

Summary
Upon influenza A virus infection of cells, a wide variety of antiviral and virus‐supportive signalling pathways are induced. Phosphatidylinositol‐3‐kinase (PI3K) is a recent addition to the growing list of signalling mediators that are activated by these viruses. Several studies have addressed the role of PI3K and the downstream effector protein kinase Akt in influenza A virus‐infected cells. PI3K/Akt signalling is activated by diverse mechanisms in a biphasic manner and is required for multiple functions during infection. While the kinase supports activation of the interferon regulatory factor‐3 during antiviral interferon induction, it also exhibits virus supportive functions. In fact, PI3K not only regulates a very early step during viral entry but also results in suppression of premature apoptosis at later stages of infection. The latter function is dependent on the expression of the viral non‐structural protein‐1 (A/NS1). It has been shown that PI3K activation occurs by direct interaction of A/NS1 with the p85 regulatory subunit and interaction sites of A/NS1 and p85 have now been mapped in detail. Here, we summarize the current knowledge on influenza virus‐induced PI3K signalling and how this pathway supports viral propagation.
Introduction
Influenza viruses belong to the genus Orthomyxoviridae and three different types (A, B and C type viruses) are known. Among these, influenza A viruses exhibit the broadest host spectrum (birds, humans, other mammals) and can evolve into highly pathogenic strains (Palese and Shaw, 2007).
To ensure efficient replication, influenza viruses hijack factors of the host cell signalling machinery that are actually activated by the cell as a defence mechanism to fight the invader (Ludwig et al., 2006; Ludwig, 2007). With regard to antiviral events, signalling pathways that control induction of antiviral cytokines such as type I interferons (IFN α/β) are induced, providing a first line of defence to viral infections. One way to counteract these responses is the expression of the viral non‐structural protein 1 (NS1) that suppresses the IFN response by both, interfering with signalling inducers and mediators (Garcia‐Sastre, 2004; Wolff et al., 2008), and by inhibiting processing of cellular pre‐mRNA (reviewed in Krug et al., 2003). Thereby, the IFNα/β and cytokine induction is kept in a tolerable limit (reviewed in Hale et al., 2008a). Based on these functions A/NS1 is primarily known as a signalling blocker. Influenza B viruses also express an NS1 (B/NS1). While A/NS1 and B/NS1 share less than 20% sequence identity, they fulfil similar but not identical functions. B/NS1 also acts as an IFN antagonist (Talon et al., 2000; Garcia‐Sastre, 2004) and inhibits the IFN inducible protein kinase R (Dauber et al., 2004; 2006). In contrast to A/NS1, B/NS1 is not capable to inhibit polyadenylation, splicing and nuclear export of cellular mRNA (Wang and Krug, 1996).
Recently, the phosphatidylinositol‐3‐kinase (PI3K) and its downstream effector Akt/protein kinase B (PKB) were identified as influenza A virus‐induced signalling mediators (Ehrhardt et al., 2006; Hale et al., 2006; Shin et al., 2007a; Zhirnov and Klenk, 2007). PI3K was initially described to act in an antiviral fashion (Sarkar et al., 2004; Ehrhardt et al., 2006). However, later it became obvious that PI3K/Akt signalling is also actively induced by the viral A/NS1 protein to support efficient replication (Ehrhardt et al., 2006; 2007a; Hale et al., 2006; Shin et al., 2007a; Zhirnov and Klenk, 2007).
The present article provides a brief introduction to PI3K biology followed by a short overview of the role of PI3K and Akt in virus‐infected cells in general. Finally, the recent findings regarding influenza virus‐induced PI3K activation and function during different stages of replication will be discussed in detail.
The PI3K
The family of PI3Ks comprises three main classes (class IA and IB, II and III) according to their lipid substrate specificity, in vitro (reviewed in Vanhaesebroeck et al., 2001). The class IA PI3Ks are heterodimeric enzymes consisting of a regulatory (p85) and an enzymatic subunit (p110). These kinases exhibit both protein kinase and lipid kinase activity (Dhand et al., 1994). In mammalian cells three class IA catalytic (p110 α, β and δ) and five regulatory (p85α, p85β, p55γ, p55α and p50α) subunits are known (Vanhaesebroeck et al., 2005). The p85 regulatory subunits can function as stabilizer and inhibitor of the p110 subunits (Vanhaesebroeck et al., 2005). PI3K is activated, for example, by binding of the regulatory subunit p85 to autophosphorylated tyrosine‐kinase receptors or G‐protein coupled receptors (reviewed in Wymann and Pirola, 1998) (Fig. 1). The major role of the kinase is to phosphorylate membrane phospholipids. Upon PI3K activation phosphatidylinositol‐3,4,5‐triphosphate (PIP3) is generated by phosphorylation of phosphatidylinositol‐4,5‐bisphosphate in the membrane, which in turn functions as a second messenger through interaction with pleckstrin homology domain‐containing proteins such as Akt/PKB and phosphoinositide‐dependent kinase (PDK)‐1 (Neri et al., 2002).
Figure 1.
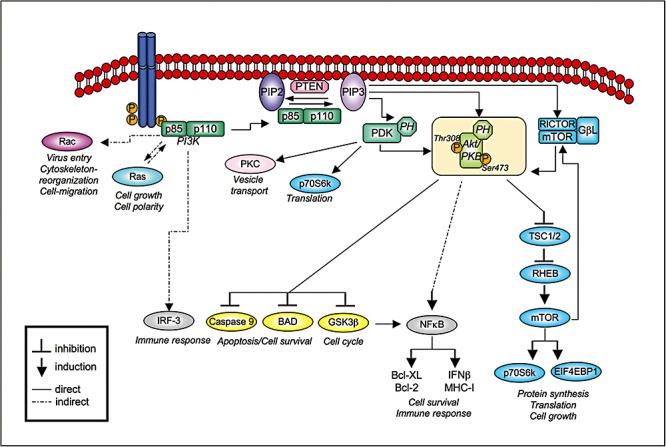
Schematic presentation of PI3K‐dependent signalling pathways. For details see the text. Abbreviations not mentioned in the text: TSC1/2, TSC 1 and 2 genes; RHEB, Ras‐homologue enriched in brain); EIFEBP1, eukaryotic initiation factor 4E binding protein 1.
