

Summary
Plus‐stranded RNA viruses replicate in the cytosol of infected cells, in membrane‐bound replication complexes containing the replicase proteins, the viral RNA and host proteins. The formation of the replication and transcription complexes (RTCs) through the rearrangement of cellular membranes is currently being actively studied for viruses belonging to different viral families. In this work, we identified double‐membrane vesicles (DMVs) in the cytoplasm of cells infected with the equine torovirus Berne virus (BEV), the prototype member of the Torovirus genus (Coronaviridae family, Nidovirales order). Using confocal microscopy and transmission electron microscopy, we observed a close relationship between the RTCs and the DMVs of BEV. The examination of BEV‐infected cells revealed that the replicase proteins colocalize with each other and with newly synthesized RNA and are associated to the membrane rearrangement induced by BEV. However, the double‐stranded RNA, an intermediate of viral replication, is exclusively limited to the interior of DMVs. Our results with BEV resemble those obtained with other related viruses in the Nidovirales order, thus providing new evidence to support the idea that nidoviruses share a common replicative structure based on the DMV arranged clusters.
In this work we identified double membrane vesicles (DMVs) in cells infected with the equine torovirus Berne virus (BEV), the prototype member of the Torovirus genus (Coronaviridae Family, Nidovirales Order). These DMVs are arranged in clusters at specific areas of the cytoplasm, and surrounded by ER membranes and mitochondria. By confocal and electron microscopy we observed a close relationship between the DMVs and the elements involved in the BEV replication and transcription processes. These results resemble those obtained with other related viruses in the Nidovirales order, thus supporting the idea that nidoviruses share a common replicative structure.
Introduction
Positive‐strand RNA viruses utilize virus‐modified cellular membranes to build their replication and transcription complexes (RTCs). On one hand, these structures serve as platforms to concentrate all the components required during replication and transcription, and on the other, they provide a physical separation to protect the viral RNA, specifically the double‐stranded RNA (dsRNA), generated as a replication intermediate, from the detection by the host antiviral defences. Many morphological, ultrastructural and biochemical studies have been performed during the last decade to characterize the RTCs from different virus families (Miller and Krijnse‐Locker, 2008; den Boon and Ahlquist, 2010; Netherton and Wileman, 2011; Harak and Lohmann, 2015).
Toroviruses are positive‐sense single‐stranded RNA viruses belonging to the Nidovirales order, which have been taxonomically classified into four viral families: Arteriviridae, Roniviridae, Mesoviridae and Coronaviridae (http://ictvonline.org/virusTaxonomy.asp). The latter encompasses viruses with the largest genome known among RNA viruses (26–32 kb) (Gorbalenya et al., 2006; Siddell et al., 2010; King et al., 2012) and includes two subfamilies called Coronaviridae (genera Alphacoronavirus, Betacoronavirus, Deltacoronavirus and Gammacoronavirus) and Torovirinae (genera Bafinivirus and Torovirus).
Toroviruses infect different animal species including humans, causing enteric disease and diarrhoea (de Groot, 2008; Hoet and Linda, 2008). Four torovirus species have been described: equine torovirus, also known as Berne virus (BEV) (Weiss et al., 1983), bovine torovirus (BToV), porcine torovirus and human torovirus (http://ictvonline.org/virusTaxonomy.asp). Only BEV, the prototype member of the genus, has been studied at the molecular level, as, for a long time, it was the only strain of torovirus that could be grown in cell culture. Nonetheless, the propagation of different BToV strains has also been reported (Kuwabara et al., 2007; Ito et al., 2010; Aita et al., 2012).
The torovirus genomic organization is similar to that of all other nidoviruses. The last third of the viral genome encodes the structural proteins: spike (S), membrane (M), haemagglutinin‐esterase (HE), whose coding gene is partially deleted in BEV (Snijder et al., 1991), and nucleocapsid (N) (Snijder and Horzinek, 1993), which are individually expressed from a nested set of subgenomic messenger RNAs. As in all nidoviruses, the first two‐thirds of the torovirus genome are occupied by two large and overlapping open reading frames (ORFs), ORF1a and ORF1b, which encode two partially overlapping replicase polyproteins, pp1a and pp1ab, of 4569 and 6857 amino acids, respectively (http://www.uniprot.org/uniprot/P0C6V7), that are translated from the genomic RNA. Expression of the pp1ab C‐terminal end is produced by a ribosomal frameshifting occurring just upstream of the ORF1a stop codon (Brierley et al., 1989; Snijder et al., 1990; den Boon et al., 1991; Siddell et al., 2010). Among the nidoviruses, the most conserved domain is found in the ORF1b, which includes key enzymes in the viral RNA replication and transcription, like the RNA‐dependent RNA polymerase (RdRp) and the helicase (Hel) among others (Gorbalenya et al., 2006). The mature non‐structural proteins (nsps) are post‐translationally released from the replicase polyproteins pp1a and pp1ab by the viral proteinases (Ziebuhr et al., 2000), the papain‐like proteinase and the main proteinase (Mpro), a chymotrypsin‐like proteinase responsible for most of the post‐translational cleavages. In BEV, this region has been poorly characterized, and thus, the presence of putative functional domains in the torovirus polyproteins has only been predicted by comparative amino acid sequence analysis with other nidoviruses (Snijder et al., 1990; Smits et al., 2006). The BEV Mpro is the only torovirus nsp that has been characterized biochemically to date (Smits et al., 2006). The BEV Mpro is a serine proteinase, like those of arteriviruses (Snijder et al., 1996; Barrette‐Ng et al., 2002) and the related bafiniviruses (Ulferts et al., 2011), but differs from the coronavirus and ronivirus Mpro (Ziebuhr et al., 2000; Hegyi et al., 2002), which are cysteine proteinase. Based on the sequence specificity, Smits et al. (2006) proposed a tentative processing scheme for the BEV pp1a/pp1ab polyproteins, where the sequence and localization of the potential cleavage sites of Mpro were shown. Also, they pointed out the conservation of the cleavage sites in the BToV sequence published by Draker et al. (2006).
A common characteristic of all known nidoviruses, which is also conserved in torovirus, is the presence of three hydrophobic domains in the pp1a/pp1ab polyproteins (Gorbalenya et al., 2006). The first domain is localized near the papain‐like proteinase, and the other two are flanking the Mpro domain. These hydrophobic domains are believed to anchor the RTCs to intracellular membranes and are proposed to be responsible for the membrane rearrangement produced during the viral infection (Snijder et al., 2001; Clementz et al., 2008; Gadlage et al., 2010; Angelini et al., 2013). In this regard, characteristic double‐membrane vesicles (DMVs) that have been related with the replication/transcription processes have been observed in the cytoplasm of cells infected with different nidoviruses. Specifically, DMVs were observed in cells infected with several betacoronaviruses, i.e. severe acute respiratory syndrome coronavirus (SARS‐CoV) (Snijder et al., 2006), mouse hepatitis virus (MHV) (Gosert et al., 2002; Ulasli et al., 2010) and Middle East respiratory syndrome coronavirus (MERS‐CoV) (de Wilde et al., 2013), as well as with members of the Alphacoronavirus (NL63‐CoV) (Orenstein et al., 2008) and Gammacoronavirus [infectious bronchitis virus (IBV)] (Maier et al., 2013) genera, and with the arterivirus equine arteritis virus (EAV) (Pedersen et al., 1999). Furthermore, the presence of other membranous structures related to the replication/transcription processes was described, such as convoluted membranes (Knoops et al., 2008; Ulasli et al., 2010; de Wilde et al., 2013) or spherules (Maier et al., 2013). Electron tomography studies performed with SARS‐CoV (Knoops et al., 2008) and EAV (Knoops et al., 2012) indicate that actually the DMVs form part of a reticulovesicular network of modified membranes derived from the endoplasmic reticulum (ER) that supports the viral replication machinery.
In this study, we describe for the first time the presence of DMVs in the cytoplasm of cells infected with torovirus, specifically with BEV. To characterize the torovirus RTCs, we generated specific sera for BEV Mpro, Hel and RdRp, three key proteins in the formation and functioning of nidovirus RTCs, that were used in confocal and transmission electron microscopy (TEM) assays to analyse the location of viral components involved in BEV replication. This work complements the knowledge about membrane rearrangements promoted by nidovirus infection by adding the characterization of these structures in torovirus, a group of viruses for which there was no available information as yet.
Results
Formation of double‐membrane vesicles during Berne virus infection
Accumulating evidences from different positive‐strand RNA viruses indicate that in this heterogeneous group of viruses, genomic RNA replication and transcription invariably occur in close association with highly modified cellular membranes of diverse origins. It has been shown that in cells infected with different nidoviruses, virus‐induced membrane reorganization produces characteristic DMVs, as well as other membranous structures, which have been related with the viral RNA replication and transcription processes. However, nothing is known about membrane rearrangements occurring in torovirus‐infected cells. Here, E.Derm cells mock infected or infected with BEV for 16 h were subjected to conventional embedding in TAAB‐812 epoxy resin and analysed by TEM. In thin sections of infected cells groups of DMVs were observed in the cell cytoplasm, frequently in the perinuclear area and surrounded by mitochondria and ER membranes (Fig. 1A and B). These membranous structures were not observed in mock‐infected cells (not shown). As shown in Fig. 1, vesicles of different sizes could be observed, ranging from 50 to 150 nm in diameter, but in some cases, large DMVs, bigger than 200 nm, were also seen. Moreover, while some DMVs appeared as empty vesicles, in others, an electron‐dense inner content could be observed. Apparently, the DMVs look as independent structures isolated from each other; however, an in‐depth inspection of the infected cells revealed the existence of connections between many of them, as well as between DMVs and the ER (Fig. 1C and D). In addition, a seeming continuity between DMVs and the mitochondrial‐associated ER membranes (MAM) could occasionally be observed (Fig. 1E and F). The MAM is a subdomain of the ER with different properties and lipid composition, which regulates ER–mitochondria communications and appears as ER tubules closely apposed to mitochondria on the electron micrographs (Raturi and Simmen, 2013). Although some virions could be seen inside rough ER‐derived vesicles close to the DMV clusters (Figs 1B and S1A and B), at this time post infection (pi), most viral particles appeared accumulated in large vesicles (Fig. S1C), usually segregated from these areas.
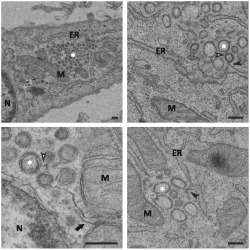
Figure 1.
Ultrastructural analysis of E.Derm cells infected with BEV embedded in epoxy resin. Electron micrographs of E.Derm cells infected with BEV for 16 h and embedded in the epoxy resin TAAB‐812 after conventional chemical fixation, where the presence of DMVs can be clearly observed.
