

Abstract
Higher order structure of protein therapeutics is an important quality attribute, which dictates both potency and safety. While modern experimental biophysics offers an impressive arsenal of state-of-the-art tools that can be used for the characterization of higher order structure, many of them are poorly suited for the characterization of biopharmaceutical products. As a result, these analyses were traditionally carried out using classical techniques that provide relatively low information content. Over the past decade, mass spectrometry made a dramatic debut in this field, enabling the characterization of higher order structure of biopharmaceuticals as complex as monoclonal antibodies at a level of detail that was previously unattainable. At present, mass spectrometry is an integral part of the analytical toolbox across the industry, which is critical not only for quality control efforts, but also for discovery and development.
Introduction:
Higher order structure as a critical quality attribute of protein therapeutics
Higher order structure of a protein therapeutic is an important determinant of its function, and a loss of native conformation even within a small subset of molecules has a negative impact on its potency. Perhaps even more important is the safety aspect associated with the integrity of higher order structure, as even a relatively small-scale loss of structure may expose neo-epitopes that can trigger an immune response to the protein drug. The integrity of higher order structure may be compromised at any stage during the product’s life cycle from production to formulation to storage to administration, necessitating the need for robust, sensitive and highly reliable methods to monitor the conformational integrity of biopharmaceutical products. While modern experimental biophysics offers an impressive arsenal of state-of-the-art tools that can be used for elucidating intimate details of protein higher order structure, many of them are poorly suited for the characterization of biopharmaceutical products. For example, high-resolution NMR still suffers from rather unforgiving molecular weight limitations which place detailed structural analysis of most protein therapeutics with natural isotopic abundance outside of its reach, limiting the scope of NMR in biopharmaceutical analysis to fingerprinting [1, 2]. The length of the typical high-resolution NMR analysis, as well as high material quantity requirements are two other important factor limiting the application of this technique in the field of biopharmaceutical analysis. As a result, until very recently the field of biophysical characterization of protein therapeutics was dominated by classical techniques, such as optical spectroscopy, light scattering, analytical ultracentrifugation, calorimetry and size exclusion chromatography [3]. While being robust and free of molecular weight limitations, these techniques rarely provide detailed structural information.
Mass spectrometry (MS) has been an indispensable tool for covalent structure analysis of recombinant proteins from the early days of biotechnology, enabling the analysis of both amino acid sequence [4] and post-translational modifications (PTMs), including tasks as challenging as glycan analysis [5] and disulfide mapping [6]. Above and beyond being an excellent tool for characterization of covalent structure of recombinant proteins and their derivatives at a variety of levels, MS has the capability to provide information on higher order structure [7, 8]. Therefore, it is not surprising that both academic laboratories and the key stake holders in industry were keen on evaluating the capabilities of MS vis-à-vis characterization of the higher order structure of biopharmaceutical products as early as mid-1990s [9], with the first reports of successful use of MS for these tasks published in the following decade [10, 11]. In the years since the publication of these initial reports, MS was demonstrated to be a robust and reliable tool capable of characterizing the higher order structure of protein therapeutics and related products at an unprecedented level of detail. It is an integral part of the analytical toolbox across the industry, which not only proved critical for quality control efforts, but is also indispensable at the discovery and development stages. In fact, the majority of regulatory filings in the field of biopharmaceuticals are now supported by MS-based characterization of the higher order structure.
Use of native MS in higher order structure characterization
The advent of electrospray ionization (ESI) MS [12, 13] provided an opportunity to obtain mass spectra of intact macromolecules directly from solution with minimal distortion of their structure. While the initial enthusiasm was understandably focused on the ability to measure masses of intact polypeptides, nucleic acids and polysaccharides, it was soon recognized that the gentle nature of ESI MS allows in many cases the higher order structure of these biopolymers to be preserved as well [14, 15], giving rise to a technique which is now known as “native MS.” This section examines in detail various aspects of the higher order structure of protein therapeutics that can be probed using this approach.
Protein ion charge state distributions: conformational integrity of monomeric proteins.
ESI as an ionization method for macromolecular analytes is unique in that it generates multiply charged ions, and the extent of multiple charging is determined by the physical dimensions of the biopolymer in solution [16] (with its solvent-accessible surface area having particularly strong correlation with the average ionic charge in ESI MS [17]). Since even partial unfolding in solution results in a significant increase of the hydrodynamic radius of the protein, a loss of conformational integrity is reflected in protein charge state distributions [18]. An example is shown in Figure 1, where a mass spectrum of a monoclonal antibody (mAb) acquired under near-native conditions in solution (physiological pH and ionic strength, no organic co-solvent) exhibits a narrow distribution of charge states (+26 through +30) in the high-m/z range, while the very same protein gives rise to a dramatically different ionic signal (charge states > +50) when placed in a strongly denaturing solution (low pH/low ionic strength and 50% of organic co-solvent by volume). It is possible in many cases to obtain more nuanced information on protein conformations from the charge state distributions, as suggested by the appearance of the mAb mass spectrum acquired under mildly denaturing conditions (pH 3.0, physiological ionic strength, see the black trace in Figure 1). A subset of ions in this distribution (m/z > 5,000) clearly resembles the mass spectrum acquired under native conditions, and likely represents the sub-population of mAb molecules maintaining the near-native fold. The extent of the multiple charging for the rest of ionic population is notably higher, indicating partial loss of the native structure. The uneven shape of this part of the charge state distributions (the presence of two local maxima at z = +47 and +37) suggests that the population of mAb molecules giving rise to the ionic signal in this m/z region is heterogeneous, and contains sub-populations with different conformations. All these states retain a significant amount of residual structure, as the extent of multiple charging displayed by these ions is still lower compared to that of mAb ions generated under strongly denaturing conditions in solution.
Figure 1.
