

Summary
Bioluminescence imaging (BLI) is a powerful new method to study virus dissemination in the live animal. Here we used this method to monitor the spatial and temporal progression of mouse hepatitis coronavirus (MHV) infection in mice using luciferase‐expressing viruses. Upon intranasal inoculation, virus replication could initially be observed in the nasal cavity and the cervical lymph nodes, after which the infection spread to the brain and frequently to the eyes. The kinetics of virus spread to and clearance from the brain appeared to depend on the inoculation dose. After intraperitoneal inoculation, virus replication was predominantly observed in the liver and occasionally in the intestines, but interestingly also in the tail and paws. BLI thus elucidated new anatomic locations of virus replication. Furthermore, MHV dissemination was shown to be critically depended on the viral spike protein, but also on the mouse strain used. Widespread dissemination was observed in mice lacking a functional type I interferon response. The importance of the type I interferon system in limiting viral spread was also demonstrated by the administration of type I interferons to mice. Our results provide new insights in coronavirus pathogenesis and demonstrate the potential of BLI to study coronavirus–host interactions in vivo.
Introduction
Current insights into infection processes of pathogens in their hosts and into the dynamics of their spread are largely based on studies using conventional methodologies. These methods have, however, many limitations. They require the experimental animals to be sacrificed in order to identify the sites of infection and to quantify the replication of the pathogens. They hence require large numbers of animals and are costly. They do not allow the real‐time monitoring of spatial and temporal progression of infection in the same animal. Important variations in host–pathogen interactions might therefore be overlooked, while dissemination of a pathogen to unexpected anatomical locations might be missed, simply because the infected tissue is not harvested and analysed (Hutchens and Luker, 2007; Luker and Luker, 2008a). As of recently many of these limitations of the conventional techniques can be overcome by powerful new methods that involve non‐invasive imaging of pathogen replication and spread in infected live animals.
As a member of the Coronavirus (CoV) family, the mouse hepatitis virus (MHV) provides a practical model system for studying CoV‐induced pathogenesis in mice. Depending on the inoculation route, the virus strain and the genetic background of the host, MHV infection can result in a variety of pathological disorders (Compton et al., 1993). The most commonly used laboratory strains primarily infect the liver and the brain, thereby providing animal models for studying encephalitis and hepatitis as well as for immune‐mediated demyelinating disease that sometimes develop during a later stage of infection (Perlman, 1998). Inoculation of susceptible mice, either intracranially or intranasally, with neurotropic MHV strains can result in a number of different outcomes, ranging from acute encephalomyelitis to chronic demyelinating disease (Houtman and Fleming, 1996). Besides a mild encephalitis, MHV strain A59 also causes enteric disease and moderate hepatitis (Lavi et al., 1986). The differences in pathogenesis between various MHV strains have been linked mainly to the spike protein which mediates virus–cell attachment and subsequent membrane fusion (Phillips et al., 1999). However, other viral genes have been shown to also significantly contribute to pathogenesis (de Haan et al., 2002; Sperry et al., 2005; Iacono et al., 2006).
The role of the immune system in response to MHV infection has been extensively studied. Both humoral and cellular immune reactions are essential to guard against MHV infections (Bergmann et al., 2001; Marten et al., 2001; Morales et al., 2001). Clearance of virus during acute infection is predominantly regulated by a typical expression pattern of proinflammatory chemokines that attract CD8+ and CD4+ T lymphocytes to sites of infection (Glass et al., 2002; Lane et al., 2006; Stiles et al., 2006). Also interferon (IFN)‐ and perforin‐mediated mechanisms are involved in the clearance of MHV from different cell types (Weiss and Navas‐Martin, 2005). The kinetics and the extent of the host immune response, which aims to limit the production of infectious virus and viral spread without inducing extensive deleterious effects, is important in determining survival of the host. Infectious virus is usually cleared from MHV‐infected mice within 2 weeks. Most animals, however, do not appear to obtain complete sterile immunity, since viral RNA is often found to persist within the central nervous system (CNS).
Recent biotechnological advances now allow the real‐time imaging of pathogen replication and spread in living animals by making use of bioluminescence imaging (BLI). To this end, light emitted by luciferase reporter proteins is detected by a cooled charge‐coupled device (CCD) camera (Contag et al., 1997; Wu et al., 2001). Important advantages of BLI are an intrinsically low background combined with a very high sensitivity for monitoring light emission in vivo (Contag and Bachmann, 2002; Hutchens and Luker, 2007). Furthermore, the substrate for firefly luciferase (FL), d‐luciferin, is able to cross cellular membranes as well as the intact blood–brain barrier, thereby allowing the imaging of any anatomic location. In addition, d‐luciferin is non‐toxic, allowing the monitoring of individual mice over time through consecutive imaging. Thus, fewer animals are typically needed to acquire statistically meaningful data at multiple time points as compared with conventional approaches (Hutchens and Luker, 2007). Initially, BLI was used to identify sites of bacterial replication in intact animals (Contag et al., 1995). Nowadays, this technique is widely used to study the formation and spread of cancer metastases and to visualize the effect of chemotherapy (Shah et al., 2004). BLI has also been used to study the dissemination of herpes simplex virus, Sindbis virus, Vaccinia virus, and Infectious hematopoietic necrosis virus (Rodriguez et al., 1988; Luker et al., 2002; Cook and Griffin, 2003; Harmache et al., 2006).
All previous studies describing MHV infections and pathogenesis in mouse models have been relying on the sacrifice of infected mice in order to determine virus distribution and titres in various organs over time. Although these conventional approaches have defined important factors that dictate replication and virulence of MHV in vivo as described above, non‐invasive whole‐body imaging of MHV infection in living mice is likely to offer new insights into virus replication, dissemination and pathogenesis. Here, we have taken advantage of BLI to study the replication and spread of FL‐expressing MHV (de Haan et al., 2003; 2005a) in mice in real‐time. We were also able to demonstrate differences in virus replication and spread, resulting either from differences in virus doses, mutations in the spike gene, differences in host susceptibility or from the administration of antiviral compounds. In addition, we identified new anatomic sites of virus replication. Our results provide new insights in CoV pathogenesis and demonstrate the potential of BLI to study CoV–host interactions in vivo.
Results
The characteristics of reporter MHVs in vitro and in vivo
In our previous studies we extensively characterized various recombinant MHVs expressing luciferase reporter genes. The insertion of an FL expression cassette at different positions in the viral RNA genome was shown not to appreciably affect virus multiplication in vitro, while the FL expression levels were demonstrated to be a reliable measure for virus replication (de Haan et al., 2003; Verheije et al., 2008). To verify the feasibility of detecting MHV infection in vivo with BLI, we compared two recombinant MHVs containing the luciferase gene at different positions in the viral genome with a recombinant wild‐type MHV‐A59 with respect to virus replication. Therefore, we prepared new stocks of MHV‐EFLM (containing the FL gene as an additional expression cassette between the E and M gene), MHV‐2aFLS (containing the FL expression cassette at the position of the haemagglutinin esterase pseudo‐gene) and wild‐type MHV‐A59 (Fig. 1A). In agreement with the previous studies we observed that the growth characteristics of the luciferase‐expressing viruses in tissue culture were highly similar to that of the parental MHV‐A59 (Fig. 1B), with the FL reporter gene being expressed at high levels for both MHV‐EFLM and MHV‐2aFLS. For the characterization of the FL‐expressing viruses in vivo we used BALB/c mice, which have been shown to be susceptible to MHV infection (Ohtsuka and Taguchi, 1997). As the inoculation route largely determines the dissemination of virus infection in an animal, we made use of both intranasal and intraperitoneal injection. The 6‐ to 8‐week‐old mice were inoculated with 106 tissue culture 50% infectious dose (TCID50) of virus or with phosphate‐buffered saline (PBS) (control) and sacrificed at 5 days post inoculation, at which day virus titres peak (MacNamara et al., 2005). As MHV‐A59 mainly targets the liver and brain (Lavi et al., 1984; Haring and Perlman, 2001), these organs were taken for analyses. Tissue homogenates were prepared and subsequently tested for virus titres, RNA levels and FL activity. In intranasally inoculated animals, we detected high virus titres in the brains of all infected mice, with no disparity between the wild‐type and FL‐expressing viruses (Fig. 2A). Infectious virus could not be recovered from the liver homogenates. Also the viral RNA levels in the brains of the animals, as determined by quantitative reverse transcriptase polymerase chain reaction (RT‐PCR), were indistinguishable between groups. However, viral RNA in the liver could only be detected in wild‐type MHV‐infected mice (Fig. 2B). All mice infected with the FL‐expressing recombinants showed high FL activity in the brain, as well as some FL activity in the liver (Fig. 2C). Although not statistically significant, the FL expression levels of MHV‐2aFLS were somewhat lower than those of MHV‐EFLM, which is consistent with the relative expression levels observed in vitro (de Haan et al., 2003). After intraperitoneal inoculation, infectious virus could only be recovered from the livers of wild‐type MHV‐A59‐infected animals (Fig. 2A). Although viral RNA was detected in the livers of all groups, the levels were approximately a 1000‐fold lower for the FL‐expressing MHV recombinants (Fig. 2B). Furthermore, only in some livers of MHV‐EFLM‐ and MHV‐2aFLS‐infected mice could FL activity above background level be detected. Overall, these data show that the introduction of the FL expression cassette into the genome of MHV‐A59 affected virus multiplication in the liver but, importantly, not in the brain of 6‐ to 8‐week‐old BALB/c mice. High levels of FL activity were measured in the brains of MHV‐EFLM‐ and MHV‐2aFLS‐infected mice at 5 days post infection, indicating that these viruses are attractive candidates to be used for BLI after intranasal inoculation.
