be more pronounced in atopic asthma compared to healthy individuals which is because of an immunological imbalance in Th1‐ and Th2‐type cytokines released during the inflammation.
This cascade of airway inflammation of the initial upper respiratory infection may result in bronchial hyperresponsiveness, (RSV‐) bronchiolitis and persistence of asthma symptoms. Even relatively mild viral infections might induce more or less severe inflammation of the airways possibly accompanied by prolonged periods of bronchial hyperreactivity 44, 57, 58. One reason for that phenomenon is the absence of protective immunological memory, another might be a genetically determined premorbid abnormality, simply maternal smoking during pregnancy or involuntary second hand smoke exposure since early years are also known promoters 59.
About half of all patients developing RSV bronchiolitis suffer from persistent episodes of wheezing in early childhood. A child born at the peak of the winter bronchiolitis season has an apparent high risk for developing asthma. Another risk factor seems to be RSV‐mediated wheezing illness in early life eventually lead to subsequent persistent wheezing and asthma when a child begins to go to school 57, 60. Various reasons have been discussed for that phenomenon:
The immaturity of the immune system at the time of initial infections resulting in slower recovery. This might even cause severe symptoms in later years although the starting infection is relatively mild.
Different genetic background and genetic susceptibility 61, 62, 63 or virus strain variability 64.
However, not all investigations have demonstrated such association. May be an additional genetic predisposition for the development of asthma in later years exists as discussed in a twin registry study from Denmark 65.
Also HRVs which trigger the common cold and respiratory tract infections can cause bronchiolitis and promote later asthma. Jackson et al calculated the risk factors for HRV or RSV‐induced wheezing illness during the first 3 years of life and the probability of asthma development at the age of 6 with odds ratio 2,6 (95% CI 1,0‐6,3) for RSV‐related wheezing and 9,8 (95% CI 4,3‐22,0) for HSV 57. HRV‐related wheezing in the first 3 years was highest when the children acquired an additional aeroallergen sensitisation 60. In contrast, sensitisation during the first year of life had no promoting effect on asthma development. The knowledge of host virus interaction after viral infection during the early phase of their life is essential to understand why some children develop asthma, but others are protected from allergic sensitisation.
Viral infection promote asthma attacks
Sensitive diagnostic tests based on polymerase chain reaction (PCR) or microarray technology revealed that respiratory viral infections are associated with up to 85%–95% of exacerbations of wheezing or acute worsening of asthma 66, 67. Rhinovirus is probably the most important pathogen in triggering asthma attacks. RSV and HRV turned out in epidemiologic studies to be the most often detected viruses depending on season and geographic areas 68, 69. HRV infections peak with asthma hospitalisations in spring and autumn, while RSV is more likely to trigger acute asthma symptoms in the winter time 70, 71. Although there is clear evidence that virus infection is a trigger for asthma exacerbations, there is no clear evidence that asthma patients have more colds than healthy individuals. But asthmatics may suffer from extended duration of illness and increased severity of lower respiratory tract symptoms. This finding suggest that the response to a viral infection but not the frequency differ between susceptible and healthy patients 72, 73. Atopy promotes more severe illnesses than non‐allergic asthma after infection with respiratory viruses 71. Delayed viral clearance and increased viral load in the upper respiratory tract are obviously not the reasons for that phenomenon. Authors suggested an abnormal antiviral activity e.g. by reduced generation of interferons to virus infections as a possible reason for increased susceptibility 73 or disruption of the innate immune host defence pathways through the antagonistic interaction between allergic inflammation and antiviral immunity 74.
Hygiene hypothesis
Epidemiological studies have demonstrated that infections within the first year of life protects against the development of atopy and allergy 75. The hygiene hypothesis is drawn from the observation that the individual susceptibility of allergic diseases is promoted by changes in the interaction between the immune system and microbes 76. The following findings hint that infections in early life may protect from atopy:
Children with more siblings have less skin‐prick test reactivity 50.
The lack of wheezing lower respiratory tract infections in early life coincide with low IgE levels in serum 77, 78.
The use of daycare centres correlates inversely with the prevalence of typical allergic symptoms 50.
Children who grew up in close proximity to farms close to livestock within the first year of life or whose parents are farmers also have fewer allergies than those who grew up under cleaner or more hygienic conditions 79. A huge list of putative protective factors have been discussed such as high exposure to endotoxins, exposure to livestock especially cattle, traditional diet, ingestions of unpasteurised milk and many more 80.
A strong Th1 response of delayed‐type hypersensitivity to mycobacterium tuberculosis also reduces atopy incidence 81.
But the hygiene hypothesis has not been unanimously accepted because epidemiologic inconsistencies exist regarding the beneficial but also diseases promoting functions of microbes 82. Many observations seem to contradict the hygiene hypothesis: For example, regardless of the lack of cleanliness in inner cities, allergic sensitisation for example against cockroach allergen increases 83, 84. A more prevailing version of the hypothesis now focuses on the protective role of intracellular mild pathogens ingested through the faecally contaminated environment (water, food) which also includes microbes 85. All of these exposures vanguard a long‐lasting immune response, and thus protecting from allergies 62, 82, 86.
The role of bacterial infections
While the importance of viral infections as a source of asthma exacerbations and chronic inflammation of the airways in children and adults is well demonstrated, the role of bacterial infections in the development of stable asthma and acute asthma exacerbations remains more controversial. Of all bacterial respiratory pathogens assumed to have a potential role in the pathogenesis of asthma, Chlamydophila pneumoniae (previously Chlamydia pneumoniae) and Mycoplasma pneumoniae are most commonly discussed in this context 68. Although the cascade of pathogenetic mechanisms between the atypical bacterial infection and airway inflammation in asthma is not yet completely understood, still enough data have been accumulated supporting such a hypothesis 87.
Detection methods of atypical bacteria
The greater limitation of confirming an association between atypical respiratory infection and asthma is the lack of standardised laboratory methods for the detection of these pathogens and the practical or even ethical difficulty of obtaining clinical samples of the lower respiratory tract in asymptomatic population 88, 89. Geographical and temporal variations in the natural history of these infections seem also to play a role. Commonly used methods like serological detection of IgG antibodies cannot distinguish between previous infection, chronic colonisation and reactivation of a chronic infection, while the prevalence of IgG antibodies against atypical bacteria is high among the general population, allowing no comparison between asthmatic patients and control group 90. On the other hand, acute infections and reinfections are not always accompanied by IgM antibodies production causing an even greater uncertainty.