A. A cluster of DMVs (white asterisk) surrounded by mitochondria and ER.
B. Enlarged view of a DMV cluster showing some viral particles inside a vesicle located in the periphery of the cluster (white arrow).
C, D. Connections between DMVs (white arrowhead) and between DMVs and the ER (black arrowhead).
E, F. DMVs located close to the nucleus showing connections between them (white arrowhead) and with the MAM (black arrow), which can be observed with more detail in the enlarged area shown in F.
M: mitochondria; ER: endoplasmic reticulum; N: nucleus. Scale bars, 200 nm.
Interestingly, similar membrane structures were also observed in human MRC5 cells infected with BEV (Fig. S2) where the DMVs were clearly present. The susceptibility of these cells to the BEV infection was previously reported in our laboratory (Maestre et al., 2011). As observed in E.Derm cells, in the MRC5 cells, some DMVs are connected between them and with the membranes of the ER (Fig. S2B) and the MAM (Fig. S2C). Also, viral particles inside small or large vesicles could be observed (Fig. S2A shows just one individual particle inside a vesicle).
To further characterize these membranous structures, infected cells were processed under different preservation conditions before sectioning. Hence, cells were either subjected to quick freezing and freeze substitution before being embedded in Lowicryl resin or processed for cryosectioning. Although the freeze substitution protocols have been previously described to improve the preservation of DMVs in other nidoviruses (Snijder et al., 2006; Knoops et al., 2012), the BEV DMVs were barely identified under these conditions (Fig. 2A and B); nonetheless, those that could be recognized showed a dense inner content. On the other hand, in cryosections from BEV‐infected E. Derm, DMVs were clearly seen (Fig. 2C and D). As observed in conventional sections of epoxy resin, the DMVs were mainly localized in the perinuclear area and were surrounded by mitochondria and ER membranes. We did not observe differences in the DMV sizes when compared with those observed in epoxy sections; however, in the cryosections, the inner content of most of the DMVs was preserved. Also, some viral particles could be observed in the vicinity of DMV clusters, but in separate areas (Fig. 2C). However, the large clusters of convoluted membranes found in several coronavirus were not observed in the case of BEV infection. Similarly, there was no evidence of spherules, like those recently described in IBV‐infected cells.
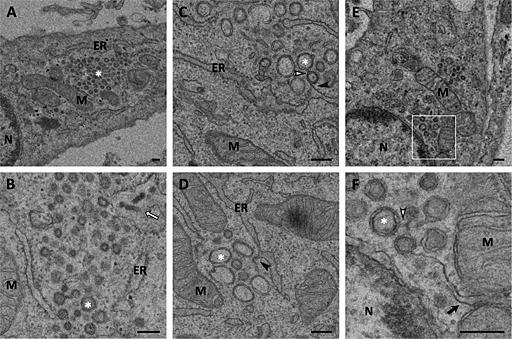
Figure 2.
Ultrastructural analysis of E.Derm cells infected with BEV processed by cryosubstitution or cryosectioning.
A–D. Electron micrographs of E.Derm cells infected with BEV for 16 h that were subjected to (A, B) freeze substitution before embedding them in Lowicryl HM20 resin or (C, D) cryosectioning as described in the Experimental Procedures section. Clusters of DMVs (white asterisks) surrounded by mitochondria and ER were observed in the infected cells. Viral particles could be visualized outside the clusters (white arrows). The boxed area in A is enlarged in B. M: Mitochondria; ER: Endoplasmic reticulum; N: nucleus. Scale bars, 200 nm.
These results show that, as described for all the nidoviruses analysed to date, BEV causes membrane rearrangements leading to the formation of characteristic DMVs.
Distribution of the viral proteins involved in Berne virus replication/transcription
In betacoronaviruses and arteriviruses, the DMVs have been described as the sites where the viral RTCs are localized (Pedersen et al., 1999; Gosert et al., 2002; van Hemert et al., 2008). To identify the torovirus RTCs, we have obtained specific sera for the BEV RdRp, Hel and Mpro, three key proteins in the formation and functioning of nidovirus RTCs.
First, we studied the subcellular location of these nsps along the infection by confocal immunofluorescence microscopy. For these, E.Derm cells infected with BEV were fixed at different times pi and processed for confocal microscopy. The assay was finished at 20 h pi because later times were considered unsuitable for the viral replication studies given that, on one hand, the release of virus progeny already reaches the plateau phase at about 16 h pi, although it is extended until 24 h pi, and on the other hand, cytopathic effects caused by BEV infection are apparent from about 21 h pi (Weiss and Horzinek, 1986) and lead to apoptosis (Maestre et al., 2011). The subcellular distribution pattern throughout the infection was almost identical for the three proteins. As exemplified in Fig. 3A for Mpro, for all of them, the first signal was observed between 4 and 6 h pi, but it increased in intensity from 8 h pi onwards. At early times pi, these proteins were localized in dispersed and discrete foci in the cells, which develop into a number of densely labelled areas, giving rise to a ring‐like signal distantly surrounding the nucleus at 10–12 h pi, but which moves towards the nucleus later in infection. The mock‐infected cells were not stained with any of these antibodies (not shown).
Figure 3.
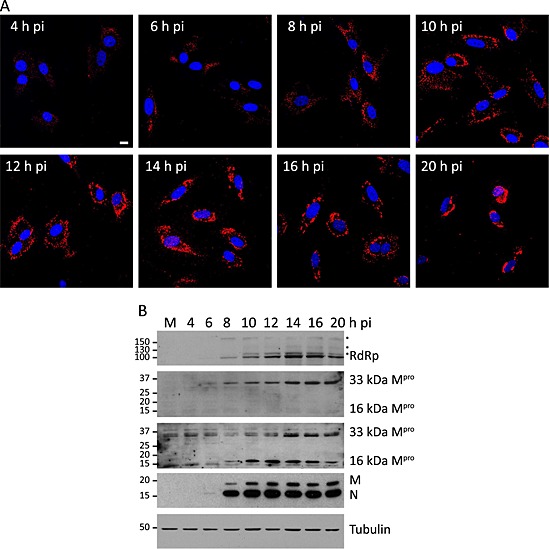
Expression and distribution along the infection of the BEV proteins involved in the replication/transcription processes.
A. Immunofluorescence analysis of E.Derm cells infected with BEV, fixed with 4% PFA in PBS at the indicated times pi and incubated with the anti‐Mpro antibody produced in rats (red). Nuclei were stained with DAPI (blue). Scale bar, 10 µm.
B. Western blot analysis of extracts of E.Derm cells mock infected (M) or infected with BEV, collected at the indicated pi times, which were incubated with the following antibodies (in descending order): rabbit anti‐RdRp, rat anti‐Mpro, rabbit anti‐Mpro, rabbit anti‐M plus rabbit anti‐N and mouse mAb anti‐tubulin. Positions and sizes in kilodalton of the molecular weight markers are shown at the left side of each panel. Asterisks indicate protein intermediate species generated during the proteolytic processing of the pp1a/pp1ab polyproteins.
The specificity of the sera for the corresponding viral proteins was checked by Western blot. As shown in Fig. 3B, the anti‐RdRp serum recognized a 100 kDa protein, which concurs with size predicted by Smits et al. (2006) for the RdRp. In addition, this serum detected three other bands (denoted by asterisks in Fig. 3B) that likely represent intermediate species of the pp1a/pp1ab precursors. However, the anti‐Hel serum did not react with any specific protein by this assay. On the other hand, the two anti‐Mpro sera obtained recognized the mature Mpro of 33 kDa characterized by Smits and co‐workers, but curiously, the serum produced in rabbit detected an additional protein of 16 kDa. Smits and co‐workers predicted an internal cleavage site in the Mpro of BEV and showed that the protein was processed at this site when expressed in bacterial cells, but they did not find evidence of that internal processing in mammalian cells infected with BEV (Smits et al., 2006).
A representative time course of nsp accumulation throughout the infection is shown in Fig. 3B, where the RdRp and the Mpro showed a progressive accumulation from 8 h pi onwards, although traces of these proteins could already be observed at 6 h pi in overexposed films. The structural proteins N and M were also observed from 6 h pi, but in contrast to the nsps, they showed an exponential expression from 6 to 10 h pi, resulting in the accumulation of high amounts of protein form 10 h pi onwards.
Colocalization of the viral proteins involved in Berne virus replication/transcription
To further determine whether the nsps indeed share the same intracellular location, the anti‐Mpro antibody generated in rats was used in double‐labelling studies with the rabbit anti‐RdRp or anti‐Hel antibodies. As shown in Fig. 4, both Hel (Fig. 4A) and RdRp (Fig. 4B) showed a high degree of colocalization with the Mpro at 8 h pi. The assay was also performed at 12 and 16 h pi, and the degree of colocalization at the three pi times was quantified by determining Pearson's correlation coefficient (r) between each pair of proteins (Fig. 4D). By definition, r values range from −1 (negative correlation) to 1 (positive correlation), and an r value of 0 indicates no correlation. Only r values higher than 0.5 were considered as true colocalization. These quantitative studies indicated a quite high degree of colocalization between the BEV nsps, with an r for the Hel‐Mpro pair of 0.78 ± 0.04 at 8 h pi, 0.76 ± 0.03 at 12 h pi and 0.65 ± 0.04 at 16 h pi. Similar values were obtained for the RdRp–Mpro pair, with an r of 0.75 ± 0.03 at 8 h pi, 0.82 ± 0.02 at 12 h pi and 0.70 ± 0.03 at 16 h pi. These results confirm that the Hel, the RdRp and the Mpro are localized in the same areas, which likely represent the sites where the viral replication and transcription occur.
Figure 4.
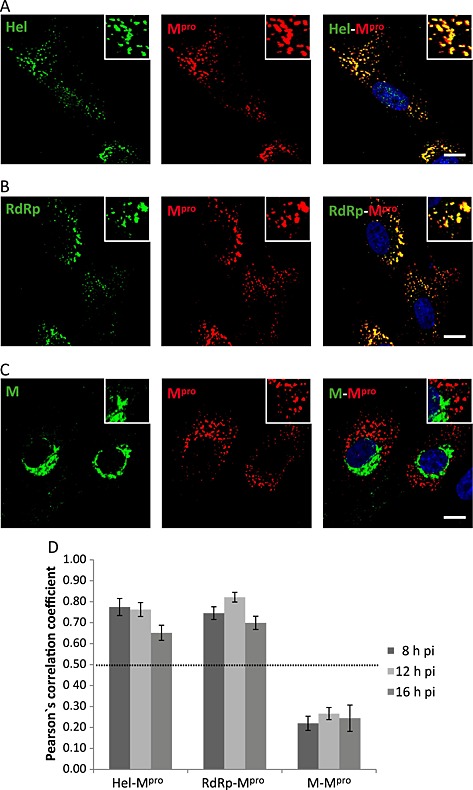
Colocalization analysis of the BEV proteins involved in the replication/transcription processes.