Activation of Akt/PKB
Akt/PKB is a major PI3K effector and its full activation requires phosphorylation at Thr308 and Ser473 (Fig. 1). Thr308 is phosphorylated by 3′‐PDK that itself is a PI3K effector. For phosphorlyation of Ser473, different kinases were discussed. Recently, the mTOR kinase associated with rictor (rapamycin‐insensitive companion of mTOR) and the small Gβ‐like protein GβL have been identified to build the mTORC2 complex that is responsible for phosphorylation at Ser473 (Sarbassov et al., 2005). Although Akt is not phoshorylated directly by PI3K, these post‐translational modifications occur strictly dependent on PI3K activity. Therefore, detection of phosphorylated Akt at Ser473 is commonly used to monitor the activation state of PI3K.
Cellular functions of PI3K – interplay with different signalling mediators
Besides the activation of the major PI3K effector Akt, PI3K further cross‐talks with a vast amount of other signalling mediators (reviewed in Carracedo and Pandolfi, 2008). In general, PI3K regulates various cellular events, such as cell metabolism, proliferation and survival (reviewed in Neri et al., 2002; Vanhaesebroeck et al., 2005). Here, only some regulatory key players involved in PI3K signalling are discussed (Fig. 1). A vivid cross‐talk between PI3K/Akt‐mTOR and the Ras/Raf/MEK/ERK signalling is crucial to regulate cell differentiation and survival (Carracedo and Pandolfi, 2008). Regulation of endocytosis and vesicular trafficking via Rac1 activation are further functions described for PI3K that are required for ligand internalization (Saeed et al., 2008). PDK1 activates, in addition to Akt, several other AGC protein kinase family members (cAMP, cGMP, Ca2+‐dependent), such as p70 S6 kinase and protein kinase C to control vesicular transport and translation (Kikani et al., 2005). Regulation of apoptosis is also an important function ascribed to PI3K/Akt. Activated Akt has been reported to inactivate proapoptotic factors such as BAD, caspase‐9 and glycogen synthase kinase‐3β (GSK‐3β) by phosphorylation. Akt also acts as a co‐regulator of transcription factors, such as NF‐κB, leading to the synthesis of antiviral acting proteins, such as IFN regulatory factor I and MHC class I proteins or anti‐apoptotic Bcl‐2 family members (Bcl‐2 and Bcl‐XL) (Cooray, 2004).
Feedback regulation of PI3K signalling
Activated PI3K is negatively regulated by phosphatases or by PI3K‐induced downstream components regulating feedback loops (Carracedo and Pandolfi, 2008). Among the phosphatases, PTEN (phosphatase of tensin homologue deleted on chromosome 10) is likely to be the most important enzyme to control the PI3K pathway activity (Fig. 1). PTEN harbours protein as well as lipid phosphatase activities. It dephosphorylates phosphoinositides in position 3′ of the inositol ring (Maehama and Dixon, 1998). The levels of PTEN and its activity are positively and negatively regulated on transcriptional, as well as on post‐translational level by phosphorylation, ubiquitylation, oxidation and acetylation (reviewed in Tamguney and Stokoe, 2007). The most prominent PI3K/Akt‐induced feedback regulator is the mTOR kinase, which forms complexes with raptor (regulatory‐associated protein of mTOR) – mTORC1 (Hara et al., 2002) and rictor (rapamycin‐insensitive companion of mTOR) – mTORC2 (Sarbassov et al., 2004). Hyperphosphorylation of mTORC1, for exmaple, by loss of PTEN, results in repression of PI3K whereas inhibition of mTORC1 leads to PI3K activation (Carracedo and Pandolfi, 2008).
The role of PI3K in virus‐infected cells
PI3K signalling in DNA virus‐infected cells
During the last decade, many viruses were found to induce PI3K/Akt signalling and thereby suppress apoptosis of the infected cells through expression of viral gene products (Cooray, 2004). The majority of reports focused on DNA tumour viruses as cell death suppression is crucial for tumour growth. Thus, the role of PI3K was studied in the context of survival during chronic and latent infections (Cooray, 2004). The viral LMP1 protein of Epstein‐Barr virus regulates B‐cell survival, the viral E5 protein of Papillomavirus functions as an apoptosis supressor in epithelial cells and the viral HBx protein of Hepatitis B viruses inhibits TGF‐β‐induced apoptosis in hepatoma cells (Cooray, 2004). All these viral proteins induce PI3K signalling that contributes to their cell death suppressing function.
PI3K signalling during chronic infection with RNA viruses – prevention of apoptosis in hepatitis C virus‐infected cells
Hepatitis C virus (HCV), an RNA virus that can cause chronic infection and cell transformation in the liver, also activates PI3K (Cooray, 2004). HCV belongs to the family of Flaviviridae. The virus has a single‐stranded RNA genome with positive orientation (Lemon et al., 2007). Initially, the non‐structural protein 5 A (NS5A) of HCV was shown to induce p85 phosphorylation by complex‐formation with Grb2‐associated binder 1 (He et al., 2002), a substrate of the epidermal growth factor receptor. These results have been extended by observations that NS5A directly binds to the src homology (SH) 3 domain of p85. Binding is mediated by a proline‐rich motif in the NS5A protein and results in activation of the kinase (Street et al., 2004). Investigation of downstream factors of Akt indicated a NS5A‐induced regulation of the Forkhead transcription factor FKHR and GSK‐3β and accumulation of cellular beta‐catenin. This results in stimulation of beta‐catenin‐responsive transcription, which is probably involved in development of hepatocellular carcinoma (Street et al., 2005). Furthermore, the induction of the PI3K/Akt‐mTOR signalling upon HCV infection was reported to control cell survival, and thereby also determines apoptosis (Mannova and Beretta, 2005).
