

Abstract
Actin is a major component of the cytoskeleton and is present as two isoforms in non‐muscle cells: β‐ and γ‐cytoplasmic actin. These isoforms are strikingly conserved, differing by only four N‐terminal amino acids. During spread from infected cells, vaccinia virus (VACV) particles induce localized actin nucleation that propel virus to surrounding cells and facilitate cell‐to‐cell spread of infection. Here we show that virus‐tipped actin comets are composed of β‐ and γ‐actin. We employed isoform‐specific siRNA knockdown to examine the role of the two isoforms in VACV‐induced actin comets. Despite the high level of similarity between the actin isoforms, and their colocalization, VACV‐induced actin nucleation was dependent exclusively on β‐actin. Knockdown of β‐actin led to a reduction in the release of virus from infected cells, a phenotype dependent on virus‐induced Arp2/3 complex activity. We suggest that local concentrations of actin isoforms may regulate the activity of cellular actin nucleator complexes.
Keywords: actin, transport, virus
Abbreviations
- CYA
cytoplasmic actin
- EEV
extracellular enveloped virus
- EV
extracellular virus
- FBS
foetal bovine serum
- GBM
glioblastoma
- GST
glutathione S‐transferase
- VACV
vaccinia virus
- WR
Western Reserve.
1. Introduction
A select group of microbial pathogens induce highly localized actin nucleation at their surfaces during the infection of host cells, through the recruitment and activation of cellular nucleation protein complexes (Gouin, Welch, & Cossart, 2005; Radtke, Dohner, & Sodeik, 2006; Welch and Way, 2013). Remodelling of the actin cytoskeleton at the microbial interface promotes pathogen survival and dispersal through a variety of mechanisms. These include intracellular motility of Listeria monocytogenes and escape of autophagic recognition (Tilney et al., 1990), adhesion of Enteropathogenic Escherichia coli to the intestinal epithelium (Shaner, Sanger, & Sanger, 2005), and nuclear entry and nonreplicative spread of Autographa californica multiple nucleopolyhedrovirus (Ohkawa, Volkman, & Welch, 2010).
One of the best‐characterized pathways of microbial cytoskeletal remodelling is the stimulation of actin nucleation by vaccinia virus (VACV; Newsome and Marzook, 2015). Replication of VACV results in the formation of wrapped viruses (WV) at the early endosome/trans‐Golgi network, which are transported to the cell periphery via microtubules in a kinesin‐1‐dependent manner (Hollinshead, Hollinshead, Rodger, Van Eijl, Law, Hollinshead, Vaux, & Smith, 2001; Rietdorf, Ploubidou, Reckmann, Holmstrom, Frischknecht, Zettl, … Way, 2001; Ward, 2004; Ward and Moss, 2001). Here, they fuse with the plasma membrane in an exocytic event leaving an extracellular WV that remains tightly associated with the cell surface. The nucleation of actin in the cytoplasm beneath WV results in comet‐like structures of F‐actin (also referred to as actin tails) that propel virus particles across the surface of the infected cell (Cudmore, Cossart, Griffiths, & Way, 1995; Frischknecht, Moreau, Rottger, Gonfloni, Reckmann, Superti‐Furga, & Way, 1999b; Moreau, Frischknecht, Reckmann, Vincentelli, Rabut, Stewart, & Way, 2000; Wolffe, Weisberg, & Moss, 1998). Infected cells typically exhibit virus‐tipped membrane protrusions that are rich in F‐actin.
Mutant strains unable to undergo actin‐based motility are attenuated in whole‐animal mouse infection models and deficient in cell‐to‐cell spread in vitro (Parkinson and Smith, 1994; Wolffe, Isaacs, & Moss, 1993; Wolffe, Katz, Weisberg, & Moss, 1997; Wolffe et al., 1998). It is likely that actin‐based motility contributes to virus spread via a variety of mechanisms. For example, the repulsion of superinfecting virions allows nonreplicative transmission across early stage‐infected cells, thereby increasing the overall rate of cell‐to‐cell spread (Doceul, Hollinshead, van der Linden, & Smith, 2010). It has also been observed that when VACV‐induced actin nucleation is abrogated, WV remain trapped at the cell surface in pits (Horsington, Lynn, Turnbull, Cheng, Braet, Diefenbach, … Newsome, 2013), indicating that actin nucleation provides a force necessary to resolve exocytosis of virus particles.
Actin nucleation is instigated by the VACV type Ib transmembrane protein A36 that resides in the outer membrane of WV. Following fusion of this outer WV membrane with the plasma membrane, A36 becomes phosphorylated at Tyr112 and Tyr132 by the action of Src‐ and Abl‐family tyrosine kinases (Frischknecht, Cudmore, Moreau, Reckmann, Rottger, & Way, 1999a; Newsome, Scaplehorn, & Way, 2004; Newsome, Weisswange, Frischknecht, & Way, 2006; Reeves, Bommarius, Lebeis, McNulty, Christensen, Swimm, … Kalman, 2005). Phosphorylation of A36 leads to the recruitment of a host of cellular proteins, the core cascade comprising of Nck, WIP/WIRE and N‐WASP, which act to stabilize and activate the Arp2/3 complex at the cytoplasmic surface of EV (Moreau et al., 2000; Snapper, Takeshima, Anton, Liu, Thomas, Nguyen, … Alt, 2001). Optimal virus‐induced actin nucleation also utilizes other host factors such as Grb2, FHOD1, casein kinase 2, clathrin, the clathrin adaptor AP2, and is accompanied by activation of the small Rho GTPases Cdc42 and Rac1 (Alvarez and Agaisse, 2013; Alvarez and Agaisse, 2014; Humphries, Dodding, Barry, Collinson, Durkin, & Way, 2012; Scaplehorn, Holmstrom, Moreau, Frischknecht, Reckmann, & Way, 2002). The critical signal output that results from A36 tyrosine phosphorylation is actin nucleation, and drug inhibition of actin dynamics during viral infection phenocopies, to a large extent, the effects of genetic ablation of A36 or substitution of the Tyr112 and Tyr132 to phenylalanines (VACV‐A36YdF) (Horsington et al., 2013).
The de novo nucleation of new actin filaments by the Arp2/3 complex, during normal cellular function or during VACV infection, stimulates robust actin polymerization by increasing the local concentration of free plus ends. Actin filaments are composed of actin monomers that are expressed from multiple loci that give rise to six highly conserved actin isoforms: two striated muscle (α‐skeletal and α‐cardiac), two smooth muscle (α‐ and γ‐) and two cytoplasmic (β‐ and γ‐) actins (Vandekerckhove and Weber, 1978b).
The muscle isoforms exhibit tissue specific expression, while the β‐ and γ‐cytoplasmic actins (CYA) are the most abundant in nonmuscle cells (Rubenstein, 1990) and often exist in 2:1 ratio in epithelial cell lines like HeLa and chicken embryo fibroblasts (Khaitlina, 2007), the cell types commonly used to study VACV in vitro and cell types commonly targeted by orthopoxviruses during pathogenesis (Johnston and McFadden, 2004; McFadden, 2005; Smith and Kotwal, 2002; Werden, Rahman, & McFadden, 2008). Despite their surprisingly similar sequence homologies [only differing by four amino acids; (Vandekerckhove and Weber, 1978a)], recent reports have revealed that β‐ and γ‐CYA have different functions and display distinct subcellular localization patterns (Artman, Dormoy‐Raclet, von Roretz, & Gallouzi, 2014; Dugina, Zwaenepoel, Gabbiani, Clement, & Chaponnier, 2009; Joseph, Srivastava, & Pfister, 2014; Pasquier, Tuset, Sinnappan, Carnell, Macmillan, & Kavallaris, 2015). β‐actin is predominantly found in basal areas of the cell, cell‐cell contacts and stress fibres, while γ‐CYA is more apically abundant, and concentrated at peripheral structures such as lamellipodia and the leading edge of motile cells (Chaponnier and Gabbiani, 2004; Dugina et al., 2009]. Differential localization may contribute to the functional diversity of these isoforms, as γ‐CYA has been shown to be specifically required for directional cell migration, while β‐CYA contributes to cell attachment and microparticle formation (Latham, Chaponnier, Dugina, Couraud, Grau, & Combes, 2013). The aim of this study was to determine if there are isoform‐specific functions of β‐ and γ‐CYA during the well‐characterized phenomenon of VACV‐induced actin nucleation.
2. Results
2.1. VACV comets comprise both β‐ and γ‐CYA
Given the differential distributions of β‐ and γ‐CYA in a cell, our first aim was to determine the actin isoform composition of VACV‐induced actin ‘comets’. Actin comets generated by a subpopulation of egressing virus particles are evident below the cell surface of VACV infected cells at 7–9 hr postinfection (hpi; Cudmore et al., 1995; Smith and Law, 2004). Immunofluorescence assays (IFA) of siRNA‐treated cells stained with anti‐β‐CYA and anti‐γ‐CYA specific antibodies revealed the localization of both CYA isoforms in the cytoskeleton (Figure 1a) and VACV‐induced actin comets in endothelial hCMEC/D3 cells (Figure 1b).
Figure 1.