However, molecular methods like PCR enabled over the recent years the detection of these pathogens with higher sensitivity, which is very important in case of chronic colonisation where the bacterial load is very low, and higher specificity, which is also important in order to avoid usually observed cross‐reactions 90, 91. The most recently emerged reverse‐transcriptase PCR based on the detection of messenger RNA allowed the discrimination between viable and non‐viable organisms resulting to a better differentiation between chronic colonisation with viable bacteria and preceding infections with non‐viable bacteria 92, 93. Additionally to the refined laboratory methods, numerous human studies have outlined the clinical relevance between atypical infections and asthma, while animal models highlighted the pathophysiological pathways by which these infections promote lung inflammation and airway remodeling.
Cellular changes due to atypical bacteria
C. pneumoniae is predominately implicated as a pathogen in community‐acquired pneumonia, but as an obligatory intracellular bacterium, it has also the potency to cause chronic infection of the affected cells. C. pneumoniae infection is associated with the production and release of several cytokines such as TNF‐α, IL‐8 and INF‐γ 94, 95. The major role of these molecules is the attraction of immune cells, like TH‐2 and macrophages, as a response against bacterial presence. However, the recruitment of immune cells within the airway wall may subsequently trigger inflammatory responses with tissue damage. The most predominant correlation between immune system activation and structural changes of the airways is mediated through MMPs. MMPs and their physiological inhibitors, tissue inhibitors of TIMPs, are produced by different structural and inflammatory cells mainly by smooth muscle cells of submucosa and stimulated macrophages recruited in the lung. The proteolytic–antiproteolytic activity of these proteins is regulated by several cytokines and thought to be significant for various physiological processes like healing and development. In the contrary, the dysfunction of this proteolytic system provoked by deregulation of their expression or change of their biological activity is involved in a wide range of lung diseases including chronic obstructive pulmonary disease, lung fibrosis, lung cancer and asthma 96, 97.
Additionally to MMPs causing airway wall remodeling (see above), several sophisticated pathophysiological mechanisms are also proposed as a linkage between atypical infection and asthma. An emerging body of molecular studies has shown that C. pneumoniae has the ability to inhibit affected cells apoptosis in order to assure the longevity of the host which is a precondition for its own viability as an obligate intracellular pathogen. The impairment of apoptotic mechanisms enhances chronic inflammation by favouring persistent C. pneumoniae infection and creates a link with chronic asthma inflammatory manifestations 98, 99. On the other hand, chronically affected cells appear to have an increased susceptibility to respiratory viruses due to impaired apoptosis which may provide evidence that atypical infections can also indirectly contribute to acute exacerbations of asthma 100.
The viral–bacterial co‐infection consists a representative example of coexisting causative factors, while further interactions are parallel under discussion even between infectious and non‐infectious agents like pollution, allergens, nutrition elements and stressful conditions 101. The most discussed theory in this context is the induction of airway sensitisation against allergens on the ground of chronic C. pneumoniae infection. Cellular and molecular studies have partly elucidated the potential role of the immune system in such bacterial–allergens interactions, but more comprehensive analysis remains to be done in order to evaluate all the involving mechanisms 102, 103.
In the contrary M. pneumoniae, being an extracellular microorganism, destroys the ciliated epithelium after attached to the bronchial mucosa and is so far associated with a wide spectrum of acute respiratory infections such as pneumonia, exacerbations of bronchitis and bronchiolitis, sinusitis and median otitis 104, 105, 106. M. pneumoniae infection is also accompanied, as similarly observed by C. pneumoniae, with increased production of IgA antibodies and greater mast cell tissue infiltration in the airways and may subsequently play a role in the chronic course of asthma 107.
Do atypical bacteria promote asthma?
All the previous described biological mechanisms, although many of them still under discussion, indicate that atypical respiratory infection may contribute to inflammation and remodeling of the airways of asthmatic patients. Regardless of this, the causative relationship between atypical bacteria and asthma remains unclear, as not all infected patients develop asthma and not all of the people with asthma are found to become infected. The main question to be answered is, whether the atypical infection makes the development of asthma more likely, or pre‐existed alterations of the airways favour the colonisation and persistence of these pathogens, leading to their increased detection in asthma as an epiphenomenon with any etiological correlation 87, 108.
However, despite the absence of definite causative direction, many controlled observational clinical studies have demonstrated the association between chronic stable asthma and atypical bacteria as infected subjects were found to have elevated markers of inflammation, increased severity of obstruction identified by FEV1, higher daytime symptom score and required high dose of inhaled corticosteroids in comparison with non‐infected controls 46, 100. A strong connection between acute exacerbations of asthma and infection with C. pneumoniae and/or M. pneumoniae was also identified in the majority of research protocols conducted for this purpose, while insufficient data allow any reliable conclusion about the role of atypical bacteria in late‐onset asthma 109, 110. It is important to note that inadequate laboratory methods used for the detection of these pathogens may influence the results of clinical studies, where no correlation was confirmed, underling the need of more standardised diagnostic techniques in this setting.
Other bacteria involved in asthma
Additionally to atypical bacteria, other respiratory and most recently non‐respiratory bacterial species have drawn attention concerning their role in asthma. Helicobacter pylori for example is one of the non‐respiratory pathogens assumed to have, except of its local complications, systemic immunomodulatory effects that may prevent the development of asthma 111, 112. An inverse prevalence between H. pylori infection and asthma is consistently confirmed in many different populations and geographical areas. It still remains unclear if this is an etiological association or an observation triggered by other lifestyle trends or even an incidentally occurred phenomenon 113. Up to now, eradication therapy is no recommended treatment strategy for asthma, as prospective studies are lacking in this field and much more research is required.
Communal bacteria of the human gut are also thought to have an immunomodulatory effect on the innate and adaptive immunity of the host by interacting with immune cells of intestinal mucosa. These interactions lead (i) to the activation of tolerogenic dendritic cells, and through these (ii) to a balanced Th1/Th2 differentiation important for the lifetime maintenance of immune tolerance of the host. If the establishment of intestinal microbiota is disrupted by early‐life events like antibiotic use, the tolerogenic immunomodulatory effects are minimised, resulting in overproduction of Th2 cytokines which is a risk factor for autoimmune and allergic diseases 114. Interventional methods with potentially probiotics (Bifidobacterium and Lactobacillus strains) aiming to create healthy microflora have been intensively tested as treatment or prevention in many allergic diseases like atopic dermatitis, allergic rhinitis and asthma, but so far available studies provide contradictory results, and the generalisation of a positive effect in all allergic conditions should be carefully considered 115, 116.