Multiple functions of PI3K signalling during acute infections with RNA viruses
Apoptosis suppression. RNA viruses also interfere with PI3K activity during acute infections. The respiratory syncytial virus (RSV) manipulates PI3K activity to ensure virus replication by prevention of apoptosis, although a viral product responsible for PI3K induction has not been specified (Cooray, 2004). First, RSV was shown to regulate NF‐κB‐transcriptional activity dependent on PI3K/Akt. In addition, RSV infection caused enhanced apoptosis upon PI3K inhibition (Thomas et al., 2002). Recent investigations showed that RSV decreases p53 expression, thereby extending survival of the infected cells (Groskreutz et al., 2007). P53 is a tumour suppressor and induces expression of pro‐apoptotic factors. The murine double minute protein 2 is a p53 antagonist, which is phosphorylated by Akt upon RSV infection and leads to p53 degradation (Groskreutz et al., 2007). In influenza virus‐infected cells p53‐dependent apoptosis is induced at late stages of infection, but murine double minute protein 2 upregulation was not detected (Zhirnov and Klenk, 2007).
In addition, coxsackie B virus (Zhang et al., 2003), SARS coronavirus (Mizutani et al., 2004), rubella virus (Cooray et al., 2005), dengue virus, Japanese encephalitis virus (Lee et al., 2005), poliovirus (Autret et al., 2008) and influenza A virus (reviewed here) have been added to the growing list of RNA viruses, which utilize PI3K signalling to prevent apoptosis during replication.
Virus entry. Besides the anti‐apoptotic function of PI3K during viral infection, the kinase also promotes the process of viral entry. A recent publication demonstrates the requirement of PI3K activity during endocytic uptake of Ebola viruses (Saeed et al., 2008) that are enveloped negative‐strand RNA viruses belonging to the family of Filoviridae (Sanchez et al. 2007). Inhibition of PI3K/Akt suppressed infection by Ebola virus at an early step during virus replication (Saeed et al., 2008). As UV‐inactivated virus also induced Akt phosphorylation shortly upon inoculation of cells, an activation mechanism independent from virus replication was suggested (Saeed et al., 2008). Examination of downstream targets of PI3K/Akt identified the small GTPase Rac1 as additional factor regulating vesicular trafficking necessary for Ebola virus entry (Saeed et al., 2008).
These results are in line with earlier observations that PI3K is crucial for uptake of influenza A virus (Ehrhardt et al., 2006), a study that is further discussed in the next paragraphs.
Activation and function of PI3K in influenza virus‐infected cells
PI3K activation by influenza viruses is a rather complex issue. The kinase is activated by diverse mechanisms and plays multiple roles during infection (see Fig. 2). Directly upon infection the kinase is activated in a short and transient fashion, followed by a second phase of sustained activation later in the infection cycle (Ehrhardt et al., 2006). While early activation is also observed upon infection with influenza B viruses, late activation only occurs with A type viruses (Ehrhardt et al., 2007b).
Figure 2.

PI3K activation and function in the influenza virus‐infected cell. For details see the text.
Antiviral functions of PI3K
Signalling profiling studies of infected cells using the PI3K inhibitor (LY294002) indicated that virus‐induced IL‐8‐ and RANTES‐secretion is dependent on PI3K (Guillot et al., 2005). Furthermore, inhibition of PI3K or blocking of its effector PIP3 results in a misphosphorylation of IRF‐3 and impaired transcriptional activation of the IFNβ promoter (Ehrhardt et al., 2006). These results corresponded with earlier findings that PI3K is involved in phosphorylation and activation of IRF‐3 in response to TLR3 engagement upon double‐stranded RNA stimulation (Sarkar et al., 2004). Interestingly, this occurred independent of the IRF‐3 kinases, Tank‐binding‐kinase‐1 or IκB kinase epsilon. Thus, it was suggested that coactivation of IRF‐3 most likely is mediated by phosphorylation of a different target phosphorylation site on IRF‐3 (Sarkar et al., 2004). Consequently, these observations indicate for an alternative PI3K‐dependent signalling mechanism besides the canonical RIG‐I/Tank‐binding‐kinase‐1/IκB kinase epsilon/IRF‐3 signalling module that is required for full activation of IRF‐3 in influenza virus‐infected cells.
In the context of in vivo infection, an indirect antiviral function of PI3K has been unravelled. Here, virus‐induced expression of the chemokine (C‐C motif) ligand 5 (Ccl5) results in induction of PI3K/Akt signalling in murine tissue macrophages via the according receptor (Tyner et al., 2005). Activated PI3K/Akt prevents apoptosis of macrophages and allows removal of virus‐infected and apoptotic cells from infected surrounding tissue (Tyner et al., 2005).
Functions of PI3K that promote virus replication
Besides the reported antiviral activities, PI3K also exhibits virus supportive functions in the context of influenza A virus infection. This became evident by significantly decreased progeny virus titers from cells where PI3K or PIP3 were inhibited (Ehrhardt et al., 2006). Thus, PI3K activity appears to be beneficial for virus propagation. In fact, the virus supportive events dominate the previously reported antiviral activity.