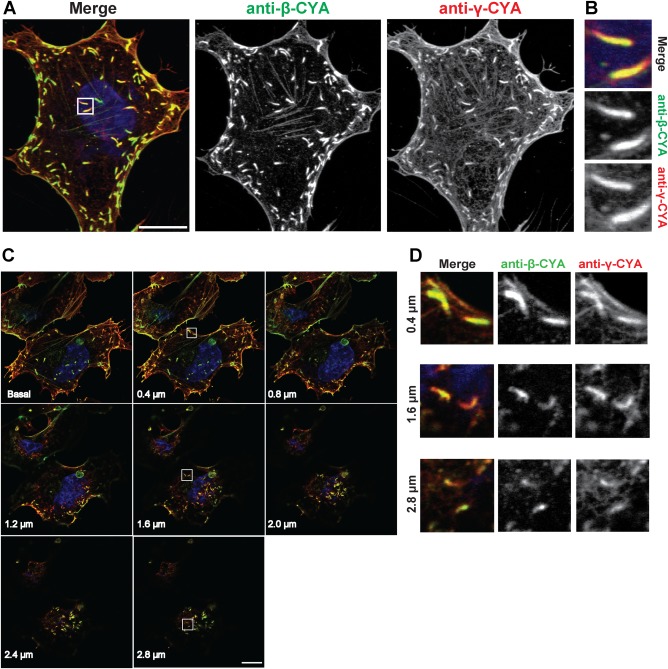
VACV comets contain both β‐ and γ‐CYA. (a) Fluorescent micrographs of hCMEC/D3 cells infected with VACV‐WR and fixed 8 hpi. Cells were stained with anti‐β‐CYA (green), anti‐γ‐CYA (red), and DAPI (blue). Scale bar is 10 μm. (b) Close‐ups of outlined sections in A. (c) Individual z‐stack planes of a single field of view for VACV‐infected cells, along with close‐ups of actin tails from three z‐planes (d). [Color figure can be viewed at http://wileyonlinelibrary.com]
VACV‐induced actin comets are readily imaged at the level of the substrate due to the low z‐axis profile of adherent cells. β‐CYA preferentially localizes to the basal region, while γ‐CYA preferentially localizes to apical regions of HSCF cells (Dugina et al., 2009). We therefore examined z‐sections of infected cells to determine if the preference of actin isoforms along the apical‐basal axis was reflected in their localization to actin comets on the apical or basal membrane. A z‐stack of images of infected cells (Figure 1c) with VACV‐induced actin comets (Figure 1d) was taken via confocal microscopy. These data led us to conclude that VACV‐associated actin comets are composed of both CYA isoforms irrespective of their basal or apical localization.
2.2. β‐ and γ‐siRNA treatment induces isoform‐specific knockdown in multiple cell lines
Since both CYAs were reported to not only have different localizations, but also different functions within a cell (Dugina et al., 2009), the next step was to validate siRNA‐mediated depletion of individual actin isoforms that could be used to assay VACV actin‐based motility. To this end, we established knockdown of β‐CYA and γ‐CYA in HeLa, GBM (glioblastoma) and hCMEC/D3 cell lines. Treatment of HeLa, GBM and hCMEC/D3 cells resulted in isoform‐specific knockdown when compared to the scrambled siRNA condition (Figure 2a). Densitometry analysis showed that β‐CYA knockdown caused a significant reduction of β‐CYA to about 31, 8, and 44% compared with levels in the scrambled siRNA control in HeLa, GBM, and hCMEC/D3 cells, respectively (p < .05, n ≥ 2; Figure 2b). γ‐CYA siRNA treatment produced a comparatively less efficient knockdown of γ‐CYA to about 52, 35, and 55% in HeLa, GBM, and hCMEC/D3 cells, respectively (p < .05, n ≥ 2; Figure 2b). Interestingly, knockdown of β‐CYA led to a corresponding significant increase in γ‐CYA in HeLa and GBM cell types. This suggests that under knockdown conditions, cells overexpress one isoform to compensate for loss of the other. This has not been observed for CYA isoforms in other studies, either using the same cocktail of isoform‐specific siRNA (Dugina et al., 2009) or using only one of those sequences (Pasquier et al., 2015). This could be due to their use of different cell types, varying doses of siRNA or incubation times, and/or the use of only a single CYA‐specific siRNA in one of the studies.
Figure 2.
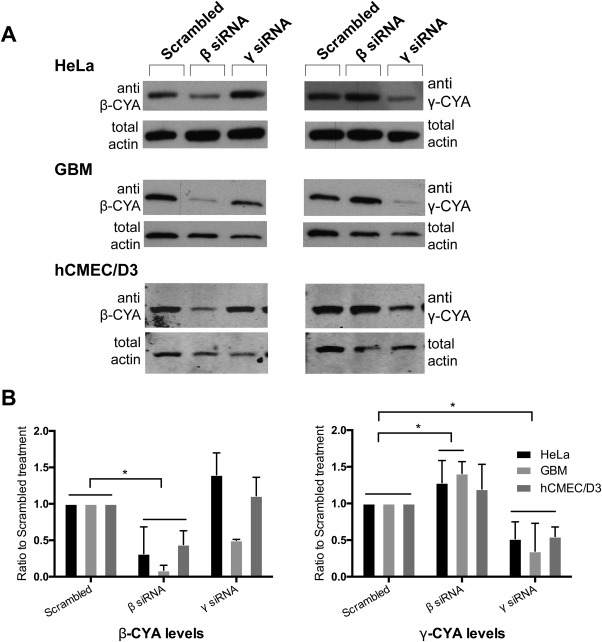
β‐ and γ‐siRNA result in respective actin isoform knockdown. (a) Immunoblot of cells treated with either β‐, γ‐, or scrambled‐siRNA. Cell lysates were probed with either mouse anti‐β‐CYA (left) or anti‐γ‐CYA (right) antibodies. Ratio of densitometry measurements of β‐CYA or γ‐CYA under specified siRNA treatments compared with a scrambled siRNA control are shown for HeLa, GBM, and hCMEC/D3 cell lines.
The use of a pan‐actin antibody which detects both cytoplasmic isoforms revealed that there is a compensatory effect of actin isoform expression during either β‐CYA or γ‐CYA knockdown in HeLa cells, as there is no significant difference in total actin levels under either knockdown condition (Supporting Information Fig. S1). This is supported by observations from other studies where neither isoform knockdown significantly affected the total level of actin in HSCF cells (Dugina et al., 2009) and A549 human lung epithelial cells (Lechuga, Baranwal, Li, Naydenov, Kuemmerle, Dugina, … Ivanov, 2014). GBM cells, however, show significantly reduced levels of total actin expression during γ‐CYA knockdown (Supporting Information Fig. S1).
2.3. β‐CYA is required for VACV‐induced actin tails
Since both CYAs were reported to not only have different localizations, but also different functions within a cell (Dugina et al., 2009), the next step was to determine if there was a distinct role for either CYA in VACV‐induced actin comets. hCMEC/D3 cells were treated with β‐ or γ‐CYA‐specific siRNA for 72 hr before being infected with VACV‐WR and fixed 8 hpi. IFA of siRNA‐treated cells stained with anti‐β‐CYA and anti‐γ‐CYA specific antibodies (Dugina et al., 2009) again showed localization of both CYA isoforms in the cytoskeleton and VACV‐induced actin comets in scrambled siRNA‐treated cells. Levels of β‐CYA and γ‐CYA were unaffected by scrambled siRNA, however both were significantly reduced in the presence of their respective siRNA, as shown by IFA (Figure 3a). While β‐ or γ‐CYA‐specific siRNA have previously been shown to cause incomplete knockdown of their respective targets, it was still sufficient to cause significant changes to cell morphology and behavior (Dugina et al., 2009). As with the control cells from Figure 1, both β‐CYA and γ‐CYA were detected in virus‐associated actin comets in the scrambled siRNA‐treated cells (Figure 1b). In γ‐CYA knockdown cells, γ‐CYA was greatly reduced and β‐CYA was concentrated at VACV‐induced actin tails (Figure 3b,c). Virus comets induced under β‐CYA knockdown accordingly exhibited a strong γ‐CYA signal with a reduction in β‐CYA. VACV‐associated actin comets present under β‐CYA knockdown conditions exhibited faint accumulations of β‐CYA at the interface of the virus particle and actin comet, which is the site of F‐actin nucleation. We compared the length of β‐CYA staining to VACV‐comet length, which revealed an average ratio of 0.68 ± 0.14 (under β‐CYA knockdown) and 0.81 ± 0.12 (scrambled control). These data indicate a correlation between the amount of β‐CYA recruited to the virus and the resulting comet length.
Figure 3.