A–C. E.Derm cells infected with BEV were fixed with 4% PFA in PBS at 8 h pi and used for dual immunolabelling with the anti‐Mpro antibody produced in rats (red) in combination with (A) rabbit anti‐Hel, (B) rabbit anti‐RdRp or (C) rabbit anti‐M (green). The nuclei were stained with DAPI (blue). The boxed areas are representative fields that are shown at higher magnification. Scale bars, 10 µm.
D. Quantitative analysis of the colocalization degree. The same experiment as described in A–C was also performed at 12 and 16 h pi, and Pearson's correlation coefficient was calculated for each pair of antibodies at the three analysed pi times, using for the analysis images (n = 5) containing an average of 20 cells each. Only the values above 0.5 were considered as indicative of colocalization.
Similar studies were performed using antibodies directed against the BEV structural M protein, a protein essential for the viral assembly process. Previously, we described that at early times pi the M protein accumulates at the perinuclear area, specifically within the ER–Golgi intermediate compartment (ERGIC) where the virus assembly is thought to occur, and at later times, the signal corresponding to the M protein spreads towards the cell surface once the protein was incorporated into the progeny virions (Garzon et al., 2006). Figure 4C shows that the M protein does not colocalize with the Mpro at 8 h pi. Both proteins showed different distributions along the infection as confirmed by the quantitative colocalization study that gave r values lower than 0.5 (0.22 ± 0.03 at 8 h pi, 0.27 ± 0.03 at 12 h pi and 0.24 ± 0.06 at 16 h pi) (Fig. 4D). The analysis was extended to the S and N structural proteins with similar results, although there could be a certain degree of colocalization between N and Mpro at an earlier time point (6 h pi) (data not shown). The lack of colocalization between the structural proteins and the nsps indicates that the sites of viral replication are separated from the sites of virus assembly.
Intracellular localization of viral RNA involved in Berne virus replication/transcription
For a variety of positive‐strand RNA viruses, the detection of RNA duplexes corresponding to replication/transcription intermediates has previously been used as a marker for viral RTCs (Lee et al., 1994; Mackenzie et al., 1996; Westaway et al., 1997; 1999; Targett‐Adams et al., 2008). To determine whether in BEV‐infected cells the nsps are located at the sites of viral RNA synthesis, we used an anti‐dsRNA monoclonal antibody (mAb). E.Derm cells infected with BEV were fixed at 8, 12 and 16 h pi and stained with the anti‐dsRNA and anti‐Mpro antibodies. The dsRNA staining was detected in bright dots, whose number increases along the infection, reaching a steady state at about 10 h pi (data not shown). We did not observe clear colocalization between Mpro and dsRNA at any time pi, as exemplified in Fig. 5A for 8 h pi and as determined by the quantitative analysis that showed r values below 0.5 at either time pi analysed in this assay (0.13 ± 0.03 at 8 h pi, 0.13 ± 0.02 at 12 h pi and 0.10 ± 0.01 at 16 h pi) (Fig. 5C). Nonetheless, although the dsRNA and Mpro signals could not be superimposed, we observed that they were very close to each other in many areas, in fact, about 50% of dsRNA dots exhibited a partial colocalization or were surrounded by the Mpro signal.
Figure 5.
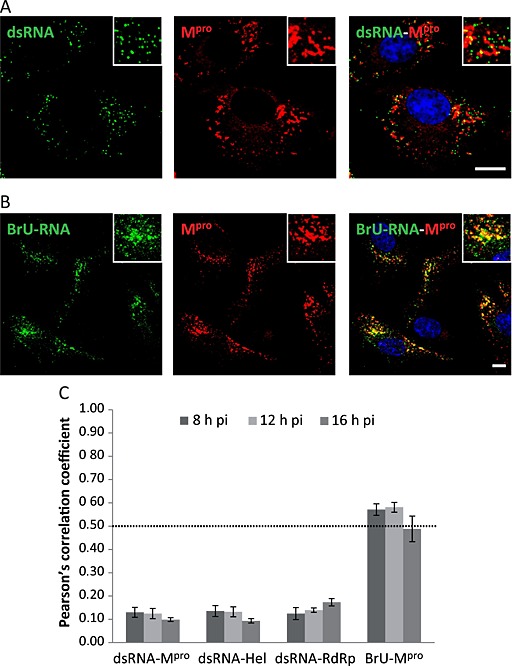
Colocalization analysis of the dsRNA and newly synthesized RNA (BrU‐RNA) with the Mpro.
A. E.Derm cells infected with BEV were fixed with 4% PFA in PBS at 8 h pi and used for dual immunolabelling with the anti‐Mpro antibody produced in rats (red) in combination with mouse mAb anti‐dsRNA (green).
B. Infected cells were incubated with 5 mM 5‐BrU in the presence of 5 mg ml−1 of actinomycin D for 1 h before fixation and permeabilization for immunofluorescence staining with the rat anti‐Mpro antibody (red) in combination with a mouse anti‐BrdU mAb (green). The nuclei were stained with DAPI (blue). Boxed areas are representative fields shown at higher magnification. Scale bars, 10 µm.
C. Quantitative analysis of the colocalization degree. The same experiment as described in A and B was also performed at 12 and 16 h pi, and Pearson's correlation coefficient was calculated for each pair of antibodies at the three analysed pi times, using for the analysis images (n = 5) containing an average of 20 cells each. Only the r values above 0.5 were considered as indicative of colocalization.
Additionally, although we have determined that the BEV nsps were located in the same compartment, as the Mpro is not a protein directly involved in the replication and transcription processes, we decided to perform additional analyses using the Hel and RdRp antibodies in combination with the anti‐dsRNA mAb. As was expected, the results obtained did not differ significantly from those obtained with the Mpro (Figs 5C and S3). These results are in agreement with those obtained with other nidoviruses, where only a partial colocalization was found between the dsRNA and nsps (Knoops et al., 2008; Hagemeijer et al., 2012; Knoops et al., 2012; Maier et al., 2013). However, for EAV and MHV, there has been described a slight colocalization between the dsRNA and nsps but only at early times pi (Hagemeijer et al., 2012; Knoops et al., 2012). Thus, we repeated the analysis at 6 h pi, but no dsRNA/Mpro overlapping was obtained (r = 0.11 ± 0.02), although the signals were clearly juxtaposed.
In view of the aforementioned results, and in order to determine if the BEV nsps were located at the sites of viral RNA synthesis, the location of newly synthesized viral RNA was analysed as an alternative approach. For this, E.Derm cells infected with BEV were treated with actinomycin D to block host cell messenger RNA synthesis and simultaneously incubated with 5‐bromouridine (5‐BrU) for 1 h before fixation, as described in the Experimental Procedures section. The nascent RNA immunolabelled with anti‐BrdU‐specific antibodies exhibited a staining pattern very similar to those of the nsps (Fig. S4). The signal associated to the newly synthesized RNA was first detected between 4 and 6 h pi, was more apparent from 8 h pi, and kept increasing along the infection until 16–18 h pi, at which time RNA synthesis was turned off in many cells (Fig. S4). This result indicates that viral RNA synthesis takes place mainly between 8 and 12 h pi and ceases at around 16 h pi. As shown in Fig. 5B, in cells stained with anti‐BrdU and anti‐Mpro specific antibodies, a clear colocalization was observed between the nascent RNA (BrU‐RNA) and Mpro at 8 h pi. The r values for the BrU‐RNA‐Mpro pair at 8 and 12 h pi were of 0.57 ± 0.02 and 0.58 ± 0.02 respectively (Fig. 5C). However, at 16 h pi we observed an r value slightly below 0.5 (0.48 ± 0.06). This low value could be explained by the shutoff of RNA synthesis in some cells at this time pi, since in those cells where the BrU‐RNA signal was still observed at 16 h pi, the r value was similar to those obtained at 8 or 12 h pi.
As shown in Fig. 5B, we observed that not all the sites positive for nascent RNA were also positive for Mpro. To further extend this observation, we performed a triple fluorescence labelling experiment in which, in addition to the Mpro and BrU‐RNA, the structural M protein was labelled. Interestingly, we observed that at 8 h pi those Mpro‐negative and BrU‐RNA‐positive sites were also positive for the structural M protein (Fig. S5). This raises the possibility that these areas might correspond to sites where the translation of structural proteins takes place.
Berne virus replication/transcription complexes are associated to double‐membrane vesicles
To study the relationship between the DMVs induced by BEV with viral replication and transcription processes, cryosections of E.Derm cells infected with BEV fixed at 16 h pi were immunolabelled with anti‐dsRNA mAb. Representative dsRNA labelling is shown in Fig. 6A (left panels), where the dsRNA is observed in the interior of the DMVs. Only some DMVs are labelled, probably as a consequence of a loss of material due to the mild fixation conditions used to preserve the antigenicity, but the signal was exclusively localized in the interior of these DMVs. A different pattern was observed when cryosections were labelled with antibodies directed against the RdRp (Fig. 6A, middle panels) or Mpro (Fig. 6A, right panels). We observed that the RdRp and Mpro polypeptides were clearly localized at DMV clusters, specifically associated to DMV membranes as well as to the adjacent membranes, while the interior of DMVs was mainly devoid of signal, although occasionally we observed some labelling within the DMV interior. When the same antibodies were used in parallel with cryosections of mock‐infected cells, only a few scattered gold particles could be observed in each case, indicating the specificity of the immunolabelling observed (Fig. S6).
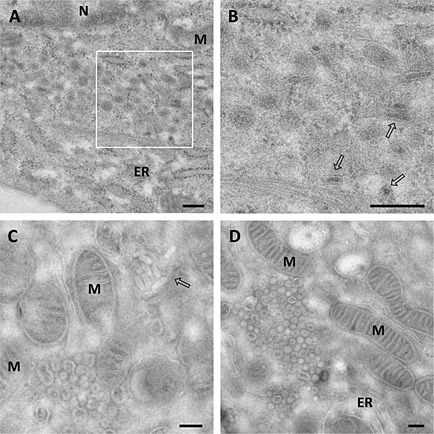
Figure 6.
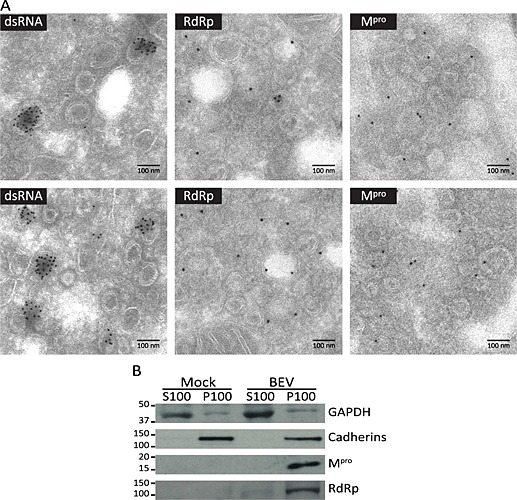
Relationship between the DMVs and the RTCs in BEV‐infected cells.