Virus entry
Further experiments revealed that PI3K targets a very early step of the viral replication cycle (Ehrhardt et al., 2006). Immunofluorescence microscopy analysis indicated that PIP3 and PI3K are involved in regulation of the virus uptake, as upon inhibition of PI3K, virus particles accumulated at the cell surface. In addition, they failed to traffic to early endosomes during receptor‐mediated endocytosis (Ehrhardt et al., 2006). Thus, PI3K and PIP3 appear to control a step that precedes endosomal transport (Ehrhardt et al., 2006), which most likely is the initial virus uptake at the membrane of cells.
Prevention of premature apoptosis
Notwithstanding PI3K's role in entry, additionally virus supportive functions at later stages of infection are required (Ehrhardt et al., 2006). Initially, it was not clear which cellular or viral components activate PI3K late in the infection cycle. First evidence on the activating mechanism resulted from observations that PI3K is barely activated at later stages of infection in cells infected with the deltaNS1 virus, a virus mutant that lacks the IFN antagonistic A/NS1 protein (Ehrhardt et al., 2006). This was a surprising finding, as signalling pathways usually were found to be hyperactivated upon infection with deltaNS1. Hence, it was suggested that the A/NS1 protein acts as an efficient and broad signalling suppressor. The absence of PI3K activation in deltaNS1‐infected cells led to the intriguing conclusion that A/NS1 itself may induce PI3K‐mediated signalling (Ehrhardt et al., 2006). This hypothesis was subsequently verified by four independent studies, which showed that the A/NS1 protein is required and sufficient for activation of the kinase (Hale et al., 2006; Ehrhardt et al., 2007a; Shin et al., 2007a; Zhirnov and Klenk, 2007). Furthermore, it was demonstrated by different technical approaches that A/NS1 is complexed with the regulatory subunit p85, an event that probably leads to activation of PI3K (Hale et al., 2006; Ehrhardt et al., 2007a; Shin et al., 2007a).
Consistent with the finding that influenza B virus infection failed to induce late activation of PI3K, B/NS1 proteins did neither bind to PI3K nor activate the kinase (Ehrhardt et al., 2007b). Thus, the inability of B/NS1 to induce PI3K unravels another exclusive function of the viral A/NS1 that is not matched by its influenza B virus counterpart.
Given the exciting finding that A/NS1 binds and efficiently activates PI3K, the question arose for which virus supportive function the kinase may be required at advanced stages of infection. Earlier examinations demonstrated that A/NS1 exhibits anti‐apoptotic activity in the context of influenza virus infection (Zhirnov et al., 2002). Several recent studies identified PI3K and Akt as molecular mediators of this process as A/NS‐1‐induced PI3K/Akt activation was shown to control phosphorylation and therefore inhibition of pro‐apoptotic factors, such as caspase‐3, caspase‐9 and GSK‐3β, which finally resulted in suppression of premature apoptosis (Ehrhardt et al., 2007a; Shin et al., 2007b; Zhirnov and Klenk, 2007). These findings provided new insights into the molecular mode of the anti‐apoptotic activity of A/NS1 that was previously thought to be exclusively due to the type I IFN antagonistic function of the protein (Zhirnov et al., 2002).
Other proposed functions of PI3K late in the infection cycle
Suppression of apoptosis might not be the only virus supportive function of PI3K during ongoing replication. One report showed that in cells treated with the PI3K inhibitor LY294002 viral RNA and protein synthesis as well as nuclear export of the RNPs were impaired (Shin et al., 2007c). Further, Zhirnov and colleagues observed a correlation between Akt phosphorylation and virus particle production, leading to the conclusion that they could not exclude an involvement of PI3K in virus assembly (Zhirnov and Klenk, 2007). Thus, there might still be other so far unidentified functions of the kinase during infection.
Activation of PI3K by the influenza virus A/NS1 protein – identification of motifs and domains responsible for interaction and activity
A number of studies focused on the precise mapping of interaction sites of A/NS1 and p85 polypeptides. The A/NS1 protein harbours two well‐characterized functional domains (reviewed in Hale et al., 2008a). An RNA binding domain is located within the N‐terminal 73 amino acids (aa) and a so‐called effector domain is located between the RNA binding domain and the last 20–30 aa in the C‐terminus of the protein (Hale et al., 2008a). Furthermore, three characteristic motifs common to well‐known binding consensus sequences have been identified. Two polyproline‐rich sequences PXXP are located at aa 164–167 and 213–216, representing putative binding motifs for SH3 domain proteins. One YXXXM motif similar to tyrosine‐bearing SH2 binding motifs (YXXM) is located at aa 89–92 (Hale et al., 2008a) (Fig. 3). SH2 and SH3 domains are common protein–protein interaction modules of signalling proteins, and are also present in the regulatory subunit p85 of PI3K. P85 harbours one tandem SH2 domain (SH2N, SH2C), which flank the inter‐SH2 domain and one N‐terminal SH3 domain (Vanhaesebroeck et al., 2005) (Fig. 3).
Figure 3.