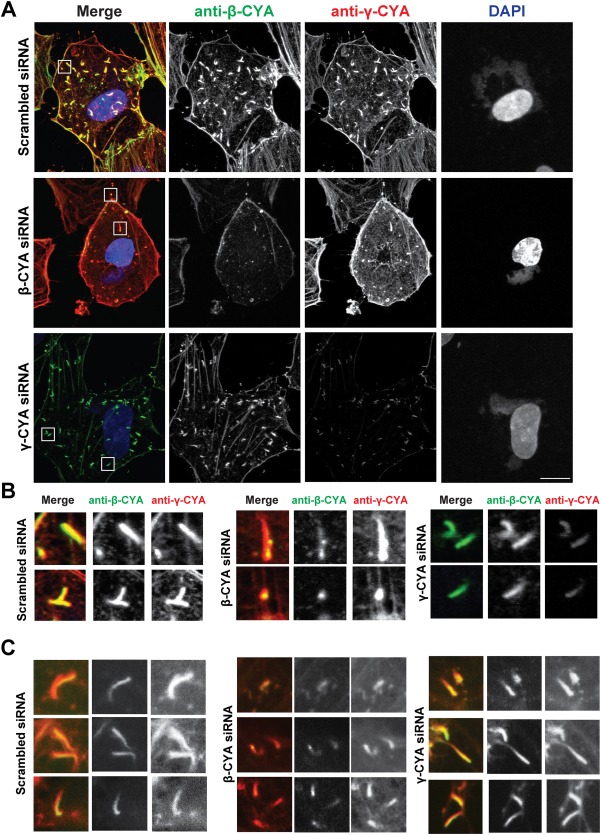
Distribution of β‐ and γ‐CYA in VACV comets under CYA knockdown. (a) Fluorescent micrographs of hCMEC/D3 cells treated with the isoform‐specific siRNA described, infected with VACV‐WR and fixed at 8 hpi. Cells were stained with anti‐β‐CYA (green), anti‐γ‐CYA (red), and DAPI (blue). (b) Close‐ups of outlined sections in A. (c) Additional close‐up micrographs from other infected cells. Scale bar is 10 μm. [Color figure can be viewed at http://wileyonlinelibrary.com]
Z‐stack images of hCMEC/D3 treated with CYA isoform‐specific siRNA and infected with VACV‐WR show that in scrambled siRNA‐ and γ‐CYA‐targeting siRNA‐treated cells (cells where actin comets were most readily seen), both γ‐ and β‐CYA isoforms comprise the VACV‐induced actin comets, irrespective of whether they were present at the basal or apical membrane (Supporting Information Fig. S2A‐B).
To further characterize the effect of β‐CYA depletion on actin comet formation, hCMEC/D3 cells were infected with VACV following treatment with respective siRNAs, and visualized by IFA. Infected cells were picked at random (by their characteristic peri‐nuclear virus factories) and of those, cells with 10 or more actin comets were counted. Treatment of cells with siRNA targeting β‐CYA resulted in a significant reduction in the percentage of infected cells containing at least 10 actin comets (Figure 4a). There was no significant difference seen in the γ‐CYA‐depleted and scrambled siRNA‐treated cells. In addition to a reduction in the number of actin comets under β‐CYA depletion, we also recorded a reduction in their length (Figure 4b), which is related to the stability of actin comets and EV release (Horsington et al., 2013; Weisswange, Newsome, Schleich, & Way, 2009). VACV‐WR was used to infect hCMEC/D3 or HeLa cells treated with siRNA specific for either β‐CYA or γ‐CYA, which were then fixed 8 hpi. In both cell types examined, VACV‐induced actin comets produced in β‐CYA knockdown cells were significantly shorter compared to those produced in the γ‐CYA knockdown or scrambled siRNA‐treated cells. There was no significant difference in comet length between the γ‐CYA knockdown and scrambled siRNA‐treated cells. Despite the decrease in comet length observed under conditions of β‐CYA depletion, live‐cell imaging of infected cells labelled with Lifeact‐GFP revealed no significant difference in the speed of virus motility (Figure 4c). Surprisingly, knockdown of γ‐CYA elicited a two‐fold increase in virus motility (0.17 μm/s compared with 0.8 μm/s). In summary, we observed that actin comets produced under β‐CYA knockdown are shorter although their speeds remain the same relative to the controls. There was no significant difference in length of actin comets produced by γ‐CYA knockdown and scrambled siRNA‐treated HeLa cells. This implies that while β‐CYA is necessary for the initiation of VACV comets, the ratio of β‐CYA to γ‐CYA in a cell may determine the speed of VACV motility, whereby the presence of γ‐CYA in the actin comet has a moderating effect on virus speed.
Figure 4.
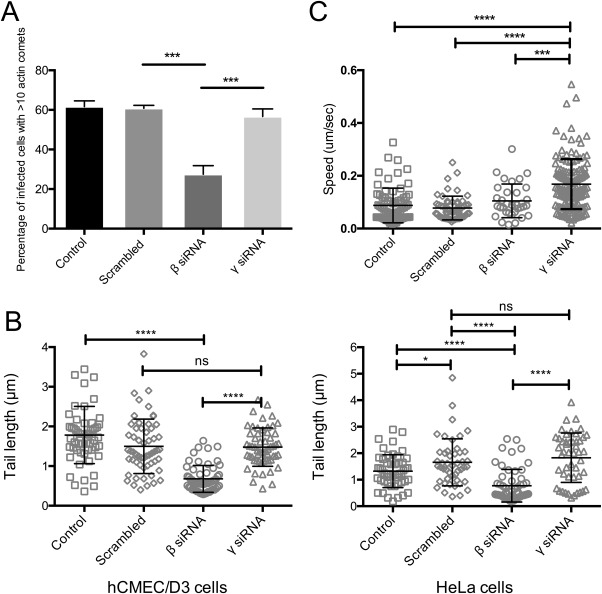
Effects of CYA knockdown on VACV comet phenotype. (a) hCMEC/D3 cells were treated with the respective siRNA for 72 hr, followed by VACV‐WR infection at an MOI > 5. Cells were fixed 8 hpi and stained for F‐actin and the number of actin tails per cell was enumerated (‘***’: p < .001; n = 40 cells for each treatment, with 3 replicate experiments performed). (b) hCMEC/D3 (D1) and HeLa (D2) cell lines were treated with the siRNA as indicated for 72 hr and infected with VACV‐WR at an MOI > 3. Cells were fixed 8 hpi, followed by staining for F‐actin and viral envelope proteins. Actin tail lengths were measured using ImageJ image‐analysing software and graphs created using GraphPad PRISM, with nonparametric t‐tests used to determine significance in differences between parameters (n = 60 tails each). (c) Speeds of VACV actin comets were calculated using FIJI (ver. 2.0.0) image‐analyzing software from live‐cell imaging experiments and statistical analyses were carried out using GraphPad PRISM (ver. 6 for Mac OSX), with one‐way ANOVA and Tukey's multiple comparison tests. “ns”: p > .05, “*”: p ≤ .05, “***”: p ≤ .001, “****”: p ≤ .0001.
2.4. The role of β‐CYA in the release and spread of VACV
Since a clear attenuation of VACV‐induced actin nucleation during β‐CYA knockdown was evident, we next determined its effect on virus release, which is dependent on actin nucleation (Horsington et al., 2013). HeLa cells under either CYA knockdown were infected with either the vaccinia reference strain VACV‐WR, or the mutant strain VACV‐A36YdF. VACV‐A36YdF contains point mutations of the two tyrosine residues in A36R involved in recruiting actin polymerisation machinery, rendering A36 incapable of being tyrosine phosphorylated. The mutant virus is therefore unable to induce actin comets (Rietdorf et al., 2001) and a subsequent reduction in release of extracellular enveloped virus (EEV) from the cell is observed (Horsington et al., 2013). Supernatants from infected cells were collected and plaque assays were performed to determine the infectious EEV titre of each condition (Figure 5a). In cells infected by VACV‐WR, EEV release into the supernatant (measured in plaque forming units or pfu/mL) was significantly reduced under β‐CYA knockdown. These results are consistent with our previous observations that disrupting actin‐based motility leads to a corresponding reduction in EEV release. Cells infected by VACV‐A36YdF showed greatly reduced EEV release when compared with VACV‐WR as has been previously reported (Horsington et al., 2013). However, no difference in EEV release of VACV‐A36YdF was observed under either CYA knockdown condition. All phenotypes associated with actin‐based motility are ablated in VACV‐A36YdF, and β‐CYA knockdown does not exacerbate defects in EEV release in a VACV‐A36YdF. This suggests that β‐CYA also disrupts EEV release through actin‐based motility rather than other WV protein interactions known to affect EEV release (Smith, Vanderplasschen, & Law, 2002).
Figure 5.