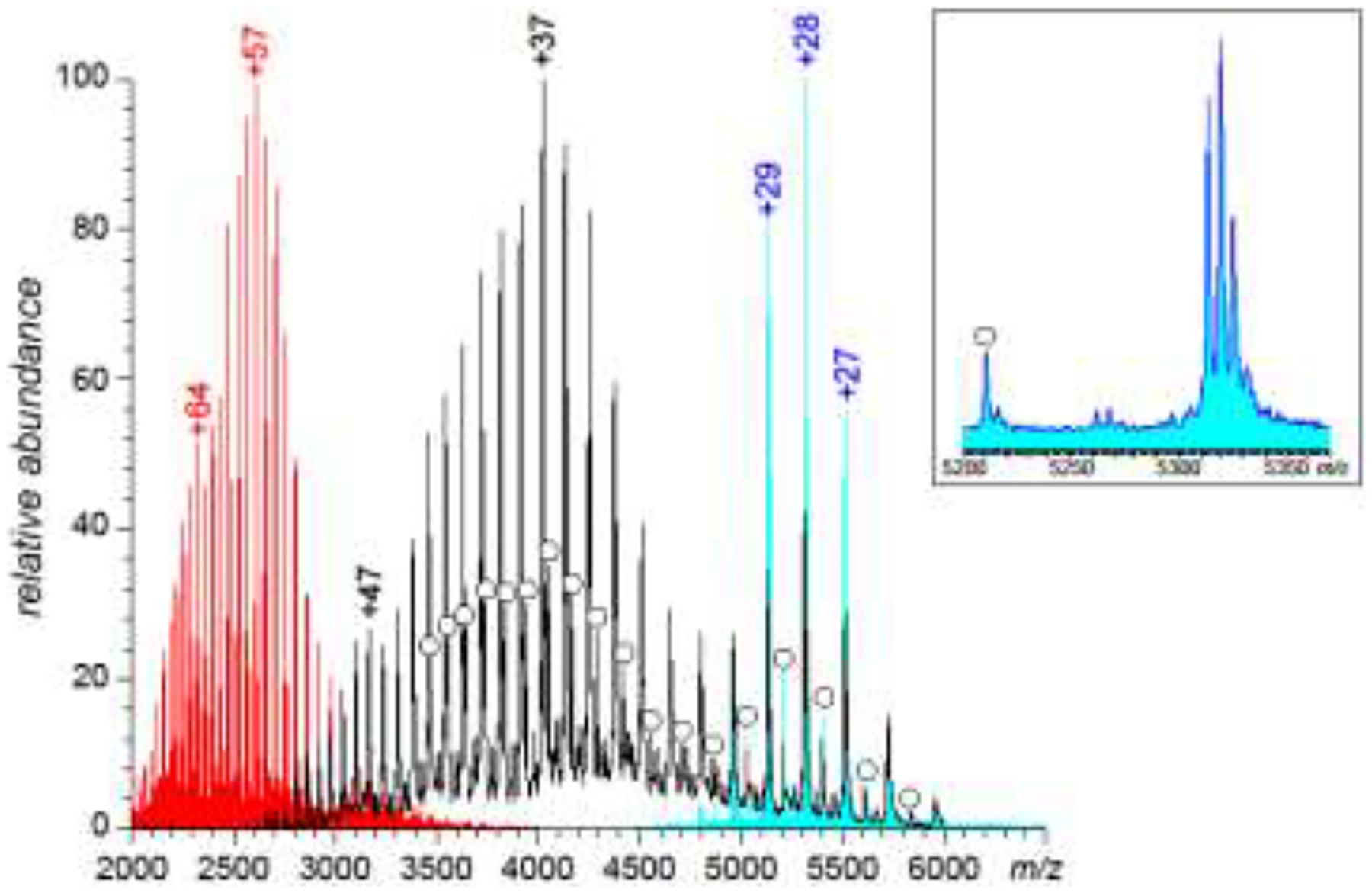
Mass spectra of a monoclonal antibody acquired under strongly denaturing (red trace), mildly denaturing (black trace) and near-native conditions (cyan trace). The inset (a zoomed view of the latter mass spectrum) shows contributions to the ionic signal form all different glycoforms of the protein, including the carbohydrate-free form (labeled with a circle).
An important feature of the charge state distribution analysis in ESI MS is that it can be readily carried out even if multiple species are present in solution, as long as a distinction can be made among them based on mass difference. For example, the mass spectra shown in Figure 1 reveal the presence of multiple mAb glycoforms, including a glycan-free species (labeled with circles in Figure 1). The charge state distribution of the a-glycosylated (glycan-free) mAb species closely mirrors that of the glycosylated molecules both under native and mildly denaturing conditions, providing a clear indication that in this particular case the missing glycan does not result in a loss of the higher order structure. Alteration of the appearance of the charge state distributions in the mass spectra of protein therapeutics triggered by stress-related PTMs can be used to determine whether a particular modification of the covalent structure compromises the protein conformation [10, 19].
Quaternary assemblies of multi-unit proteins and interactions of protein drugs with therapeutic targets and physiological partners.
A unique feature of native MS is its ability to preserve a large variety of non-covalent assemblies of proteins and other biopolymers. Above and beyond the characterization of multi-unit protein therapeutics (for which the integrity of the non-covalent quaternary assemblies can be evaluated under a variety of conditions [20]), native MS provides clear advantages in the studies of protein drug interactions with its therapeutic targets and physiological partners [21]. Continuous improvements in MS hardware have allowed meaningful measurements to be made for therapeutically relevant assemblies exceeding 1 MDa, such as mAb/antigen complexes associated with complement component C1 [22, 23]. However, it must be remembered that non-covalent assemblies remain stable in the gas phase only if they are maintained primarily by local electrostatic interactions (salt bridges) and hydrogen bonding. The protein/protein interactions driven primarily by hydrophobic forces become highly unstable in the absence of solvent, which in most cases causes dissociation of these assemblies in the gas phase. Fortunately, in the vast majority of cases the gas phase dissociation proceeds via a very distinct mechanism (asymmetric charge partitioning [24, 25]) which generates an unambiguous signature in the mass spectrum and allows data misinterpretation to be avoided [26], although the existence of multiple dissociation pathways may complicate the data analysis.
An example is shown in Figure 2, where mAb incubation with its antigen (the former presented in a molar excess) followed by the acquisition of a mass spectrum with native ESI gives rise to a very weak signal of the antigen/antibody (Ag·IgG) complex on the background of abundant Ag and IgG ions. A fraction of the free Ag ions exhibits a significant extent of multiple charging (i.e., appear at an m/z below 2,000 in the mass spectrum shown in Figure 2), which would be typically indicative of gas phase dissociation proceeding via asymmetric charge state partitioning [26]. At the same time, a significant population of the antigen ions show a modest extent of multiple charging; this makes it difficult to conclude whether the low abundance of Ag·IgG ions (and the absence of Ag2IgG) in the native ESI mass spectrum is due to a low binding affinity in solution or due to the complex dissociation in the gas phase. In such situations, a combination of size exclusion chromatography (SEC) with on-line detection by native MS [27, 28] is likely to provide a reliable answer. In the case of the Ag/IgG interaction, an SEC chromatogram of the mixture (with UV detection) reveals the presence of two species with retention times shorter than that of IgG, consistent with the notion of Ag/IgG binding taking place in solution. On-line MS detection confirms that Ag is a constituent of both of these species, which are assigned as Ag·IgG and Ag2IgG complexes (the presence of free Ag ions at early elution time can be only explained by the dissociation of these complexes in the gas phase). Free Ag ions are also detected during the elution of a low-abundance, high molecular weight (early-eluting) species, which may be indicative of the occurrence of Ag aggregation, although in this case a definitive conclusion can be made only by obtaining information on the molecular weight of these species using an orthogonal method of analysis, such as light scattering.
Figure 2.