Figure 1.
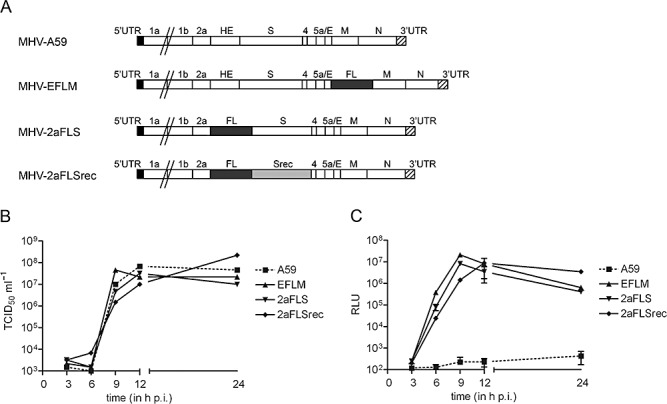
Recombinant viruses with an FL expression cassette at different genomic locations. A. The genomic organization of the recombinant wild‐type MHV (MHV‐A59) and of the recombinant viruses carrying an FL expression cassette. The numbers designate the genes encoding the non‐structural proteins, while genes encoding the spike (S) protein, envelope (E) protein, membrane (M) protein, haemagglutinin‐esterase (HE) protein, and nucleocapsid (N) protein are indicated by their abbreviations. Srec indicates the S gene carrying the mutations that are responsible for the extended host range. The 5′‐ and 3′‐untranslated regions (UTR) are also designated. B. The growth kinetics of the MHV recombinants. LR7 cells were infected with each recombinant MHV at a multiplicity of infection of 1. Viral infectivity in the cell culture media at different times post infection was determined by a quantal assay on LR7 cells, and TCID50 values were calculated. C. In parallel, the intracellular expression of luciferase in relative light units (RLU) was determined as described in the Experimental procedures. Standard deviations (n = 3) are indicated.
Figure 2.
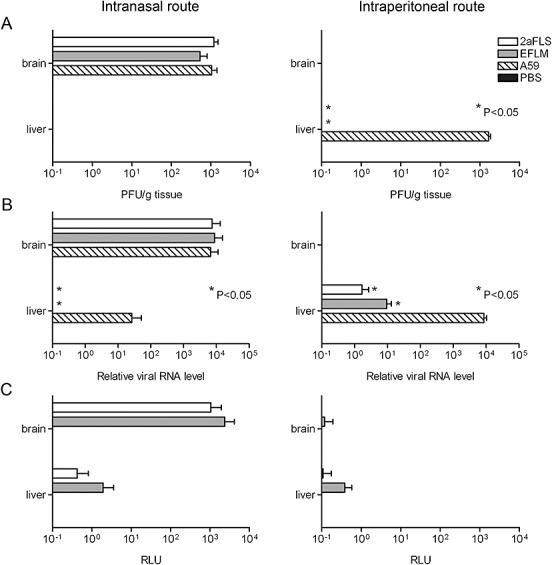
Replication of MHV‐EFLM and MHV‐2aFLS in vivo. The 6‐ to 8‐week‐old female BALB/c mice were inoculated either intranasally or intraperitoneally with PBS or with 106 TCID50 of wild‐type MHV‐A59, MHV‐EFLM or MHV‐2aFLS. At 5 days post inoculation, all mice were sacrificed and brains and livers were collected. Homogenates were prepared as described in the Experimental procedures. A. Viral infectivity in the homogenates was determined by performing a plaque assay on LR7 cells. The viral titres are expressed as plaque‐forming units per gram (PFU g−1) tissue. B. The amounts of viral genomic RNA relative to total RNA, isolated from the liver and brain homogenates, were determined by quantitative Taqman RT‐PCR. C. Luciferase activities (in RLU) in each of the homogenates were determined by using a luminometer as described above. Standard deviations (n = 4) are indicated in all graphs.
Bioluminescence imaging of MHV‐EFLM‐ and MHV‐2aFLS‐infected mice
Thus, we applied whole‐body BLI to study the replication and spread of the two FL‐expressing MHVs. To this end, BALB/c mice were inoculated intranasally with 106 TCID50 of virus after which virus replication was followed over time in individual mice. In general, very similar patterns of virus dissemination were observed for both viruses. At 2 days post inoculation, a strong signal was observed, apparently emanating from the nasal cavity, when the mice were imaged at their dorsal side (Fig. 3A). When imaged from the ventral side, replication additionally could be observed in two distinct spots, probably representing the cervical lymph nodes of the animal. At this time point, no signal coming from the brain could yet be detected. Three days later, dissemination of virus replication to the brain was apparent, while replication in the cervical lymph nodes was no longer observed. At 7 days post inoculation, the signal coming from the brain appeared more dispersed, while its intensity was clearly declining. At 9 days post inoculation, signal from the brain was scarcely detectable in most mice, while it could no longer be detected at day 12 (data not shown). With BLI, it is important to note that the measurable photon flux decreases with increasing depth of the target tissue (Luker and Luker, 2008b). Thus, the signal coming from the brain is significantly attenuated when compared with the signal from the olfactory epithelium, because photons emitted from brain cells must penetrate considerably more tissue to be detected (Fig. 3B). As a consequence, signal intensities should only be compared when photons emanate from the same tissue. Photon quantification of the head regions demonstrated that mice infected with MHV‐EFLM showed significantly higher signal intensities than mice infected with MHV‐2aFLS at 2 days post inoculation (Fig. 3B). In conclusion, with BLI the temporal and spatial spread of MHV replication could be readily visualized in living mice. While mice infected with different luciferase‐expressing viruses displayed very similar patterns of virus dissemination, BLI allowed the quantitative detection of relatively small differences in FL‐expression levels between MHV‐EFLM and MHV‐2aFLS, which is consistent with our earlier observations (de Haan et al., 2003).
Figure 3.
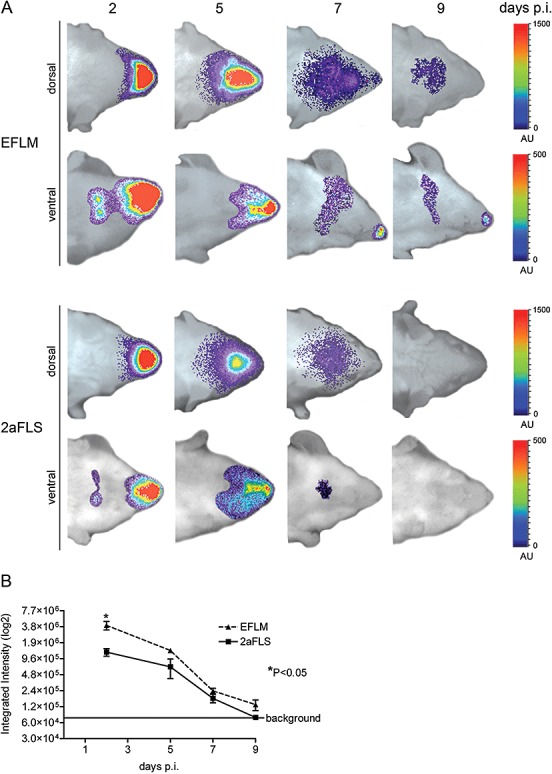
BLI of MHV‐EFLM and MHV‐2aFLS: spatial and temporal progression of infection. The 6‐ to 8‐week‐old female BALB/c mice were inoculated intranasally with 106 TCID50 of either MHV‐EFLM or MHV‐2aFLS. At the indicated times post inoculation (days p.i.), mice were processed for BLI as described in the Experimental procedures. A. Dorsal and ventral images of a representative mouse, infected either with MHV‐EFLM or with MHV‐2aFLS, are shown. The emitted photons were measured by a CCD camera and are displayed as a heat map in an overlay image. Note that the scaling [in arbitrary units (AU)] differs between the dorsal and ventral images. B. The total amounts of emitted photons from the dorsal head regions at the different time points were quantified and are expressed as integrated intensities on a log2 scale with standard deviations (n = 4).