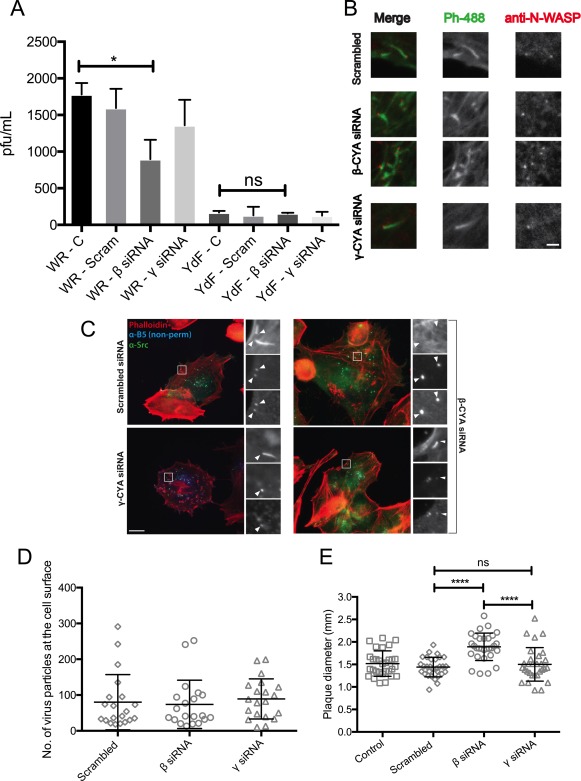
Effect of β‐CYA depletion on VACV signalling and spread. (a) HeLa cells were treated with the specified siRNA and then infected with the specified VACV (WR strain or VACV‐A36YdF, depicted as YdF), at an MOI of 0.1. Supernatants were collected at 16 hpi and used to perform plaque assays on BSC‐1 cells to estimate viral titre (“ns”: p > .05, “*”: p ≤ .05, n = 3). (b) VACV‐infected cells under CYA knockdown were fixed 8 hpi and stained for N‐WASP (red) and phalloidin (green). Scale bar is 1 μm. (c) VACV‐infected cells under CYA knockdown were fixed 8 hpi and stained for virus envelope protein B5 (nonpermeablized, blue), phallodin (red), and anti‐Src (green). Scale bar is 10 μm. (d) HeLa cells were treated with CYA siRNA and infected with VACV‐WR at an MOI > 3. Cells were fixed 8 hpi, followed by staining with anti‐B5 prior to permeabilization. The number of virus particles at the cell surface was counted in each case (n = 10 cells each, in 2 experimental replicates). (e) GBM cells were treated with the specified CYA siRNA and infected with VACV‐WR at an MOI of 0.1. Cells were fixed 3 dpi and stained to measure plaque diameters. (“****”: p ≤ .0001, n = 30 plaques each, with 2 experimental replicates). [Color figure can be viewed at http://wileyonlinelibrary.com]
To rule out the possibility that depleting β‐CYA would affect motility of virus particles to the cell surface, and therefore indirectly cause a reduction in EEV release, we assessed the signalling upstream and downstream of A36 phosphorylation as well as the number of WV reaching the cell surface. We visualized N‐WASP and Src in VACV‐infected cells treated for CYA siRNA knockdown (Figure 5b,c). Depletion of β‐CYA or γ‐CYA did not lead to overt changes in the recruitment of Src or N‐WASP to virus particles, suggesting that the phenotypes we have observed in virus motility are downstream of these critical signalling molecules.
Cell‐associated extracellular virus was enumerated by staining cells with anti‐B5 antibody (against viral envelope protein B5) prior to permeabilizing the cells, such that only enveloped virus particles at the cell surface would be labelled. There was no significant difference in the number of anti‐B5‐stained virus particles at the cell surface between all three siRNA treatment conditions (Figure 5d). Finally, since actin‐based motility in VACV enhances cell‐to‐cell spread, with VACV‐A36YdF producing a reduced plaque phenotype compared with parental strains (Horsington et al., 2013; Rodger and Smith, 2002; Zhang, Wilcock, & Smith, 2000], we aimed to test how β‐CYA knockdown in cells might affect VACV plaque size or morphology. GBM cells (selected over HeLa due to their amenability to forming monolayers) were subjected to either CYA knockdown and allowed to near confluence, at which point a plaque assay was conducted with VACV‐WR. Three days post infection, plaque assays were fixed and the diameter of plaques was measured (Figure 5e). Confounding expectations, plaques produced by VACV‐WR treated with β‐CYA knockdown were significantly larger than those compared to plaques produced in cells under γ‐CYA knockdown or treated with scrambled siRNA. These results indicate that knockdown of β‐CYA produces a complex phenotype and that other mechanisms may compensate for the expected reduction in plaque size elicited by the reduction in actin‐based motility.
2.5. The VCA domain of N‐WASP does not show specificity for one actin isoform
Having uncovered a strict requirement for β‐CYA for the efficient formation of VACV‐induced actin comets, we wanted to further define the mechanism. N‐WASP is an actin nucleation‐promoting factor that is recruited to the site of actin comet formation during VACV egress (Frischknecht et al., 1999b; Moreau et al., 2000). The VCA domain of N‐WASP recruits the Arp2/3 complex and two actin monomers, branching off from an existing actin filament. If the VCA domain were to have a preference for the β‐CYA isoform, that would explain the dependence on β‐CYA for actin‐based virus transport. Two bacterial expression plasmids were constructed: one expressing a glutathione S‐transferase (GST)‐tagged VCA domain and another expressing a GST‐tagged VCA domain containing two arginine‐alanine substitutions in its actin‐binding region, VCA‐R410A/R438A (called VCA‐RA/RA), which effectively abrogate actin binding (Co, Wong, Gierke, Chang, & Taunton, 2007). GST‐tagged VCA proteins were expressed in bacteria, purified and used to enrich for actin by passing cell lysates over the bound, immobilized protein and probing for actin isoforms. As expected, actin bound to GST‐VCA and did not bind to the GST‐VCA RA/RA (Figure 6a). The next step was to examine whether the actin that bound to the VCA domain was specifically one CYA isoform. Both anti‐β‐CYA and anti‐γ‐CYA antibodies bound to an immunoblot of cell lysates passed over GST‐VCA protein (Figure 6b), suggesting that the VCA domain does not specifically bind to one CYA over the other.
Figure 6.
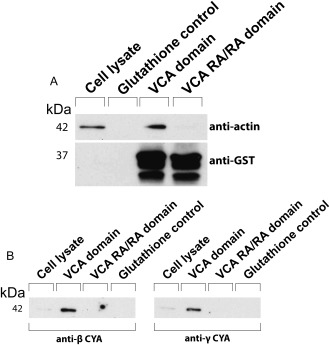
GST pull‐down assays to determine binding preferences for β‐or γ‐CYA. Immunoblots of cell lysate bound to GST‐VCA or GST‐VCA RA/RA on glutathione‐containing Sepharose beads. Immunoblots were probed with either anti‐actin and anti‐GST antibodies (a) or antibodies specific to β‐CYA or γ‐CYA (b).
3. Discussion
There has been increasing interest in the roles of the two CYA isoforms, β‐CYA and γ‐CYA, as two distinct proteins with differing localizations and functions despite their high homology and overlapping expression. For example, while β‐CYA localises to stress fibres and cell‐cell contacts, γ‐CYA is abundant in lamellipodial structures and is important for cell motility (Dugina et al., 2009). Studies have identified isoform‐specific functions during multiple cellular processes (Artman et al., 2014; Hajkova, Nyman, Lindberg, & Karlsson, 2000; Joseph et al., 2014; Pasquier et al., 2015), as well as distinct requirements in the replication cycle of viruses like coronaviruses (Wang, Fang, Xiao, Chen, Tam, & Liu, 2009) and the classical swine fever virus (He, Ling, Liao, Li, Han, Zhao, … Qiu, 2014). While these reports revealed a role for β‐CYA in the early stages of viral infection, from viral gene expression and replication to assembly, our study supports a requirement for β‐CYA in the initiation of actin comets induced by VACV during the late stages of infection.
Depletion of β‐CYA quelled VACV‐induced actin nucleation, implicating a specific requirement for this isoform rather than a general reduction in actin expression. As is typical for siRNA‐mediated knockdown, the loss of β‐CYA expression was not complete, with a reduction to approximately 20% (on average for both cell types tested) of control levels, which suggests a threshold at which reduced levels no longer supports actin nucleation. Levels of β‐CYA recruited to VACV particles correlated with comet length, even under β‐CYA knockdown, and at least some accumulation of β‐CYA was evident at the viral surface in all cases. Indeed due to the variability of knockdown at the cellular level, we could observe cells with low β‐CYA expression that gave rise to few virus‐associated comets. These results are consistent with an absolute, albeit dosage‐sensitive, requirement for β‐CYA in VACV actin nucleation and actin‐based motility. Previous studies have also demonstrated significant morphological and functional changes in various cell types with incomplete knockdown of CYA isoforms, such as taking on a less or a more contractile phenotype, or an increase/decrease in lamellipodial structures, stress fibres, and cell motility (Dugina et al., 2009; Pasquier et al., 2015; Shum, Pasquier, Po'uha, O'Neill, Chaponnier, Gunning, & Kavallaris, 2011).