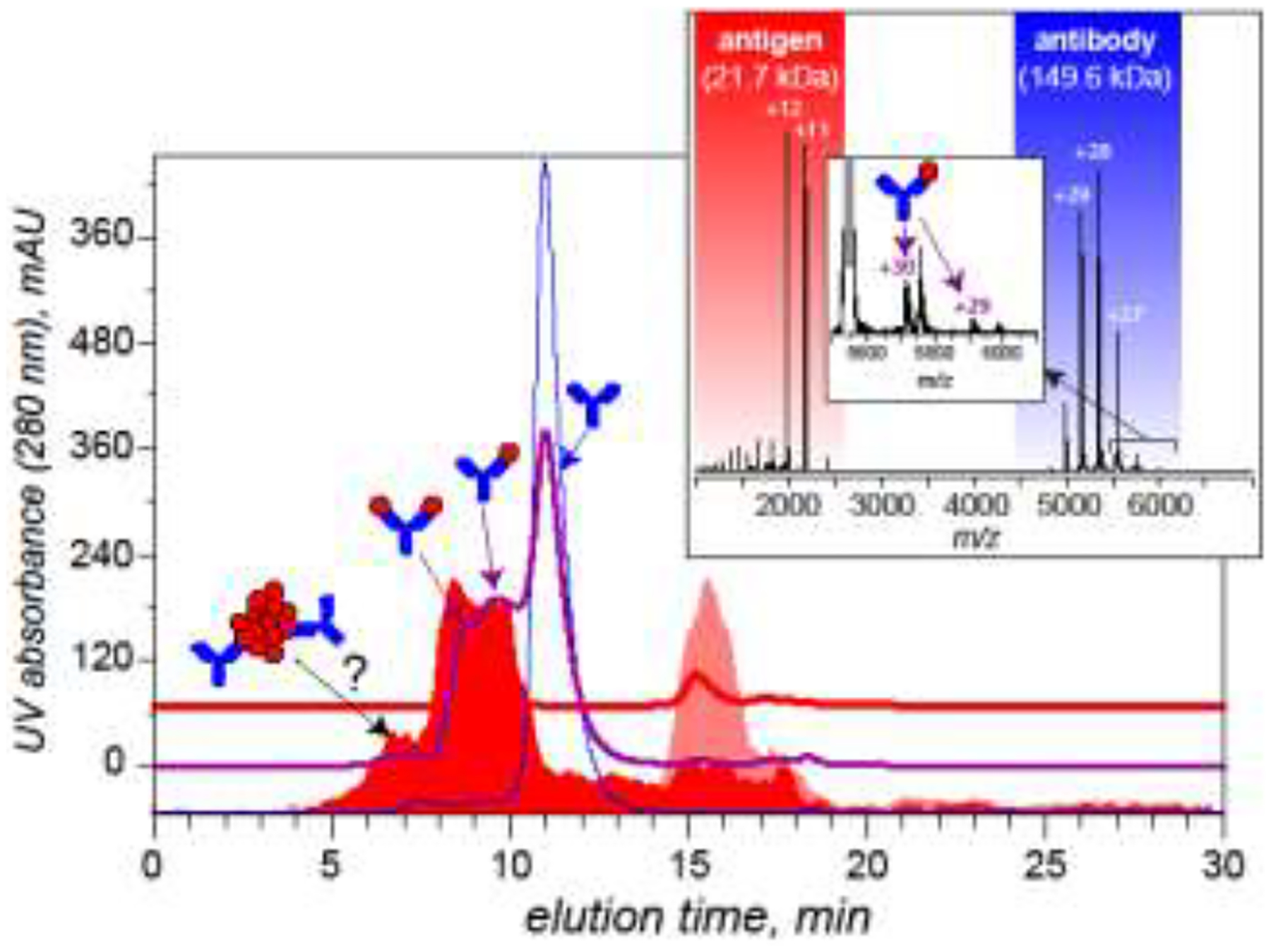
Interaction of a monoclonal antibody with its antigen characterized by SEC/MS and direct infusion (off-line) native ESI MS (inset). The three traces on the main panel show SEC elution profiles of Ag (red), mAb (blue) and their mixture at 1:1 molar ratio (purple). The color-filled curves show extracted ion chromatograms of Ag ions in SEC/MS analysis of Ag alone (pink) and Ag/mAb mixture (red). While SEC/MS reveals formation of both 2:1 and 1:1 Ag/IgG complexes when the two proteins are mixed at a 1:1 molar ratio, only low-abundance 1:1 species are detected by native ESI MS in the off-line mode due to facile dissociation of the complexes in the gas phase. Adapted and used with permission [28].
Characterization of highly heterogeneous systems with native MS.
One problem that is frequently encountered in native MS of protein therapeutics is the significant extent of structural heterogeneity (e.g., due to extensive glycosylation and/or presence of other PTMs), which gives rise to broad, poorly resolved peaks in the mass spectra. In extreme situations the extent of the peak broadening could be so high that even individual charge states cannot be discerned from the ionic signal, rendering the native MS data meaningless. Protein analysis in such cases can be greatly assisted by a recently introduced technique of limited charge reduction [29]. The use of this technique is illustrated in Figure 3, where a mass spectrum of cross-linked oligomers of an 80 kDa protein (black trace) shows a near-continuous distribution of ionic signal in the high-m/z region, which prevents confident mass assignment based on the native MS data alone. Mass selection of narrow ionic populations within this region followed by their charge reduction (induced by allowing these polycations react with either electrons or small anions in the gas phase) generates resolved charge state ladders (blue and red) that can be used to determine the ionic masses with high confidence and identify all protein species in the sample.
Figure 3.
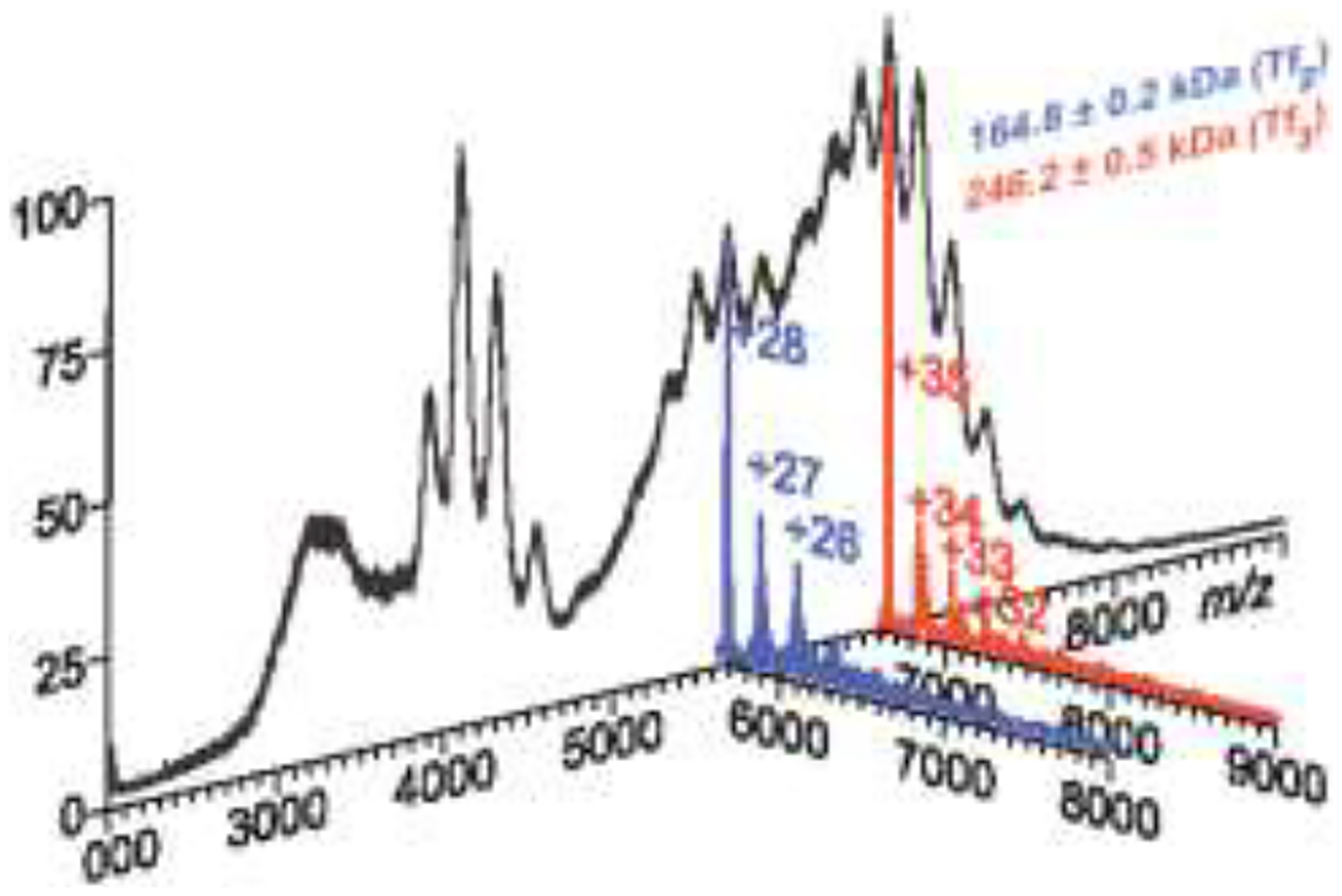
Use of limited charge reduction for identification of cross-linked transferrin oligomers in native MS. A high degree of structural heterogeneity causes ion peaks at different charge states to overlap (black trace). Mass selection of narrow ionic populations followed by their charge reduction via a gas-phase electron transfer from an anion to the protein polycation generates resolved charge ladders (blue and red) that can be used to identify all protein species in the sample (transferrin dimers and trimers, respectively).
