

Abstract
The masking of nucleoside phosphate and phosphonate groups by an aryl motif and an amino acid ester, nowadays known as the ‘ProTide’ technology, has proven to be effective in the discovery of nucleotide therapeutics. Indeed, this technology, which was invented by Chris McGuigan in the early 1990s, has inspired the discovery of two FDA‐approved antiviral nucleotide drugs, and many more are currently undergoing (pre)clinical development. The usefulness of this technology in the discovery of nucleotide therapeutics is showcased in this Highlight by discussing the ProTides development and the various ProTides that have reached clinical trials.
Keywords: nucleosides, nucleotides, phosphorylation, prodrugs, protides
Nucleotide therapeutics: The phosphate and phosphonate ‘ProTide’ technology, which was invented by the McGuigan lab in the early 1990s, has proven to be effective in the discovery of nucleotide therapeutics. Impressively, it has already inspired the discovery of two FDA‐approved drugs with many more in (pre)clinical development.
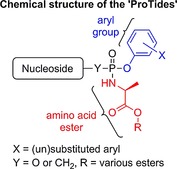
Phosphate prodrug strategies have been successfully used in the discovery of various nucleotide therapeutics that are currently in daily clinical use for the treatment of viral infections, in particular, hepatitis B and C (HBV and HCV, respectively) and HIV.1 Over the last few years, the ‘ProTide’ technology—pioneered by Chris McGuigan (Cardiff University, UK)—has emerged as a powerful strategy in the discovery of nucleotide therapeutics.2 Indeed, this technology was used in the successful discovery and development of two FDA‐approved ProTides, Sovaldi™ and tenofovir alafenamide. The pipelines of several pharma and biotech companies seem to be stocked with a selection of ProTides, some of which are currently in phase III clinical trials, while there is an increasing number of reports on ProTides undergoing preclinical studies. Collectively, these reflect a ProTides boom that could, in the future, be translated into the approval of more ProTides to treat viral infections and cancer.
The in vivo activation of many antiviral and anticancer nucleosides involves phosphorylation into their active di‐ or triphosphate counterparts.3 Among the three kinase‐dependent phosphorylation steps required for the bioactivation of this class of therapeutics, the first phosphorylation step that converts nucleosides into their 5′‐O‐monophosphate derivatives is often found to be the rate‐limiting step.4 To overcome this, numerous strategies that deliver nucleoside 5′‐O‐monophosphates into cells have been developed.1 Nowadays, one of the most applied phosphate prodrug approaches is the ProTide technology. The development2 of this technology started by masking the 5′‐O‐monophosphate groups of therapeutic nucleosides with simple dialkyl and then haloalkyl groups. However, these attempts did not lead to improved biological activity, most likely due to the inability of these masking groups to be hydrolyzed in vivo to release the nucleoside monophosphate, which can be subsequently further phosphorylated to the active species. Next, McGuigan and co‐workers synthesized alkyloxy and haloalkyl phosphoramidate prodrugs, and these showed better activities than their parent nucleosides.5 This was the first breakthrough in the development of ProTides and provided evidence that masking of phosphate groups with biocleavable motifs may yield an effective prodrug system for the delivery of therapeutic nucleoside monophosphates. These initial studies identified l‐alanine as a superior amino acid in the alkyloxy phosphoramidates, an observation that has informed recent drug discovery programs that yielded FDA‐approved drugs.5b Encouraged by this, the phosphate group was masked with two amino acid esters. Back then, this was found not to be beneficial as the prodrugs were largely inactive. Diaryl phosphates were studied next, and these showed very good activity. Hence, the McGuigan lab combined the amino acid ester from the alkyloxy and haloalkyl phosphoramidates and the aryl masking group to generate aryloxy triester phosphoramidates.6 These were found to be superior in the delivery of therapeutic nucleoside 5′‐O‐monophosphates. Since then, the masking of the phosphate group with an amino acid ester and an aryl motif, nowadays known as the ProTide technology, has become an approved prodrug strategy in the discovery of nucleotide therapeutics. The mechanism by which the ProTides are metabolized in vivo to release the nucleoside monophosphate is believed to proceed through the action of two enzymes: an esterase, such as cathepsin A,7 and a phosphoramidase‐type enzyme, such as hint‐1,8 as illustrated in Scheme 1.2
Scheme 1.
