

Calmodulin (CaM) is a calcium-binding protein that can directly inhibit cardiac ryanodine receptor calcium release channels (ryanodine receptor 2 [RyR2]) (1). CaM mutations can cause an autosomal-dominant form of catecholaminergic polymorphic ventricular tachycardia (CPVT), a syndrome characterized by exercise- and/or emotional stress-induced ventricular arrhythmia and sudden death (2). We previously reported that CPVT-linked mutant CaMs (N54I and N98S) had either no or a slight stimulating effect on the activity of sheep RyR2, whereas wild-type (wt)-CaM inhibited sheep RyR2 by approximately 40% and inhibited sarcoplasmic reticulum (SR) calcium release in permeabilized mouse cardiomyocytes (3). Although the mutant CaMs failed to inhibit mouse and sheep RyR2, they bound even more tightly to RyR2 than wild-type (wt)-CaM (3), providing an explanation for why CPVT mutant CaM could have a dominant effect on RyR2 in the presence of excess wt-CaM. To date, data are lacking on CaM regulation of human RyR2. To address this question, we investigated the action of wt and CPVT mutant CaMs on the single channel activity of RyR2 isolated from hearts of healthy human donors.
Human left ventricular tissues were obtained from the Human Heart Tissue Repository at the University of Sydney with approval from the human research ethics committees of the University of Newcastle (approval number H-2009–0369) and the University of Sydney (#09–2009-12146). SR membranes containing RyR2 were isolated from these tissues and incorporated into artificial lipid bilayers (4). RyR2 channel gating was measured by single-channel recording (4) in the presence and absence of physiological CaM concentrations (o.i nmol/l). CaM was added and removed from the RyR2 complex by using continuous local perfusion via a tube placed close to the bilayer, enabling solution changes within 1 s. Because CaM readily dissociates from RyR2 in 25 s (1,3), channels that are incorporated into artificial lipid bilayers are devoid of CaM unless exogenous CaM is applied. The effect of CaM was measured by repeatedly applying CaM for 1 min interleaved by l-min periods of washout. Surprisingly, neither wt-CaM nor CPVT mutant CaMs (N54I and N98S) had any effect on the activity of human RyR2 (Figure 1A). The lack of inhibition of human RyR2 by wt-CaM was not anticipated because CaM is inhibitory in all animal models investigated so far (sheep, dog, and mouse [1,3]). This common lack of efficacy of wt and mutant CaMs on human RyR2 activity necessitates a re-examination of our hypothesis for the role of CaM regulation of RyR2 in CPVT.
FIGURE 1. The Effect of wt-CaM and CPVT CaM Mutants on Activity of Human RyR2.
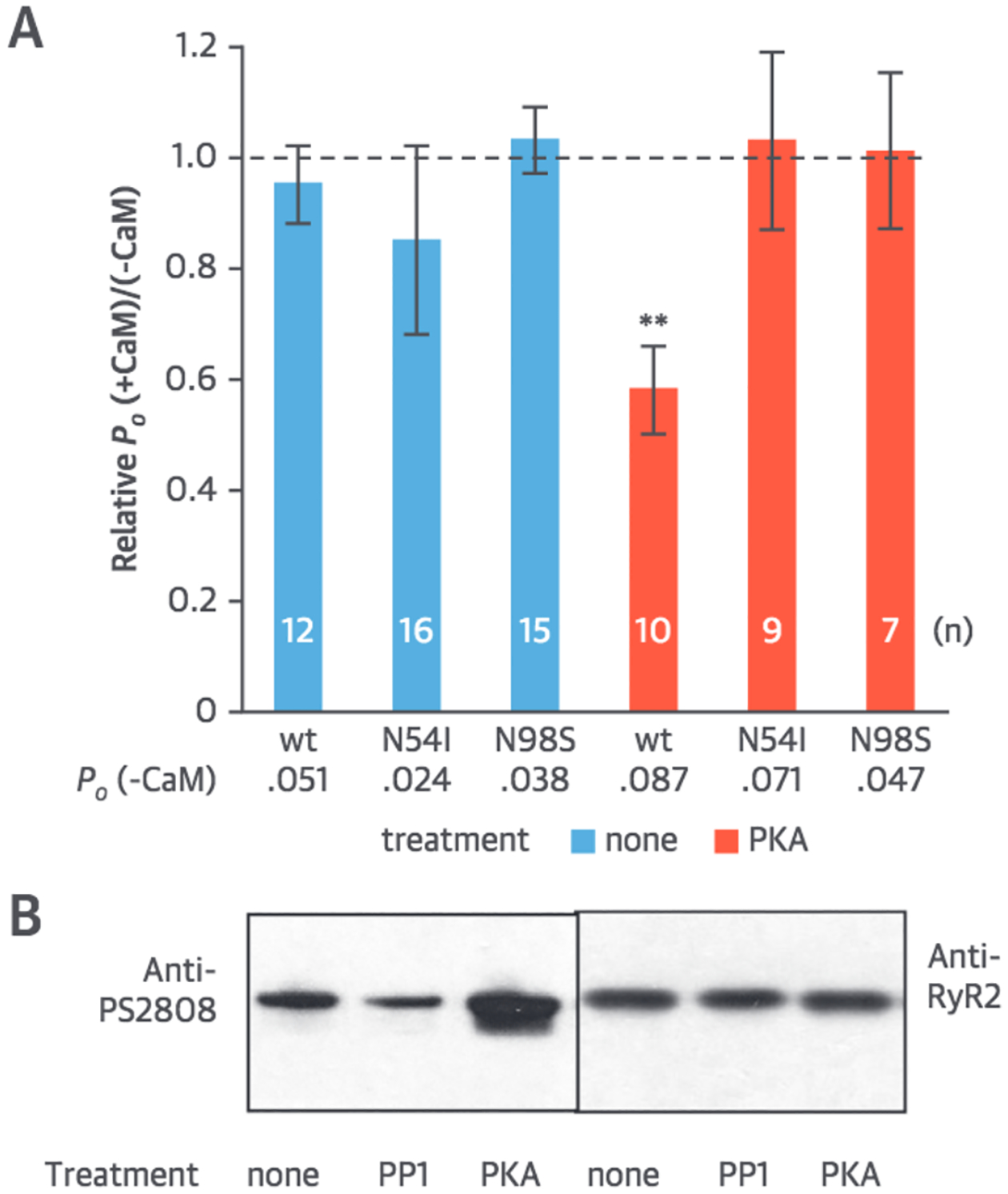
RyR2 is in the presence of 100 nmol/l cytoplasmic Ca2+ and 2 mmol/l ATP and 0.1 mmoL/l Luminal [Ca2+] with membrane potential −4 0 mV. (A) Relative effect of wt-CaM on RyR2 open probability (P0) compared with that of N54I and N9BS (mean ± SEM with replicate numbers in each bar). P0, (−CaM) denotes mean absolute P0 before wt or mutant CaM application. **Significant difference to 1 (p < 0.01). (B) Western blots of RyR2 subjected to incubations in PP1 (15 min) or PKA (5 min) at 30°C probed with antibodies for PS2808 (left), stripped and reprobed with antibodies for RyR2 (right). Ca2+ = calcium ion; CaM = calmodulin; CPVT = catecholaminergic polymorphic ventricular tachycardia; PKA = protein kinase A; PP1 = protein phosphatase 1; RyR2 = ryanodine receptor 2; wt = wild-type.