While the example shown in Figure 3 illustrates the use of limited charge reduction to determine masses of covalently linked protein oligomers, it can be readily applied to non-covalent assemblies of structurally heterogeneous biopolymers as well. For example, a combination of native ESI MS with limited charge reduction in the gas phase can be used to study non-covalent assemblies ranging from large (> 0.5 MDa) ordered quaternary assemblies [20] to small soluble protein aggregates [30]. Limited charge reduction can be used in the on-line format in combination with either SEC or ion-exchange chromatography (IXC). This provides additional benefits by allowing simultaneous detection and characterization of various PTMs (e.g., glycosylation, PEGylation and deamidation) to be achieved at the whole protein level, as was recently demonstrated for PEGylated interferon β1a [31].
Evaluation of thermal stability of protein therapeutics with temperature-controlled MS.
A key environmental parameter that affects protein conformation is temperature, and heat stress is frequently used to assess the stability of protein therapeutics [32]. Aggregation propensity of mAbs has a strong correlation with their thermal stability [33], and differential scanning calorimetry (DSC) is frequently used to provide quantitative data on the latter [3]. Unfortunately, DSC does not provide much information on the mechanistic aspects of heat-induced conformational transitions, which could be useful not only for understanding the aggregation pathways, but also for designing strategies to delay the onset of such processes. While the potential of ESI MS to probe behavior of biopolymers as a function of solution temperature had been recognized soon after the introduction of this ionization technique [34], various technical issues prevented wide adaptation of temperature-controlled ESI MS measurements for at least a decade. One of the problems that is commonly encountered in such temperature-resolved studies is fast cooling of the protein solution just prior to spraying (when it flows through an unheated small-diameter capillary/needle). The fast solution cooling/temperature drop in this region (due to a large surface-to-volume ratio) frequently results in protein refolding, effectively erasing the memory of all/most reversible unfolding events. While several approaches can be used to avoid this problem [35, 36], a great deal of care must be taken to ensure that the measurements are not affected by the heat loss in the ESI interface (e.g., by obtaining melting curves of well-studied biopolymers whose reversible unfolding or dissociation had been previously documented using orthogonal techniques [35]).
An example of using temperature-controlled ESI MS to study the behavior of a protein therapeutic under the heat-stress condition is presented in Figure 4. In this case glucocerebrosidase (GCase, a protein used in enzyme replacement therapy treatment of Gaucher disease [37]) displays a narrow charge state distribution in the high m/z region when the protein solution is kept at room temperature, and little changes are evident in the mass spectra as the temperature is increased up to 46 °C. However, ramping up the solution temperature just one degree over the DSC-determined melting point for this protein gives rise to a dramatic change in the appearance of the ionic signal (see the panel labeled “50 °C” in Figure 4). The charge state distribution of the GCase ions has a clearly bimodal character (indicative of partial unfolding in solution), and the prominent ionic signal at m/z above 4,500 shows the presence of protein dimers and trimers. Further increase of the solution temperature (from 50 °C to 52 °C) results in a dramatic increase in the abundance of ionic species representing protein aggregates (dimers, trimers and tetramers), and the size of the protein aggregates continues to grow as the solution temperature is continuously increased (see the panels labeled “60 °C” and “70 °C” in Figure 4). While the value of the melting temperature of GCase reported by DSC is in agreement with the behavior revealed by the temperature-controlled ESI MS, the latter offers much higher informational content, allowing both unfolding of the protein monomers and ensuing formation/evolution of the aggregates to be directly visualized as a function of the solution temperature. Recent reports show that temperature-controlled ESI MS can be used to study multi-stage transitions, such as sequential domain unfolding in mAbs [38].
Figure 4.