While our data support a role for β‐CYA knockdown in the release of EEV through actin nucleation, we also found that it did not disrupt the intracellular movement of enveloped virus particles to the cell periphery. This is as expected, as both the generation of intracellular enveloped VACV, as well as their transport to the cell periphery, is independent from release of EEV particles (Horsington et al., 2013; Lynn, Horsington, Ter, Han, Chew, Diefenbach, … Newsome, 2012; Payne and Kristensson, 1982; Ward and Moss, 2001). The translocation of enveloped virus particles to the cell surface was performed by quantifying B5‐positive extracellular virus. β‐CYA knockdown resulted in a higher percentage of multinucleated cells compared with scrambled siRNA and γ‐CYA knockdown in both infected and uninfected HeLa cells (results not shown). This has also been previously observed in uninfected HaCaT cells (Dugina et al., 2009). This made it challenging to find singular infected cells to quantify the number of virus particles reaching the surface and/or number of actin comets produced per cell. While the depletion of γ‐CYA by siRNA can suppress microtubule dynamics in SH‐EP cells expressing GFP‐labelled tubulin (Po'uha, Honore, Braguer, & Kavallaris, 2013), we did not observe any difference in the number of virus particles either being released or being transported to the cell periphery under γ‐CYA depletion. As the major effects observed by Po'uha et al (2013) were a decrease in microtubule shortening rates and a delayed metaphase‐anaphase transition, this may have been insufficient to grossly perturb the microtubule‐based transport of VACV. Reducing levels of γ‐CYA did lead to increased speed of VACV undergoing actin‐based transport but this did not lead to additional phenotypes in virus release or cell‐to‐cell spread. The knockdown of γ‐CYA in HeLa cells causes a compensatory increase in the levels of β‐CYA (Figure 2b), which indicates that comets with greater speeds observed in γ‐CYA‐depleted cells may be caused by a shift in the ratio of CYAs in favor of β‐CYA. This observation is in agreement with the previously shown ability of β‐CYA to polymerize at faster rates than γ‐CYA in a reconstituted system (Bergeron, Zhu, Thiem, Friderici, & Rubenstein, 2010).
Virus‐associated actin comets were fewer and shorter when levels of β‐CYA were reduced. Reductions in actin comet length have been observed previously, either due to mutations in the viral A36 envelope protein, or mutations in cellular N‐WASP, which abrogates its ability to bind to actin monomers and reduce its stability during actin polymerization (Horsington et al., 2013; Weisswange et al., 2009). As we also observed a significant reduction in the length of actin comets during β‐CYA depletion, we hypothesized that altered binding, such as a preference for β‐CYA by N‐WASP, may contribute to the actin nucleation phenotype. To test this model, we expressed GST‐tagged fusions of the VCA domain of N‐WASP (its actin‐binding region of N‐WASP; see Takenawa and Suetsugu, 2007 for a review), or a mutated version of the domain with abrogated actin‐binding ability (VCA‐RA/RA; Co et al., 2007). The ability of these tagged proteins to bind to actin was examined, and no preference for either CYA isoform was observed by immunoblot. Additionally, phosphorylation of the N‐WASP VCA domain has been shown to enhance actin polymerization activity (Cory, Cramer, Blanchoin, & Ridley, 2003), a post‐translational modification that may not be present in bacterially‐expressed GST‐VCA domains. Nonetheless, our results do not support a specific binding affinity of the WH2 domains of N‐WASP for β‐CYA as the underlying mechanism that results in a requirement for this isoform in VACV actin‐based motility. A recent genome‐wide siRNA screen performed during infection of HeLa cells with the bacterial pathogen Listeria, also known to employ actin‐based motility for infection (Lambrechts, Gevaert, Cossart, Vandekerckhove, & Van Troys, 2008), showed that siRNA‐mediated knockdown of β‐CYA, but not γ‐CYA, impaired infection (Kuhbacher et al., 2015). These findings suggest that specificity for actin isoforms for actin nucleation might be at the level of the Arp2/3 complex, rather than with its activator N‐WASP.
Our most surprising observation was the increase in VACV plaque size during β‐CYA knockdown in GBM cells. γ‐CYA is a known regulator of Rho‐associated kinase (ROCK)‐mediated cell migration, and the knockdown of γ‐CYA has been shown to reduce cell migration (Dugina et al., 2009; Shum et al., 2011). Additionally, we observed a significant increase in γ‐CYA expression during β‐CYA knockdown in GBM cells, which may have had an additive effect, contributing to increased viral plaque size. An enhancement of directional cell migration during β‐CYA depletion, as well as a reduction in migration during γ‐CYA depletion, has also been observed previously (Dugina et al., 2009). This increase in motility during β‐CYA knockdown could also be playing a role in VACV virus‐induced cell motility, contributing to an increased plaque size. The VACV protein F11 is also responsible for virus‐induced cell migration, through its inhibition of RhoA signalling (Cordeiro, Guerra, Arakawa, Dodding, Esteban, & Way, 2009; Valderrama, Cordeiro, Schleich, Frischknecht, & Way, 2006). This may explain why we have not observed a corresponding reduction in plaque size during γ‐CYA knockdown. Interestingly, β‐CYA synthesis has also been associated with Rho signalling (Latham, Yu, Tullio, Adelstein, & Singer, 2001) and the use of a Rho kinase inhibitor can cause the selective disorganization of β‐CYA bundles while leaving γ‐CYA undisturbed (Dugina et al., 2009). Clearly there is a strong link between β‐CYA and Rho signalling, such that the depletion of β‐CYA may have a significant effect on Rho and its downstream signalling targets. The use of ROCK inhibitors under conditions of β‐ and γ‐CYA knockdown may, in future studies, clarify the complex networks involved in this process. It is evident, however, that cell motility is an important determinant of VACV plaque size.
This work contributes to the emerging paradigm that not all actin isoforms are equivalent. Studies have shown a specific interaction of β‐CYA with viral membrane proteins like the classical swine fever virus E2 protein (He et al., 2014) and the coronavirus M protein (Wang et al., 2009). While we have not observed a physical interaction between a specific actin isoform and a VACV protein, our results indicate that β‐CYA plays a role in the N‐WASP‐mediated nucleation of actin beneath VACV particles.
The evidence of isoform‐specific functions of actin during microbial pathogenesis suggests that the most basic of cellular processes may potentially be therapeutically targetable. Clearly blocking all actin functions would be highly detrimental to a cell. Our results indicate that specific pathogenic mechanisms can be successfully targeted, even for such a fundamental process as actin nucleation, by inhibiting one isoform of a highly conserved group of proteins. Our data was unable to uncover the precise mechanism by which β‐CYA actin is required during VACV‐induced actin nucleation, but they suggest a link between the activity of a particular nucleator, N‐WASP, and an actin isoform, β‐CYA. This raises the tantalizing possibility that nucleators may be subject to regulation by isoform availability, which is consistent with the highly localized distributions of β‐CYA and actin‐CYA at the subcellular level.
4. Materials and methods
4.1. Cells and viruses
Cell lines used in this study were HeLa (human cervical cancer cell line, ATCC CCL‐2), GBM A‐172 (glioblastoma cell line, ATCC CRL‐1620), and hCMEC/D3 cells (human cerebral microvascular endothelial cell line, gifted by Prof. C.O. Couraud, reviewed in Weksler Romero, & Couraud, 2013). Cells, unless otherwise indicated, were cultured in Dulbecco?s Modified Eagle's Medium (DMEM; Invitrogen) supplemented with 5% foetal bovine serum (FBS) and 292 µg/ml L‐ glutamine, 100 units/ml penicillin and 100 µg/ml streptomycin at 37°C in 5% CO2 (DMEM‐FBSG). hCMEC/D3 cells were cultured in supplemented EBM‐2 medium (Lonza), as described by Latham et al. (Latham et al. 2013). Viruses used in this study were the VACV strain Western Reserve (VACV‐WR, ATCC VR‐1354), the WR strain where the gene A36 contains two point mutations (VACV‐A36YdF, (Scaplehorn et al., 2002), and WR VACV‐Lifeact‐GFP, which is a VACV‐WR strain carrying a Lifeact‐GFP transgene expressed through a synthetic early/late VACV promoter. Lifeact is a 17‐amino acid peptide that is used to visualize actin (Riedl, Crevenna, Kessenbrock, Yu, Neukirchen, Bista, … Wedlich‐Soldner, 2008), and when bound to GFP enables visualization of the F‐actin cytoskeleton in live cells. Lifeact‐GFP was cloned and inserted between VACV genes C10L and C11R by homologous recombination as previously described (Marzook et al., 2014).
4.2. Antibodies and plasmids
Mouse antibodies against β‐CYA and γ‐CYA were a gift (Dugina et al., 2009). A mouse anti‐pan‐actin antibody (clone C4; MAB1501, Merck Millipore) was used to detect total actin, a mouse anti‐Src antibody (clone 327; ab16885, Abcam) for Src, a rabbit anti‐N‐WASP antibody (ab126626, Abcam) for N‐WASP, and a monoclonal rat anti‐GST antibody (Sigma‐Aldrich, clone GST‐R 6G9) for GST‐bound VCA or VCA‐RA/RA proteins were used. Secondary rabbit anti‐mouse antibodies conjugated to HRP (Merck Millipore) were used in western blots. For IFA distinguishing the two CYAs, cyanine dye‐tagged secondary antibodies (Jackson Immunotech, AffiniPure goat anti‐mouse IgG, Fcγ Subclasses 1‐CY2 and 2b‐CY3; catalogue numbers 115‐225‐205 and 115‐165‐207) were used concurrently. For visualizing enveloped VACV, rat monoclonal antibodies against viral envelope protein B5 (19C2, 1/200) were used, along with goat anti‐rat secondary antibodies conjugated to either AlexaFluor©‐488 or ‐568 (Invitrogen, 1/200). Alexa Fluor® 488‐Phalloidin (ThermoFisher Scientific) and DAPI were used to detect the actin cytoskeleton and nuclei, respectively, in IFA.