Sufficient evidence is available suggesting that exposure to Staphylococcus aureus and/or its enterotoxins function as an environmental risk factor for the development and severity of asthma. The local or systemic released enterotoxins show superantigen activity and may provoke Th2 and eosinophilic stimulation with multi‐clonal IgE production leading to deterioration of upper and lower respiratory tract atopic diseases 117. Specific IgE antibodies against S. aureus enterotoxins are more likely to be found in patients with nasal polyps, allergic rhinitis, chronic rhinosinusitis and asthma, and may indicate a future therapeutic target 118.
The role of other respiratory bacteria like Haemophilus influenzae, Moraxella catarrhalis and Streptococcus pneumoniae was mainly discussed in the framework of hygiene hypothesis (see above) where infections in early years was claimed to have a protective effect against asthma and atopy development. In contrast of being protective, these pathogens more often cause severe persistent wheeze in preschool children, and this group may significantly benefit from long‐term antibiotic treatment 119. It was also found that neonates colonised in hypopharyngeal region are under increased risk for recurrent wheeze and asthma within the first 5 years of life 120. Particularly evident is the association of these pathogens with a subset of stable asthma, known as neutrophilic asthma, where inflammation is primarily mediated by neutrophils and less by eosinophils. H. influenzae was isolated from the airways of neutrophilic people with asthma, and infection‐induced inflammation was mediated by IL‐17 expression 121.
The role of antibacterial therapy in the control of asthma
Asthma is a complex disease with a great heterogeneity in its clinical features and even greater heterogeneity in treatment responses as a result of individual environmental and genetic pressures. Therapeutic measurements cannot always adequately cover all different subtypes of the disease and even patients having the same clinical phenotype may not benefit equally. The complexity increases as more than one pathogen is involved in the pathogenesis. An accumulating number of new therapies are emerging as highly efficient, especially for patients with persistent asthma. This benefits optimised health care but it is also a challenge for the selection of the right target group for each compound. The opportunity of phenotype‐driven treatment options may open the way for personalised health care in asthma 122, 123.
Antibiotics and especially macrolides (erythromycin, clarithromycin, roxithromycin, azithromycin) were tested in this context aiming against persistent atypical infection in chronic asthma or in acute exacerbations caused by atypical bacteria. Their effects were recognised as time as well as dose dependent, but the underlying mechanisms of their action is not completely clarified. Macrolides, except of their obvious antimicrobial properties, are shown to exert anti‐inflammatory and immunomodulatory activity by suppressing the production of cytokines in inflamed tissue 124, 125. They have been reported to reduce airway hyperresponsiveness, improve pulmonary function and sometimes soften the symptoms, but it still remains a matter of debate if this is an antimicrobial effect resulting to the limitation of the bacterial load or just an anti‐inflammatory action of the macrolide, or both as the reduction of bacteria is subsequently followed by decreased cytokine expression 126. Much additional research is required to define the right agents for the right patient, the optimum dosage and duration of therapy and the adequate treatment until macrolide treatment can be recommended as a long‐term treatment option for asthma patients 127.
In the contrary, the antibiotic exposure during pregnancy or within the first years of life is thought to be associated with increased risk of childhood asthma through incomplete establishment of microflora or modified natural exposure in pathogens described by the hygiene hypothesis. Studies conducted to clarify this matter confirmed an increased risk of developing asthma in later years, but many factors confound this relationship 128, 129.
The role of parasitic infections
Epidemiologic data suggest that helminth infections may be protective against asthma, as allergic diseases are rarely observed in populations where prevalence of intestinal parasites is high. It is thought, that the immunomodulatory effect of the parasites which is supposed to influence atopic responses is mediated by regulation of Th2 differentiation and IgE production 130. However, a meta‐analysis of epidemiologic studies did not confirm this general conclusion, as only infection with hookworm was found to be protective, while Ascaris lumbricoides was associated with increased risk of asthma, and no significant effect was found with other species. Trichuris trichiura in early life are associated with less positive prick tests in childhood, while Toxocara infection increases the risk of wheeze in some populations 47. Apparently, the positive or negative effect of helminths in allergy and asthma differ between different species, and it is now suggested that many more factors can also be causative for this interaction as the chronicity, the timing and the parasitic burden of the disease 113, 131.
What can we learn from this?
The relationship between inflammation and remodeling and the pathogenic role of viral and bacterial infection in the airways of asthmatic patients is still highly debatable and incompletely understood. The presence of airway inflammation does not always translate into irreversible changes in many cases, and there is not always a clear‐cut relationship between the degree of inflammation and the level of remodeling (Fig. 1). Remodeling processes in paediatric population raises further questions about the meaning of chronic inflammation and the genetic susceptibility that needs to be explored. This area is certainly challenging in regard to the development of new therapeutic compounds aiming at the inhibition of specific inflammatory pathways including those triggered by viruses and bacteria. Finally, identification of biomarkers identifying remodeling is essential to study these processes in detail: (i) to identify patients being particularly at risk to develop those irreversible changes, and (ii) to clarify the effect of anti‐inflammatory treatment regimes.
Figure 1.
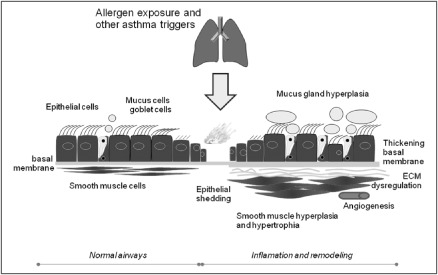
Chronic inflammation in the airways leads to remodeling in the airways (right side). ECM, extracellular matrix.
Conflict of interest
The authors have stated explicitly that there are no conflicts of interest in connection with this article.
Authorship and contributorship
Adrian Gillissen and Maria Paparoupa have equally contributed to the writing of this paper. Adrian Gillissen made all the final corrections.