Virus inoculation dose strongly affects intensity and spread of the infection
We next analysed whether we could visualize virus inoculation dose‐dependent effects. To this end, BALB/c mice were infected with two different doses of MHV‐EFLM, a high dose of 5 × 106 and a low dose of 2 × 103 TCID50, and progression of infection was subsequently monitored by BLI at different time points post inoculation (Fig. 4A). As expected, photon quantification of the head regions at 2 days post inoculation demonstrated a clear correlation between the virus inoculation dose and the total signal produced from the olfactory epithelium (Fig. 4B). In addition, spread of virus replication from the nasal cavity into the CNS was more rapidly observed for the mice inoculated with the higher dose. This effect was quantified by dividing the head region into two sections (i.e. anterior versus posterior) after which the relative amount of photons emitted from these sections was determined (Fig. 4C). With a high virus inoculation dose, approximately 50% of the total amount of photons was coming from the posterior part at day 5, while with the low virus dose this degree of dissemination was delayed until around day 7. Consistently, while mice that received the high dose showed virus replication in the cervical lymph nodes after 2 days, this was apparent in the mice inoculated with the low dose only after 5 days (Table S1A). Interestingly, mice infected with the higher dose appeared to clear the virus infection faster than animals infected with the low dose. MHV replication was easily detectable at day 9 in mice inoculated with the low dose, whereas the luciferase signal of the high dose‐inoculated group was almost undetectable at this time point. In some mice infected with a high virus dose, we could additionally visualize infection of the liver and lungs after 2 days, whereas at later time points (9 days), strong signals from the eyes were occasionally detectable. For a summary of the results see Table S1A. In conclusion, virus dissemination was shown to differ in mice infected with a low or a high dose. In mice that received the low dose virus replication was initially restricted to the nasal cavity, while much faster dissemination of virus infection was observed after inoculation with a high dose. Strikingly, and counter intuitively, this rapid dissemination appeared to come at a cost, as these mice were able to clear the infection faster than the mice that received the low dose.
Figure 4.
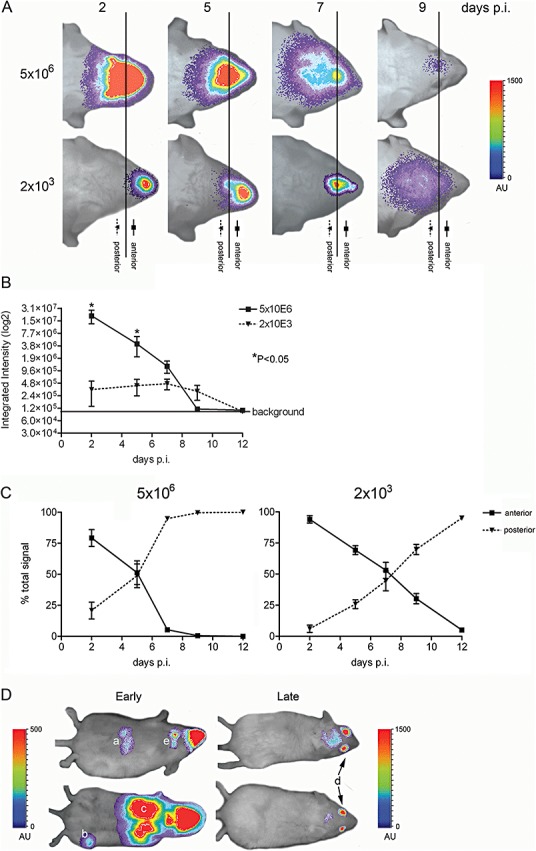
The inoculation dose affects virus dissemination. BALB/c mice were inoculated intranasally with 5 × 106 or 2 × 103 TCID50 of MHV‐EFLM. At the indicated times post inoculation (days p.i.), mice were processed for BLI. A. Dorsal images of a representative mouse are portrayed. The emitted photons were measured at the indicated time points and are displayed as a heat map in an overlay representation. Scaling [in arbitrary units (AU)] is similar in all depicted images. The vertical line in all panels represents the arbitrary border between the anterior (nose) and posterior (brain) regions of the head, which were selected for photon quantification. B. The total amounts of emitted photons from the complete head regions at the different time points were quantified and are expressed as integrated intensities on a log2 scale with standard deviations (n = 4). C. The relative amounts of emitted photons from the anterior and posterior regions of the head (i.e. brain versus nasal epithelium) at the different time points were quantified and are expressed as percentage of the total signal, with standard deviations for both virus doses (n = 4). D. Interesting phenotypes of mice at early (i.e. 2 days; ventral images portrayed) or late (i.e. 9 days; dorsal images portrayed) times post inoculation with 5 × 106 TCID50 of MHV‐EFLM are shown: (a) liver, (b) paw, (c) lung, (d) eye and (e) cervical lymph nodes. Note that the scaling [in arbitrary units (AU)] differs between the ventral and dorsal images.
In vitro adaptation of MHV to heparan sulfate reduces viral replication and spread in vivo
We next analysed the essential contribution of the S protein to virus dissemination by using BLI. The S protein of MHV is a major determinant of pathogenesis and tropism (Phillips et al., 1999). It mediates virus–cell attachment and fusion via binding of the MHV receptor CEACAM1a both in vitro and in vivo (Blau et al., 2001; Hemmila et al., 2004). Schickli et al. (1997) have described a limited set of mutations in the S protein of an MHV‐A59 variant that had been acquired after extensive passaging in cell culture. We have recently shown that these mutations enable the virus to use heparan sulfate as an additional attachment/entry factor. As a result, viruses carrying these mutations appeared to have acquired an extended host range as they were capable of entering cells also in a CEACAM1a‐independent manner (Schickli et al., 1997; de Haan et al., 2005b). The physiological consequences of this adaptation in vivo, however, remain to be elucidated. Here, we examined the infection of mice by an MHV with such an extended host range. Therefore, we made use of a previously described recombinant virus (de Haan et al., 2005b), which contains the mutant spike gene (Srec) and the FL reporter gene. Thus, this recombinant virus MHV‐2aFLSrec only differs from its control virus MHV‐2aFLS in its spike protein. Both viruses replicated to comparable titres in murine LR7 cells as demonstrated by the one‐step growth curve shown in Fig. 1B and displayed similar FL expression kinetics (Fig. 1C). Next, BALB/c mice were inoculated via the intranasal route with these viruses. Clearly, MHV‐2aFLSrec replicated to a lower extent than the parental virus MHV‐2aFLS at all time points measured (Fig. 5A and B). Although the luciferase signal exhibited by the virus with the extended host range increased until 5 days post infection, the infection did not progress towards the CNS as was observed for the control virus. Apparently, the acquisition of a heparan sulfate‐dependent tropism significantly attenuated the ability of the virus to replicate and spread in vivo.
Figure 5.
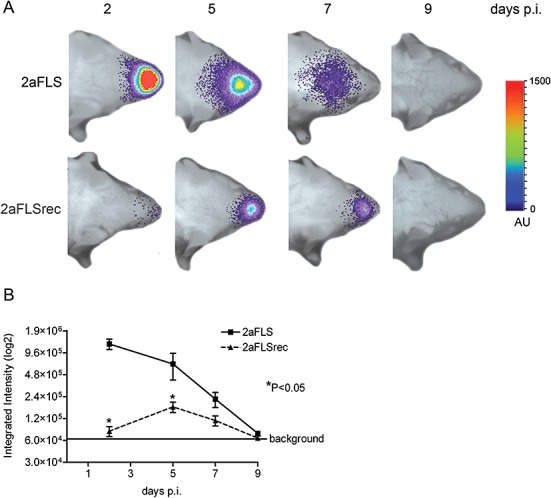
Adaptation to heparan sulfate affects virus replication and spread. The 6‐ to 8‐week‐old female BALB/c mice were inoculated intranasally with 106 TCID50 of MHV‐2aFLS or MHV‐2aFLSrec. At the indicated times post inoculation (days p.i.), mice were processed for BLI. A. The dorsal side of the head region of a representative mouse, infected with either MHV‐2aFLS or MHV‐2aFLSrec, is shown. The emitted photons were measured at the indicated time points and are displayed as a heat map in an overlay image. B. The total amounts of emitted photons from the head regions at the different time points were quantified and are expressed as integrated intensities on a log2 scale with standard deviations (n = 4).
Replication and spread of MHV in mice of different genetic background
Replication of viruses in vivo is not only dependent on the genetic make‐up of the virus, but also that of its host. It has been observed that the susceptibility of inbred mouse strains to different virus infections can vary significantly (Parker et al., 1978; Jubelt et al., 1991; Thach et al., 2000). In order to comparatively evaluate the MHV infection process in mice, we infected three commonly used mouse strains with MHV‐EFLM. To this end, BALB/c, C57BL/6 and 129SvEv mice were inoculated in parallel each with 106 TCID50 via the intranasal route after which virus replication was monitored over time. Clearly, at 5 days post inoculation BALB/c mice displayed the highest signal in the brain, followed by C57BL/6 mice (Fig. 6A and B). Interestingly, 129SvEv mice were significantly less susceptible to MHV‐EFLM infection. In contrast to BALB/c and C57BL/6 mice, virus replication was no longer detectable from 7 days post inoculation in these animals (data not shown). To confirm these observations, we inoculated mice also with MHV‐A59 wild‐type virus and determined the viral RNA load in the brain by quantitative RT‐PCR at 5 days post inoculation. In agreement with the BLI results, BALB/c mice showed the highest viral RNA levels, while the brains from C57BL/6 and 129SvEv mice contained considerably less viral RNA (Fig. 6B).