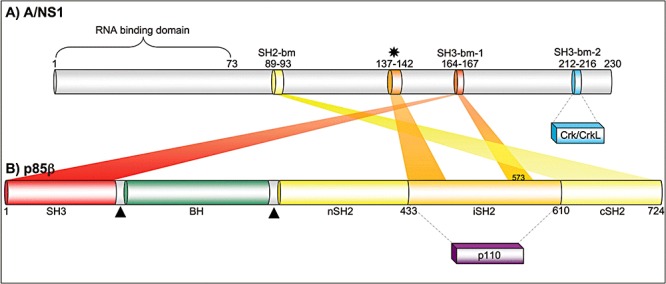
Schematic illustration of the binding domain structure of A/NS1 and p85β. A. Influenza A virus NS1 encompasses up to 237 aa, depending on the strain. The RNA binding domain is located within the first 73 aa and three putative binding motifs (bm) for SH domains have been identified (SH2‐bm, SH3‐bm‐1, SH3‐bm‐2) (presumed motifs for A/NS1 interaction with p85β or Crk/CrkL are depicted). The asterisk indicates the binding sequence responsible for interaction with the inter‐SH2 domain (iSH2) domain of p85β. B. The regulatory subunit p85β encompasses 724 aa and harbours one N‐terminal SH3 domain, two SH2 domains flanking the iSH2, one BH (Bcr/Rac GAP homology) domain and two proline‐rich motifs (triangle) (Vanhaesebroeck et al., 2005). Interaction of the N‐terminal SH3 domain of p85β with A/NS1 was shown to be stronger than with the C‐terminal SH2 domain. Recently, the iSH2 domain has been identified as additional interaction site for A/NS1, whereby the Valine 573 seems to be the most important aa responsible for interaction.
The SH2 motif of A/NS1
Mutational analysis revealed that A/NS1 directly binds to the β isoform but not the α isoform of p85 involving a conserved tyrosine residue at aa 89 and a methionine residue at aa 93 that are most likely recognized by the SH2 domain of the regulatory PI3K subunit (Hale et al., 2006; Shin et al., 2007a). The viruses mutated at position tyrosine 89 (Y89F) formed smaller plaques and showed reduced virus propagation (Hale et al., 2006).
The SH3 motifs of A/NS1
Shin et al. (2007b) demonstrated by GST‐pull down assays that besides the SH2 binding motif at Y89, also the SH3 binding motif of NS1 is required for p85 binding. In a subsequent study recombinant virus strains with mutations in the SH3 motif of A/NS1 were generated. This included proline replacements in the first SH3 binding domain at aa 164 and 167 and/or in the second domain at aa 212, 213 and 216 of A/NS1 (Shin et al., 2007b). The mutant viruses displayed decreased replication fitness, smaller plaque size and the lack of Akt activation in the case of the aa 164–167 mutant (Shin et al., 2007b). Further studies showed that the interaction is mainly mediated by the first SH3 motif in the A/NS1 protein (Shin et al., 2007a).
Taken together, these studies highlight the importance of both the N‐terminal SH2 binding motifs and the first SH3 binding motif of A/NS1 (aa 164–167) for p85 binding.
The different A/NS1 binding domains of p85β
In a recent study, A/NS1 was found to coimmunoprecipitate both p85β and p85α (Ehrhardt et al., 2007a). However, complex‐formation of p85α and A/NS1 was only observed in infected but not in transfected cells (Ehrhardt et al., 2007a), suggesting either an indirect association of the viral protein with p85α or a requirement of cofactors in infected cells. Thus, p85β is most likely the only direct binding subunit of A/NS1. Within p85β, its SH3 domain and to a lesser extent the C‐terminal SH2 domain appeared to be required for interaction with the viral protein (Shin et al., 2007a). Recent reports revealed an additional interaction of A/NS1 with the inter‐SH2 domain of p85β around Valine 573 (Hale et al., 2008b; Li et al., 2008). While this domain also interacts with the catalytic subunit p110 of PI3K, A/NS1 did not compete with p110 for binding (Hale et al., 2008b; Li et al., 2008).
The multiple sites of A/NS1 and p85β that were reported to contribute to their interaction are summarized in Fig. 3.
Interaction of the adaptor proteins Crk/CrkL with A/NS1
Recently, the adaptor proteins Crk and CrkL have been identified as binding partners for A/NS1 proteins (Heikkinen et al., 2008). A/NS1 binds to Crk/CrkL via a specific class II SH3 domain binding motif 2 (PPLPPK) located at aa 212–217 (Heikkinen et al., 2008). This second SH3 binding motif is conserved in NS1 proteins of a number of avian influenza A viruses but with a few exceptions, not in human influenza A virus strains. Binding‐disruption of Crk and A/NS1 resulted in reduced PI3K activity (Heikkinen et al., 2008), indicating that Crk or CrkL are crucial cofactors for A/NS1‐mediated PI3K activation, at least in the case of avian influenza strains. While the formation of a large ‘signalosome’ of Crk/CrkL‐p110/p85‐A/NS1 was hypothesized (Heikkinen et al., 2008), the precise role of the complex‐formation between avian A/NS1 and Crk/CrkL in PI3K induction is not resolved and PI3K‐independent functions of the interaction should also be considered (Hale et al., 2008a).
Conclusion
A number of evidence indicated that PI3K activation is induced by influenza A virus infection by diverse mechanisms at different time points during replication. The kinase fulfils several functions in the infected cell (summarized in Fig. 2). An early and transient peak of activity is observed during viral attachment to cells. Later in the infection cycle the kinase is activated in a more persistent manner due to expression of the viral A/NS1 protein. With regard to the virus supportive function of PI3K, there is an early requirement of the kinase for efficient viral entry. Similar mechanisms have already been observed for Ebola virus infection (Saeed et al., 2008). At later stages, PI3K seems to suppress premature apoptosis, most likely mediated by binding to the A/NS1 protein. Finally, the kinase also exhibits antiviral functions by regulating the IRF‐3 transcription factor during type I IFN induction.
While it was a surprising finding that the viral A/NS1 protein, actually known as a suppressor of signalling, induces PI3K activity, similar observations have been made for a HCV protein. The viral NS5A protein induces PI3K activity via direct binding to the regulatory subunit (Street et al., 2004), like A/NS1 (Hale et al., 2006; Ehrhardt et al., 2007a; Shin et al., 2007a). The currently proposed model of A/NS1‐induced PI3K activity suggests a complex‐formation of p110/p85β‐A/NS1, whereby A/NS1 disrupts the inhibiting interaction interface of p110 and p85β (Hale et al., 2008b; Li et al., 2008). Nonetheless, the different interaction sites from A/NS1 and p85β described above (Fig. 3) are involved in PI3K induction, too (Hale et al., 2006; Shin et al., 2007a;b).