References
- 1. GINA Executive and Science Committee , editor. Global Initiative for Asthma (GINA). Global Strategy for Asthma Management and Prevention. Bethesda, USA, US Department of Health and Human Services, 2012. [Google Scholar]
- 2. Koziol‐White CJ, Panettieri RA Jr. Airway smooth muscle and immunomodulation in acute exacerbations of airway disease. Immunol Rev. 2011;242: 178–185. [DOI] [PubMed] [Google Scholar]
- 3. Green RH, Brightling CE, McKenna S, Hargadon B, Parker D, Bradding P, Wardlaw AJ, Pavord ID. Asthma exacerbations and sputum eosinophil counts: a randomized controlled trial. Lancet. 2002;360: 1715–1721. [DOI] [PubMed] [Google Scholar]
- 4. Bjermer L. History and future perspectives of treating asthma as a systemic and small airways disease. Respir Med. 2001;95: 703–719. [DOI] [PubMed] [Google Scholar]
- 5. Jeffery PK. Remodelling and inflammation of bronchi in asthma and chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2004;1: 176–183. [DOI] [PubMed] [Google Scholar]
- 6. Hashimoto T, Akiyama K, Kawaguchi H, Maeda Y, Taniguchi M, Kobayashi N, Mori A. Correlation of allergen‐induced IL‐5 and IL‐13 production by peripheral blood T cells of asthma patients. Int Arch Allergy Appl Immunol. 2004;134: 7–11. [DOI] [PubMed] [Google Scholar]
- 7. Zangrilli JG, Peters SP. Cytokines in allgeric airway disease. In: Busse W, Holgate ST, editors. Asthma and Rhinitis. Cambridge, Blackwell Science Inc., 1999: 426–436. [Google Scholar]
- 8. Chuang‐Stein C. Sample size and the probability of a successful trial. Pharm Stat. 2006;5: 305–309. [DOI] [PubMed] [Google Scholar]
- 9. Sabroe I, Lloyd CM, Whyte MKB, Dower SK, Williams TJ, Pease JE. Chemokines, innate and adaptive immunity, and respiratory disease. Eur Respir J. 2002;19: 350–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dahlén S‐E. Cysteinyl leukotrienes as common mediators of asthma and allergic disease. Clin Exp Allergy Rev. 2003;3: 69–73. [Google Scholar]
- 11. Zhu Y, Chen L, Huang Z, Alkan S, Bunting KD, Wen R, Wang D, Huang H. Cutting edge: IL‐5 primes the Th2 cytokine‐producing capacity in eosinophils through a STAT5‐dependent mechanism. J Immunol. 2004;173: 2918–2922. [DOI] [PubMed] [Google Scholar]
- 12. Numata T, Konno A, Yamakoshi T, Hanazawa T, Terada N, Nagata H. Comparative role of peptide leukotrienes and histamine in the development of nasal mucosal swelling in nasal allergy. Ann Otol Rhinol Laryngol. 1999;108: 467–473. [DOI] [PubMed] [Google Scholar]
- 13. Dweik RA, Sorkness RL, Wenzel S, et al. Use of exhaled nitric oxide measurement to identify a reactive, at‐risk phenotype among patients with asthma. Am J Respir Crit Care Med. 2010;181: 1033–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Boman G, Ludviksdottir D, Janson C, Högman M, Hedenström H, Björnsson E, Boman G. Exhaled nitric oxide and its relationship to airway responsiveness and atopy in asthma. Respir Med. 2008;93: 552–556. [DOI] [PubMed] [Google Scholar]
- 15. Dressel H, de la Motte D, Reichert J, Ochmann U, Petru R, Angerer P, Holz O, Nowak D, Jörres RA. Exhaled nitric oxide: independent effects of atopy, smoking, respiratory tract infection, gender and height. Respir Med. 2008;102: 962–969. [DOI] [PubMed] [Google Scholar]
- 16. Monneret G, Gravel S, Diamond M, Rokach J, Powell WS. Prostaglandin D2 is a potent chemoattractant for human eosinophils that acts via a novel DP receptor. Blood. 2001;98: 1942–1948. [DOI] [PubMed] [Google Scholar]
- 17. Mauad T, Bel EH, Sterk PJ. Asthma therapy and airway remodeling. J Allergy Clin Immunol. 2011;120: 997–1009. [DOI] [PubMed] [Google Scholar]
- 18. Jeffery PK. Inflammation and remodeling in the adult and child with asthma. Pediatr Pulmonol Suppl. 2001;21: 3–16. [PubMed] [Google Scholar]
- 19. Palmans E, Kips JC, Pauwels RA. Prolonged allergen exposure induces structural airway changes in sensitized rats. Am J Respir Crit Care Med. 2000;161: 627–635. [DOI] [PubMed] [Google Scholar]
- 20. Chetta A, Foresi A, Del Donno M, Bertorelli G, Pesci A, Olivieri D. Airways remodeling is a distinctive feature of asthma and is related to severity of disease. Chest. 1997;111: 852–857. [DOI] [PubMed] [Google Scholar]
- 21. Girodet PO, Ozier A, Bara I, Tunon de Lara JM, Marthan R, Berger P. Airway remodeling in asthma: new mechanisms and potential for pharmacological intervention. Pharmacol Ther. 2011;130: 325–337. [DOI] [PubMed] [Google Scholar]
- 22. Bergeron C, Tulic MK, Hamid Q. Airway remodelling in asthma: from benchside to clinical practice. Can Respir J. 2010;17: e85–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Elias JA, Zhu Z, Chupp G, Homer RJ. Airway remodeling in asthma. Proc Am Thorac Soc. 2009;6: 301–305.19387034 [Google Scholar]
- 24. Murdoch JR, Lloyd CM. Chronic inflammation and asthma. Mutat Res. 2010;690: 24–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Payne DN, Rogers AV, Adelroth E, Bandi V, Guntupalli KK, Bush A, Jeffery PK. Early thickening of the reticular basement membrane in children with difficult asthma. Am J Respir Crit Care Med. 2003;167: 78–82. [DOI] [PubMed] [Google Scholar]
- 26. Milanese M, Crimi E, Scordamaglia A, Riccio A, Pellegrino R, Canonica GW, Brusasco V. On the functional consequences of bronchial basement membrane thickening. J Appl Physiol. 2001;91: 1035–1040. [DOI] [PubMed] [Google Scholar]
- 27. Laprise C, Laviolette M, Boutet M, Boulet L‐P. Asymptomatic airway hyperresponsiveness: relationships with airway inflammation and remodelling. Eur Respir J. 1999;14: 63–73. [DOI] [PubMed] [Google Scholar]
- 28. Durant LR, Makris S, Voorburg CM, Loebbermann J, Johansson C, Openshaw PJ. Regulatory T cells prevent Th2 immune responses and pulmonary eosinophilia during RSV infection in mice. J Virol. 2013;87: 10946–10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Robinson D. Regulatory T cells and asthma. Clin Exp Allergy. 2009;39: 1314–1323. [DOI] [PubMed] [Google Scholar]