A. Cryosections of E.Derm cells infected with BEV and fixed at 16 h pi were immunogold labelled with a mAb anti‐dsRNA (left panel) or with antibodies against the RdRp (middle panel) and Mpro (right panel) and analysed by electron microscopy. Scale bars, 100 nm.
B. Immunodetection of Mpro and RdRp in a membrane‐enriched cell fraction analysed by Western blot. Mock‐infected and BEV‐infected (16 h) E.Derm cells were fractionated by differential centrifugation into cytosolic (S100) and membranous (P100) fractions and analysed by Western blot with anti‐Mpro and anti‐RdRp antibodies. As control of the fractionation procedure, antibodies against the cellular cytosolic GAPDH and membrane‐associated cadherins were also used. The positions and sizes in kilodalton of the molecular weight markers are shown at the left side of each picture.
The replicase subunits of BEV are probably embedded in a membrane‐associated complex, as expected based on the prediction of the hydrophobic regions in nsp1, nsp2 and nsp4. To analyse if the Mpro and the RdRp of BEV are membrane‐associated proteins, E.Derm cells mock infected or infected with BEV were homogenized and subjected to differential ultracentrifugation such that cellular membranes were pelleted (P100) and separated from the cytosolic fraction (S100). Both fractions were analysed by Western blot with antibodies against Mpro and RdRp, and against specific markers for cytosolic proteins [glyceraldehyde 3‐phosphate dehydrogenase (GAPDH)] or membrane proteins (p‐cadherins). As shown in Fig. 6B, both proteins, the Mpro and RdRp, were found within the membrane fraction.
Clearly, the pattern observed in immunolabelled cryosections reveals that the dsRNA is located inside the DMVs, surrounded by the nsps anchored to the adjacent membranes. However, this pattern was less clear in the immunofluorescence assay shown previously in Fig. 5A. For this reason, sequential z‐sections with a step size optimized were captured to generate three‐dimensional (3D) images of dsRNA and Mpro using confocal images (Fig. 7 and Movie S1). The 3D reconstruction generated (Fig. 7B) shows that part of the dsRNA is localized inside the volume formed by the Mpro (Fig. 7C).
Figure 7.

Three‐dimensional reconstruction from images obtained by confocal microscopy: Mpro‐dsRNA. A series of consecutive images with a z‐step optimized were taken by confocal microscopy from E.Derm cells infected with BEV, fixed at 8 h pi and immunostained with the anti‐Mpro (red) and anti‐dsRNA (green) antibodies as described in the legend to Fig. 5A. The nuclei were stained with DAPI (blue).
A. Projection in the z‐axis of the captured images of an individual cell.
B. General view of the 3D reconstruction generated from the confocal images using the imaris software (Bitplane AG). Scale bars, 5 µm.
C. Enlarged area from the 3D reconstruction. (C1) Frontal view. (C2) Frontal view of a vertical section that shows the dsRNA inside the structure labelled with anti‐Mpro antibody. (C3) Top view. (C4) Top view of a cross section. Scale bars, 0.5 µm.
Membranous origin of the double‐membrane vesicles induced by Berne virus
A wide panel of antibodies was used to identify the donor organelle from which the DMVs are formed. For that, E.Derm cells infected with BEV were fixed at 10 h pi and immunolabelled with the anti‐Mpro serum and antibodies against organelle markers such as TGN46, a marker for the trans‐Golgi network (Fig. 8A); ERGIC p53, a marker for the intermediate compartment (Fig. 8B); calnexin or protein disulfide isomerase (PDI), which are markers for ER (Fig. 8C and D); sigma‐1 receptor (S1R) for MAM (Fig. 8F); or EEA1 for early endosomes (Fig. 8H). Also, the ectopic expression of GFP‐Lamp1 (Fig. 8G) was used as marker of lysosomes, and the mitochondrion compartment was stained with MitoTracker (Fig. 8E). All the cellular markers used in this study were previously tested in mock‐infected cells, and with the exception of ERGIC p53, none of the proteins showed a differential pattern between infected and non‐infected cells. As previously mentioned, the ERGIC is the compartment where the virus assembly is thought to occur (Garzon et al., 2006).
Figure 8.
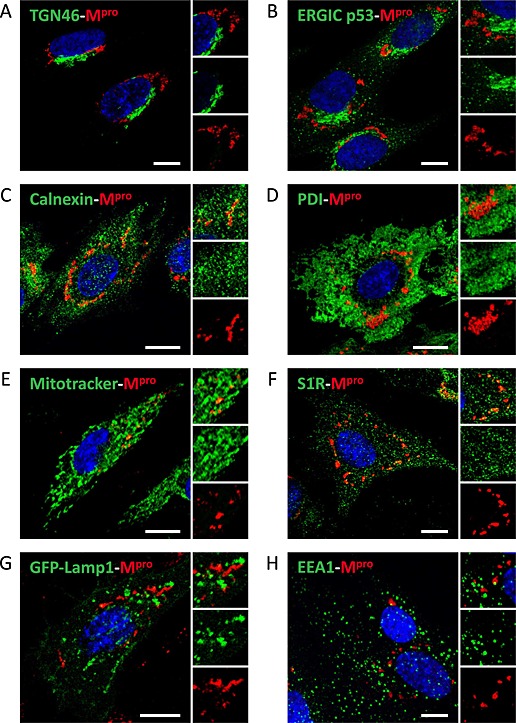
Origin of the membranes of the DMVs induced by BEV. E.Derm cells infected with BEV and fixed at 10 h pi were immunolabelled with the rat anti‐Mpro antibody (red) in combination with antibodies against different membranous cellular compartments or with MitoTracker reagent for mitochondrial staining (green). In addition, E.Derm cells transfected with a plasmid expressing the GFP‐Lamp1 fusion protein (as described in the Experimental Procedures section) were used for lysosome detection. Nuclei were stained with DAPI (blue). Each panel shows a general image of the labelling and, at the right side, an enlarged image of a representative area labelled with both antibodies (upper panel), with the antibody to the cellular marker (middle panel) or with the anti‐Mpro antibody (lower panel). Scale bars, 10 µm.
A. Anti‐TGN46 for Golgi.
B. Anti‐ERGIC p53 for intermediate compartment.
C. Anti‐calnexin for ER.
D. Anti‐PDI for ER.
E. MitoTracker for mitochondria.
F. Anti‐S1R for MAM.
G. GFP‐Lamp1 expression for lysosome.
H. Anti‐EEA1 for early endosomes.
As shown in Fig. 8, none of the markers used in this assay showed colocalization with the Mpro protein, but, apparently, the signals of PDI, calnexin, S1R and MitoTracker are surrounding or partially overlapping the Mpro signal. Although the quantitative analysis showed r values below 0.5, the values for PDI (0.36 ± 0.03), calnexin (0.30 ± 0.04), S1R (0.37 ± 0.04) or MitoTracker (0.38 ± 0.05) were higher than those for EEA1 (0.03 ± 0.01), ERGIC (0.13 ± 0.05), TGN46 (0.10 ± 0.02) or Lamp1 (0.05 ± 0.03), reinforcing the idea of a partial overlapping. These results are in concordance with observations made during the TEM study (Fig. 1), where the DMVs were surrounded by ER and mitochondria, but only in few cases could we observe connections between the DMVs and the ER or the MAM.
Again, the results are in agreement with those obtained with other nidoviruses, where the membrane origin of the DMVs is questioned because the conventional organelle markers do not colocalize or only partially colocalize with viral replicase proteins (Shi et al., 1999; Ivanov et al., 2004; Snijder et al., 2006; Knoops et al., 2010; Ulasli et al., 2010).
Discussion
In this study, the RTCs of a member of the torovirus genus are described for the first time, and a preliminary characterization has been performed. First, we carried out an ultrastructural study to analyse the cellular membrane rearrangements elicited upon torovirus infection. The presence of characteristic DMVs within the cytoplasm of cells infected with BEV was observed both in equine E.Derm cells and in human MRC5 cells, treated under different preservation conditions, such as conventional chemical fixation or cryofixation. However, the BEV DMVs were poorly visualized under the cryofixation and freeze substitution conditions used in this study. A cryofixation and freeze substitution method was previously used by Snijder et al. (2006) to improve the preservation of DMVs in SARS‐CoV‐infected cells for their examination by TEM, and later utilized for electron tomography studies, with some minor changes (Knoops et al., 2008). The same group described the presence of DMVs in EAV‐infected cells using a similar protocol; however, for the tomography assays, they included high‐pressure freezing before freeze substitution (HPF‐FS) to further improve DMV preservation (Knoops et al., 2012). In our assay, the membranes appeared poorly contrasted by this technique, probably owing to differences in the cryofixation and embedding conditions with regard to the protocols used for SARS‐CoV and EAV. Although the HPF‐FS has been recognized as an efficient technique for preserving DMVs (Limpens et al., 2011; Knoops et al., 2012; Romero‐Brey et al., 2012; Maier et al., 2013), the embedding in epoxy resin TAAB‐812 protocol used in these assays provided a high preservation of the cellular structures as well as of the DMVs, which exhibit the spherical shape similar to that previously observed in other nidoviruses. Moreover, the great ultrastructural preservation achieved in this study allows us to clearly distinguish the two tightly apposed membranes of the DMVs, most of which show an electron‐dense inner content, as well as to visualize membranous connections among different DMVs and between DMVs and ER membranes.
Apparently, the DMVs arranged during BEV infection look like independent structures separated from each other; however, as mentioned earlier, an in‐depth inspection of the infected cells revealed the existence of connections between many of them and between them and ER membranes. This suggests that, as has been described to occur with the related SARS‐CoV (Knoops et al., 2008) and EAV (Knoops et al., 2012), the BEV‐induced DMVs form part of a network of modified membranes derived from the ER. Nonetheless, a more sophisticated ultrastructural analysis would be required to confirm the existence of this network. An apparent difference with SARS‐CoV (Knoops et al., 2008) or EAV (Knoops et al., 2012) was the absence of ribosomes associated to the outer membranes of the BEV DMVs. We observed ribosomes attached to ER membrane but not in association with the DMVs.
In this ultrastructural study, we did not observe other membranous structures different from the DMVs induced by BEV infection. The large convoluted membrane clusters found in SARS‐CoV‐infected cells (van Hemert et al., 2008), and later described in cells infected with MHV (Ulasli et al., 2010) and Middle East respiratory syndrome coronavirus (de Wilde et al., 2013), were not observed in the case of BEV. The absence of convoluted membranes in torovirus, as well as in cells infected with EAV (van Hemert et al., 2008) or IBV (Maier et al., 2013), supports the idea that these structures are specific to betacoronaviruses. Likewise, the spherules recently described for IBV (Maier et al., 2013) were not observed in our study. Nonetheless, we cannot rule out the presence of these structures in the BEV‐infected cells because their appearance in two‐dimensional ultrastructural studies is very similar to that of the DMVs, and, therefore, the application of 3D studies is essential to elucidate the presence or the absence of spherules in BEV‐infected cell.