Figure 6.
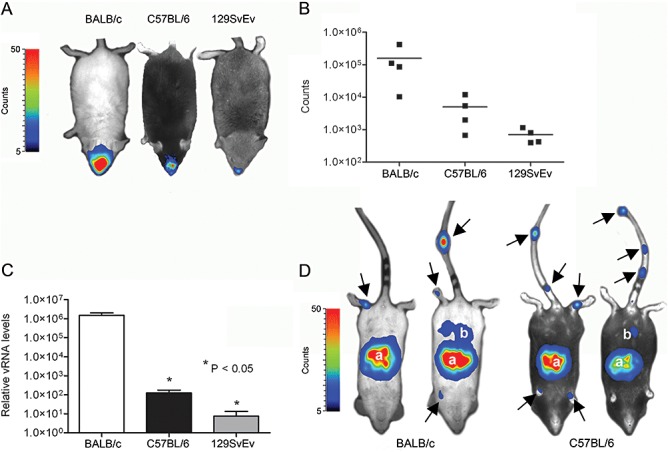
Replication of MHV‐EFLM in mice of different genetic background. The 6‐ to 8‐week‐old female BALB/c, C57BL/6 and 129SvEv mice were inoculated intranasally with 106 TCID50 of MHV‐EFLM. A. BLI was performed at 5 days post inoculation as described in the Experimental procedures. Here, the Biospace photon imager was used as detector for reporter gene expression. The emitted photon counts were measured and are displayed as a heat map in an overlay image. A representative dorsal image is shown for the different mouse strains. B. The emitted photons from the head regions of each individual mouse were quantified and are expressed as the total number of counts measured within the 10 min imaging period. Data are corrected for the background values (signal from uninfected mice). C. Mice infected with wild‐type MHV‐A59 were sacrificed at day 5 post infection, after which the individual brains were collected. Total RNA was isolated and Taqman RT‐PCR was performed targeting MHV genomic RNA sequences. The relative viral RNA (vRNA) levels and standard deviations are depicted (n = 4). D. The 4‐week‐old BALB/c and C57BL/6 mice were inoculated intraperitoneally with 106 TCID50 of MHV‐EFLM. BLI was performed at 2 days post inoculation. Also here, the Biospace photon imager was used. The emitted photons were measured and are displayed as a heat map in an overlay image. Two representative ventral images are shown for the two different mouse strains. The anatomic locations displaying virus replication are indicated and include: (a) liver and (b) intestine. The arrows point to sites of virus replication in the tail and paws of the mice.
Next we studied virus replication and spread of the FL‐expressing virus after intraperitoneal inoculation of BALB/c and C57BL/6 mice. To this end 4‐week‐old mice were inoculated intraperitoneally with 106 TCID50 of MHV‐EFLM (Fig. 5C). At 2 days post inoculation we could visualize virus replication of the liver, with no apparent difference between the two mouse strains. In some mice, infection of the intestine was also observed. Interestingly, in all mice we could observe infection of the tail and paws, hitherto unidentified sites of infection. The signal rapidly decreased in time, with infection no longer being visible at 5–7 days post inoculation (data not shown). These results show that although replication of FL gene‐containing viruses is decreased in 6‐ to 8‐week‐old mice (Fig. 2), which hardly enabled the detection of MHV replication by BLI (data not shown), the infection could easily be monitored in younger mice, which are known to be more susceptible to infection. In addition, the technology allowed the identification of new anatomic sites of MHV replication that had been missed with the conventional techniques (Table S1B).
Enhanced spread of MHV in IFNAR–/– mice
Next, BLI was used to investigate the importance of the antiviral type I IFN system in controlling acute MHV infection. To this end, the effect of IFN alpha receptor knock‐out (IFNAR–/–) was studied using 129SvEv mice. Wild‐type and knock‐out mice were inoculated via the intranasal route with 106 TCID50 of MHV‐EFLM and the infection process was monitored as before. Already at 2 days post infection a significant difference between the IFNAR–/– and wild‐type mice was apparent when the animals were examined from the ventral side (Fig. 7A). A much higher signal was emitted from the nasal cavity in the IFNAR–/– mice compared with the wild‐type mice while, in addition, infection of the cervical lymph nodes was only detected in the knock‐out mice. At later time points, infection was rapidly cleared from the wild‐type mice, whereas dissemination to other organs, including liver and intestine, was manifest in the IFNAR–/– mice. Virus dissemination was accompanied by severe clinical signs, which included a significant decrease in body weight (Fig. 7B). Mice had to be euthanized at 7 days post infection. Interestingly, virus replication in the tail and paws could again be detected in the IFNAR–/– mice, similar to what we had observed after inoculation of BALB/c and C57BL/6 mice via the intraperitoneal route (Fig. 5C). Ex vivo imaging of the abdominal region of the IFNAR–/– mice clearly identified the liver as the major site of MHV‐EFLM replication (Fig. 7C), the organ showing a focal pattern of luciferase expression. This observation was confirmed by the histological analysis of liver sections, as focal lesions were only observed in IFNAR–/– mice, but not in the control mice (Fig. 7D). Overall, these results indicate that mice lacking a functional type I IFN response exhibit disseminated infection after intranasal inoculation with MHV‐A59, which could be readily visualized using BLI. In view of the increasing availability of all kinds of mutant mice, BLI is an attractive approach to investigate the role of a particular host protein or pathway in virus replication and dissemination in vivo.
Figure 7.
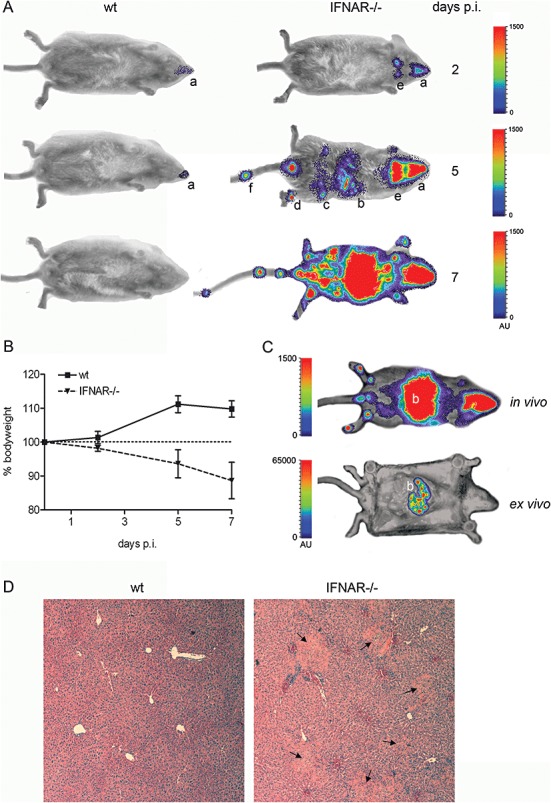
IFNAR–/– mice are highly susceptible to MHV‐EFLM infection. IFNAR–/– or the parental 129SvEv mice were inoculated intranasally with 1 × 106 TCID50 of MHV‐EFLM. At the indicated times post inoculation (days p.i.), mice were processed for BLI. A. Ventral images of a representative mouse are shown. The emitted photons were measured at the indicated time points and are displayed as a heat map in an overlay image: (a) nasal cavity, (b) liver, (c) intestine, (d) paws, (e) cervical lymph nodes and (f) tail. B. The average body weight of both groups of mice is shown as the percentage relative to the initial weight at the beginning of the experiment. Standard deviations are indicated (n = 4). C. In vivo versus ex vivo imaging of an IFNAR–/– mouse infected with MHV‐EFLM at 7 days post inoculation. After the standard BLI procedure, this mouse was sacrificed and immediately processed for imaging of the internal organs. Note that the scaling [in arbitrary units (AU)] in both images is different in order to visualize the focal infection pattern in the liver after ex vivo imaging. D. The liver of the IFNAR–/– mouse in C was subsequently processed for histology as described in the Experimental procedures. As a control, a liver section of a MHV‐EFLM‐infected 129SvEv wild‐type mouse is shown. Typical focal lesions resulting from infection with MHV are indicated by the black arrows.
Recombinant IFNα/β blocks virus spread
As apparently uncontrolled virus dissemination was observed in mice lacking a functional type I IFN response, we next evaluated the anti‐coronaviral effect of exogenous administration of type I IFN. Thus, a cocktail of IFNα/β was applied to BALB/c mice intranasally. It has been established before that IFN can bypass the blood–brain barrier after intranasal delivery (Ross et al., 2004). Following subsequent intranasal inoculation with 2 × 103 TCID50 of MHV‐EFLM, mice were processed for BLI at the indicated time points as described above (Fig. 8A). At day 2, replication of MHV was not significantly affected by the application of IFN. However, while virus replication spread to the brains of the mock‐treated animals, this was not observed in the IFN‐treated mice. Rather, the luciferase signal was lost by day 7. These observations were confirmed by the photon quantification of the head regions of the individual mice (Fig. 8B). Thus, exogenous delivery of type I IFN did not prevent the initial replication in the nose, but prevented dissemination of virus infection to the CNS. The results also indicate that BLI is a promising technique for the non‐invasive screening of antiviral compounds, the major advantage being that this system allows the detection of virus replication and spread after application of an antiviral compound over time within the same animal.