- 30. Eum S‐Y, Maghni K, Hamid Q, Eidelman DH, Martin JG. Involvement of eotaxin and IL‐5 in the development of antigen‐induced airway eosinophilia but not in airway hyperresponsiveness in mice. Am J Respir Crit Care Med. 1999;159: A228. [Google Scholar]
- 31. Cieslewicz G, Tomkinson A, Adler A, Duez C, Schwarze J, Takeda K, Larson KA, Lee JJ, Irvin CG, Gelfand EW. The late, but not early, asthmatic response is dependent on IL‐5 and correlates with eosinophil infiltration. J Clin Invest. 1999;104: 301–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chiba Y, Sato S, Misawa M. Upregulation of geranylgeranyltransferase I in bronchial smooth muscle of mouse experimental asthma: its inhibition by lovastatin. J Smooth Muscle Res. 2010;46: 57–64. [DOI] [PubMed] [Google Scholar]
- 33. James AL, Bai TR, Mauad T, Abramson MJ, Dolhnikoff M, McKay KO, Maxwell PS, Elliot JG, Green FH. Airway smooth muscle thickness in asthma is related to severity but not duration of asthma. Eur Respir J. 2009;34: 1040–1045. [DOI] [PubMed] [Google Scholar]
- 34. Hollins F, Kaur D, Yang W, Cruse G, Saunders R, Sutcliffe A, Berger P, Ito A, Brightling CE, Bradding P. Human airway smooth muscle promotes human lung mast cell survival proliferation, and constitutive activation: cooperative roles for CADM1, stem cell factor, and IL‐6. J Immunol. 2008;181: 2772–2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Slats AM, Janssen LJ, Schadewijk VA, et al. Expression of smooth muscle and extracellular matrix proteins in relation to airway function in asthma. J Allergy Clin Immunol. 2008;121: 1196–1292. [DOI] [PubMed] [Google Scholar]
- 36. Sarir H, Henricks PAJ, Houwelingen AH, Nijkamp FP, Folkerts G. Cells, mediators and Toll‐like receptors in COPD. Eur J Pharmacol. 2008;585: 346–353. [DOI] [PubMed] [Google Scholar]
- 37. Camateros P, Tamaoka M, Hassan M, Marina R, Moisan J, Marion D, Guiot MC, Martin JG, Radzioch D. Chronic asthma‐induced airway remodeling is prevented by Toll‐like receptor‐7/8 ligand S28463. Am J Respir Crit Care Med. 2007;175: 1241–1249. [DOI] [PubMed] [Google Scholar]
- 38. Lien E, Ingalls RR. Toll‐like receptors. Crit Care Med. 2002;30: S1–11. [PubMed] [Google Scholar]
- 39. Westergren‐Thorsson G, Chakir J, Lafreniere‐Allard MJ, Boulet LP, Tremblay GM. Correlation between airway responsiveness and proteoglycan production by bronchial fibroblasts from normal and asthmatic subjects. Int J Biochem Cell Biol. 2002;34: 1256–1267. [DOI] [PubMed] [Google Scholar]
- 40. Postma DS, Timens W. Remodeling in asthma and chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2006;3: 434–439. [DOI] [PubMed] [Google Scholar]
- 41. Suzuki R, Kato T, Miyazaki Y, Iwata M, Noda Y, Takagi K, Nakashima N, Torii K. Matrix metalloproteinases and tissue inhibitors of matrix metalloproteinases in sputum from patients with bronchial asthma. J Asthma. 2001;38: 477–484. [DOI] [PubMed] [Google Scholar]
- 42. Takeyama K, Fahy JV, Nadel JA. Relationship of epidermal growth factors to goblet cell production in human bronchi. Am J Respir Crit Care Med. 2001;163: 511–516. [DOI] [PubMed] [Google Scholar]
- 43. Holgate ST. The airway epithelium is central to the pathogenesis of asthma. Allergol Int. 2008;57: 1–10. [DOI] [PubMed] [Google Scholar]
- 44. Wu P, Dupont WD, Griffin MR, Carroll KN, Mitchel EF, Gebretsadik T, Hartert TV. Evidence of a causal role of winter virus infection during infancy in early childhood asthma. Am J Respir Crit Care Med. 2008;178: 1123–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hogg JC. Childhood viral infection and the pathogenesis of asthma and chronic obstructive lung disease. Am J Respir Crit Care Med. 1999;160: S26–28. [DOI] [PubMed] [Google Scholar]
- 46. Papadopoulos NG, Christodoulou I, Rohde G, et al. Viruses and bacteria in acute asthma exacerbations – a GA2LEN‐DARE systematic review. Allergy. 2011;66: 458–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cooper PJ. Interactions between helminth parasites and allergy. Curr Opin Allergy Clin Immunol. 2009;9: 29–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Leonardi‐Bee J, Pritchard D, Britton J. Asthma and current intestinal parasite infection: systematic review and meta‐analysis. Am J Respir Crit Care Med. 2006;174: 514–523. [DOI] [PubMed] [Google Scholar]
- 49. Illi S, von Mutius E, Lau S, Niggemann B, Grüber C, Wahn U. Perennial allergen sensitisation early in life and chronic asthma in children: a birth cohort study. Lancet. 2006;368: 763–770. [DOI] [PubMed] [Google Scholar]
- 50. Nickel R, Lau S, Niggemann B, Gruber C, von Mutius E, Illi S, Kulig M, Wahn U. Messages from the German multicentre allergy study. Pediatr Allergy Immunol. 2003;13: 7–10. [DOI] [PubMed] [Google Scholar]
- 51. Nicholson KG, Kent J, Ireland DC. Respiratory viruses and exacerbations of asthma in adults. BMJ. 1993;310: 1225–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Talbot TR, Hartert TV, Mitchel E, Halasa NB, Arbogast PG, Poehling KA, Schaffner W, Craig AS, Griffin MR. Asthma as a risk factor for invasive pneumococcal disease. N Engl J Med. 2005;352: 2082–2090. [DOI] [PubMed] [Google Scholar]
- 53. Escobar GJ, Masaquel AS, Li S X, Walsh EM, Kipnis P. Persistent recurring wheezing in the fifth year of life after laboratory‐confirmed, medically attended respiratory syncytial virus infection in infancy. BMC Pediatr. 2013;13: 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Papadopoulos NG, Johnston SL. Rhinoviruses as pathogens of the lower respiratory tract. Can Respir J. 2000;7: 409–414. [DOI] [PubMed] [Google Scholar]