A common characteristic of all the nidoviruses studied to date is that the DMVs are always surrounded by ER and mitochondria. Concerning this, we observed clusters of DMVs with apparent continuity with ER tubules closely apposed to mitochondria, which might represent the areas of communication between these two organelles, known as MAMs. Recently, Friesland et al. (2013) have suggested that the membranes of the MAM could serve as platforms for initial RNA replication during hepatitis C virus infection, a virus that induces DMVs with similar characteristics to those produced by nidoviruses. The interaction between virus replication factories and MAMs is probably due to the fact that viruses need the energy produced by the mitochondria for their processes, and hence, the formation of their replicative platforms from MAMs could be advantageous to obtain this energy. Moreover, MAMs play a major role in lipid metabolism, a function that could be essential for the membrane remodelling process induced by the virus. However, biochemical assays are indispensable to confirm the implication of MAMs with the DMVs of BEV.
To investigate whether torovirus DMVs are the sites where replication and transcription occur, we obtained specific sera for the BEV Mpro, Hel and RdRp, three key proteins in the formation and functioning of RTCs. All showed a high colocalization with each other and with newly synthesized RNAs. However, the dsRNA, widely used as a marker of RTC in several positive‐strand RNA viruses, only showed a partial colocalization with the nsps. A 3D reconstruction of confocal images showed that part of the dsRNA is localized inside the volume occupied by the Mpro. Additionally, the immunolabelling assays on cryosections indicated that the nsps are associated to DMV membranes or to the adjacent membranes, while the dsRNA was exclusively detected inside DMVs. An apparent separation between the bulk of the nsps and most of the dsRNA was previously described for other nidoviruses (Knoops et al., 2008; 2012; Maier et al., 2013). Therefore, in nidoviruses, the relationship between the sites of dsRNA accumulation and RTCs remains an open question. The apparent absence of pores connecting the interior of DMVs with the cytoplasm and the presence of dsRNA inside of these DMVs, as well as the lack of colocalization with replicase proteins, suggested the idea that many DMVs are not actively involved in RNA synthesis and might instead represent shielding structures that protect the dsRNA molecules from the detection by the innate immune sensors (Hagemeijer et al., 2012). Other authors consider that a huge excess of nsps may be produced in virus‐infected cells, with only a fraction of these molecules actively participating in viral RNA synthesis (Knoops et al., 2008). We believe that the DMVs formed in torovirus‐infected cells are the sites where the replication and transcription occur. By confocal microscopy, we observed a clear colocalization between the newly synthesized RNA and the nsps, but attempts to perform similar studies by immunoelectronmicroscopy were unsuccessful. On the other hand, it has to be considered that the colocalization between the newly synthesized RNA and nsps does not necessarily imply that those are areas of active replication exclusively; they can also represent sites of protein translation, as it probably occurs in the regions that are positive for the nascent RNA and the M protein (Fig. S5). Colocalization studies between the dsRNA and the newly synthesized RNA could help to resolve this question. Our attempts to perform these assays have failed because of the unavailability of an adequate combination of antibodies for the double labelling. Similar assays were performed in MHV‐infected cells, where the dsRNA and an analogue of uridine, EU, were placed at closely adjacent locations, but only at an early time pi (Hagemeijer et al., 2012). Similar results were described for EAV (Knoops et al., 2012), where a limited colocalization between the nsps and dsRNA was described at an early time pi. In this regard, we did not observe colocalization of the BEV nsps characterized here with the dsRNA at any time of infection, although the signals observed by confocal microscopy corresponding to nsps and dsRNA were closely juxtaposed in many areas at all pi times analysed.
Additionally, another fact observed in this work is the separation between the RTC and the site of virus assembly. In our confocal assays, we observed a complete exclusion between the Mpro and the structural protein M during the infection with BEV, which was also supported by the separation of ERGIC p53 and the Mpro. Furthermore, the TEM assays show a high amount of viral particles enclosed in vesicles physically separated from the sites where the DMVs are localized. Similarly, in SARS‐CoV‐infected cells, the separation between the RTC factories and the sites of virus assembly has been described (Snijder et al., 2006).
Finally, we have attempted to determine the membranous origin of the DMVs using specific organelle markers. None of the markers used in these confocal microscopy assays, to identify early endosomes, Golgi, ERGIC, lysosomes, ER, mitochondria or MAM, showed a clear colocalization with the Mpro protein. However PDI, calnexin, MitoTracker and S1R, markers of ER, mitochondria and MAM respectively, were found surrounding the Mpro signal or even partially colocalizing with it. These results together with the fact that the TEM assays showed that the DMVs are connected with the ER, and probably with the MAM, lead us to believe that the membranes of this organelle are the donors for the DMV formation. The absence of colocalization between the traditional ER markers and nsps may be due to the high accumulation of BEV nsps in this region, which could promote the exclusion of these cellular proteins. The origin of the rearranged membranes produced by the studied coronaviruses for their replication is also in question. From the TEM assays, it appears that coronaviruses subvert the ER membrane to establish their replication factories. However, the absence of conventional ER markers in virus‐induced membranous structures (Snijder et al., 2006; Ulasli et al., 2010) makes it difficult to undoubtedly establish their origin. Only colocalization with the ER protein sec61 and a partial colocalization with PDI were reported (Knoops et al., 2010). Some studies indicated that the origin is found in specific organelles derived from the ER, like the autophagosome. By their very nature, DMVs resemble cellular autophagosomes generated during the cellular autophagy process. Nonetheless, the relationship between DMV formation and autophagy is not clear, as although it has been shown that some coronaviruses induce autophagy (Prentice et al., 2004; Snijder et al., 2006), this process is not required for virus replication (Zhao et al., 2007; Schneider et al., 2012). Furthermore, it has been described that the coronavirus nsp6 protein generates autophagosomes from the ER via an omegasome intermediate (Cottam et al., 2011). The relationship between BEV and the autophagy pathway is as yet unknown, but we have some ongoing work to clarify this issue. Recent reports described an autophagy‐independent role for LC3 in several nidovirus infections, where the ER‐derived membranes of EDEMosomes may be the platform for virus replication, showing that the RTCs of MHV and EAV colocalize with markers for cellular EDEMosomes, as EDEM1 and OS‐9. Our attempts to explore this possibility in BEV‐infected cells were inconclusive.
Overall, our results show a close relationship between the replicative structures of BEV and those of the related viruses in the Nidovirales order. The formation of DMVs in torovirus‐infected cells, as well as the similar characteristic of the RTCs, provides a new evidence to support the idea that nidoviruses have a common replicative structure based on the DMVs arranged in clusters at specific areas of the cytoplasm and surrounded by ER membranes and mitochondria. Further analysis will be required to define the biogenesis of BEV‐induced replicative compartment. This work can serve as a starting point to understand the remodelling of cellular membranes induced by BEV infection to build its RTC factories, a field of research that remains almost unexplored as yet.
Experimental procedures
Cells and viruses
Equine dermal (E.Derm) cells (NBL‐6; ATCC CCL‐57) and human fetal lung fibroblasts (MRC‐5) (ATTC CCL‐171) were cultured in Dulbecco's modified Eagle's medium (Gibco) supplemented with 15% and 10% fetal calf serum (FCS) respectively. The equine torovirus, strain Berne P138/72 (BEV), was used to infect monolayers of E.Derm and MRC‐5 cells as described previously (Maestre et al., 2011). A multiplicity of infection of 2.5 plaque‐forming units per cell was used in all experiments.
Antibodies
The BEV nucleocapsid (N), membrane (M) and spike (S) structural proteins were detected by using specific antisera previously described (Garzon et al., 2006; Maestre et al., 2011). Specific antisera against the predicted proteins from the replicase complex, Mpro, Hel and RdRp were generated for this study. The anti‐Hel and anti‐RdRp sera were obtained by immunizing rabbits (New Zealand White) with Escherichia coli‐expressed histidine‐tagged recombinant proteins harbouring the predicted BEV helicase domain (residues Ala‐5411 to Gln‐5968 from the replicase pp1ab polyprotein) or a fragment of the BEV RdRp (residues Ala‐4576 to Ala‐4828 of the replicase pp1ab polyprotein) respectively. The construction of expression plasmids, the production and purification of the recombinant antigens and the rabbit immunization protocol are described in the supporting information. Also, two anti‐Mpro sera were obtained by subcutaneous immunization of rabbits or rats with the previously described synthetic peptide N‐KPLQYFHVPSFWQPFKKQ (Smits et al., 2006) coupled to keyhole limpet haemocyanin.
Viral RNA replication intermediates were visualized by using an anti‐dsRNA mAb (clone K1; English and Scientific Consulting Kft). To visualize newly synthesized viral RNA, infected cells were incubated in the presence of BrU and an anti‐BrdU mAb (clone UI‐4; Invitrogen) was used for detection. The primary antibodies used to label cellular organelles were rabbit polyclonal anti‐calnexin, mouse mAb anti‐PDI (clone 1D3) and anti‐ERGIC p53 (clone G1/93) (Enzo Life Science), sheep anti‐TGN‐46 (AbD Serotec), mouse mAb anti‐EEA1 (BD Biosciences) and mouse mAb anti‐S1R1 (clone F‐5; Santa Cruz). The MitoTracker® Red FM (Invitrogen) dye was also used. As control for the subcellular fractionation experiments, the rabbit polyclonal anti‐p‐cadherin (Sigma‐Aldrich) and the mAb anti‐GAPDH (clone 14C10; Cell Signaling) antibodies were used. Also, for loading control, a rabbit mAb anti‐α‐tubulin (Cell Signaling) was used.
SDS‐PAGE and Western blot
Infected cells were lysed at the indicated times pi directly in Laemmli loading buffer with 5% β‐mercaptoethanol. Protein lysates were subjected to SDS‐PAGE and transferred to nitrocellulose membranes (Bio‐Rad). The membranes were blocked for 1 h in Tris‐buffered saline with 0.05% Tween 20 (TBS‐T) containing 5% non‐fat dry milk and then were incubated with the primary antibodies diluted in the same buffer at 4°C overnight. The incubation with the mAb anti‐GAPDH was carried out in TBS‐T containing 5% BSA. Then, the membranes were washed with TBS‐T, incubated with appropriate dilutions of the corresponding horseradish peroxidase‐labelled secondary antibodies (Sigma‐Aldrich) for 1 h at room temperature and washed again. Proteins were detected using an enhanced chemiluminescence Western blot detection kit (ECL, GE Health Care).