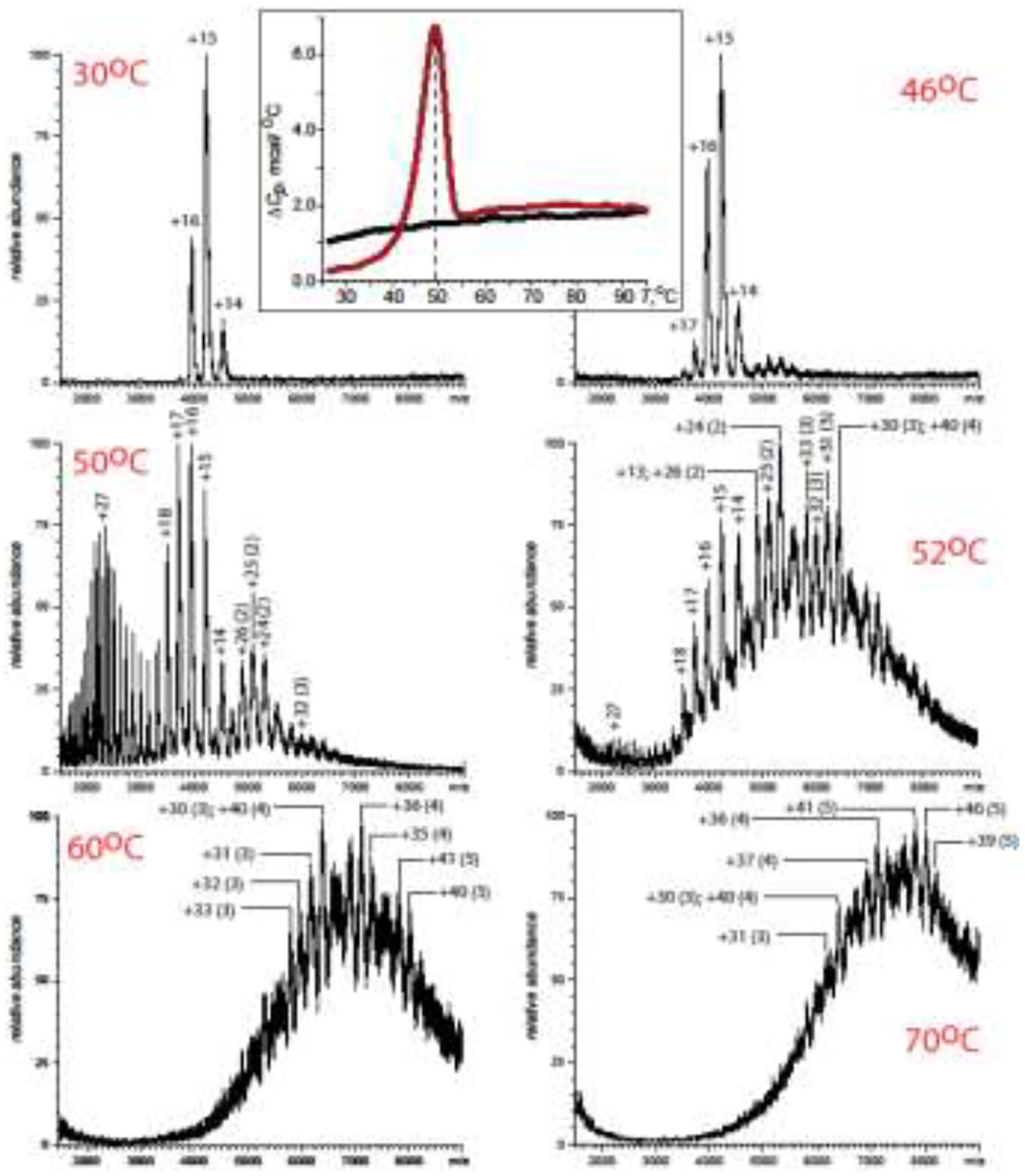
ESI mass spectra of GCase (1 μM in 20 mM ammonium acetate, pH 4.7) recorded at various solution temperatures (as indicated on each panel). The numbers without parentheses indicate charge states of the GCase monomers; the numbers in parentheses indicate the size of the protein associations (the number of monomeric units in a single oligomer). Inset at the top: the DSC profile of GCase (1 μM in 20 mM ammonium acetate, pH 4.7) showing a transition at 49 °C (brown trace); the black trace represents a rerun of the DSC experiment for the GCase sample that already went through a single cycle of DSC measurements. Adapted and used with permission [35].
The role of ion mobility measurements in characterization of protein therapeutics.
Ion mobility (IM) spectroscopy is a sister technique to MS, and can be used on a stand-alone basis. In IM ions are separated and distinguished from one another based on the differences in collisional cross-sections. While this information may provide an important dimension in characterization of biomolecules that is orthogonal to MS, mobility measurements alone are not sufficient for identification (let alone meaningful characterization) of biopolymer ions. Consequently, in the field of biomolecular analysis IM is almost always used in combination with (and in fact is considered to be either an extension of or a front and for) MS analysis [39]. Above and beyond the ability to separate isobaric ions based on differences in their shapes in the gas phase, IM measurements allow collisional cross-sections (CCSs) to be determined. The CCS values of biopolymer ions reflect their gas-phase conformations, naturally leading to suggestions that the higher order structure analysis of proteins in general and protein therapeutics in particular should include IM in its arsenal [40]. However, application of IM-based methods to characterize protein conformations raises a range of questions (summarized in a recent review [41]), which undermine the value of the information deduced from such studies. The most serious issue is related to the phenomenon known as a conformational collapse or compaction in the gas phase. Non-globular proteins (such as antibodies and other proteins containing flexible hinge regions) are especially prone to the gas phase compaction. This phenomenon leads to a significant underestimation of the physical size of solution-phase conformations when relying solely on the CCS values derived from IM measurements [41]. Another potential caveat in using IM to assess the integrity of native (solution-phase) higher order structures relates to the fact that these measurements are not sensitive to the secondary structure, and compactness of protein ions in the gas phase does not guarantee their assuming correct (native) conformations [41].
In contrast to this mixed record as a conformational analysis tool, IM enjoyed great success as a means of ion separation in the analysis of complex and heterogeneous systems by enabling separation that is orthogonal to both MS measurements and conventional LC [41]. Availability of this unique separation dimension also proved very useful in facilitating other methods of analysis of highly heterogeneous macromolecular medicines, such as limited charge reduction (vide supra), allowing the scope of the latter technique to be dramatically expanded to include macromolecules as heterogeneous as heparin [42, 43]. IM-MS has also been shown to provide a significant benefit vis-à-vis monitoring transitions occurring within monoclonal antibodies upon their thermal denaturation in solution [44].
Hydrogen/deuterium exchange to characterize higher order structure and dynamics of therapeutic proteins
While native ESI MS is a powerful technique that can be used very effectively as a means of detecting an offset of protein unfolding/aggregation, as well as monitoring the interactions of protein drugs with their physiological partners and therapeutic targets, it usually provides little structural information (e.g., what segments of the protein are affected by unfolding processes or participate in ligand binding). Answers to these questions can be provided in many cases by hydrogen/deuterium exchange (HDX) with MS detection. HDX as a biophysical technique has a long history, which long predates the advent of biological MS (as reviewed by Englander [45]). Another important advantage of HDX MS is its tolerance to a range of solvent systems that cannot be used in native ESI MS measurements (the latter are routinely carried out using volatile electrolyte systems). This allows the protein behavior to be studied under conditions that mimic either the physiological environment or the drug formulation very closely. If executed properly, HDX MS measurements can provide a wealth of information on protein higher order structure and dynamics, and it is not surprising that in a mere decade that followed the first use of HDX MS to characterize a protein therapeutic [10] this technique had become widely adopted in the biopharmaceutical industry and is now routinely used not only in exploratory studies [46], but also in filings with regulatory agencies [47].
Global HDX MS measurements to monitor conformational integrity of protein therapeutics.
In HDX MS experiments the protein higher order structure is probed by measuring the exchange rates of labile hydrogen atoms with the solvent. Protons that are involved in hydrogen bonding or sequestered from the solvent in the protein interior cannot be exchanged readily, unless they become exposed to the solvent molecules via dynamic events that affect the protein conformation either locally (e.g., via structural fluctuations) or globally (e.g., via transient unfolding events). The exchange reactions are usually initiated by diluting the protein solution in appropriately buffered D2O, and quenched after a certain period of time by quickly acidifying the protein solution to pH 2.5–3.0 and lowering its temperature to 0 – 4 °C (Figure 5). Such conditions result in a dramatic deceleration of the exchange reactions of the backbone amide hydrogen atoms with the solvent, while the exchange rate for other labile hydrogen atoms remains relatively high. As a result, the isotope labels acquired prior to the quench step are kept exclusively at the backbone amides (despite the loss of the higher order structure - and protection - that is inevitable for nearly all proteins in the acidic environment). This provides a single reporter for each amino acid residue in the protein sequence with the exception of the N-terminal residue and all proline residues. Since each individual exchange event (substitution of hydrogen with deuterium) results in a mass increase of 1.01 Da, the total increase of the protein mass can be equated to the total number of amide groups that acquired an isotopic label prior to the quench step. Measuring the protein mass increase as a function of the time interval between the dilution step (initiation of the HDX reactions) and the quench step allows the kinetics of the exchange reactions to be followed at the intact protein level (see the “Global HDX” box in Figure 5). Such measurements provide a relatively straightforward way to detect changes in the stability of the higher order structure as a result of certain modifications of the protein covalent structure [10].