Figure 8.
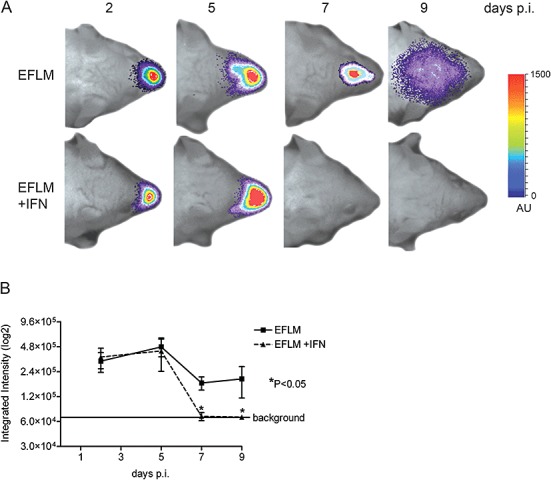
Recombinant IFNα/β inhibits spread of MHV‐EFLM. The 6‐ to 8‐week‐old female BALB/c mice were given either 1000 U of recombinant mouse IFN‐α/β in PBS or PBS alone on each of days −2, −1, 0, 1 and 2 relative to inoculation with MHV‐EFLM. At the indicated times post inoculation, mice were processed for BLI. A. Dorsal images of a representative mouse, inoculated with 2 × 103 TCID50 MHV‐EFLM, are indicated. The emitted photons were measured at the indicated time points and are displayed as a heat map in an overlay image. B. The total amounts of emitted photons from the head regions at the different time points were quantified and are expressed as integrated intensities on a log2 scale with standard deviations (n = 4).
Discussion
Mouse models are essential for defining factors that regulate replication and virulence of viruses. However, the conventional approaches for studying viral infections in mouse models are frequently limited by the need to sacrifice large numbers of animals to quantify viral titres, and establish the complete pattern of virus dissemination. The application of non‐invasive imaging methods for the monitoring of virus infections in living animals can significantly facilitate studies on determinants of viral spread and pathogenesis (Hutchens and Luker, 2007). We used BLI for the real‐time monitoring of MHV infection by using recombinant MHV viruses that express FL reporter proteins. We confirm and extend previous observations by demonstrating that MHV dissemination is critically dependent on the virus inoculation route and dose, the viral spike protein, the genetic background of the host, and the type I IFN system. In addition, the technology provided insights into the kinetics and dynamics of the infection process under these different conditions. Furthermore, BLI revealed an animal‐to‐animal variation in viral spread and elucidated new anatomic locations of virus replication.
Despite its many attractive features for studying MHV replication and spread, the BLI technology has also some inherent limitations. First of all, photon transmission is affected by hair and organ pigmentation. Thus, photon flux from the surface of an animal will be superior to that from an internal organ, because light is attenuated about 10‐fold for every centimetre of tissue through which it has to pass (Contag and Bachmann, 2002). As a consequence it is impossible to quantitatively compare bioluminescent signals derived from different anatomic locations. This phenomenon also explains why the photon flux from the head region at 2 days post infection is much higher than at later time points as the signal initially originates from the nasal cavity but thereafter increasingly derives from the CNS, where it is much more shielded by surrounding tissue. To partly overcome this limitation, we routinely imaged the mice from both the ventral and the dorsal side. Second, the introduction of a foreign gene into the viral genome may affect virus replication in vivo. Interestingly, no significant differences could be observed between the replication of wild‐type and FL‐expressing viruses in the brain of intranasally infected mice, indicating that introduction of the foreign gene did not affect replication in vivo per se. However, after intraperitoneal application the replication of reporter gene expressing viruses in the liver was clearly much lower. The cause of this apparent discrepancy is currently unknown. Importantly, however, FL expression was found at all the known sites of MHV replication (Robbins et al., 1990; Holmes and Lai, 1996; Komurasaki et al., 1996; McIntosh, 1996; Perlman et al., 1999; MacNamara et al., 2008).
The MHV spike protein is an important determinant of pathogenesis (Phillips et al., 1999). Using BLI, we monitored virus replication and spread of a recombinant virus that carried mutations in the spike gene shown to result in an extended host range in vitro due to the virus' ability to enter cells in a CEACAM1a‐independent but heparan sulfate‐dependent manner (Schickli et al., 1997; de Haan et al., 2005b; 2006). Although it has been suggested that host range variants might arise in vivo during persistent infection of tissues that express low levels of the native MHV receptor (Schickli et al., 1997; 2004), this particular host range extension was obtained after serial passaging in vitro. Clearly, the mutant virus replicated to a much lower extent in vivo and was not able to spread to the brain. Many viruses adapt to heparan sulfate as an entry receptor upon propagation of the virus in cell culture (Olmsted et al., 1984; Mandl et al., 2001; Lee et al., 2004). Often, but not always (Liu and Thorp, 2002; de Haan et al., 2008), this adaptation results in a reduction of virulence in living animals, consistent with our results.
Inbred mouse strains are known to differ in their susceptibility to many virus infections (Parker et al., 1978; Jubelt et al., 1991; Compton et al., 1993; Thach et al., 2000). In our experiments we monitored the replication of MHV in three different mouse strains. C57BL/6 and 129SvEv mice appeared to be less susceptible to infection compared with BALB/c mice after intranasal inoculation. Similar results were obtained when mice were infected with wild‐type MHV‐A59 and viral RNA load monitored using quantitative RT‐PCR. Strikingly, MHV replicated to approximately the same extent in BALB/c and C57BL/6 mice after intraperitoneal inoculation. Previously, mouse susceptibility to MHV infection was found to be linked to the viral receptor genotype (Ohtsuka and Taguchi, 1997). However, quantitative RT‐PCR analysis of the CEACAM1a expression levels in brain and liver revealed only relatively small differences in the MHV receptor levels between the different mice strains, which did not correlate with the observed differences in viral replication (data not shown). Apparently, other yet unknown host factors contribute to or limit the dissemination of MHV. The resistance of 129SvEv mice to infection is not restricted to MHV, because also vesicular stomatitis virus replicated much more efficiently in BALB/c than in 129SvEv mice after intranasal inoculation (Durbin et al., 2002). The difference in MHV replication between BALB/c and C57BL/6 mice might be associated with the mouse strain variation in immune responses which have been described for other virus infections (Scalzo et al., 1992; Brenner et al., 1994; Thach et al., 2000; Leipner et al., 2004; Weinberg et al., 2004). In general, C57BL/6 mice have a propensity to elicit a predominant T helper cell 1 response, while BALB/c mice have a tendency to elicit a predominant T helper cell 2 response.
Our results confirm and extend previous observations that dissemination of MHV infection is determined both by the route and by the dose of inoculation. Interestingly, clear differences in the spatio‐temporal dissemination of the infection could be observed after intranasal inoculation with different virus doses. As expected, the virus inoculation dose correlated with the luminescent signal at 2 days post infection. However, inoculation with a low virus dose resulted in delayed spreading of the infection to the CNS and subsequent delayed clearance. This result suggests that a certain level of replication in the nasal epithelium is required for subsequent dissemination. Strikingly, at 9 days post infection higher levels of virus replication could be observed after inoculation with the low dose compared with the high dose. Thus after inoculation with the high dose, virus was cleared faster from the brain. Apparently, clearance of the virus is triggered by the extent of virus replication in the brain. Probably high levels of virus replication more effectively induce an antiviral innate immune response. These results are corroborated by the observation that the expression levels of type I IFN and other cytokines positively correlate with the viral load in the brain of MHV‐infected mice (M. Raaben, unpublished results).
Type I IFN is a fundamental component of the innate immune response, which upon induction elicits an important antiviral signalling cascade that controls and orchestrates the outcome of numerous virus infections (Zuniga et al., 2007). Here, we analysed the full extent of MHV dissemination in mice lacking a functional type I IFN response. These mice appeared to be highly susceptible to MHV and showed severe spread of the infection throughout the body when analysed with BLI after intranasal inoculation. In contrast, the parental 129SvEv animals only showed some low levels of viral replication in the nasal cavity. These results confirm and extend recent studies in which MHV‐A59‐infected IFNAR–/– mice were also shown to be highly susceptible to infection, both after intraperitoneal (Cervantes‐Barragan et al., 2007) and after intracranial inoculation (Roth‐Cross et al., 2008). We additionally demonstrated the importance of the type I IFN system in controlling MHV dissemination by administration of type I IFNs. Intranasal delivery of recombinant type I IFNs inhibited the spread of the infection to the brain, consistent with a previous study using the MHV strain JHM (Minagawa et al., 1987). It was interesting to observe that, while the spread to the brain was affected, the initial replication in the nasal epithelium was not decreased by the intranasally administered IFNs. The experiments demonstrate the power of the BLI technology for studying the effects of antiviral agents on virus replication and dissemination in animal models as was also illustrated earlier (Luker et al., 2002; 2005).