- 55. Becker S, Reed W, Henderson FW, Noah TL. RSV infection of human airway epithelial cells causes production of the beta‐chemokine RANTES. Am J Physiol. 1997;273: L512–520. [DOI] [PubMed] [Google Scholar]
- 56. Rochlitzer S, Hoymann HG, Muller M, Braun A. No exacerbation but impaired anti‐viral mechanisms in a rhinovirus‐chronic allergic asthma mouse model. Clin Sci (Lond). 2014;126: 55–65. [DOI] [PubMed] [Google Scholar]
- 57. Jackson DJ, Gangnon RE, Evans MD, et al. Wheezing rhinovirus illnesses in early life predict asthma development in high‐risk children. Am J Respir Crit Care Med. 2008;178: 667–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Gern JE, Lemanske RF Jr, Busse WW. Early life origins of asthma. J Clin Invest. 1999;104: 837–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Tager IB. The effects of second‐hand and direct exposure to tobacco smoke on asthma and lung function in adolescence. Paediatr Respir Rev. 2008;9: 29–37. [DOI] [PubMed] [Google Scholar]
- 60. Kusel MM, Klerk NH, Kebadze T, Vohma V, Holt PG. Early‐life respiratory viral infections, atopic sensitization, and risk of subsequent development of persistent asthma. J Allergy Clin Immunol. 2007;119: 1105–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Holt PG, Macaubas C, Stumbles PA, Sly PD. The role of allergy in the development of asthma. Nature. 1999;402: B12–17. [DOI] [PubMed] [Google Scholar]
- 62. Holt PG. Infection and the development of allergic disease. Allergy. 2011;66: 13–15. [DOI] [PubMed] [Google Scholar]
- 63. Martin JG, Siddigui S, Hassan M. Immune response to viral infections: relevance for asthma. Paediatr Respir Rev. 2006;7: S125–127. [DOI] [PubMed] [Google Scholar]
- 64. Kimpen JLL. Viral infections and childhood asthma. Am J Respir Crit Care Med. 2000;162: S108–112. [DOI] [PubMed] [Google Scholar]
- 65. Thomsen SF, van der Sluis S, Stensballe LG, Posthuma D, Skytthe A, Kyvik KO, Duffy DL, Backer V, Bisgaard H. Exploring the association between severe respiratory syncytial virus infection and asthma: a registry‐based twin study. Am J Respir Crit Care Med. 2009;179: 1091–1097. [DOI] [PubMed] [Google Scholar]
- 66. Jackson DJ, Johnston SL. The role of viruses in acute exacerbations of asthma. J Allergy Clin Immunol. 2010;125: 1178–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Murray CS, Poletti G, Kebadze T, Morris J, Woodcock A, Johnston SL, Custovic A. Study of modifiable risk factors for asthma exacerbations: virus infection and allergen exposure increase the risk of asthma hospital admissions in children. Thorax. 2006;61: 376–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Leung TF, Chan PK, Wong GW, Fok TF, Ng PC. Respiratory viruses and atypical bacteria triggering severe asthma exacerbation in children. Hong Kong Med J. 2013;19(Suppl. 4): 11–14. [PubMed] [Google Scholar]
- 69. O'Callaghan‐Gordo C, Bassat Q, Diez‐Padrisa N, Morais L, Machevo S, Nhampossa T, Quinto L, Alonso PL, Roca A. Lower respiratory tract infections associated with Rhinovirus during infancy and increased risk of wheezing during childhood. A cohort study. PLoS ONE. 2013;8: e69370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Johnston SL, Pattemore PK, Sanderson G, et al. The relationship between upper respiratory infections and hospital admissions for asthma: a time‐trend analysis. Am J Respir Crit Care Med. 1996;154: 654–660. [DOI] [PubMed] [Google Scholar]
- 71. Heymann PW, Carper HT, Murphy DD, et al. Viral infections in relation to age, atopy, and season of admission among children hospitalized for wheezing. J Allergy Clin Immunol. 2004;114: 239–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Corne JM, Marshall C, Smith S, Schreiber J, Sanderson G, Holgate ST, Johnston SL. Frequency, severity, and duration of rhinovirus infections in asthmatic and non‐asthmatic individuals: a longitudinal cohort study. Lancet. 2002;359: 831–834. [DOI] [PubMed] [Google Scholar]
- 73. Sachs AP, Sachs A, van Loon AM, Haarman M, van de Vijver DA, Kimman TG, Zuithoff P, Schipper PJ, Verheij TJ, Nijhuis M. Enhanced severity of virus associated lower respiratory tract disease in asthma patients may not be associated with delayed viral clearance and increased viral load in the upper respiratory tract. J Clin Virol. 2011;41: 116–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Gill MA, Bajwa G, George TA, Dong CC, Dougherty I, Jiang N, Gan VN, Gruchalla RS. Counterregulation between the Fc epsilon RI pathway and antiviral responses in human plasmacytoid dendritic cells. J Immunol. 2010;184: 5999–6006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Kuo CH, Kuo HF, Huang CH, Yang SN, Lee MS, Hung CH. Early life exposure to antibiotics and the risk of childhood allergic diseases: an update from the perspective of the hygiene hypothesis. J Microbiol Immunol Infect. 2013;46: 320–329. [DOI] [PubMed] [Google Scholar]
- 76. Okada H, Kuhn C, Feillet H, Bach J‐F. The ‘hygiene hypothesis’ for autoimmune and allergic diseases: an update. Clin Exp Immunol. 2010;160: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Martinez F, Wright A, Taussig L, Holberg C, Halonen M, Morgan WJ. Asthma and wheezing in the first six years of life. N Engl J Med. 1995;332: 133–138. [DOI] [PubMed] [Google Scholar]
- 78. Martinez FD, Stern DA, Wright AL, Taussig LM, Halonen M. Association of non‐wheezing lower respiratory tract illnesses in early life with persistently diminished serum IgE levels. Thorax. 1994;50: 1067–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Schaub B, Liu J, Höppler S, Schleich I, Hühn J, Olek S, Wieczorek G, Illi S, von Mutius E. Maternal farm exposure modulates neonatal immune mechanisms through regulatory T cells. J Allergy Clin Immunol. 2009;123: 774–782. [DOI] [PubMed] [Google Scholar]