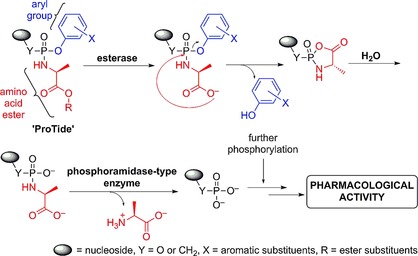
Postulated mechanism of the in vivo metabolism of the ProTides to release nucleoside monophosphates.
ProTides as clinical candidates and drugs
To date, at least ten ProTides have reached clinical trials and have been investigated as treatments for viral infections and cancer (Figure 1). The McGuigan research group discovered and developed the anti‐HCV agent INX‐189, a ProTide of 6‐O‐methyl‐2′‐C‐methyl guanosine (1).9 At that time, this compound was the most potent inhibitor of HCV replication in cell‐based replication assays (EC50=0.01 μm, EC90=0.04 μm, CC50=7 μm). Critically, it generated significantly higher levels of the 6‐O‐methyl‐2′‐C‐methyl guanosine triphosphate than the parent nucleoside and had a half‐life of >24 h. Given the excellent pharmacokinetics (PK) and pharmacodynamics (PD) profiles of INX‐189, it was chosen as a clinical candidate that was then developed by Inhibitex Inc.10 Following successful early clinical results, INX‐189 was acquired by Bristol‐Myers Squibb (BMS) and studied in phase III clinical trials in combination with daclatasvir, another anti‐HCV experimental drug of BMS. However, cardiotoxicity was observed, and further development was suspended.11
Figure 1.
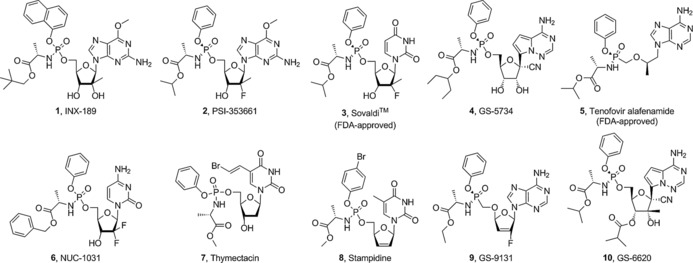
Structures of a selection of ProTides that have reached clinical trials and those that have been approved by the FDA.
Other ProTides for the treatment of HCV, particularly PSI‐353661 (2)12 and PSI‐7977 (3),13 proceeded to clinical evaluation. PSI‐353661 is a ProTide of 6‐O‐methyl‐2′‐deoxy‐2′‐fluoro‐2′‐C‐methylguanosine, whereas PSI‐7977 is a ProTide of 2′‐deoxy‐2′‐fluoro‐2′‐C‐methyluridine. Both compounds showed potent anti‐HCV activity through efficient delivery of the parent nucleosides 5′‐O‐monophosphates. As a result of the impressive early data from the clinical trials of these two agents, they were acquired by Gilead Sciences, Inc. Out of the two compounds, PSI‐7977, which subsequently became GS‐7977, successfully completed clinical evaluations and became known as sofosbuvir (SovaldiTM), the first ProTide approved for clinical use against HCV.
GS‐5734 (4)14 is a C‐nucleoside ProTide, which is currently undergoing phase I clinical trials for the treatment of Ebola.14 Preclinical data showed that GS‐5734 exerts potent antiviral activity against variants of the Ebola virus (EBOV). The ProTide showed potent inhibition of EBOV replication (EC50=0.06 to 0.14 μm), while the parent C‐nucleoside was not as effective (EC50: 0.77 to >20 μm). Impressively, in rhesus monkeys infected with EBOV, once‐daily intravenous administration of GS‐5734 led to significant suppression of EBOV replication and 100 % protection of infected animals against lethal disease. GS‐5734 also showed promising inhibition of the replication of other pathogenic RNA viruses, such as arenaviruses, filoviruses, and coronaviruses, indicating the wide‐spectrum of activity of this ProTide.