Figure 5.
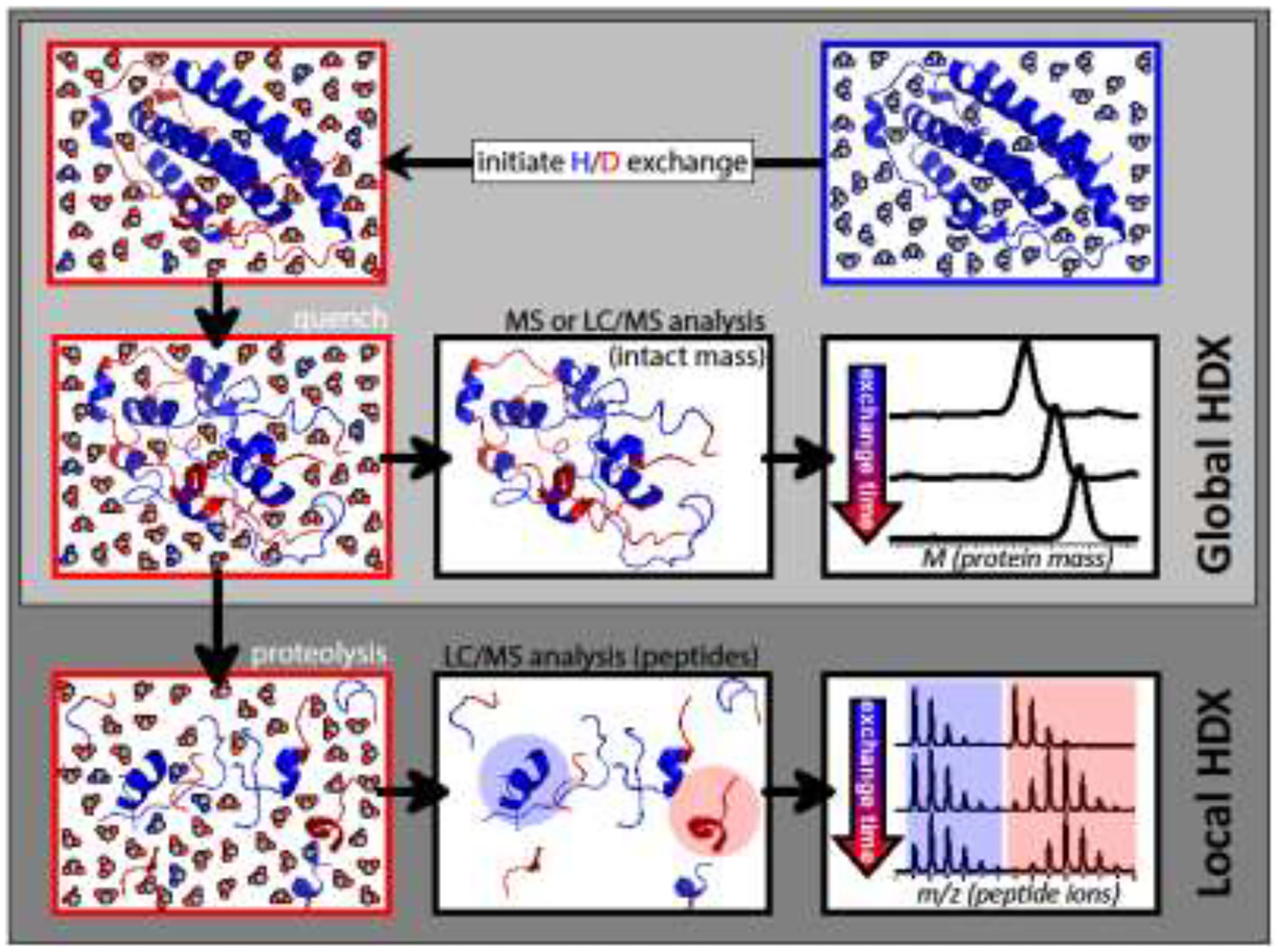
A schematic diagram of the workflows for global and local HDX MS measurements.
Site-specific HDX MS measurements to identify instability hot-spots and binding interfaces.
The ability to minimize the exchange of backbone amide hydrogen atoms under conditions when they are not afforded any protection by the protein higher order structure also allows the distribution of the labile isotopic labels along the polypeptide backbone to be determined by carrying out proteolysis prior to MS analysis (see the “Local HDX” box in Figure 5). Pepsin is one of a handful of proteases that is active within the pH range 2.5–3.0, and is most commonly used to probe backbone amide protection following the quench of the exchange reactions [45, 48]. However, the slow exchange of the backbone amide hydrogen atoms continues even under the quench conditions, and the proteolytic step must be relatively fast in order to avoid the excessive loss of the deuterium labels (the so-called “back exchange”). The efficiency of the proteolytic step carried out under such sub-optimal conditions can be enhanced by reducing the disulfide bonds prior to the protein digestion. The need to maintain the slow-exchange conditions during the disulfide reduction places significant restrictions on the available repertoire of the reducing agents (with TCEP being the most common reagent used in such applications [49]). The proteolysis efficiency can also be enhanced by a judicial use of strong chaotropes, such as guanidinium chloride. Elimination of all ESI-incompatible components from the protein digest solution prior to the MS analysis is usually achieved during a quick LC separation step with on-line MS analysis of the deuterium content of all produced peptic fragments [50].
An example of using HDX MS to localize unstable elements within a protein is shown in Figure 6. Here the evolution of the isotopic distribution of a peptic fragment (88–102) derived from intact (unmodified) interferon β1a reveals a rather anemic uptake of deuterium atoms within this segment of the protein, a hallmark of a stable conformation. However, the behavior of the same peptide derived from the protein bearing a single covalent modification (at a cysteine residue that is distal to this peptide in the protein sequence, but proximal in the three-dimensional structure) is markedly different. The bimodal shape of the isotopic distribution of the peptide ions in this case is indicative of the presence of two conformations in solution, one of which appears to lack significant backbone protection (red traces in Figure 6). In contrast, most other peptic fragments do not reveal a difference between the two forms of the protein, suggesting that the covalent modification does not compromise conformational integrity of the protein uniformly across the entire polypeptide chain. Instead, only several segments within the protein are affected, but the presence of even a small number of such instability hot spots results in a significant increase of the aggregation propensity [51].
Figure 6.