Our observations show that virus dissemination in an organism is a multifactorial process in which the genetic make‐up of pathogen and host, the inoculation dose and route, and the status of the immune system play important roles. In addition, through the application of BLI it has now become evident that, besides the typical, reproducible dissemination characteristics of MHV in mice, clear animal‐to‐animal variation also occurs. Thus, while the infection invariably targets organs like brain, cervical lymph nodes and liver, depending on the inoculation route/dose, other anatomical sites of replication are less frequently detected (see Table S1 for an overview). Occasionally, MHV replication was measured in the lungs, in the intestine or in the eyes. Quite frequently infection was also detected in the tail and paws, body parts not described earlier as sites of MHV replication. These results suggest that MHV can spread to and replicate in bone marrow in vivo, consistent with earlier in vitro studies demonstrating infection of bone marrow‐derived macrophages and dendritic cells (Zhou and Perlman, 2006; Roth‐Cross et al., 2008). Strikingly, MHV replication in tail and paws was never observed throughout the entire length of these extremities, but occurred only in distinct sites that differed between animals. In this respect, the dissemination of MHV is reminiscent of cancer metastases, which often also occur at distinct anatomical locations (Molloy and van't Veer, 2008). Similar to the importance of micro‐environmental host factors in determining the non‐random pattern of tumour localization, replication of MHV may also be enabled by local conditions in a specific micro‐environment.
Experimental procedures
Cells and viruses
LR7 mouse fibroblast cells (Kuo et al., 2000) were maintained in Dulbecco's modified Eagle's medium (Cambrex Bio Science) containing 10% (v/v) fetal calf serum (Bodinco B.V), 100 U ml−1 penicillin, and 100 µg ml−1 streptomycin, supplemented with geneticin G418 (250 µg ml−1). MHV strain A59 and the derivates expressing the FL reporter gene (MHV‐EFLM, MHV‐2aFLS and MHV‐2aFLSrec) have been described previously (de Haan et al., 2003; 2005b). All viruses were grown in LR7 cells. Virus stocks were concentrated by pelleting through sucrose‐cushion centrifugation, and subsequently resuspended in PBS.
Mice
BALB/c and C57BL/6 mice at different ages were obtained from Charles River, while type I IFN receptor knock‐out mice (IFNAR–/–) (Muller et al., 1994) and the parental 129SvEv mice were obtained from B&K Universal Ltd. Mice were inoculated either intraperitoneally or intranasally with various doses of the different viruses. Infected mice were either processed for BLI, or sacrificed at the indicated time points for organ dissection. When indicated, mice were (pre‐)treated with 1000 U of a cocktail of recombinant mouse IFN alpha/beta (IFN‐α/β; Sigma‐Aldrich). The cytokines were applied intranasally on each of days −2, −1, 0, 1 and 2 relative to the inoculation with MHV‐EFLM. Control animals were treated with PBS.
Tissue homogenization and histology
Whole brains and livers were dissected from the MHV‐infected and control mice. The tissues were added to Lysing Matrix D tubes (MP Biomedical), containing 1 ml of PBS, and processed using a FastPrep instrument (MP Biomedical). The tissues were homogenized at 6000 r.p.m. for 40 s and immediately placed on ice. Subsequently, the homogenates were centrifuged at 14 000 r.p.m. for 10 min at 4°C and supernatants were harvested, aliquoted and stored at −80°C. The aliquots were processed for the different assays as described below. When indicated, whole livers from MHV‐infected mice were harvested, fixed in phosphate‐buffered formalin, embedded in paraffin, sectioned and stained with haematoxylin.
Total RNA isolation and quantitative RT‐PCR
Total RNA was isolated from brain and liver homogenates using the TRIzol reagent (Invitrogen) according to the manufacturer's protocol. RNA was further purified using the RNeasy mini‐kit with subsequent DNaseI treatment on the column (Qiagen). The relative amounts of viral genomic RNA were determined by quantitative Taqman RT‐PCR as described before (de Haan et al., 2004).
Virus titration
Titration of virus stocks was performed by determination of the TCID50 in LR7 cells (Verheije et al., 2006). The viral titres in tissue homogenates were determined by plaque assays. Briefly, LR7 cells in 6 well microplates were inoculated with fourfold serial dilutions of the homogenates. At 1 h post inoculation, medium was replaced with an agar overlay. At approximately 48 h post inoculation, the LR7 cell monolayers were fixed and stained with crystal violet, after which the total amount of plaques was counted. The viral titres are expressed as plaque‐forming units per gram (PFU g−1) tissue.
Luciferase assays
Monolayers of MHV‐infected LR7 cells were lysed with 1× Passive Lysis Buffer (Promega). Luciferase expression was measured according to the manufacturer's instructions, and relative light units (RLU) were determined in a Turner Designs TD‐20/20 luminometer. In case of the tissue homogenates, 40 µl was mixed with 40 µl 2× Passive Lysis Buffer and incubated for 5 min at room temperature after which the luciferase expression was measured as described above.
Bioluminescence imaging
When indicated, MHV replication in mice was assessed by in vivo BLI with a highly sensitive, CCD camera (VersArray 1300B, Roper Scientific Inc.) mounted in a light‐tight imaging chamber (Roper Scientific Inc.). Imaging and quantification of signals was controlled by the acquisition software MetaVue (Universal Imaging Corporation). Prior to imaging, mice were anaesthetized by intraperitoneal injection of KXA: ketamine hydrochloride (100 mg kg−1; Vétoquinol BV) plus xylazine (10 mg kg−1; Eurovet Animal Health BV) and atropine (0.05 mg kg−1; Pharmachemie BV). The substrate d‐luciferin sodium salt (Synchem Laborgemeinschaft OHG) dissolved in PBS was injected intraperitoneal at a dose of ∼125 mg kg−1. Mice were positioned in a specially designed box and placed onto the stage inside the light‐tight camera box. Four mice were imaged simultaneously exactly 5 min after the injection of d‐luciferin. The integrated light intensity of a stack of sequential 1 min exposures (10 from the dorsal side and 10 from the ventral side) was used to calculate the amount of emitted light from the animals. A low‐intensity visible light image was made and used to create overlay images. Images were further analysed with Metamorph Imaging software (Universal Imaging Corporation). In some BLI experiments, the detection of emitted photons was performed by the equally sensitive photon imager from Biospace Laboratory. BLI images obtained with the Biospace CCD camera were analysed by Photovision software (Biospace Laboratory).
Supporting information
Table S1A. Anatomical sites of MHV‐EFLM infection in BALB/c mice inoculated with different doses (i.e. 2 × 103 and 5 × 106 TCID50). The numbers of mice from a group of four which showed infection at the indicated anatomical sites are indicated at the different time points post infection (days p.i.).
Table S1B. Anatomical sites of infection in MHV‐EFLM infected mice of different genetic background (i.e. BALB/c, C57BL/6, 129SvEv and IFNAR–/–). Note that mice were inoculated intranasally (i.n.) or intraperitoneally (i.p.) with 106 TCID50 of MHV‐EFLM and subsequently imaged at different time points post inoculation. The numbers of mice from a group of eight or four animals which showed infection at the indicated anatomical sites at any time point post inoculation are indicated.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Acknowledgements
This work was supported by grants from the M.W. Beijerinck Virology Fund, Royal Netherlands Academy of Arts and Sciences, and the Netherlands Organization for Scientific Research (NWO‐VIDI‐700.54.421) to C.A.M. de Haan. We thank Monique Oostra, Marne Hagemeijer and Mijke Vogels for stimulating discussions.