4.3. siRNA and knockdown treatment
siRNAs specific to either β‐CYA (Hs_ACTB_8, SI04205306 and Hs_ACTB_9, SI04287759), or γ‐CYA (Hs_ACTG1_8, SI04155480; Hs_ACTG1_9, SI04361007 and Hs_ACTG1_10, SI04364871) were ordered from QIAGEN. A scrambled siRNA (SI03650325) was also ordered as a negative control. Cells were transfected with the respective siRNA with Lipofectamine® 2000 (Thermo Fisher Scientific) in OptiMEM (Invitrogen) for six hours, followed by replacement with the respective complete media. Knockdown was allowed to proceed for 72 hr before prior to analysis by immunoblot, immunofluorescence or plaque assay.
4.4. Immunoblotting
Cells treated with siRNA were lysed after 72 hr in sodium dodecyl sulfate (SDS)‐reducing sample buffer (62.5 mM Tris‐HCl, 0.25 M glycerol, 2% SDS, 0.01% [wt/vol] bromophenol blue, 12.5% [vol/vol] β‐mercaptoethanol) and boiled at 95°C for 5 min. Proteins were separated by SDS‐polyacrylamide gel electrophoresis (SDS‐PAGE) (resolving gel of 10% acrylamide‐Bis solution [37.5:1], 0.375 M Tris‐HCl, pH 8.8, 0.1% [wt/vol] SDS, 0.1% ammonium persulfate (APS), and 0.1% N,N,N′,N′‐tetramethylethylenediamine (TEMED); stacking gel of 4% to 30% acrylamide‐Bis solution [37.5:1], 0.375 M Tris‐HCl, pH 6.8, 0.1% [wt/vol] SDS, 0.1% APS, and 0.1% TEMED). Proteins resolved by SDS‐PAGE were transferred to nitrocellulose membranes (Hybond‐C Extra; Amersham Biosciences) and probed with primary antibodies diluted in PBST‐milk (5% [wt/vol] skim milk in PBS with 0.1% Tween 20), followed by washing three times in PBST‐milk. The membrane was then probed with secondary antibodies conjugated with horseradish peroxidase. Immunoreactive protein bands were visualized with ECL Western blotting reagent (GE Healthcare) and developed using a CP1000 photographic film developer. Blots were scanned using the GS‐900 calibrated densitometer (Bio‐Rad), densities were measured using FIJI image processing software, and statistical analysis was done using GraphPad PRISM.
4.5. Immunofluorescence assays
hCMEC/D3, HeLa or GBM cells were grown on glass coverslips to about 80% confluency (72 hr post‐siRNA treatment) and infected with VACV‐WR at an MOI > 5.
Cells were fixed 8 hpi, the optimum time to observe VACV‐associated actin comets (Cudmore et al., 1995), in 3% paraformaldehyde in cytoskeleton buffer (CB; 10 mM 2‐(N‐morpholino) ethanesulfonic acid buffer, 0.15 M NaCl, 5 mM ethylene glycol tetraacetic acid, 5 mM MgCl2, 50 mM glucose, pH 6.1) for 10 min at room temperature. For the visualization of both CYA isoforms in actin comets, hCMEC/D3 cells were permeabilized for 5 min in cold methanol and blocked for 1 hr in 2% BSA. Coverslips were incubated in mouse primary antibodies (anti‐β‐CYA at 1:50 and anti‐γ‐CYA at 1:100) diluted in BSA for 1 hr, followed by washing in PBS. Secondary antibodies were added for 30 min. Samples were counterstained with DAPI and mounted in Prolong Gold anti‐fade. For the visualization of F‐actin in comets or enveloped virus particles, hCMEC/D3 or HeLa cells were blocked in blocking buffer (1% BSA, 2% FBS in CB) for 20 min, followed by antibodies (all diluted in CB) against VACV envelope protein B5 (rat 19C2) for 40 min and goat anti‐rat secondary antibodies for 20 min, or against F‐actin with Phalloidin‐488 for 20 min, and DAPI. Cells were stained without permeabilization to only visualise EEV particles, and their associated actin comets. The coverslips were mounted on a glass slide with anti‐fade 0.3–1% (w/v) P‐phenylenediamine (Sigma‐Aldrich) in mowiol mounting media [10% (w/v) Polyvinyl Alcohol 4–88 (Sigma‐Aldrich), 25% (w/v) glycerol, 0.1 M Tris, pH 8.5]. Cells were imaged with an Olympus microscope BX51 with filter sets 31,001, 31,002, and 31,013v2 (Chroma) at 40× or 100× (with glycerol) magnification at room temperature. AnalySIS LS image capture software was used to acquire the images with an F‐view monochrome fluorescence camera. Z‐stack images were collected with the Zeiss LSM 510 confocal microscope at 63× magnification with 1.4 NA at room temperature. The resulting images were processed with Photoshop CS3 (Adobe; to separate RGB color channels and crop images), and analysed using FIJI (for measuring actin comet length and plaque diameter) and GraphPad PRISM (for data visualization and statistical measurements).
4.6. Measuring virus speed
Cells infected with VACV‐WR Lifeact‐GFP were imaged using the Nikon Eclipse Ti‐E inverted microscope system at 40× magnification in a chamber maintained at 37°C in a 5% CO2‐enriched atmosphere. Images were captured every 4 s over a period of 5 min using NIS‐Elements AR (v4.51.00) image capture software. Maximal intensity projections for 1 min intervals over the 5 min time course were created, and lengths of actin comets in these projections were measured using FIJI (ver. 2.0.0‐rc‐43). Speed was calculated as length of comets over the time interval of that projection (1 min in this case).
4.7. GST‐VCA pull‐down experiments
A plasmid expressing GST‐VCA was created by PCR from a plasmid containing rat N‐WASP O08816 (forward primer containing a Not restriction site: 5′ GGCGGCCGCGACCATCAAGTTCCAGC 3′, and reverse primer containing an Eco restriction site: 5′ GAATTCTCAGTCTTCCCACTCATC 3′), creating a 333 kb oligonucleotide. This was ligated into a pMW T7 vector containing GST, as described previously by (Newsome et al., 2006; using the NotI – EcoRI restriction sites). The GST‐VCA‐RA/RA plasmid was created using the plasmid previously described, where a 333 kb VCA RA/RA oligonucleotide (containing mutations corresponding to positions R410A and R438A in the full‐length rat N‐WASP) was generated synthetically (Integrated DNA Technologies), and ligated into the backbone using the same restriction sites (NotI – EcoRI). Plasmids expressing either GST‐VCA or GST‐VCA‐RA/RA were transformed into bacterial BL21 competent E. coli (NEB) and grown at 37°C until they reached an OD of around 0.5. Bacterial cells were lysed in lysis buffer [1% Triton‐X (v/v) and 200 uM phenylmethylsulfonyl fluoride (PMSF) in PBS] by sonication, and passed over glutathione‐containing Sepharose beads (GE Healthcare), primed in lysis buffer not containing PMSF. Three rounds of washing in lysis buffer enriched for GST‐bound expressed protein from the bacterial cells. HeLa cell lysates (prepared with the same lysis buffer) were then passed over the beads, followed by washing three times. Beads were then boiled in SDS reducing sample buffer (described above) and separated by SDS‐PAGE, followed by immunoblotting.
4.8. Plaque assays
Following siRNA treatment for 72 hr, GBM cells that had reached confluency were infected by VACV‐WR at an MOI of 0.1. 1 hpi, cells were washed and over‐laid with modified Eagle medium (MEM; Invitrogen) supplemented with 5% FBS, 292 μg/ml l‐glutamine, 100 units/ml penicillin, 100 μg/ml streptomycin, and 1.5% carboxymethyl cellulose. Plaques were allowed to form for 3 days before being fixed with 1% crystal violet in methanol. Cell culture plates were scanned using the GS‐900 densitometer (Bio‐Rad) and plaque diameters from two experiment replicates were measured using FIJI image processing software.
4.9. EEV release assays
Following siRNA treatment for 72 hr, cells HeLa cells were infected by the designated virus (either VACV‐WR or VACV‐A36YdF), at an MOI of 0.1 for 1 hr. Cells were then washed twice and overlaid with DMEM‐FBSG. Supernatants were collected at 16 hpi and plaque assays using 10‐fold serial dilutions of the supernatant were conducted on BSC‐1 cells, as described above. Plaques were enumerated from three experimental replicates and statistical analysis was carried out with GraphPad PRISM software.
Supporting information
Supporting Information Figure 1
Supporting Information Figure 2
Supporting Information Figure 3
Acknowledgments
The authors would like to thank Prof. C. Chaponnier for the anti‐β‐ and anti‐γ‐actin antibodies and Prof. C. O. Couraud for the kind contribution of hCMEC/D3 cells. They additionally acknowledge the Advanced Microscopy Facility within the Bosch Institute at The University of Sydney, Australia. The authors declare that they have no conflict of interests.