It is currently not clear how the described antiviral functions of the kinase, namely coactivation of IRF‐3 in the antiviral type I IFN induction, fits into the picture. It may be hypothesized that PI3K is one of the factors that are activated by the virus‐infected cell in a vRNA‐dependent fashion to fight the invader, but is redirected by the pathogen to support replication via its A/NS1 protein. This may include uncoupling of PI3K from the antiviral RNA signalling response by keeping the kinase under control of a viral protein.
It will be interesting to elucidate in which cellular compartment the interaction of p85 and A/NS1 takes place. A/NS1 is located predominantly in the nucleus and only accumulates in the cytoplasm at very late stages of the infection cycle. Nevertheless, cytoplasmic activities of A/NS1 have been described, including inhibition of the RNA sensor RIG‐I or binding to the eukaryotic translation initiation factor 4GI (Hale et al., 2008a). In this context it is crucial to elucidate the sorting mechanisms of A/NS1. Other analysis may comprise additional interaction partners of A/NS1, such as Crk/CrkL and their regulation. A major challenge is also to unravel the activation mechanism and function of PI3K during virus entry. These approaches may help to expand our knowledge regarding the delicate balance between replication enhancing and host cell defence mechanisms.
Acknowledgements
We apologize to those authors whose papers were not cited due to space limitations. We thank E.R. Hrincius, T. Eierhoff, D. Demirov, V. Wixler, W. Nacken, M. Scheelhaas and T. Wolff for helpful discussions and comments on this manuscript. This work was supported by grants from the fund ‘Innovative Medical Research’ of the University of Muenster Medical School, by the Deutsche Forschungsgemeinschaft (Grant EH‐235/1‐1) and by the Fonds der Chemischen Industrie.
References
- Autret, A. , Martin‐Latil, S. , Brisac, C. , Mousson, L. , Colbere‐Garapin, F. , and Blondel, B. (2008) Early phosphatidylinositol 3‐kinase/Akt pathway activation limits poliovirus‐induced JNK‐mediated cell death. J Virol 82: 3796–3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carracedo, A. , and Pandolfi, P.P. (2008) The PTEN‐PI3K pathway: of feedbacks and cross‐talks. Oncogene 27: 5527–5541. [DOI] [PubMed] [Google Scholar]
- Cooray, S. (2004) The pivotal role of phosphatidylinositol 3‐kinase‐Akt signal transduction in virus survival. J Gen Virol 85: 1065–1076. [DOI] [PubMed] [Google Scholar]
- Cooray, S. , Jin, L. , and Best, J.M. (2005) The involvement of survival signaling pathways in rubella‐virus induced apoptosis. Virol J 2: 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dauber, B. , Heins, G. , and Wolff, T. (2004) The influenza B virus nonstructural NS1 protein is essential for efficient viral growth and antagonizes beta interferon induction. J Virol 78: 1865–1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dauber, B. , Schneider, J. , and Wolff, T. (2006) Double‐stranded RNA binding of influenza B virus nonstructural NS1 protein inhibits protein kinase R but is not essential to antagonize production of alpha/beta interferon. J Virol 80: 11667–11677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhand, R. , Hiles, I. , Panayotou, G. , Roche, S. , Fry, M.J. , Gout, I. , et al. (1994) PI 3‐kinase is a dual specificity enzyme: autoregulation by an intrinsic protein‐serine kinase activity. Embo J 13: 522–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrhardt, C. , Marjuki, H. , Wolff, T. , Nurnberg, B. , Planz, O. , Pleschka, S. , and Ludwig, S. (2006) Bivalent role of the phosphatidylinositol‐3‐kinase (PI3K) during influenza virus infection and host cell defence. Cell Microbiol 8: 1336–1348. [DOI] [PubMed] [Google Scholar]
- Ehrhardt, C. , Wolff, T. , Pleschka, S. , Planz, O. , Beermann, W. , Bode, J.G. , et al. (2007a) Influenza A virus NS1 protein activates the PI3K/Akt pathway to mediate antiapoptotic signaling responses. J Virol 81: 3058–3067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrhardt, C. , Wolff, T. , and Ludwig, S. (2007b) Activation of phosphatidylinositol 3‐kinase signaling by the nonstructural NS1 protein is not conserved among type A and B influenza viruses. J Virol 81: 12097–12100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia‐Sastre, A. (2004) Identification and characterization of viral antagonists of type I interferon in negative‐strand RNA viruses. Curr Top Microbiol Immunol 283: 249–280. [DOI] [PubMed] [Google Scholar]
- Groskreutz, D.J. , Monick, M.M. , Yarovinsky, T.O. , Powers, L.S. , Quelle, D.E. , Varga, S.M. , et al. (2007) Respiratory syncytial virus decreases p53 protein to prolong survival of airway epithelial cells. J Immunol 179: 2741–2747. [DOI] [PubMed] [Google Scholar]