References
- Bergmann, C.C. , Marten, N.W. , Hinton, D.R. , Parra, B. , and Stohlman, S.A. (2001) Cd8 T cell mediated immunity to neurotropic MHV infection. Adv Exp Med Biol 494: 299–308. [DOI] [PubMed] [Google Scholar]
- Blau, D.M. , Turbide, C. , Tremblay, M. , Olson, M. , Letourneau, S. , Michaliszyn, E. et al. (2001) Targeted disruption of the Ceacam1 (Mhvr) gene leads to reduced susceptibility of mice to mouse hepatitis virus infection. J Virol 75: 8173–8186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner, G.J. , Cohen, N. , and Moynihan, J.A. (1994) Similar immune response to nonlethal infection with herpes simplex virus‐1 in sensitive (Balb/C) and resistant (C57bl/6) strains of mice. Cell Immunol 157: 510–524. [DOI] [PubMed] [Google Scholar]
- Cervantes‐Barragan, L. , Zust, R. , Weber, F. , Spiegel, M. , Lang, K.S. , Akira, S. , et al. (2007) Control of coronavirus infection through plasmacytoid dendritic‐cell‐derived type I interferon. Blood 109: 1131–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compton, S.R. , Barthold, S.W. , and Smith, A.L. (1993) The cellular and molecular pathogenesis of coronaviruses. Lab Anim Sci 43: 15–28. [PubMed] [Google Scholar]
- Contag, C.H. , and Bachmann, M.H. (2002) Advances in in vivo bioluminescence imaging of gene expression. Annu Rev Biomed Eng 4: 235–260. [DOI] [PubMed] [Google Scholar]
- Contag, C.H. , Contag, P.R. , Mullins, J.I. , Spilman, S.D. , Stevenson, D.K. , and Benaron, D.A. (1995) Photonic detection of bacterial pathogens in living hosts. Mol Microbiol 18: 593–603. [DOI] [PubMed] [Google Scholar]
- Contag, C.H. , Spilman, S.D. , Contag, P.R. , Oshiro, M. , Eames, B. , Dennery, P. , et al. (1997) Visualizing gene expression in living mammals using a bioluminescent reporter. Photochem Photobiol 66: 523–531. [DOI] [PubMed] [Google Scholar]
- Cook, S.H. , and Griffin, D.E. (2003) Luciferase imaging of a neurotropic viral infection in intact animals. J Virol 77: 5333–5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durbin, R.K. , Mertz, S.E. , Koromilas, A.E. , and Durbin, J.E. (2002) Pkr protection against intranasal vesicular stomatitis virus infection is mouse strain dependent. Viral Immunol 15: 41–51. [DOI] [PubMed] [Google Scholar]
- Glass, W.G. , Chen, B.P. , Liu, M.T. , and Lane, T.E. (2002) Mouse hepatitis virus infection of the central nervous system: chemokine‐mediated regulation of host defense and disease. Viral Immunol 15: 261–272. [DOI] [PubMed] [Google Scholar]
- De Haan, C.A. , Masters, P.S. , Shen, X. , Weiss, S. , and Rottier, P.J. (2002) The group‐specific murine coronavirus genes are not essential, but their deletion, by reverse genetics, is attenuating in the natural host. Virology 296: 177–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Haan, C.A. , Van Genne, L. , Stoop, J.N. , Volders, H. , and Rottier, P.J. (2003) Coronaviruses as vectors: position dependence of foreign gene expression. J Virol 77: 11312–11323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Haan, C.A. , Stadler, K. , Godeke, G.J. , Bosch, B.J. , and Rottier, P.J. (2004) Cleavage inhibition of the murine coronavirus spike protein by a furin‐like enzyme affects cell‐cell but not virus‐cell fusion. J Virol 78: 6048–6054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Haan, C.A. , Haijema, B.J. , Boss, D. , Heuts, F.W. , and Rottier, P.J. (2005a) Coronaviruses as vectors: stability of foreign gene expression. J Virol 79: 12742–12751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Haan, C.A. , Li, Z. , Te Lintelo, E. , Bosch, B.J. , Haijema, B.J. , and Rottier, P.J. (2005b) Murine coronavirus with an extended host range uses heparan sulfate as an entry receptor. J Virol 79: 14451–14456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Haan, C.A. , Te Lintelo, E. , Li, Z. , Raaben, M. , Wurdinger, T. , Bosch, B.J. , and Rottier, P.J. (2006) Cooperative involvement of the S1 and S2 subunits of the murine coronavirus spike protein in receptor binding and extended host range. J Virol 80: 10909–10918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Haan, C.A. , Haijema, B.J. , Schellen, P. , Schreur, P.W. , Te Lintelo, E. , Vennema, H. , and Rottier, P.J. (2008) Cleavage of group 1 coronavirus spike proteins: how furin cleavage is traded off against heparan sulfate binding upon cell culture adaptation. J Virol 82: 6078–6083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haring, J. , and Perlman, S. (2001) Mouse hepatitis virus. Curr Opin Microbiol 4: 462–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmache, A. , LeBerre, M. , Droineau, S. , Giovannini, M. , and Bremont, M. (2006) Bioluminescence imaging of live infected salmonids reveals that the fin bases are the major portal of entry for novirhabdovirus. J Virol 80: 3655–3659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemmila, E. , Turbide, C. , Olson, M. , Jothy, S. , Holmes, K.V. , and Beauchemin, N. (2004) Ceacam1a‐/‐ mice are completely resistant to infection by murine coronavirus mouse hepatitis virus A59. J Virol 78: 10156–10165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes, K.V. , and Lai, M.M.C. (1996) Coronaviruses. In Fields Virology, 3rd edn Fields B.N., Knipe D.M., and Howley P.M. (eds). Philadelphia, PA: Lippincott‐Raven Publishers, pp. 1075–1094. [Google Scholar]
- Houtman, J.J. , and Fleming, J.O. (1996) Pathogenesis of mouse hepatitis virus‐induced demyelination. J Neurovirol 2: 361–376. [DOI] [PubMed] [Google Scholar]
- Hutchens, M. , and Luker, G.D. (2007) Applications of bioluminescence imaging to the study of infectious diseases. Cell Microbiol 9: 2315–2322. [DOI] [PubMed] [Google Scholar]
- Iacono, K.T. , Kazi, L. , and Weiss, S.R. (2006) Both spike and background genes contribute to murine coronavirus neurovirulence. J Virol 80: 6834–6843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jubelt, B. , Ropka, S.L. , Goldfarb, S. , Waltenbaugh, C. , and Oates, R.P. (1991) Susceptibility and resistance to poliovirus‐induced paralysis of inbred mouse strains. J Virol 65: 1035–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komurasaki, Y. , Nagineni, C.N. , Wang, Y. , and Hooks, J.J. (1996) Virus Rna persists within the retina in coronavirus‐induced retinopathy. Virology 222: 446–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo, L. , Godeke, G.J. , Raamsman, M.J. , Masters, P.S. , and Rottier, P.J. (2000) Retargeting of coronavirus by substitution of the spike glycoprotein ectodomain: crossing the host cell species barrier. J Virol 74: 1393–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane, T.E. , Hardison, J.L. , and Walsh, K.B. (2006) Functional diversity of chemokines and chemokine receptors in response to viral infection of the central nervous system. Curr Top Microbiol Immunol 303: 1–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavi, E. , Gilden, D.H. , Highkin, M.K. , and Weiss, S.R. (1984) Persistence of mouse hepatitis virus A59 Rna in a slow virus demyelinating infection in mice as detected by in situ hybridization. J Virol 51: 563–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavi, E. , Gilden, D.H. , Highkin, M.K. , and Weiss, S.R. (1986) The organ tropism of mouse hepatitis virus A59 in mice is dependent on dose and route of inoculation. Lab Anim Sci 36: 130–135. [PubMed] [Google Scholar]
- Lee, E. , Hall, R.A. , and Lobigs, M. (2004) Common E protein determinants for attenuation of glycosaminoglycan‐binding variants of japanese encephalitis and west nile viruses. J Virol 78: 8271–8280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leipner, C. , Grun, K. , Schneider, I. , Gluck, B. , Sigusch, H.H. , and Stelzner, A. (2004) Coxsackievirus B3‐induced myocarditis: differences in the immune response of C57bl/6 and Balb/C mice. Med Microbiol Immunol 193: 141–147. [DOI] [PubMed] [Google Scholar]
- Liu, J. , and Thorp, S.C. (2002) Cell surface heparan sulfate and its roles in assisting viral infections. Med Res Rev 22: 1–25. [DOI] [PubMed] [Google Scholar]
- Luker, G.D. , and Luker, K.E. (2008b) Optical imaging: current applications and future directions. J Nucleic Med 49: 1–4. [DOI] [PubMed] [Google Scholar]
- Luker, G.D. , Bardill, J.P. , Prior, J.L. , Pica, C.M. , Piwnica‐Worms, D. , and Leib, D.A. (2002) Noninvasive bioluminescence imaging of herpes simplex virus type 1 infection and therapy in living mice. J Virol 76: 12149–12161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luker, K.E. , and Luker, G.D. (2008a) Applications of bioluminescence imaging to antiviral research and therapy: multiple luciferase enzymes and quantitation. Antiviral Res 78: 179–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luker, K.E. , Hutchens, M. , Schultz, T. , Pekosz, A. , and Luker, G.D. (2005) Bioluminescence imaging of vaccinia virus: effects of interferon on viral replication and spread. Virology 341: 284–300. [DOI] [PubMed] [Google Scholar]