- 80. Kabesch M, Lauener RP. Why Old McDonald had a farm but no allergies: genes, environments, and the hygiene hypothesis. J Leukoc Biol. 2004;75: 383–387. [DOI] [PubMed] [Google Scholar]
- 81. Shirakawa T, Enomoto T, Shimazu S, Hopkin J. The inverse association between tuberculin response and atopic disorder. Science. 1997;275: 77–79. [DOI] [PubMed] [Google Scholar]
- 82. Matricardi PM. 99th Dahlem conference on infection, inflammation and chronic inflammatory disorders: controversial aspects of the ‘hygiene hypothesis’. Clin Exp Immunol. 2010;160: 98–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Platts‐Mills TA, Woodfolk JA, Sporik RB. The increase in asthma cannot be ascribed to cleanliness. Am J Respir Crit Care Med. 2001;164: 1107–1108. [DOI] [PubMed] [Google Scholar]
- 84. Matricardi PM, Bouygue GR, Tripodi S. Inner‐city asthma and the hygiene hypothesis. Ann Allergy Asthma Immunol. 2002;89: 69–74. [DOI] [PubMed] [Google Scholar]
- 85. Kramer A, Bekeschus S, Broker BM, Schleibinger H, Razavi B, Assadian O. Maintaining health by balancing microbial exposure and prevention of infection: the hygiene hypothesis versus the hypothesis of early immune challenge. J Hosp Infect. 2013;83(Suppl. 1): S29–34. [DOI] [PubMed] [Google Scholar]
- 86. Holt PG, Sly PD. Prevention of allergic respiratory disease in infants: current aspects and future perspectives. Curr Opin Allergy Clin Immunol. 2007;7: 547–555. [DOI] [PubMed] [Google Scholar]
- 87. Starkey MR, Nguyen DH, Kim RY, Nair PM, Brown AC, Essifie AT, Horvat JC, Hansbro PM. Programming of the lung in early life by bacterial infections predisposes to chronic respiratory disease. Clin Obstet Gynecol. 2013;56: 566–576. [DOI] [PubMed] [Google Scholar]
- 88. Metz G, Kraft M. Effects of atypical infections with mycoplasma and chlamydia on asthma. Immunol Allergy Clin North Am. 2010;30: 575–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Tompkins LS, Schachter J, Boman J, et al. Collaborative multidisciplinary workshop report: detection, culture, serology, and antimicrobial susceptibility testing of Chlamydia pneumoniae. J Infect Dis. 2000;181: S460–461. [DOI] [PubMed] [Google Scholar]
- 90. Ferrari M, Poli A, Olivieri M, Verlato G, Tardivo S, Nicolis M, Campello C. Respiratory symptoms, asthma, atopy and chlamydia pneumoniae IgG antibodies in a general population sample of young adults. Infection. 2002;30: 203–207. [DOI] [PubMed] [Google Scholar]
- 91. Johansson N, Kalin M, Tiveljung‐Lindell‐Giske CG, Hedlund J. Etiology of community‐acquired pneumonia: increased microbiological yield with new diagnostic methods. Clin Infect Dis. 2010;50: 202–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Kern DG, Neill MA, Schachter J. A seroepidemiologic study of chlamydia pneumoniae in Rhode Island. Evidence of serologic cross‐reactivity. Chest. 1993;104: 208–213. [DOI] [PubMed] [Google Scholar]
- 93. Goldschmidt P, Rostane H, Sow M, Goépogui A, Batellier L, Chaumeil C. Detection by broad‐range real‐time PCR assay of Chlamydia species infecting human and animals. Br J Ophthalmol. 2006;90: 1425–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Patel KK, Vicencio AG, Du Z, Tsirilakis K, Salva PS, Webley WC. Infectious chlamydia pneumoniae is associated with elevated interleukin‐8 and airway neutrophilia in children with refractory asthma. Pediatr Infect Dis. 2010;29: 1093–1098. [DOI] [PubMed] [Google Scholar]
- 95. Franco RR, Bodanese LC, Repetto G, Piccoli Ida C, Wiehe M, Bonato C, Duarte MM, Duarte T. Inflammatory markers and antichlamydial antibodies in patients with metabolic syndrome. Arq Bras Cardiol. 2011;96: 134–139. [DOI] [PubMed] [Google Scholar]
- 96. Guerders MM, Foidart JM, Noel A, Cataldo DD. Matrix metalloproteinases (MMPs) and tissue inhibitors of MMPs in the respiratory tract: potential implications in asthma and other lung diseases. Eur J Pharmacol. 2006;533: 133–144. [DOI] [PubMed] [Google Scholar]
- 97. Demedts IK, Brusselle GG, Bracke KR, Vermaelen KY, Pauwels RA. Matrix metalloproteinases in asthma and COPD. Curr Opin Pharmacol. 2005;5: 257–263. [DOI] [PubMed] [Google Scholar]
- 98. Olivares‐Zavaleta N, Carmody A, Messer R, Whitmire WM, Caldwell HD. Chlamydia pneumoniae inhibits activated human T lymphocyte proliferation by the induction of apoptotic and pyroptotic pathways. J Immunol. 2011;186: 7120–7126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Kern JM, Maass V, Maass M. Chlamydia pneumoniae adversely modulates vascular cell properties by direct interaction with signalling cascades. Thromb Haemost. 2009;102: 1064–1070. [DOI] [PubMed] [Google Scholar]
- 100. Johnston SL, Martin RJ. Chlamydophila pneumoniae and mycoplasma pneumoniae: a role in asthma pathogenesis? Am J Respir Crit Care Med. 2005;172: 1078–1089. [DOI] [PubMed] [Google Scholar]
- 101. Jackson DJ, Sykes A, Mallia P, Johnston SL. Asthma exacerbations: origin, effect, and prevention. J Allergy Clin Immunol. 2011;128: 1165–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Horvat JC, Starkey MR, Kim RY, Beagley KW, Preston JA, Gibson PG, Foster PS, Hansbro PM. Chlamydial respiratory infection during allergen sensitization drives neutrophilic allergic airways disease. J Immunol. 2010;184: 4159–4169. [DOI] [PubMed] [Google Scholar]
- 103. Crother TR, Schröder NW, Karlin J, Chen S, Shimada K, Slepenkin A, Alsabeh R, Peterson E, Arditi M. Chlamydia pneumoniae infection induced allergic airway sensitization is controlled by regulatory T‐cells and plasmacytoid dendritic cells. PLoS ONE. 2011;6: e20784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Klapdor B, Ewig S, Pletz MW, Rohde G, Schütte H, Schaberg T, Welte T. Community‐acquired pneumonia in the younger is an entity of its own. Eur Respir J. 2012;39: 1156–1161. [DOI] [PubMed] [Google Scholar]