Because CPVT is an arrhythmia brought on by β-adrenergic-induced RyR2 phosphorylation, we simulated adrenergic stress by incubating SR vesicles containing RyR2 with protein kinase A (PKA) before incorporation into bilayers. Western blots using phospho-specific antibodies (Figure 1B) confirmed an increased phosphorylation at RyR2 residue S2808, an accepted PKA consensus site. We found that wt and CPVT-mutant CaMs had clearly divergent effects on RyR2 when S2808 phosphorylation levels were increased by PKA (Figure 1A). wt-CaM caused approximately 40% inhibition of RyR2, whereas N54I and W98S had no effect. Taken together, our results support the hypothesis that wt-CaM inhibition blunts RyR2 activity after β-adrenergic stimulation, whereas CPVT mutant CaMs fail to do so. Hence, mutant CaMs facilitate pathological overactivation of RyR2 during periods of stress and exercise. These results indicate that, in humans, CaM functions to protect against aberrant SR Ca2+ release during periods of adrenergic stress where RyR2 is highly phosphorylated. The failure to inhibit phosphorylated RyR2 likely explains the pathogenic role of mutant CaM in CPVT.
Acknowledgments
Please note: Dr. Chazin was supported in part by a grant from the National Institutes of Health (R35 GMU8O89). Dr. Laver was supported b y a Project Grant (APP1082204) from the National Health and Medical Research Council, Canberra, Australian Capital Territory, Australia, and by an infrastructure grant from the Hunter Medical Research Institute, Newcastle, New South wales, Australia. Dr. Knollmann was supported in part by grants from the National Institutes of Health (R01H L128044, R01HL124935, R01H L108173) and by a Project Grant (APP1082204) from the National Health and Medical Research Council, Canberra. All other authors have reported that they have no relationships relevant to the contents of this paper to disclose. The authors wish to thank Mr. Paul Johnson for his assistance with the experiments.
REFERENCES
- 1.Xu L, Meissner G. Mechanism of calmodulin inhibition of cardiac sarcoplasmic reticulum Ca2+ release channel (ryanodine receptor). Biophys J 2004;86:797–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gomez-Hurtado N, Boczek NJ, Kryshtal DO, et al. Novel CPVT-associated calmodulin mutation in CALM3 (CALM3-A103V) activates arrhythmogenic Ca waves and sparks. Circ Arrhythm Electrophysiol 2016;9;e004161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hwang HS, Nitu FR, Yang Y, et al. Divergent regulation of ryanodine receptor 2 calcium release channels by arrhythmogenic human calmodulin missense mutants. Circ Res 2014;114:1114–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Walweel K, Li J, Molenaar P, et al. Differences in the regulation of RyR2 from human, sheep, and rat by Ca2+ and Mg2+ in the cytoplasm and in the lumen of the sarcoplasmic reticulum. J Gen Physiol 2014;144:263–71. [DOI] [PMC free article] [PubMed] [Google Scholar]