GS‐7340 (5),15 a ProTide of the acyclic nucleoside phosphonate tenofovir, of which the oral prodrug, tenofovir disoproxil, is FDA‐approved for HIV therapy, exhibited improved anti‐HIV activity and better in vivo stability than tenofovir. Impressively, the ProTide GS‐7340 generated 10‐ to 30‐fold higher levels of tenofovir and its phosphorylated metabolites following incubation of peripheral blood mononuclear cells compared with tenofovir disoproxil and tenofovir, respectively.16 Although GS‐7340 is similar to the FDA‐approved tenofovir disoproxil, it is more potent, and thus in phase III clinical studies therapeutic effects were achieved at lower doses with fewer incidences of side effects. In late 2015, GS‐7340, now known as tenofovir alafenamide, was approved in combination with other anti‐HIV agents for the treatment of HIV‐1 infection.
NUC‐1031 (6)17 is an anticancer ProTide that was discovered by the McGuigan group and is currently being developed by NuCana Biomed Ltd. It is a prodrug of the FDA‐approved anticancer drug gemcitabine (GemzarTM). This compound overcame three resistance mechanisms that limit the efficacy of the parent nucleoside gemcitabine. Results from phase I/II clinical trials showed that NUC‐1031 is effective against a wide range of cancers and was well‐tolerated by patients. Impressively, five out of 68 patients achieved tumor shrinkage of ≥30 %, while an additional 33 patients had achieved stable disease. NuCana is also pushing forward the clinical development of NUC‐3373,18 a ProTide of 5‐fluoro‐2′‐deoxyuridine (FUDR) [structure not shown].
At least a further four ProTides were reported to have undergone clinical evaluation, that is, thymectacin (7) for cancer, stampidine (8) and GS‐9131 (9) for HIV; and GS‐6620 (10) for HCV.
The plethora of ProTides that have been, are still undergoing, or have successfully completed clinical trials clearly highlights the effectiveness of this technology to deliver nucleotide therapeutics. Coupling this to the large number of ProTides that are currently undergoing preclinical evaluation, it is safe to say that more ProTides will progress into clinical studies, increasing the chances of more ProTides being approved in the future to treat viral infections and cancer. This has been made possible by the pioneering work of Prof. Chris McGuigan that started in the early 1990s, and which years later yielded the ProTide technology as we know it today. Since its development, the ProTide technology has been widely adopted and adapted by academic research groups, as well as small and large pharmaceutical companies to discover new medicines that have already improved treatment outcomes and consequently the quality of life of many patients.
Y. Mehellou, ChemMedChem 2016, 11, 1114.
References
- 1. Hecker S. J., Erion M. D., J. Med. Chem. 2008, 51, 2328–2345. [DOI] [PubMed] [Google Scholar]
- 2. Mehellou Y., Balzarini J., McGuigan C., ChemMedChem 2009, 4, 1779–1791. [DOI] [PubMed] [Google Scholar]
- 3. Jordheim L. P., Durantel D., Zoulim F., Dumontet C., Nat. Rev. Drug Discovery 2013, 12, 447–464. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Stein D. S., Moore K. H., Pharmacotherapy 2001, 21, 11–34; [DOI] [PubMed] [Google Scholar]
- 4b. Gao W. Y., Agbaria R., Driscoll J. S., Mitsuya H., J. Biol. Chem. 1994, 269, 12633–12638. [PubMed] [Google Scholar]
- 5.