Immunofluorescence
E.Derm cells grown on glass coverslips were infected with BEV, fixed at the indicated times pi with 4% paraformaldehyde (PFA) in PBS and processed for immunofluorescence using a standard protocol. Briefly, after washing with PBS, the cells were permeabilized with 0.5% Triton X‐100 in PBS and blocked with PBS containing 20% FCS. Then, the cells were incubated with a mixture of primary antibodies diluted in PBS with 20% FCS in a humidified chamber for 1 h at 37°C, washed with PBS and again incubated with the secondary antibodies conjugated with Alexa Fluor 488 or 594, and 4,6‐diamidino‐2‐phenylindole (DAPI) (Invitrogen) for nuclei staining. For the triple fluorescence labelling experiment, we used secondary antibodies conjugated with Alexa Fluor 488, 546 and 647. Cells were washed with PBS and mounted on microscope slides using the ProLong® Gold anti‐fade reagent (Invitrogen). Images were acquired with a Leica TSC SP5 confocal laser scanning microscope with a step size of about 0.5 µm. The colocalization analyses were performed with the imagej software by using the JACoP plug‐in (Bolte and Cordelières, 2006), and Pearson's correlation coefficient between each pair of signals was calculated. Image volumes were reconstructed from sequential z‐sections with an optimized step size of 0.13 µm using imaris software (Bitplane AG).
Bromouridine labelling of de novo synthesized viral RNA
Infected cells on glass coverslips were incubated with 5 mM 5‐BrU (Sigma‐Aldrich) and 5 mg ml−1 of actinomycin D (Sigma‐Aldrich) for 1 h before fixation and permeabilization for immunofluorescence staining.
Plasmid transfections
The pEGFP‐Lamp1 expression construct (kindly provided by J. Lippincott‐Schwartz, NICHD, NIH, Bethesda, EEUU) (Patterson and Lippincott‐Schwartz, 2002) was transfected into E.Derm cells grown on coverslips by using Lipofectamine 2000 (Invitrogen) as recommended by the manufacturer. At 24 h post‐transfection, cells were infected with BEV and processed for immunofluorescence staining as described earlier.
Subcellular fractionation
E.Derm cells either mock infected or infected with BEV for 16 h were lysed and subjected to differential centrifugation as previously described (Sims et al., 2000; Brockway et al., 2003). Briefly, cells were scraped in PBS, collected by low‐speed centrifugation and resuspended in cold sucrose–Tris buffer [10 mM Tris (pH 7.2), 200 mM sucrose] with protease inhibitors (cOmplete Protease Inhibitor Cocktail; Roche). The cell suspension was homogenized in a Dounce homogenizer and centrifuged at 1000 × g for 5 min to pellet the nuclei. The post‐nuclear supernatant was then centrifuged at 100 000 × g for 15 min in a Beckman tabletop ultracentrifuge, using a Beckman rotor (TLA‐120) to pellet the membranes. The final supernatant was saved as the cytosol material (S100), and the pellet was resuspended in the same volume of sucrose–Tris buffer (P100).
Transmission electron microscopy
Embedding in epoxy resin TAAB‐812
For conventional TEM, E.Derm or MRC5 cells infected for 16 h with BEV were fixed in situ for 2 h at room temperature with a mixture of 4% PFA and 2% glutaraldehyde in 0.1 M Sörensen phosphate buffer, pH 7.4. After several washes, the cells were scraped and collected in the same buffer, centrifuged and the cell pellets were processed for embedding in the epoxy resin TAAB‐812 (TAAB Laboratories, Berkshire, England) according to standard procedures previously described (DeDiego et al., 2007; Maestre et al., 2011). Ultrathin sections (70–80 nm) of the cell samples were stained with 2% uranyl acetate solution in water and lead Reynols citrate and examined at 80 kV in a Jeol JEM‐1010 (Tokyo, Japan) electron microscope. Pictures were taken with a TemCam‐F416 (4 K × 4 K) digital camera (TVIPS, Gauting, Germany).
Quick freezing and freeze substitution
E.Derm cells infected with BEV for 16 h were fixed in situ with 4% PFA and 0.1% glutaraldehyde in 0.1 M Sörensen phosphate buffer pH 7.4 for 2 h at room temperature, followed by an overnight incubation at 4°C in the same fixative solution without glutaraldehyde. After several washes, the cells were scraped, centrifuged and the cell pellets were embedded in a solution of 10% gelatin in PBS. The cells were then cryoprotected with glycerol at 4°C (15 min with 15% glycerol in PBS and another incubation of 15 min in 30% glycerol in PBS). Cells were subsequently subjected to plunge freezing in liquid propane using a KF80 (Leica) cryofixation equipment. Freeze substitution was carried out in an automated freeze substitution system (Leica) in pure methanol containing 0.5% uranyl acetate. Samples were kept at −85°C for 54 h, after which the temperature was raised by 5°C h−1 to −50°C. After being washed with pure methanol several times, the cells were embedded in Lowicryl HM20 (Polysciences Europe GmbH, Germany) at −45°C following the manufacturer's instructions. Ultrathin sections (70–80 nm) were obtained as described earlier.
Cryosections
To obtain ultrathin cryosections (Tokuyasu method) (Slot and Geuze, 2007), E.Derm cells infected with BEV were fixed in situ for 2 h at room temperature in a solution of 4% PFA and 1% glutaraldehyde in PHEM buffer (240 mM Pipes, 100 mM Hepes, 8 mM MgCl2, 40 mM ethylene glycol tetraacetic acid, pH 6.9), followed by an overnight incubation at 4°C in the same fixative solution. After several washes, the cells were pelleted, embedded in 12% gelatin and cryoprotected by overnight incubation in 2.3 M sucrose in PHEM buffer. Mounted gelatin blocks were frozen in liquid nitrogen. Ultrathin cryosections (70–80 nm) were obtained at –120°C by using a Leica UCT ultramicrotome, equipped with an FCS cryochamber, and were picked up in a 1:1 mixture of 2.3 M sucrose : 2% methylcellulose and deposited on Formvar‐coated grids. Ultrathin cryosections maintained in the sucrose : methylcellulose mixture were incubated in 2% gelatin in PBS at 37°C for 20 min. Then, sections were washed with bidistilled water and finally stained and embedded with a mix of 1.8% methylcellulose and 0.4% uranyl acetate.
Immunogold labelling
Ultrathin cryosections were obtained as described earlier with a slight modification in the fixation to preserve the antigen–antibody reactivity. In this case, the infected cells were fixed by adding an equal volume of a 2× fixative solution (8% PFA in PHEM buffer) to the culture medium. After 30 min at room temperature, the solution was replaced by a 1× fixative solution (4% PFA in PHEM buffer) and cells were incubated for an additional hour. After, the cells were incubated overnight at 4°C in a solution with 8% PFA in PHEM buffer and were processed for cryosections as described earlier. Ultrathin cryosections maintained in the sucrose : methylcellulose mixture were incubated in 2% gelatin in PBS at 37°C for 20 min. The sections were then incubated with 0.15% glycine in PBS, with four changes of 2 min each and blocked by incubation in PBS with 10% FCS for 10 min. Incubation with the primary antibody diluted in PBS containing 5% FCS was performed for 1 h at room temperature. After three washes in PBS with 0.2% FCS, sections were incubated for 1 h with protein A coupled to 10 nm gold particles (CMC Laboratory, Utrecht University, Utrecht, the Netherlands). Sections were treated with 1% glutaraldehyde in PBS for 5 min, washed with bidistilled water and finally stained and embedded with a mix of 1.8% methylcellulose and 0.4% uranyl acetate.
Conflicts of interest
The authors declare that there are no conflicts of interest.
Supporting information
Fig. S1. Close view of BEV viral particles observed in sections of BEV‐infected E.Derm cells embedded in epoxy resin TAAB‐812. Electron micrographs of E.Derm cells infected with BEV for 16 h and embedded in the epoxy resin TAAB‐812 after conventional chemical fixation, where the presence of rod‐shaped viral particles can be clearly observed. Individual particles A) or small groups of particles inside rough ER derived‐vesicles B), as well as large groups of particles packed in large vesicles C) can be observed at this pi time. Viral particles were sectioned in different orientations: longitudinal section (black arrow) or cross‐section (black arrowhead). M: Mitochondria; ER: Endoplasmic reticulum. Scale bars, 100 nm.
Fig. S2. Ultrastructural analysis of MRC5 cells infected with BEV embedded in epoxy resin TAAB‐812. Electron micrographs of sections of MRC5 cells infected with BEV for 24 h that were embedded in the epoxy resin TAAB‐812, after conventional chemical fixation, showing the presence of characteristic spherical DMVs. A) Cluster of DMVs (white asterisk) surrounded by mitochondria and ER. A particle inside a vesicle can be distinguished at the periphery of the cluster (white arrow). B‐C) DMVs connected between them (white arrowhead) and with the ER (black arrowhead) or with the MAM (black arrow). M: Mitochondria; ER: Endoplasmic reticulum. Scale bars, 200 nm.
Fig. S3. Colocalization analysis of the RdRp and Hel proteins with the dsRNA. E.Derm cells infected with BEV were fixed with 4% PFA in PBS at 8 h pi and used for immunolabeling with the mouse mAb anti‐dsRNA (green) in combination with A) rabbit anti‐RdRp or B) anti‐Hel antibodies (red). Nuclei were stained with DAPI (blue). The boxed areas are representative fields shown at higher magnification. Scale bars, 10 μm.
Fig. S4. Distribution of newly synthesized viral RNA (BrU‐RNA) along the infection with BEV. E.Derm cells infected with BEV were incubated with 5 mM BrU in the presence of 5 mg ml−1 of actinomycine D for 1 h before fixation. Cells were fixed at different pi times as indicated in each panel, and processed for immunofluorescence staining with a mouse anti‐BrdU mAb (green). The nuclei were stained with DAPI (blue). The boxed areas are representative fields that are shown at higher magnification. Scale bar, 10 μm.
Fig. S5. Partial colocalization of the newly synthesized RNA (BrU‐RNA) and the M structural protein. BEV‐Infected cells treated as described in the legend to Fig. S3 were fixed at 8 h pi. A) Triple colocalizaton assay using the rabbit anti‐M (green), mouse anti‐BrdU mAb (red) and rat anti‐Mpro (blue) antibodies. The square encloses a representative area that is shown at higher magnification in panels B‐G. B) Signal obtained with the anti‐M antibody; C) signal corresponding to the newly synthesized RNA (BrU‐RNA); D) signal corresponding to the Mpro; E) overlapping of the signals corresponding to the M protein and the BrU‐RNA; F) overlapping of the signals corresponding to the Mpro protein and the BrU‐RNA; G) overlapping of the signals corresponding to the M and the Mpro proteins. The arrow indicates the area of colocalization between the M protein and the BrU‐RNA. Scale bar, 10 μm.