How to cite this article: Bishara Marzook N, Latham SL, Lynn H, et al. Divergent roles of β‐ and γ‐actin isoforms during spread of vaccinia virus. Cytoskeleton. 2017;74:170–183. 10.1002/cm.21356
Monitoring Editor: Bruce Goode
References
- Alvarez, D. E. , & Agaisse, H. (2013). The formin FHOD1 and the small GTPase Rac1 promote vaccinia virus actin‐based motility. The Journal of Cell Biology, 202, 1075–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez, D. E. , & Agaisse, H. (2014). A role for the small GTPase Rac1 in vaccinia actin‐based motility. Small GTPases, 5, e29038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artman, L. , Dormoy‐Raclet, V. , von Roretz, C. , & Gallouzi, I. E. (2014). Planning your every move: the role of beta‐actin and its post‐transcriptional regulation in cell motility. Seminars in Cell and Developmental Biology, 34, 33–43. [DOI] [PubMed] [Google Scholar]
- Bergeron, S. E. , Zhu, M. , Thiem, S. M. , Friderici, K. H. , & Rubenstein, P. A. (2010). Ion‐dependent polymerization differences between mammalian beta‐ and gamma‐nonmuscle actin isoforms. The Journal of Biological Chemistry, 285, 16087–16095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaponnier, C. , & Gabbiani, G. (2004). Pathological situations characterized by altered actin isoform expression. Journal of Pathology, 204, 386–395. [DOI] [PubMed] [Google Scholar]
- Co, C. , Wong, D. T. , Gierke, S. , Chang, V. , & Taunton, J. (2007). Mechanism of actin network attachment to moving membranes: barbed end capture by N‐WASP WH2 domains. Cell, 128, 901–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordeiro, J. V. , Guerra, S. , Arakawa, Y. , Dodding, M. P. , Esteban, M. , & Way, M. (2009). F11‐mediated inhibition of RhoA signalling enhances the spread of vaccinia virus in vitro and in vivo in an intranasal mouse model of infection. PLoS One, 4, e8506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cory, G. O. , Cramer, R. , Blanchoin, L. , & Ridley, A. J. (2003). Phosphorylation of the WASP‐VCA domain increases its affinity for the Arp2/3 complex and enhances actin polymerization by WASP. Molecular Cell, 11, 1229–1239. [DOI] [PubMed] [Google Scholar]
- Cudmore, S. , Cossart, P. , Griffiths, G. , & Way, M. (1995). Actin‐based motility of vaccinia virus. Nature, 378, 636–638. [DOI] [PubMed] [Google Scholar]
- Doceul, V. , Hollinshead, M. , van der Linden, L. , & Smith, G. L. (2010). Repulsion of superinfecting virions: A mechanism for rapid virus spread. Science, 327, 873–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugina, V. , Zwaenepoel, I. , Gabbiani, G. , Clement, S. , & Chaponnier, C. (2009). Beta and gamma‐cytoplasmic actins display distinct distribution and functional diversity. Journal of Cell Science, 122, 2980–2988. [DOI] [PubMed] [Google Scholar]
- Frischknecht, F. , Cudmore, S. , Moreau, V. , Reckmann, I. , Rottger, S. , & Way, M. (1999a). Tyrosine phosphorylation is required for actin‐based motility of vaccinia but not Listeria or Shigella. Current Biology, 9, 89–92. [DOI] [PubMed] [Google Scholar]
- Frischknecht, F. , Moreau, V. , Rottger, S. , Gonfloni, S. , Reckmann, I. , Superti‐Furga, G. , & Way, M. (1999b). Actin‐based motility of vaccinia virus mimics receptor tyrosine kinase signalling. Nature, 401, 926–929. [DOI] [PubMed] [Google Scholar]
- Gouin, E. , Welch, M. D. , & Cossart, P. (2005). Actin‐based motility of intracellular pathogens. Current Opinion in Microbiology, 8, 35–45. [DOI] [PubMed] [Google Scholar]
- Hajkova, L. , Nyman, T. , Lindberg, U. , & Karlsson, R. (2000). Effects of cross‐linked profilin:beta/gamma‐actin on the dynamics of the microfilament system in cultured cells. Experimental Cell Research, 256, 112–121. [DOI] [PubMed] [Google Scholar]
- He, F. , Ling, L. , Liao, Y. , Li, S. , Han, W. , Zhao, B. , … Qiu, H. J. (2014). Beta‐actin interacts with the E2 protein and is involved in the early replication of classical swine fever virus. Virus Research, 179, 161–168. [DOI] [PubMed] [Google Scholar]
- Hollinshead, M. , Rodger, G. , Van Eijl, H. , Law, M. , Hollinshead, R. , Vaux, D. J. , & Smith, G. L. (2001). Vaccinia virus utilizes microtubules for movement to the cell surface. Journal of Cell Biology, 154, 389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horsington, J. , Lynn, H. , Turnbull, L. , Cheng, D. , Braet, F. , Diefenbach, R. J. , … Newsome, T. P. (2013). A36‐dependent actin filament nucleation promotes release of vaccinia virus. PLoS Pathogens, 9, e1003239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphries, A. C. , Dodding, M. P. , Barry, D. J. , Collinson, L. M. , Durkin, C. H. , & Way, M. (2012). Clathrin potentiates vaccinia‐induced actin polymerization to facilitate viral spread. Cell Host & Microbe, 12, 346–359. [DOI] [PubMed] [Google Scholar]
- Johnston, J. B. , & McFadden, G. (2004). Technical knockout: understanding poxvirus pathogenesis by selectively deleting viral immunomodulatory genes. Cellular Microbiology, 6, 695–705. [DOI] [PubMed] [Google Scholar]
- Joseph, R. , Srivastava, O. P. , & Pfister, R. R. (2014). Downregulation of beta‐actin and its regulatory gene HuR affect cell migration of human corneal fibroblasts. Molecular Vision, 20, 593–605. [PMC free article] [PubMed] [Google Scholar]
- Khaitlina, S. (2007). Mechanisms of spatial segregation of actin isoforms. Tsitologiia, 49, 345–354. [PubMed] [Google Scholar]
- Kuhbacher, A. , Emmenlauer, M. , Ramo, P. , Kafai, N. , Dehio, C. , Cossart, P. , & Pizarro‐Cerda, J. (2015). Genome‐Wide siRNA Screen Identifies Complementary Signaling Pathways Involved in Listeria Infection and Reveals Different Actin Nucleation Mechanisms during Listeria Cell Invasion and Actin Comet Tail Formation. MBio, 6, e00598–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambrechts, A. , Gevaert, K. , Cossart, P. , Vandekerckhove, J. , & Van Troys, M. (2008). Listeria comet tails: The actin‐based motility machinery at work. Trends in Cell Biology, 18, 220–227. [DOI] [PubMed] [Google Scholar]
- Latham, S. L. , Chaponnier, C. , Dugina, V. , Couraud, P. O. , Grau, G. E. , & Combes, V. (2013). Cooperation between beta‐ and gamma‐cytoplasmic actins in the mechanical regulation of endothelial microparticle formation. FASEB Journal, 27, 672–683. [DOI] [PubMed] [Google Scholar]
- Latham, V. M. , Yu, E. H. , Tullio, A. N. , Adelstein, R. S. , & Singer, R. H. (2001). A Rho‐dependent signaling pathway operating through myosin localizes beta‐actin mRNA in fibroblasts. Current Biology, 11, 1010–1016. [DOI] [PubMed] [Google Scholar]
- Lechuga, S. , Baranwal, S. , Li, C. , Naydenov, N. G. , Kuemmerle, J. F. , Dugina, V. , … Ivanov, A. I. (2014). Loss of gamma‐cytoplasmic actin triggers myofibroblast transition of human epithelial cells. Molecular Biology of the Cell, 25, 3133–3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynn, H. , Horsington, J. , Ter, L. K. , Han, S. , Chew, Y. L. , Diefenbach, R. J. , … Newsome, T. P. (2012). Loss of cytoskeletal transport during egress critically attenuates ectromelia virus infection in vivo. Journal of Virology, 86, 7427–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzook, N. B. , Procter, D. J. , Lynn, H. , Yamamoto, Y. , Horsington, J. , & Newsome, T. P. (2014). Methodology for the efficient generation of fluorescently tagged vaccinia virus proteins. Journal of Visualized Experiments, (83), e51151. doi: 10.3791/51151(83):e51151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFadden, G. (2005). Poxvirus tropism. Nature Reviews Microbiology, 3, 201–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreau, V. , Frischknecht, F. , Reckmann, I. , Vincentelli, R. , Rabut, G. , Stewart, D. , & Way, M. (2000). A complex of N‐WASP and WIP integrates signalling cascades that lead to actin polymerization. Nature Cell Biology, 2:441–8. [DOI] [PubMed] [Google Scholar]
- Newsome, T. P. , & Marzook, N. B. (2015). Viruses that ride on the coat‐tails of actin nucleation. Seminars in Cell & Developmental Biology, 46, 155–163. doi: 10.1016/j.semcdb.2015.10.008. [DOI] [PubMed] [Google Scholar]
- Newsome, T. P. , Scaplehorn, N. , & Way, M. (2004). SRC mediates a switch from microtubule‐ to actin‐based motility of vaccinia virus. Science, 306, 124–129. [DOI] [PubMed] [Google Scholar]