- 5a. Devine K. G., McGuigan C., O′Connor T. J., Nicholls S. R., Kinchington D., AIDS 1990, 4, 371–373; [PubMed] [Google Scholar]
- 5b. McGuigan C., Devine K. G., O′Connor T. J., Gaplin S. A., Jeffries D. J., Kinchington D., Antiviral Chem. Chemother. 1990, 1, 107–113. [Google Scholar]
- 6. McGuigan C., Pathirana R. N., Mahmood N., Devine K. G., Hay A. J., Antiviral Res. 1992, 17, 311–321. [DOI] [PubMed] [Google Scholar]
- 7. Birkus G., Wang R., Liu X., Kutty N., MacArthur H., Cihlar T., Gibbs C., Swaminathan S., Lee W., McDermott M., Antimicrob. Agents Chemother. 2007, 51, 543–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Brenner C., Biochemistry 2002, 41, 9003–9014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. McGuigan C., Madela K., Aljarah M., Gilles A., Brancale A., Zonta N., Chamberlain S., Vernachio J., Hutchins J., Hall A., Ames B., Gorovits E., Ganguly B., Kolykhalov A., Wang J., Muhammad J., Patti J. M., Henson G., Bioorg. Med. Chem. Lett. 2010, 20, 4850–4854. [DOI] [PubMed] [Google Scholar]
- 10. Vernachio J. H., Bleiman B., Bryant K. D., Chamberlain S., Hunley D., Hutchins J., Ames B., Gorovits E., Ganguly B., Hall A., Kolykhalov A., Liu Y., Muhammad J., Raja N., Walters C. R., Wang J., Williams K., Patti J. M., Henson G., Madela K., Aljarah M., Gilles A., McGuigan C., Antimicrob. Agents Chemother. 2011, 55, 1843–1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gentile I., Buonomo A. R., Zappulo E., Borgia G., Expert Opin. Invest. Drugs 2015, 24, 239–251. [DOI] [PubMed] [Google Scholar]
- 12. Chang W., Bao D., Chun B. K., Naduthambi D., Nagarathnam D., Rachakonda S., Reddy P. G., Ross B. S., Zhang H. R., Bansal S., Espiritu C. L., Keilman M., Lam A. M., Niu C., Steuer H. M., Furman P. A., Otto M. J., Sofia M. J., ACS Med. Chem. Lett. 2011, 2, 130–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sofia M. J., Bao D., Chang W., Du J., Nagarathnam D., Rachakonda S., Reddy P. G., Ross B. S., Wang P., Zhang H. R., Bansal S., Espiritu C., Keilman M., Lam A. M., Steuer H. M., Niu C., Otto M. J., Furman P. A., J. Med. Chem. 2010, 53, 7202–7218. [DOI] [PubMed] [Google Scholar]
- 14. Warren T. K., Jordan R., Lo M. K., Ray A. S., Mackman R. L., Soloveva V., Siegel D., Perron M., Bannister R., Hui H. C., Larson N., Strickley R., Wells J., Stuthman K. S., Van Tongeren S. A., Garza N. L., Donnelly G., Shurtleff A. C., Retterer C. J., Gharaibeh D., Zamani R., Kenny T., Eaton B. P., Grimes E., Welch L. S., Gomba L., Wilhelmsen C. L., Nichols D. K., Nuss J. E., Nagle E. R., Kugelman J. R., Palacios G., Doerffler E., Neville S., Carra E., Clarke M. O., Zhang L., Lew W., Ross B., Wang Q., Chun K., Wolfe L., Babusis D., Park Y., Stray K. M., Trancheva I., Feng J. Y., Barauskas O., Xu Y., Wong P., Braun M. R., Flint M., McMullan L. K., Chen S. S., Fearns R., Swaminathan S., Mayers D. L., Spiropoulou C. F., Lee W. A., Nichol S. T., Cihlar T., Bavari S., Nature 2016, 531, 381–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lee W. A., He G. X., Eisenberg E., Cihlar T., Swaminathan S., Mulato A., Cundy K. C., Antimicrob. Agents Chemother. 2005, 49, 1898–1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Eisenberg E. J., He G. X., Lee W. A., Nucleosides Nucleotides Nucleic Acids 2001, 20, 1091–1098. [DOI] [PubMed] [Google Scholar]
- 17. Slusarczyk M., Lopez M. H., Balzarini J., Mason M., Jiang W. G., Blagden S., Thompson E., Ghazaly E., McGuigan C., J. Med. Chem. 2014, 57, 1531–1542. [DOI] [PubMed] [Google Scholar]
- 18. McGuigan C., Murziani P., Slusarczyk M., Gonczy B., Vande Voorde J., Liekens S., Balzarini J., J. Med. Chem. 2011, 54, 7247–7258. [DOI] [PubMed] [Google Scholar]