Fig. S6. Immunolabeling assay on mock‐infected cells. A) Cryosections of mock‐infected E.Derm cells were fixed and immunogold‐labeled with a mAb anti‐dsRNA (left panel), or with antibodies against the RdRp (middle panel) and Mpro (right panel), and analyzed by electron microscopy. Scale bars, 100 nm.
Movie S1. 3D reconstruction presented in the Fig.7, panels B‐C.
Supporting info item
Acknowledgments
We thank Paco Rodriguez for helpful discussions and critical reading of the manuscript. We also thank Sylvia Gutierrez for her expert technical assistance with confocal microscopy, Milagros Guerra for her excellent work processing the samples for the electron microscopy and Susana Plazuelo for her excellent technical assistance with tissue culture. This work was supported by grant AGL2010‐15495 to D. R. from the Spanish Ministry of Economy and Competitiveness. G. A. was a recipient of an FPU fellowship from the Spanish Ministry of Education, Culture and Sport.
Ávila‐Pérez, G. , Rejas, M. T. , and Rodríguez, D. (2016) Ultrastructural characterization of membranous torovirus replication factories. Cellular Microbiology, 18: 1691–1708. doi: 10.1111/cmi.12620.
References
- Aita, T. , Kuwabara, M. , Murayama, K. , Sasagawa, Y. , Yabe, S. , Higuchi, R. , et al. (2012) Characterization of epidemic diarrhea outbreaks associated with bovine torovirus in adult cows. Arch Virol 157: 423–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelini, M.M. , Akhlaghpour, M. , Neuman, B.W. , and Buchmeier, M.J. (2013) Severe acute respiratory syndrome coronavirus nonstructural proteins 3, 4, and 6 induce double‐membrane vesicles. mBio 4: e00524–00513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrette‐Ng, I.H. , Ng, K.K. , Mark, B.L. , van Aken, D. , Cherney, M.M. , Garen, C. , et al. (2002) Structure of arterivirus nsp4: the smallest chymotrypsin‐like proteinase with an α/β C‐terminal extension and alternate conformations of the oxyanion hole. J Biol Chem 277: 39960–39966. [DOI] [PubMed] [Google Scholar]
- Bolte, S. , and Cordelières, F.P. (2006) A guided tour into subcellular colocalization analysis in light microscopy. J Microsc 224: 213–232. [DOI] [PubMed] [Google Scholar]
- den Boon, J.A. , and Ahlquist, P. (2010) Organelle‐like membrane compartmentalization of positive‐strand RNA virus replication factories. Annu Rev Microbiol 64: 241–256. [DOI] [PubMed] [Google Scholar]
- den Boon, J.A. , Snijder, E.J. , Chirnside, E.D. , de Vries, A.A. , Horzinek, M.C. , and Spaan, W.J. (1991) Equine arteritis virus is not a togavirus but belongs to the coronaviruslike superfamily. J Virol 65: 2910–2920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brierley, I. , Digard, P. , and Inglis, S.C. (1989) Characterization of an efficient coronavirus ribosomal frameshifting signal: requirement for an RNA pseudoknot. Cell 57: 537–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockway, S.M. , Clay, C.T. , Lu, X.T. , and Denison, M.R. (2003) Characterization of the expression, intracellular localization, and replication complex association of the putative mouse hepatitis virus RNA‐dependent RNA polymerase. J Virol 77: 10515–10527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clementz, M.A. , Kanjanahaluethai, A. , O'Brien, T.E. , and Baker, S.C. (2008) Mutation in murine coronavirus replication protein nsp4 alters assembly of double membrane vesicles. Virology 375: 118–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cottam, E.M. , Maier, H.J. , Manifava, M. , Vaux, L.C. , Chandra‐Schoenfelder, P. , Gerner, W. , et al. (2011) Coronavirus nsp6 proteins generate autophagosomes from the endoplasmic reticulum via an omegasome intermediate. Autophagy 7: 1335–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Groot, R.J. (2008) Chapter 9: Molecular biology and evolution of torovirus In Nidoviruses. Perlman S., Gallagher T., and Snijder E.J. (eds). Washington, DC: ASM Press, pp. 133–146. [Google Scholar]
- De Diego, M.L. , Alvarez, E. , Almazan, F. , Rejas, M.T. , Lamirande, E. , Roberts, A. , et al. (2007) A severe acute respiratory syndrome coronavirus that lacks the E gene is attenuated in vitro and in vivo . J Virol 81: 1701–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draker, R. , Roper, R.L. , Petric, M. , and Tellier, R. (2006) The complete sequence of the bovine torovirus genome. Virus Res 115: 56–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friesland, M. , Mingorance, L. , Chung, J. , Chisari, F.V. , and Gastaminza, P. (2013) Sigma‐1 receptor regulates early steps of viral RNA replication at the onset of hepatitis C virus infection. J Virol 87: 6377–6390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadlage, M.J. , Sparks, J.S. , Beachboard, D.C. , Cox, R.G. , Doyle, J.D. , Stobart, C.C. , and Denison, M.R. (2010) Murine hepatitis virus nonstructural protein 4 regulates virus‐induced membrane modifications and replication complex function. J Virol 84: 280–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garzon, A. , Maestre, A.M. , Pignatelli, J. , Rejas, M.T. , and Rodriguez, D. (2006) New insights on the structure and morphogenesis of Berne virus. Adv Exp Med Biol 581: 175–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbalenya, A.E. , Enjuanes, L. , Ziebuhr, J. , and Snijder, E.J. (2006) Nidovirales: evolving the largest RNA virus genome. Virus Res 117: 17–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosert, R. , Kanjanahaluethai, A. , Egger, D. , Bienz, K. , and Baker, S.C. (2002) RNA replication of mouse hepatitis virus takes place at double‐membrane vesicles. J Virol 76: 3697–3708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagemeijer, M.C. , Vonk, A.M. , Monastyrska, I. , Rottier, P.J. , and de Haan, C.A. (2012) Visualizing coronavirus RNA synthesis in time by using click chemistry. J Virol 86: 5808–5816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harak, C. , and Lohmann, V. (2015) Ultrastructure of the replication sites of positive‐strand RNA viruses. Virology 479–480: 418–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegyi, A. , Friebe, A. , Gorbalenya, A.E. , and Ziebuhr, J. (2002) Mutational analysis of the active centre of coronavirus 3C‐like proteases. J Gen Virol 83: 581–593. [DOI] [PubMed] [Google Scholar]
- van Hemert, M.J. , van den Worm, S.H. , Knoops, K. , Mommaas, A.M. , Gorbalenya, A.E. , and Snijder, E.J. (2008) SARS‐coronavirus replication/transcription complexes are membrane‐protected and need a host factor for activity in vitro . PLoS Pathog 4: e1000054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoet, E.A. , and Linda, J.F. (2008) Chapter 23: Torovirus pathogenesis and immune response In Nidoviruses. Perlman S., Gallagher T., and Snijder E.J. (eds). Washington, DC: ASM Press, pp. 351–359. [Google Scholar]
- Ito, T. , Katayama, S. , Okada, N. , Masubuchi, K. , Fukuyama, S. , and Shimizu, M. (2010) Genetic and antigenic characterization of newly isolated bovine toroviruses from Japanese cattle. J Clin Microbiol 48: 1795–1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov, K.A. , Thiel, V. , Dobbe, J.C. , van der Meer, Y. , Snijder, E.J. , and Ziebuhr, J. (2004) Multiple enzymatic activities associated with severe acute respiratory syndrome coronavirus helicase. J Virol 78: 5619–5632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King, A.M.Q. , Adams, M.J. , Carstens, E.B. , and Lefkowitz, E.J. (2012) Order – Nidovirales In Virus Taxonomy. King A.M.Q., Adams M.J., Carstens E.B., and Lefkowitz E.J. (eds). San Diego: Elsevier, pp. 784–794. [Google Scholar]
- Knoops, K. , Kikkert, M. , Worm, S.H.E. , Zevenhoven‐Dobbe, J.C. , van der Meer, Y. , Koster, A.J. , et al. (2008) SARS‐coronavirus replication is supported by a reticulovesicular network of modified endoplasmic reticulum. PLoS Biol 6: e226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoops, K. , Swett‐Tapia, C. , van den Worm, S.H.E. , te Velthuis, A.J.W. , Koster, A.J. , Mommaas, A.M. , et al. (2010) Integrity of the early secretory pathway promotes, but is not required for, severe acute respiratory syndrome coronavirus RNA synthesis and virus‐induced remodeling of endoplasmic reticulum membranes. J Virol 84: 833–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoops, K. , Barcena, M. , Limpens, R.W. , Koster, A.J. , Mommaas, A.M. , and Snijder, E.J. (2012) Ultrastructural characterization of arterivirus replication structures: reshaping the endoplasmic reticulum to accommodate viral RNA synthesis. J Virol 86: 2474–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwabara, M. , Wada, K. , Maeda, Y. , Miyazaki, A. , and Tsunemitsu, H. (2007) First isolation of cytopathogenic bovine torovirus in cell culture from a calf with diarrhea. Clin Vaccine Immunol 14: 998–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, J.Y. , Marshall, J.A. , and Bowden, D.S. (1994) Characterization of rubella virus replication complexes using antibodies to double‐stranded RNA. Virology 200: 307–312. [DOI] [PubMed] [Google Scholar]
- Limpens, R.W. , van der Schaar, H.M. , Kumar, D. , Koster, A.J. , Snijder, E.J. , van Kuppeveld, F.J.M. , and Bárcena, M. (2011) The transformation of enterovirus replication structures: a three‐dimensional study of single‐ and double‐membrane compartments. mBio 2: e00166–00111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie, J.M. , Jones, M.K. , and Young, P.R. (1996) Immunolocalization of the dengue virus nonstructural glycoprotein NS1 suggests a role in viral RNA replication. Virology 220: 232–240. [DOI] [PubMed] [Google Scholar]
- Maestre, A.M. , Garzón, A. , and Rodríguez, D. (2011) Equine torovirus (BEV) induces caspase‐mediated apoptosis in infected cells. PLoS One 6: e20972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier, H.J. , Hawes, P.C. , Cottam, E.M. , Mantell, J. , Verkade, P. , Monaghan, P. , et al. (2013) Infectious bronchitis virus generates spherules from zippered endoplasmic reticulum membranes. mBio 4: e00801–00813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, S. , and Krijnse‐Locker, J. (2008) Modification of intracellular membrane structures for virus replication. Nat Rev Micro 6: 363–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Netherton, C.L. , and Wileman, T. (2011) Virus factories, double membrane vesicles and viroplasm generated in animal cells. Curr Opin Virol 1: 381–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orenstein, J.M. , Banach, B. , and Baker, S.C. (2008) Morphogenesis of coronavirus HCoV‐NL63 in cell culture: a transmission electron microscopic study. Open Infect Dis J 2: 52–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson, G.H. , and Lippincott‐Schwartz, J. (2002) A photoactivatable GFP for selective photolabeling of proteins and cells. Science 297: 1873–1877. [DOI] [PubMed] [Google Scholar]