- 105. Pientong C, Ekalaksanan T, Teeratakulpisam J, Tanuwattanachai S, Kongyingyoes B, Limwattananon C. Atypical bacterial pathogen infection in children with acute bronchiolitis in northeast Thailand. J Microbiol Immunol Infect. 2011;44: 95–100. [DOI] [PubMed] [Google Scholar]
- 106. Neumark T, Ekblom M, Brudin L, Groth A, Eliasson I, Mölstad S, Petersson AC, Törngren A. Spontaneously draining acute otitis media in children: an observational study of clinical findings, microbiology and clinical course. Scand J Infect Dis. 2011;43: 891–898. [DOI] [PubMed] [Google Scholar]
- 107. Niang M, Diallo M, Cisse O, Kone M, Doucoure M, Roth JA, Balcer‐Rodrigues V, Dedieu L. Pulmonary and serum antibody responses elicited in zebu cattle experimentally infected with Mycoplasma mycoides subsp. mycoides SC by contact exposure. Vet Res. 2006;37: 733–744. [DOI] [PubMed] [Google Scholar]
- 108. Esposito S, Principi N. Asthma in children: are chlamydia or mycoplasma involved? Paediatr Drugs. 2001;3: 159–168. [DOI] [PubMed] [Google Scholar]
- 109. Cosentini R, Tarsia P, Canett C, Graziadei G, Brambilla AM, Aliberti S, Pappalettera M, Tantardini F, Blais F. Severe asthma exacerbation: role of acute chlamydophila pneumoniae and mycoplasma pneumoniae infection. Respir Res. 2008;30: 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Martin RJ. Infections and asthma. Clin Chest Med. 2006;27: 87–98. [DOI] [PubMed] [Google Scholar]
- 111. Tsai HF, Hsu PN. Interplay between helicobacter pylori and immune cells in immune pathogenesis of gastric inflammation and mucosal pathology. Cell Mol Immunol. 2010;7: 255–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Arnold IC, Dehzad N, Reuter S, Martin H, Becker B, Taube C, Müller A. Helicobacter pylori infection prevents allergic asthma in mouse models through the induction of regulatory T cells. J Clin Invest. 2011;121: 3088–3093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Sevin CM, Peebles RS Jr. Infections and asthma: new insights into old ideas. Clin Exp Allergy. 2010;40: 1142–1154. [DOI] [PubMed] [Google Scholar]
- 114. McLoughlin RM, Mills KH. Influence of gastrointestinal commensal bacteria on the immune responses that mediate allergy and asthma. J Allergy Clin Immunol. 2010;127: 1097–1107. [DOI] [PubMed] [Google Scholar]
- 115. El‐Outob López D. New methods of prevention and treatment of allergic diseases. Recent Pat Inflamm Allergy Drug Discov. 2012;6: 46–64. [DOI] [PubMed] [Google Scholar]
- 116. Vanderhoof JA, Mitmesser SH. Probiotics in the management of children with allergy and other disorders of intestinal inflammation. Benef Microbes. 2012;1: 351–356. [DOI] [PubMed] [Google Scholar]
- 117. Pastacaldi C, Lewis P, Howarth P. Staphylococci and staphylococcal superantigens in asthma and rhinitis: a systematic review and meta‐analysis. Allergy. 2011;66: 549–555. [DOI] [PubMed] [Google Scholar]
- 118. Bachert C, Gevaert P, Zhang N, Zele VT, Perez‐Novo C. Role of staphylococcal superantigens in airway disease. Chem Immunol Allergy. 2007;93: 214–236. [DOI] [PubMed] [Google Scholar]
- 119. Schwerk N, Brinkmann F, Soudah B, Kabesch M, Hansen G. Wheeze in preschool age is associated with pulmonary bacterial infection and resolves after antibiotic therapy. PLoS ONE. 2011;6: e27913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Bisgaard H, Hermansen MN, Buchvald F, et al. Childhood asthma after bacterial colonization of the airway in neonates. N Engl J Med. 2007;357: 1487–1495. [DOI] [PubMed] [Google Scholar]
- 121. Essilfie AT, Simpson JL, Horvat JC, Preston JA, Dunkley ML, Foster PS, Gibson PG, Hansbro PM. Haemophilus influenzae infection drives IL‐17‐mediated neutrophilic allergic airways disease. PLoS Pathog. 2011;7: e100224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Busse WW. Asthma diagnosis and treatment: filling in the information gaps. J Allergy Clin Immunol. 2011;128: 740–750. [DOI] [PubMed] [Google Scholar]
- 123. Gonem S, Desai D, Siddiqui S, Brightling CC. Evidence for phenotype‐driven treatment in asthmatic patients. Curr Opin Allergy Clin Immunol. 2011;11: 381–385. [DOI] [PubMed] [Google Scholar]
- 124. Zarogoulides P, Papanas N, Kioumis I, Chatzaki E, Maltezos E, Zarogoulidis K. Macrolides: from in vitro anti‐inflammatory and immunomodulatory properties to clinical practice in respiratory diseases. Eur J Clin Pharmacol. 2012;68: 479–503. [DOI] [PubMed] [Google Scholar]
- 125. Friedlander AL, Albert RK. Macrolides: from in vitro anti‐inflammatory and immunomodulatory properties to clinical practice in respiratory diseases. Chest. 2010;138: 1202–1212. [DOI] [PubMed] [Google Scholar]
- 126. Good JT Jr, Rollins DR, Martin RJ. Macrolides in the treatment of asthma. Curr Opin Pulm Med. 2012;18: 76–84. [DOI] [PubMed] [Google Scholar]
- 127. Hernando‐Sastre V. Macrolide antibiotics in the treatment of asthma: an update. Allergol Immunopathol (Madr). 2010;38: 92–98. [DOI] [PubMed] [Google Scholar]
- 128. Jedrychowski W, Perera F, Maugeri U, Mroz E, Flak E, Perzanowski M, Majewska R. Wheezing and asthma may be enhanced by broad spectrum antibiotics used in early childhood. Concept and results of a pharmacoepidemiology study. J Physiol Pharmacol. 2011;62: 189–195. [PMC free article] [PubMed] [Google Scholar]
- 129. Murk W, Risnes KR, Bracken MB. Prenatal or early‐life exposure to antibiotics and risk of childhood asthma: a systematic review. Pediatrics. 2011;127: 1125–1138. [DOI] [PubMed] [Google Scholar]
- 130. Lundy SK, Lukacs NW. Chronic schistosome infection leads to modulation of granuloma formation and systemic immune suppression. Front Immunol. 2013;4: 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Flohr C, Quinnell RJ, Britton J. Do helminth parasites protect against atopy and allergic disease? Clin Exp Allergy. 2009;39: 20–32. [DOI] [PubMed] [Google Scholar]