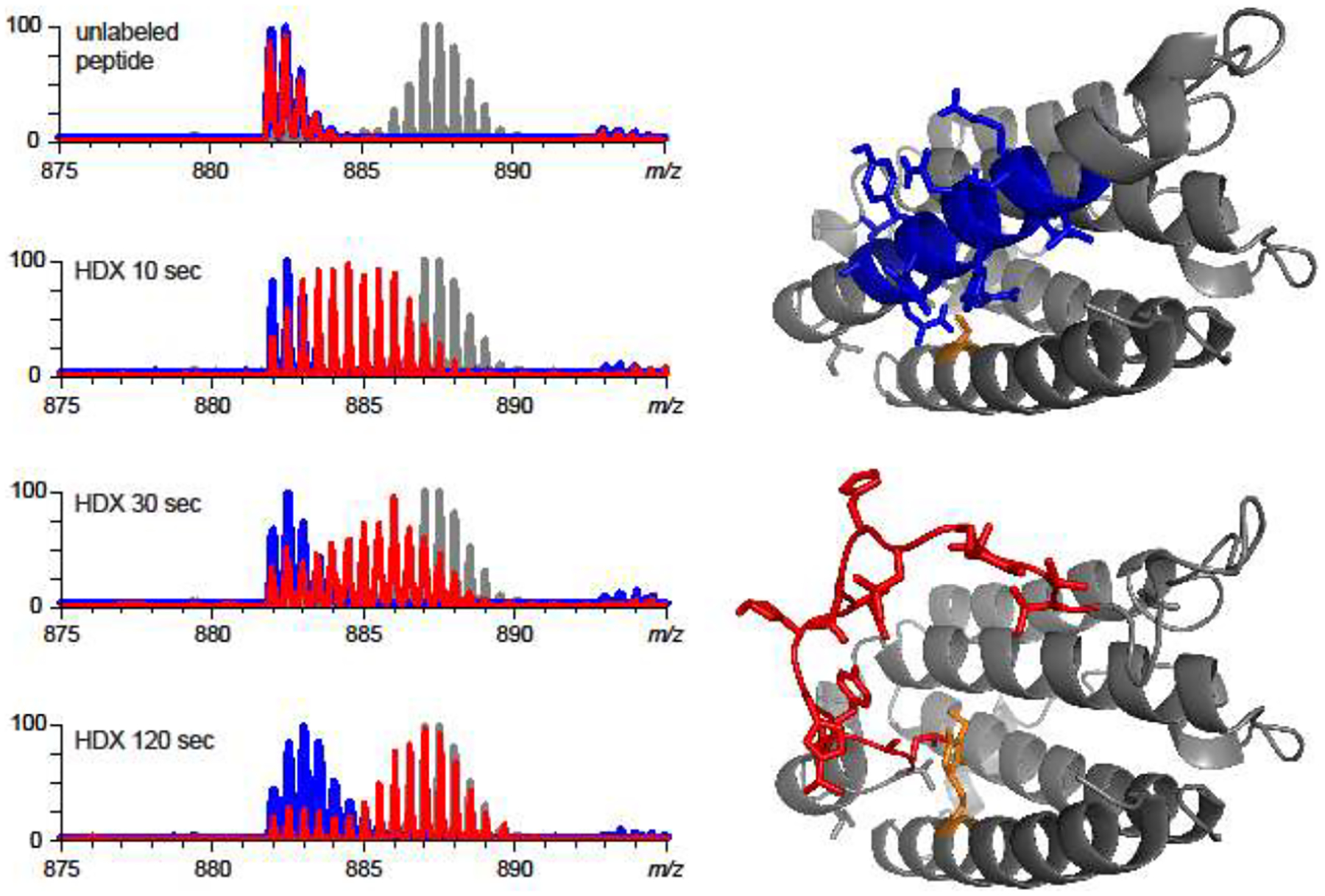
Localization of an instability hotspot within the covalently modified (NEM-alkylated) interferon β1a using HDX MS. The left-hand side panel shows the evolution of isotopic distributions of peptic fragments (88–102) derived from the intact (blue) and modified (red) proteins as a function of the exchange time in solution. The endpoint of the exchange reaction (taking into account the back-exchange) is indicated with a gray trace (isotopic distribution of a fully exchanged peptide). The panels on the right-hand side show location of the (88–102) segment (colored in blue) within the crystal structure of the intact protein (1AU1). Alkylation of Cys-17 (orange) inevitably leads to steric clashes within the native structure, which can be removed by unfolding the helical element containing the [88–102] segment (colored in red). Adapted and used with permission [51].
In the example considered in the preceding paragraph, the covalent modification was introduced intentionally and targeted a specific amino acid residue within the protein. However, this strategy can be also applied to detect/localize the instability regions that arise due to stress-related non-enzymatic PTMs targeting multiple sites within the protein, such as oxidation [19]. Above and beyond the detection and localization of instability hotspots, HDX MS can be used to map binding interfaces for interactions between biopharmaceutical products as complex as mAbs with their therapeutic targets [52, 53] and physiological partners [54].
The measurements described in this section produce segment-specific information on deuterium distribution along the backbone, and it is the length of the proteolytic fragments that limits the spatial resolution. At least some improvement in the spatial resolution can be achieved by analyzing the deuterium content of overlapping proteolytic fragments, but the residue-level resolution remains out of reach for the majority of biopharmaceutical products. Supplementing the experimental scheme depicted in Figure 5 with the gas-phase fragmentation of the peptide ions as a means of localizing deuterium atoms is another strategy that may in some cases reveal the backbone protection patterns at a single residue level [55, 56]. Unfortunately, the choice of ion fragmentation techniques that can be used for such a task remains very limited due to the need to avoid hydrogen scrambling during the ion activation process [57, 58]. Consequently, the progress in this field has not moved much beyond feasibility studies carried out with small model proteins that are of little relevance vis-à-vis the needs of the biopharmaceutical sector.
Top-down HDX MS: a useful tool in biopharma?
An alternative approach to obtaining site-specific information on deuterium distribution along the polypeptide backbone utilizes the so-called “top-down” strategy, where proteolytic degradation is completely eliminated from the experimental work flow and is replaced with fragmentation of intact protein ions in the gas phase [59]. Once again, the need to suppress/eliminate hydrogen scrambling during the ion activation process places a very stringent limitation as far as the type of ion fragmentation techniques that can be employed in such measurements. Both electron capture [60] and electron transfer [61] dissociation can be used for this purpose, as long as any collisional activation of protein ions (which is frequently used to increase the dissociation yield) is completely avoided. More recently, ultra-violet photo-dissociation had also been shown to be a viable choice for the top-down HDX MS work [62].
An example of using top-down HDX MS to generate the protein backbone protection patterns is shown in Figure 7, where the deuterium content for a set of c-ions of varying length reveals the highly localized regions with elevated flexibility (such as segments (11–16) and (32–38), which exhibit complete loss of the initial deuterium label upon short exposure to the H2O-based solvent). This example also highlights a challenge that must be dealt with when top-down HDX MS is used for characterization of protein higher order structure and dynamics. A high degree of spatial resolution can only be achieved when abundant fragment ions corresponding to dissociation of a large number of the N-Cα bonds can be generated (e.g., the gaps in the histogram in Figure 7 are due to missing c-ions). Since most fragment ions are produced at more than a single charge state, the resulting mass spectra become very crowded even for proteins of a relatively modest size. The severity of this problem is illustrated in the right-hand side panel in Figure 7, where an m/z window as narrow as 1 unit contains contributions from five different fragment ions! Such extreme spectral crowding places a very stringent requirement on the type of instruments that can be used in top-down HDX MS work (mass analyzers with high resolution, such as Fourier transform ion cyclotron resonance MS and Orbitrap MS). Nevertheless, extensive efforts invested in this field in the past decade appear to begin bearing fruit, with several reports on using top-down HDX MS to characterize proteins as large and complex as mAbs now available [63, 64]. Another recently introduced approach that may solve the spectral crowding/complexity issue (dubbed “middle-down” HDX MS) takes advantage of the restricted use of pepsin (known to act specifically on antibodies to separate Fc from Fab regions) prior to gas phase dissociation of the resulting large proteolytic fragments [65].