- Pedersen, K.W. , van der Meer, Y. , Roos, N. , and Snijder, E.J. (1999) Open reading frame 1a‐encoded subunits of the arterivirus replicase induce endoplasmic reticulum‐derived double‐membrane vesicles which carry the viral replication complex. J Virol 73: 2016–2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prentice, E. , Jerome, W.G. , Yoshimori, T. , Mizushima, N. , and Denison, M.R. (2004) Coronavirus replication complex formation utilizes components of cellular autophagy. J Biol Chem 279: 10136–10141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raturi, A. , and Simmen, T. (2013) Where the endoplasmic reticulum and the mitochondrion tie the knot: the mitochondria‐associated membrane (MAM). Biochimic et Biophys Acta (BBA) – Mol Cell Res 1833: 213–224. [DOI] [PubMed] [Google Scholar]
- Romero‐Brey, I. , Merz, A. , Chiramel, A. , Lee, J.Y. , Chlanda, P. , Haselman, U. , et al. (2012) Three‐dimensional architecture and biogenesis of membrane structures associated with hepatitis C virus replication. PLoS Pathog 8: e1003056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider, M. , Ackermann, K. , Stuart, M. , Wex, C. , Protzer, U. , Schatzl, H.M. , and Gilch, S. (2012) Severe acute respiratory syndrome coronavirus replication is severely impaired by MG132 due to proteasome‐independent inhibition of M‐calpain. J Virol 86: 10112–10122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi, S.T. , Schiller, J.J. , Kanjanahaluethai, A. , Baker, S.C. , Oh, J.W. , and Lai, M.M.C. (1999) Colocalization and membrane association of murine hepatitis virus gene 1 products and de novo‐synthesized viral RNA in infected cells. J Virol 73: 5957–5969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddell, S.G. , Ziebuhr, J. , and Snijder, E.J. (2010) Coronaviruses, toroviruses, and arteriviruses In Topley & Wilson's Microbiology and Microbial Infections. Mahy B.W.J., and ter Meulen V. (eds). London: Hodder Arnold. [Google Scholar]
- Sims, A.C. , Ostermann, J. , and Denison, M.R. (2000) Mouse hepatitis virus replicase proteins associate with two distinct populations of intracellular membranes. J Virol 74: 5647–5654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slot, J.W. , and Geuze, H.J. (2007) Cryosectioning and immunolabeling. Nat Protocols 2: 2480–2491. [DOI] [PubMed] [Google Scholar]
- Smits, S.L. , Snijder, E.J. , and de Groot, R.J. (2006) Characterization of a torovirus main proteinase. J Virol 80: 4157–4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snijder, E.J. , and Horzinek, M.C. (1993) Toroviruses: replication, evolution and comparison with other members of the coronavirus‐like superfamily. J Gen Virol 74: 2305–2316. [DOI] [PubMed] [Google Scholar]
- Snijder, E.J. , den Boon, J.A. , Bredenbeek, P.J. , Horzinek, M.C. , Rijnbrand, R. , and Spaan, W.J. (1990) The carboxyl‐terminal part of the putative Berne virus polymerase is expressed by ribosomal frameshifting and contains sequence motifs which indicate that toro‐ and coronaviruses are evolutionarily related. Nucleic Acids Res 18: 4535–4542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snijder, E.J. , den Boon, J.A. , Horzinek, M.C. , and Spaan, W.J. (1991) Comparison of the genome organization of toro‐ and coronaviruses: evidence for two nonhomologous RNA recombination events during Berne virus evolution. Virology 180: 448–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snijder, E.J. , Wassenaar, A.L.M. , van Dinten, L.C. , Spaan, W.J.M. , and Gorbalenya, A.E. (1996) The arterivirus nsp4 protease is the prototype of a novel group of chymotrypsin‐like enzymes, the 3C‐like serine proteases. J Biol Chem 271: 4864–4871. [DOI] [PubMed] [Google Scholar]
- Snijder, E.J. , van Tol, H. , Roos, N. , and Pedersen, K.W. (2001) Non‐structural proteins 2 and 3 interact to modify host cell membranes during the formation of the arterivirus replication complex. J Gen Virol 82: 985–994. [DOI] [PubMed] [Google Scholar]
- Snijder, E.J. , van der Meer, Y. , Zevenhoven‐Dobbe, J. , Onderwater, J.J. , van der Meulen, J. , Koerten, H.K. , and Mommaas, A.M. (2006) Ultrastructure and origin of membrane vesicles associated with the severe acute respiratory syndrome coronavirus replication complex. J Virol 80: 5927–5940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Targett‐Adams, P. , Boulant, S. , and McLauchlan, J. (2008) Visualization of double‐stranded RNA in cells supporting hepatitis C virus RNA replication. J Virol 82: 2182–2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulasli, M. , Verheije, M.H. , de Haan, C.A. , and Reggiori, F. (2010) Qualitative and quantitative ultrastructural analysis of the membrane rearrangements induced by coronavirus. Cell Microbiol 12: 844–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulferts, R. , Mettenleiter, T.C. , and Ziebuhr, J. (2011) Characterization of Bafinivirus main protease autoprocessing activities. J Virol 85: 1348–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss, M. , and Horzinek, M.C. (1986) Morphogenesis of Berne virus (proposed family Toroviridae). J Gen Virol 67: 1305–1314. [DOI] [PubMed] [Google Scholar]
- Weiss, M. , Steck, F. , and Horzinek, M.C. (1983) Purification and partial characterization of a new enveloped RNA virus (Berne virus). J Gen Virol 64: 1849–1858. [DOI] [PubMed] [Google Scholar]
- Westaway, E.G. , Mackenzie, J.M. , Kenney, M.T. , Jones, M.K. , and Khromykh, A.A. (1997) Ultrastructure of Kunjin virus‐infected cells: colocalization of NS1 and NS3 with double‐stranded RNA, and of NS2B with NS3, in virus‐induced membrane structures. J Virol 71: 6650–6661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westaway, E.G. , Khromykh, A.A. , and Mackenzie, J.M. (1999) Nascent Flavivirus RNA colocalized in situ with double‐stranded RNA in stable replication complexes. Virology 258: 108–117. [DOI] [PubMed] [Google Scholar]
- de Wilde, A.H. , Raj, V.S. , Oudshoorn, D. , Bestebroer, T.M. , van Nieuwkoop, S. , Limpens, R.W. , et al. (2013) MERS‐coronavirus replication induces severe in vitro cytopathology and is strongly inhibited by cyclosporin A or interferon‐alpha treatment. J Gen Virol 94: 1749–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, Z. , Thackray, L.B. , Miller, B.C. , Lynn, T.M. , Becker, M.M. , Ward, E. , et al. (2007) Coronavirus replication does not require the autophagy gene ATG5. Autophagy 3: 581–585. [DOI] [PubMed] [Google Scholar]
- Ziebuhr, J. , Snijder, E.J. , and Gorbalenya, A.E. (2000) Virus‐encoded proteinases and proteolytic processing in the Nidovirales. J Gen Virol 81: 853–879. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Close view of BEV viral particles observed in sections of BEV‐infected E.Derm cells embedded in epoxy resin TAAB‐812. Electron micrographs of E.Derm cells infected with BEV for 16 h and embedded in the epoxy resin TAAB‐812 after conventional chemical fixation, where the presence of rod‐shaped viral particles can be clearly observed. Individual particles A) or small groups of particles inside rough ER derived‐vesicles B), as well as large groups of particles packed in large vesicles C) can be observed at this pi time. Viral particles were sectioned in different orientations: longitudinal section (black arrow) or cross‐section (black arrowhead). M: Mitochondria; ER: Endoplasmic reticulum. Scale bars, 100 nm.
Fig. S2. Ultrastructural analysis of MRC5 cells infected with BEV embedded in epoxy resin TAAB‐812. Electron micrographs of sections of MRC5 cells infected with BEV for 24 h that were embedded in the epoxy resin TAAB‐812, after conventional chemical fixation, showing the presence of characteristic spherical DMVs. A) Cluster of DMVs (white asterisk) surrounded by mitochondria and ER. A particle inside a vesicle can be distinguished at the periphery of the cluster (white arrow). B‐C) DMVs connected between them (white arrowhead) and with the ER (black arrowhead) or with the MAM (black arrow). M: Mitochondria; ER: Endoplasmic reticulum. Scale bars, 200 nm.
Fig. S3. Colocalization analysis of the RdRp and Hel proteins with the dsRNA. E.Derm cells infected with BEV were fixed with 4% PFA in PBS at 8 h pi and used for immunolabeling with the mouse mAb anti‐dsRNA (green) in combination with A) rabbit anti‐RdRp or B) anti‐Hel antibodies (red). Nuclei were stained with DAPI (blue). The boxed areas are representative fields shown at higher magnification. Scale bars, 10 μm.
Fig. S4. Distribution of newly synthesized viral RNA (BrU‐RNA) along the infection with BEV. E.Derm cells infected with BEV were incubated with 5 mM BrU in the presence of 5 mg ml−1 of actinomycine D for 1 h before fixation. Cells were fixed at different pi times as indicated in each panel, and processed for immunofluorescence staining with a mouse anti‐BrdU mAb (green). The nuclei were stained with DAPI (blue). The boxed areas are representative fields that are shown at higher magnification. Scale bar, 10 μm.
Fig. S5. Partial colocalization of the newly synthesized RNA (BrU‐RNA) and the M structural protein. BEV‐Infected cells treated as described in the legend to Fig. S3 were fixed at 8 h pi. A) Triple colocalizaton assay using the rabbit anti‐M (green), mouse anti‐BrdU mAb (red) and rat anti‐Mpro (blue) antibodies. The square encloses a representative area that is shown at higher magnification in panels B‐G. B) Signal obtained with the anti‐M antibody; C) signal corresponding to the newly synthesized RNA (BrU‐RNA); D) signal corresponding to the Mpro; E) overlapping of the signals corresponding to the M protein and the BrU‐RNA; F) overlapping of the signals corresponding to the Mpro protein and the BrU‐RNA; G) overlapping of the signals corresponding to the M and the Mpro proteins. The arrow indicates the area of colocalization between the M protein and the BrU‐RNA. Scale bar, 10 μm.
Fig. S6. Immunolabeling assay on mock‐infected cells. A) Cryosections of mock‐infected E.Derm cells were fixed and immunogold‐labeled with a mAb anti‐dsRNA (left panel), or with antibodies against the RdRp (middle panel) and Mpro (right panel), and analyzed by electron microscopy. Scale bars, 100 nm.
Movie S1. 3D reconstruction presented in the Fig.7, panels B‐C.
Supporting info item