- Guillot, L. , Le Goffic, R. , Bloch, S. , Escriou, N. , Akira, S. , Chignard, M. , and Si‐Tahar, M. (2005) Involvement of toll‐like receptor 3 in the immune response of lung epithelial cells to double‐stranded RNA and influenza A virus. J Biol Chem 280: 5571–5580. [DOI] [PubMed] [Google Scholar]
- Hale, B.G. , Jackson, D. , Chen, Y.H. , Lamb, R.A. , and Randall, R.E. (2006) Influenza A virus NS1 protein binds p85beta and activates phosphatidylinositol‐3‐kinase signaling. Proc Natl Acad Sci USA 103: 14194–14199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale, B.G. , Randall, R.E. , Ortin, J. , and Jackson, D. (2008a) The multifunctional NS1 protein of influenza A viruses. J Gen Virol 89: 2359–2376. [DOI] [PubMed] [Google Scholar]
- Hale, B.G. , Batty, I.H. , Downes, C.P. , and Randall, R.E. (2008b) Binding of influenza A virus NS1 protein to the inter‐SH2 domain of p85 suggests a novel mechanism for phosphoinositide 3‐kinase activation. J Biol Chem 283: 1372–1380. [DOI] [PubMed] [Google Scholar]
- Hara, K. , Maruki, Y. , Long, X. , Yoshino, K. , Oshiro, N. , Hidayat, S. , et al. (2002) Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell 110: 177–189. [DOI] [PubMed] [Google Scholar]
- He, Y. , Nakao, H. , Tan, S.L. , Polyak, S.J. , Neddermann, P. , Vijaysri, S. , et al. (2002) Subversion of cell signaling pathways by hepatitis C virus nonstructural 5A protein via interaction with Grb2 and P85 phosphatidylinositol 3‐kinase. J Virol 76: 9207–9217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heikkinen, L.S. , Kazlauskas, A. , Melen, K. , Wagner, R. , Ziegler, T. , Julkunen, I. , and Saksela, K. (2008) Avian and 1918 Spanish influenza a virus NS1 proteins bind to Crk/CrkL Src homology 3 domains to activate host cell signaling. J Biol Chem 283: 5719–5727. [DOI] [PubMed] [Google Scholar]
- Kikani, C.K. , Dong, L.Q. , and Liu, F. (2005) ‘New’‐clear functions of PDK1: beyond a master kinase in the cytosol? J Cell Biochem 96: 1157–1162. [DOI] [PubMed] [Google Scholar]
- Krug, R.M. , Yuan, W. , Noah, D.L. , and Latham, A.G. (2003) Intracellular warfare between human influenza viruses and human cells: the roles of the viral NS1 protein. Virology 309: 181–189. [DOI] [PubMed] [Google Scholar]
- Lee, C.J. , Liao, C.L. , and Lin, Y.L. (2005) Flavivirus activates phosphatidylinositol 3‐kinase signaling to block caspase‐dependent apoptotic cell death at the early stage of virus infection. J Virol 79: 8388–8399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemon, S.M. , Walker, C. , Alter, M.J. , and Yi, M. (2007) Hepatitis C Virus In Fields Virology. Knipe D.M., Howley P.M., and Griffin D.E. (eds). Philadelphia: Lippincott Willams and Willams, pp. 1253–1304. [Google Scholar]
- Li, Y. , Anderson, D.H. , Liu, Q. , and Zhou, Y. (2008) Mechanism of influenza A virus NS1 protein interaction with the p85beta, but not the p85alpha, subunit of phosphatidylinositol 3‐kinase (PI3K) and up‐regulation of PI3K activity. J Biol Chem 283: 23397–23409. [DOI] [PubMed] [Google Scholar]
- Ludwig, S. (2007) Exploited defense: How influenza viruses take advantage of antiviral signalling responses. Future Virol 2: 91–100. [Google Scholar]
- Ludwig, S. , Pleschka, S. , Planz, O. , and Wolff, T. (2006) Ringing the alarm bells: signalling and apoptosis in influenza virus infected cells. Cell Microbiol 8: 375–386. [DOI] [PubMed] [Google Scholar]
- Maehama, T. , and Dixon, J.E. (1998) The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5‐trisphosphate. J Biol Chem 273: 13375–13378. [DOI] [PubMed] [Google Scholar]
- Mannova, P. , and Beretta, L. (2005) Activation of the N‐Ras‐PI3K‐Akt‐mTOR pathway by hepatitis C virus: control of cell survival and viral replication. J Virol 79: 8742–8749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizutani, T. , Fukushi, S. , Saijo, M. , Kurane, I. , and Morikawa, S. (2004) Importance of Akt signaling pathway for apoptosis in SARS‐CoV‐infected Vero E6 cells. Virology 327: 169–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neri, L.M. , Borgatti, P. , Capitani, S. , and Martelli, A.M. (2002) The nuclear phosphoinositide 3‐kinase/AKT pathway: a new second messenger system. Biochim Biophys Acta 1584: 73–80. [DOI] [PubMed] [Google Scholar]
- Palese, P. , and Shaw, M.L. (2007) Orthomyxoviridae: the viruses and their replication In Fields Virology. Knipe D.M., Howley P.M. and Griffin D.E. (eds). Philadelphia: Lippencott Willams and Willams, pp. 1647–1689. [Google Scholar]
- Saeed, M.F. , Kolokoltsov, A.A. , Freiberg, A.N. , Holbrook, M.R. , and Davey, R.A. (2008) Phosphoinositide‐3 kinase‐Akt pathway controls cellular entry of Ebola virus. PLoS Pathog 4: e1000141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez, A. , Geisbert, T.W. , and Feldmann, H. (2007) Filoviridae: marburg and ebola viruses In Fields Virology. Knipe D.M., Howley P.M., and Griffin D.E. (eds). Philadelphia: Lippincott Willams and Willams, pp. 1409–1448. [Google Scholar]