- Newsome, T. P. , Weisswange, I. , Frischknecht, F. , & Way, M. (2006). Abl collaborates with Src family kinases to stimulate actin‐based motility of vaccinia virus. Cellular Microbiology, 8, 233–241. [DOI] [PubMed] [Google Scholar]
- Ohkawa, T. , Volkman, L. E. , & Welch, M. D. (2010). Actin‐based motility drives baculovirus transit to the nucleus and cell surface. Journal of Cell Biology, 190, 187–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkinson, J. E. , & Smith, G. L. (1994). Vaccinia virus gene A36R encodes a M(r) 43‐50 K protein on the surface of extracellular enveloped virus. Virology, 204, 376–390. [DOI] [PubMed] [Google Scholar]
- Pasquier, E. , Tuset, M. P. , Sinnappan, S. , Carnell, M. , Macmillan, A. , & Kavallaris, M. (2015). gamma‐Actin plays a key role in endothelial cell motility and neovessel maintenance. Vascular Cell, 7, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne, L. G. , & Kristensson, K. (1982). The effect of cytochalasin D and monensin on enveloped vaccinia virus release. Archives of Virology, 74, 11–20. [DOI] [PubMed] [Google Scholar]
- Po'uha, S. T. , Honore, S. , Braguer, D. , & Kavallaris, M. (2013). Partial depletion of gamma‐actin suppresses microtubule dynamics. Cytoskeleton (Hoboken), 70, 148–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radtke, K. , Dohner, K. , & Sodeik, B. (2006). Viral interactions with the cytoskeleton: a hitchhiker's guide to the cell. Cellular Microbiology, 8, 387–400. [DOI] [PubMed] [Google Scholar]
- Reeves, P. M. , Bommarius, B. , Lebeis, S. , McNulty, S. , Christensen, J. , Swimm, A. , … Kalman, D. (2005). Disabling poxvirus pathogenesis by inhibition of Abl‐family tyrosine kinases. Nature Medicine, 11, 731–739. [DOI] [PubMed] [Google Scholar]
- Riedl, J. , Crevenna, A. H. , Kessenbrock, K. , Yu, J. H. , Neukirchen, D. , Bista, M. , … Wedlich‐Soldner, R. (2008). Lifeact: A versatile marker to visualize F‐actin. Nature Methods, 5, 605–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rietdorf, J. , Ploubidou, A. , Reckmann, I. , Holmstrom, A. , Frischknecht, F. , Zettl, M. , … Way, M. (2001). Kinesin‐dependent movement on microtubules precedes actin‐based motility of vaccinia virus. Nature Cell Biology, 3, 992–1000. [DOI] [PubMed] [Google Scholar]
- Rodger, G. , & Smith, G. L. (2002). Replacing the SCR domains of vaccinia virus protein B5R with EGFP causes a reduction in plaque size and actin tail formation but enveloped virions are still transported to the cell surface. Journal of General Virology, 83, 323–332. [DOI] [PubMed] [Google Scholar]
- Rubenstein, P. A. (1990). The functional importance of multiple actin isoforms. Bioessays, 12, 309–315. [DOI] [PubMed] [Google Scholar]
- Scaplehorn, N. , Holmstrom, A. , Moreau, V. , Frischknecht, F. , Reckmann, I. , & Way, M. (2002). Grb2 and Nck act cooperatively to promote actin‐based motility of vaccinia virus. Current Biology, 12, 740–745. [DOI] [PubMed] [Google Scholar]
- Shaner, N. C. , Sanger, J. W. , & Sanger, J. M. (2005). Actin and alpha‐actinin dynamics in the adhesion and motility of EPEC and EHEC on host cells. Cell Motility and the Cytoskeleton, 60, 104–120. [DOI] [PubMed] [Google Scholar]
- Shum, M. S. , Pasquier, E. , Po'uha, S. T. , O'Neill, G. M. , Chaponnier, C. , Gunning, P. W. , & Kavallaris, M. (2011). Gamma‐Actin regulates cell migration and modulates the ROCK signaling pathway. FASEB Journal, 25, 4423–4433. [DOI] [PubMed] [Google Scholar]
- Smith, G. L. , & Law, M. (2004). The exit of vaccinia virus from infected cells. Virus Research, 106, 189–197. [DOI] [PubMed] [Google Scholar]
- Smith, G. L. , Vanderplasschen, A. , & Law, M. (2002). The formation and function of extracellular enveloped vaccinia virus. Journal of General Virology, 83, 2915–2931. [DOI] [PubMed] [Google Scholar]
- Smith, S. A. , & Kotwal, G. J. (2002). Immune response to poxvirus infections in various animals. Critical Reviews in Microbiology, 28, 149–185. [DOI] [PubMed] [Google Scholar]
- Snapper, S. B. , Takeshima, F. , Anton, I. , Liu, C. H. , Thomas, S. M. , Nguyen, D. , … Alt, F. W. (2001). N‐WASP deficiency reveals distinct pathways for cell surface projections and microbial actin‐based motility. Nature Cell Biology, 3, 897–904. [DOI] [PubMed] [Google Scholar]
- Takenawa, T. , & Suetsugu, S. (2007). The WASP‐WAVE protein network: Connecting the membrane to the cytoskeleton. Nature Reviews. Molecular Cell Biology, 8, 37–48. [DOI] [PubMed] [Google Scholar]
- Tilney, L. G. , Connelly, P. S. , & Portnoy, D. A. (1990). Actin filament nucleation by the bacterial pathogen, Listeria monocytogenes. Journal of Cell Biology, 111, 2979–2988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valderrama, F. , Cordeiro, J. V. , Schleich, S. , Frischknecht, F. , & Way, M. (2006). Vaccinia virus‐induced cell motility requires F11L‐mediated inhibition of RhoA signaling. Science, 311, 377–381. [DOI] [PubMed] [Google Scholar]
- Vandekerckhove, J. , & Weber, K. (1978a). Mammalian cytoplasmic actins are the products of at least two genes and differ in primary structure in at least 25 identified positions from skeletal muscle actins. Proceedings of the National Academy of Sciences of the United States of America, 75, 1106–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandekerckhove, J. , & Weber, K. (1978b). At least six different actins are expressed in a higher mammal: An analysis based on the amino acid sequence of the amino‐terminal tryptic peptide. Journal of Molecular Biology, 126, 783–802. [DOI] [PubMed] [Google Scholar]
- Wang, J. , Fang, S. , Xiao, H. , Chen, B. , Tam, J. P. , & Liu, D. X. (2009). Interaction of the coronavirus infectious bronchitis virus membrane protein with beta‐actin and its implication in virion assembly and budding. PLoS One, 4, e4908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward, B. M. ( 2004). Ch 16 Vaccinia Virus and Poxvirology In Isaacs S. N. (Ed.), Methods in Molecular Biology (Vol. 269; pp. 205–18). New York City: Humana Press. [DOI] [PubMed] [Google Scholar]
- Ward, B. M. , & Moss, B. (2001). Vaccinia virus intracellular movement is associated with microtubules and independent of actin tails. Journal of Virology, 75, 11651–11663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisswange, I. , Newsome, T. P. , Schleich, S. , & Way, M. (2009). The rate of N‐WASP exchange limits the extent of ARP2/3‐complex‐dependent actin‐based motility. Nature, 458, 87–91. [DOI] [PubMed] [Google Scholar]
- Weksler, B. , Romero, I. A. , & Couraud, P. O . (2013). The hCMEC/D3 cell line as a model of the human blood brain barrier. Fluids and Barriers of the CNS, 10(1), 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch, M. D. , & Way, M. (2013). Arp2/3‐mediated actin‐based motility: A tail of pathogen abuse. Cell Host & Microbe, 14, 242–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werden, S. J. , Rahman, M. M. , & McFadden, G. (2008). Poxvirus host range genes. Advances in Virus Research, 71, 135–171: [DOI] [PubMed] [Google Scholar]
- Wolffe, E. J. , Isaacs, S. N. , & Moss, B. (1993). Deletion of the vaccinia virus B5R gene encoding a 42‐kilodalton membrane glycoprotein inhibits extracellular virus envelope formation and dissemination. Journal of Virology, 67, 4732–4741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolffe, E. J. , Katz, E. , Weisberg, A. , & Moss, B. (1997). The A34R glycoprotein gene is required for induction of specialized actin‐containing microvilli and efficient cell‐to‐cell transmission of vaccinia virus. Journal of Virology, 71, 3904–3915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolffe, E. J. , Weisberg, A. S. , & Moss, B. (1998). Role for the vaccinia virus A36R outer envelope protein in the formation of virus‐tipped actin‐containing microvilli and cell‐to‐cell virus spread. Virology, 244, 20–26. [DOI] [PubMed] [Google Scholar]
- Zhang, W. H. , Wilcock, D. , & Smith, G. L. (2000). Vaccinia virus F12L protein is required for actin tail formation, normal plaque size, and virulence. Journal of Virology, 74, 11654–11662. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Figure 1
Supporting Information Figure 2
Supporting Information Figure 3