- McIntosh, K. (1996) Coronaviruses. In Fields Virology, 3rd edn Fields B.N., Knipe D.M., and Howley P.M. (eds). Philadelphia, PA: Lippincott‐Raven Publishers, pp. 401–430. [Google Scholar]
- MacNamara, K.C. , Chua, M.M. , Phillips, J.J. , and Weiss, S.R. (2005) Contributions of the viral genetic background and a single amino acid substitution in an immunodominant Cd8+ T‐cell epitope to murine coronavirus neurovirulence. J Virol 79: 9108–9118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacNamara, K.C. , Bender, S.J. , Chua, M.M. , Watson, R. , and Weiss, S.R. (2008) Priming of Cd8+ T cells during central nervous system infection with a murine coronavirus is strain dependent. J Virol 82: 6150–6160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandl, C.W. , Kroschewski, H. , Allison, S.L. , Kofler, R. , Holzmann, H. , Meixner, T. , and Heinz, F.X. (2001) Adaptation of tick‐borne encephalitis virus to Bhk‐21 cells results in the formation of multiple heparan sulfate binding sites in the envelope protein and attenuation in vivo . J Virol 75: 5627–5637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marten, N.W. , Stohlman, S.A. , and Bergmann, C.C. (2001) MHV infection of the CNS: mechanisms of immune‐mediated control. Viral Immunol 14: 1–18. [DOI] [PubMed] [Google Scholar]
- Minagawa, H. , Takenaka, A. , Mohri, S. , and Mori, R. (1987) Protective effect of recombinant murine interferon beta against mouse hepatitis virus infection. Antiviral Res 8: 85–95. [DOI] [PubMed] [Google Scholar]
- Molloy, T. , and Van't Veer, L.J. (2008) Recent advances in metastasis research. Curr Opin Genet Dev 18: 35–41. [DOI] [PubMed] [Google Scholar]
- Morales, S. , Parra, B. , Ramakrishna, C. , Blau, D.M. , and Stohlman, S.A. (2001) B‐cell‐mediated lysis of cells infected with the neurotropic JHM strain of mouse hepatitis virus. Virology 286: 160–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller, U. , Steinhoff, U. , Reis, L.F. , Hemmi, S. , Pavlovic, J. , Zinkernagel, R.M. , and Aguet, M. (1994) Functional role of type i and type II interferons in antiviral defense. Science 264: 1918–1921. [DOI] [PubMed] [Google Scholar]
- Ohtsuka, N. , and Taguchi, F. (1997) Mouse susceptibility to mouse hepatitis virus infection is linked to viral receptor genotype. J Virol 71: 8860–8863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olmsted, R.A. , Baric, R.S. , Sawyer, B.A. , and Johnston, R.E. (1984) Sindbis virus mutants selected for rapid growth in cell culture display attenuated virulence in animals. Science 225: 424–427. [DOI] [PubMed] [Google Scholar]
- Parker, J.C. , Whiteman, M.D. , and Richter, C.B. (1978) Susceptibility of inbred and outbred mouse strains to sendai virus and prevalence of infection in laboratory rodents. Infect Immun 19: 123–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlman, S. (1998) Pathogenesis of coronavirus‐induced infections. Review of pathological and immunological aspects. Adv Exp Med Biol 440: 503–513. [PubMed] [Google Scholar]
- Perlman, S.R. , Lane, T.E. , and Buchmeier, M.J. (1999) Coronaviruses: hepatitis, hepatitis, and central nervous system disease. Effects Microbes Immune System 1: 331–348. [Google Scholar]
- Phillips, J.J. , Chua, M.M. , Lavi, E. , and Weiss, S.R. (1999) Pathogenesis of chimeric MHV4/MHV‐A59 recombinant viruses: the murine coronavirus spike protein is a major determinant of neurovirulence. J Virol 73: 7752–7760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins, S.G. , Detrick, B. , and Hooks, J.J. (1990) Retinopathy following intravitreal injection of mice with MHV strain JHM. Adv Exp Med Biol 276: 519–524. [DOI] [PubMed] [Google Scholar]
- Rodriguez, J.F. , Rodriguez, D. , Rodriguez, J.R. , McGowan, E.B. , and Esteban, M. (1988) Expression of the firefly luciferase gene in vaccinia virus: a highly sensitive gene marker to follow virus dissemination in tissues of infected animals. Proc Natl Acad Sci USA 85: 1667–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross, T.M. , Martinez, P.M. , Renner, J.C. , Thorne, R.G. , Hanson, L.R. , and Frey, W.H. , 2nd (2004) Intranasal administration of interferon beta bypasses the blood–brain barrier to target the central nervous system and cervical lymph nodes: a non‐invasive treatment strategy for multiple sclerosis. J Neuroimmunol 151: 66–77. [DOI] [PubMed] [Google Scholar]
- Roth‐Cross, J.K. , Bender, S.J. , and Weiss, S.R. (2008) Murine coronavirus mouse hepatitis virus is recognized by mda5 and induces type I interferon in brain macrophages/microglia. J Virol 82: 9829–9838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scalzo, A.A. , Fitzgerald, N.A. , Wallace, C.R. , Gibbons, A.E. , Smart, Y.C. , Burton, R.C. , and Shellam, G.R. (1992) The effect of the Cmv‐1 resistance gene, which is linked to the natural killer cell gene complex, is mediated by natural killer cells. J Immunol 149: 581–589. [PubMed] [Google Scholar]
- Schickli, J.H. , Zelus, B.D. , Wentworth, D.E. , Sawicki, S.G. , and Holmes, K.V. (1997) The murine coronavirus mouse hepatitis virus strain A59 from persistently infected murine cells exhibits an extended host range. J Virol 71: 9499–9507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schickli, J.H. , Thackray, L.B. , Sawicki, S.G. , and Holmes, K.V. (2004) The N‐terminal region of the murine coronavirus spike glycoprotein is associated with the extended host range of viruses from persistently infected murine cells. J Virol 78: 9073–9083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah, K. , Jacobs, A. , Breakefield, X.O. , and Weissleder, R. (2004) Molecular imaging of gene therapy for cancer. Gene Ther 11: 1175–1187. [DOI] [PubMed] [Google Scholar]
- Sperry, S.M. , Kazi, L. , Graham, R.L. , Baric, R.S. , Weiss, S.R. , and Denison, M.R. (2005) Single‐amino‐acid substitutions in open reading frame (Orf) 1b‐Nsp14 and Orf 2a proteins of the coronavirus mouse hepatitis virus are attenuating in mice. J Virol 79: 3391–3400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiles, L.N. , Hardison, J.L. , Schaumburg, C.S. , Whitman, L.M. , and Lane, T.E. (2006) T cell antiviral effector function is not dependent on Cxcl10 following murine coronavirus infection. J Immunol 177: 8372–8380. [DOI] [PubMed] [Google Scholar]
- Thach, D.C. , Kimura, T. , and Griffin, D.E. (2000) Differences between C57bl/6 and Balb/Cby mice in mortality and virus replication after intranasal infection with neuroadapted sindbis virus. J Virol 74: 6156–6161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verheije, M.H. , Wurdinger, T. , Van Beusechem, V.W. , De Haan, C.A. , Gerritsen, W.R. , and Rottier, P.J. (2006) Redirecting coronavirus to a nonnative receptor through a virus‐encoded targeting adapter. J Virol 80: 1250–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verheije, M.H. , Raaben, M. , Mari, M. , Te Lintelo, E.G. , Reggiori, F. , Van Kuppeveld, F.J. , et al. (2008) Mouse hepatitis coronavirus rna replication depends on Gbf1‐mediated Arf1 activation. PLoS Pathog 4: e1000088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg, J.B. , Lutzke, M.L. , Alfinito, R. , and Rochford, R. (2004) Mouse strain differences in the chemokine response to acute lung infection with a murine gammaherpesvirus. Viral Immunol 17: 69–77. [DOI] [PubMed] [Google Scholar]
- Weiss, S.R. , and Navas‐Martin, S. (2005) Coronavirus pathogenesis and the emerging pathogen severe acute respiratory syndrome coronavirus. Microbiol Mol Biol Rev 69: 635–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, J.C. , Sundaresan, G. , Iyer, M. , and Gambhir, S.S. (2001) Noninvasive optical imaging of firefly luciferase reporter gene expression in skeletal muscles of living mice. Mol Ther 4: 297–306. [DOI] [PubMed] [Google Scholar]
- Zhou, H. , and Perlman, S. (2006) Preferential infection of mature dendritic cells by mouse hepatitis virus strain JHM. J Virol 80: 2506–2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuniga, E.I. , Hahm, B. , and Oldstone, M.B. (2007) Type I interferon during viral infections: multiple triggers for a multifunctional mediator. Curr Top Microbiol Immunol 316: 337–357. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1A. Anatomical sites of MHV‐EFLM infection in BALB/c mice inoculated with different doses (i.e. 2 × 103 and 5 × 106 TCID50). The numbers of mice from a group of four which showed infection at the indicated anatomical sites are indicated at the different time points post infection (days p.i.).
Table S1B. Anatomical sites of infection in MHV‐EFLM infected mice of different genetic background (i.e. BALB/c, C57BL/6, 129SvEv and IFNAR–/–). Note that mice were inoculated intranasally (i.n.) or intraperitoneally (i.p.) with 106 TCID50 of MHV‐EFLM and subsequently imaged at different time points post inoculation. The numbers of mice from a group of eight or four animals which showed infection at the indicated anatomical sites at any time point post inoculation are indicated.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item