- Sarbassov, D.D. , Ali, S.M. , Kim, D.H. , Guertin, D.A. , Latek, R.R. , Erdjument‐Bromage, H. , et al. (2004) Rictor, a novel binding partner of mTOR, defines a rapamycin‐insensitive and raptor‐independent pathway that regulates the cytoskeleton. Curr Biol 14: 1296–1302. [DOI] [PubMed] [Google Scholar]
- Sarbassov, D.D. , Guertin, D.A. , Ali, S.M. , and Sabatini, D.M. (2005) Phosphorylation and regulation of Akt/PKB by the rictor‐mTOR complex. Science 307: 1098–1101. [DOI] [PubMed] [Google Scholar]
- Sarkar, S.N. , Peters, K.L. , Elco, C.P. , Sakamoto, S. , Pal, S. , and Sen, G.C. (2004) Novel roles of TLR3 tyrosine phosphorylation and PI3 kinase in double‐stranded RNA signaling. Nat Struct Mol Biol 11: 1060–1067. [DOI] [PubMed] [Google Scholar]
- Shin, Y.K. , Liu, Q. , Tikoo, S.K. , Babiuk, L.A. , and Zhou, Y. (2007a) Influenza A virus NS1 protein activates the phosphatidylinositol 3‐kinase (PI3K)/Akt pathway by direct interaction with the p85 subunit of PI3K. J Gen Virol 88: 13–18. [DOI] [PubMed] [Google Scholar]
- Shin, Y.K. , Li, Y. , Liu, Q. , Anderson, D.H. , Babiuk, L.A. , and Zhou, Y. (2007b) SH3 binding motif 1 in influenza A virus NS1 protein is essential for PI3K/Akt signaling pathway activation. J Virol 81: 12730–12739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin, Y.K. , Liu, Q. , Tikoo, S.K. , Babiuk, L.A. , and Zhou, Y. (2007c) Effect of the phosphatidylinositol 3‐kinase/Akt pathway on influenza A virus propagation. J Gen Virol 88: 942–950. [DOI] [PubMed] [Google Scholar]
- Street, A. , Macdonald, A. , Crowder, K. , and Harris, M. (2004) The Hepatitis C virus NS5A protein activates a phosphoinositide 3‐kinase‐dependent survival signaling cascade. J Biol Chem 279: 12232–12241. [DOI] [PubMed] [Google Scholar]
- Street, A. , Macdonald, A. , McCormick, C. , and Harris, M. (2005) Hepatitis C virus NS5A‐mediated activation of phosphoinositide 3‐kinase results in stabilization of cellular beta‐catenin and stimulation of beta‐catenin‐responsive transcription. J Virol 79: 5006–5016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talon, J. , Salvatore, M. , O'Neill, R.E. , Nakaya, Y. , Zheng, H. , Muster, T. , et al. (2000) Influenza A and B viruses expressing altered NS1 proteins: a vaccine approach. Proc Natl Acad Sci USA 97: 4309–4314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamguney, T. , and Stokoe, D. (2007) New insights into PTEN. J Cell Sci 120: 4071–4079. [DOI] [PubMed] [Google Scholar]
- Thomas, K.W. , Monick, M.M. , Staber, J.M. , Yarovinsky, T. , Carter, A.B. , and Hunninghake, G.W. (2002) Respiratory syncytial virus inhibits apoptosis and induces NF‐kappa B activity through a phosphatidylinositol 3‐kinase‐dependent pathway. J Biol Chem 277: 492–501. [DOI] [PubMed] [Google Scholar]
- Tyner, J.W. , Uchida, O. , Kajiwara, N. , Kim, E.Y. , Patel, A.C. , O'Sullivan, M.P. , et al. (2005) CCL5–CCR5 interaction provides antiapoptotic signals for macrophage survival during viral infection. Nat Med 11: 1180–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanhaesebroeck, B. , Leevers, S.J. , Ahmadi, K. , Timms, J. , Katso, R. , Driscoll, P.C. , et al. (2001) Synthesis and function of 3‐phosphorylated inositol lipids. Annu Rev Biochem 70: 535–602. [DOI] [PubMed] [Google Scholar]
- Vanhaesebroeck, B. , Ali, K. , Bilancio, A. , Geering, B. , and Foukas, L.C. (2005) Signalling by PI3K isoforms: insights from gene‐targeted mice. Trends Biochem Sci 30: 194–204. [DOI] [PubMed] [Google Scholar]
- Wang, W. , and Krug, R.M. (1996) The RNA‐binding and effector domains of the viral NS1 protein are conserved to different extents among influenza A and B viruses. Virology 223: 41–50. [DOI] [PubMed] [Google Scholar]
- Wolff, T. , Zielecki, F. , Abt, M. , Voss, D. , Semmler, I. , and Matthaei, M. (2008) Sabotage of antiviral signaling and effectors by influenza viruses. Biol Chem 389: 1299–1305. [DOI] [PubMed] [Google Scholar]
- Wymann, M.P. , and Pirola, L. (1998) Structure and function of phosphoinositide 3‐kinases. Biochim Biophys Acta 1436: 127–150. [DOI] [PubMed] [Google Scholar]
- Zhang, H.M. , Yuan, J. , Cheung, P. , Luo, H. , Yanagawa, B. , Chau, D. , et al. (2003) Overexpression of interferon‐gamma‐inducible GTPase inhibits coxsackievirus B3‐induced apoptosis through the activation of the phosphatidylinositol 3‐kinase/Akt pathway and inhibition of viral replication. J Biol Chem 278: 33011–33019. [DOI] [PubMed] [Google Scholar]
- Zhirnov, O.P. , and Klenk, H.D. (2007) Control of apoptosis in influenza virus‐infected cells by up‐regulation of Akt and p53 signaling. Apoptosis 12: 1419–1432. [DOI] [PubMed] [Google Scholar]
- Zhirnov, O.P. , Konakova, T.E. , Wolff, T. , and Klenk, H.D. (2002) NS1 protein of influenza A virus down‐regulates apoptosis. J Virol 76: 1617–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]