Figure 7.
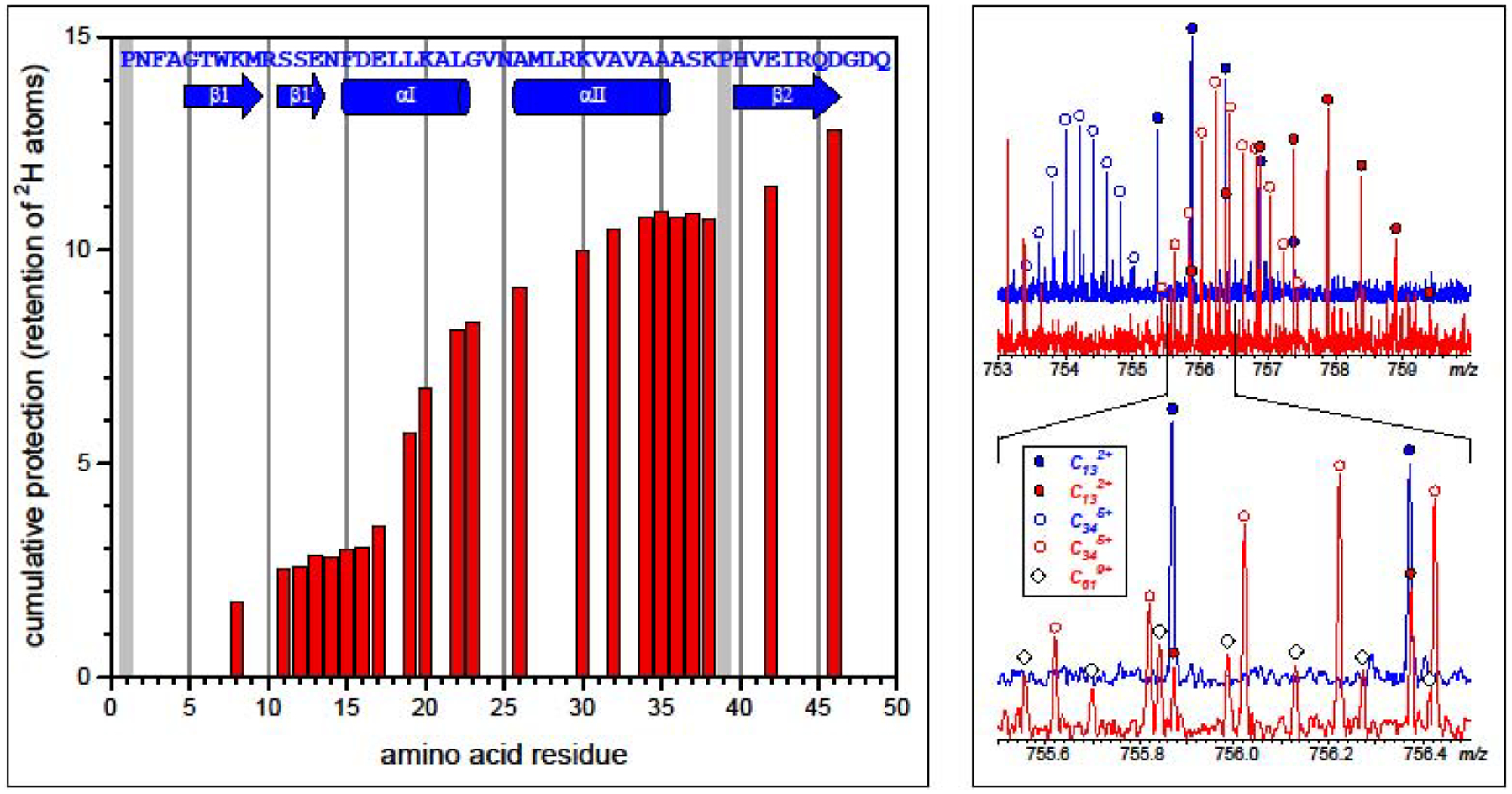
Left: backbone protection pattern of an 18 kDa protein (retinoic acid transporter) deduced from the top-down HDX MS/MS measurements (electron transfer dissociation of intact protein ions). The exchange reactions were initiated by exposing the fully deuterated protein to 1H2O/CH3CO2N1H4 for 5 min. followed by rapid quenching and acquisition of the MS/MS data. Right: an example of raw HDX MS/MS data used to generate the protection plot shown on the left-hand side diagram. Isotopic distributions of c13 and c34 fragments derived from the protein subjected to 5 min. HDX exchange in solution (red trace) and protein at the HDX end-point (blue trace) were used to calculate the bar heights at n = 12 and 35. Adapted and used with permission [59].
Other MS-based methods for higher order structure characterization
HDX MS has become a commonly accepted tool for characterization of therapeutic proteins in recent years, but the search for alternative/complementary methods of analysis of protein higher order structure continues. One of the most annoying limitations of HDX is the labile character of the deuterium label both in the solution phase (where the need to minimize the back exchange dictates the solution conditions that must be used for protein processing prior to MS detection – frequently leading to sub-optimal proteolysis, disulfide reduction, peptide separation, etc.) and in the gas phase (where the specter of hydrogen scrambling precludes the use of highly efficient methods of peptide and protein ion fragmentation). The realization that the ability to label the unprotected/solvent-exposed segments of the protein irreversibly would be a boon to the field incentivizes the relentless search for experimental strategies based on chemical labeling techniques.
Chemical labeling and cross-linking: what limits their use in characterization of biopharmaceutical products?
Chemical labeling [66] and cross-linking [67] are among the oldest biophysical techniques that remain in continuous use in studies aimed at characterization of protein (and, more generally, biopolymer) higher order structure. Unfortunately, these techniques suffer from a very significant limitation that makes the vast majority of them much less attractive compared to HDX MS vis-à-vis characterization of protein therapeutics. Most existing labeling protocols rely on extensive modification of the protein molecules in the sample in order to achieve the yields sufficient for MS characterization, and in most situations this results in placing multiple labels on a single protein molecule. However, chemical modification even of a single site within the protein molecule can result in a significant change of local conformation as well as the global conformational stability. Therefore, placement of each additional label would not necessarily correlate with the solvent accessibility within the context of the native state, but rather reflect the compromised conformational integrity of the covalently modified protein. In fact, exactly this behavior was seen in the example presented in Figure 6, where alkylation of a single residue within interferon β1a had resulted in a dramatic decrease of conformational stability within several (but not all) segments of the protein. These changes were readily detected by HDX MS, and there is no doubt that the local unfolding triggered by a single covalent modification would result in a greater accessibility of the affected segment(s) to other chemical probes as well. This example is by no means unique, which is the reason why the classical chemical labeling techniques have failed so far to achieve any notable level of prominence in the analysis of the higher order structure of biopharmaceutical products (and while the exceptions to that do exist [68], they remain extremely rare). Chemical cross-linking is affected by this problem to even a more significant extent, as placing even a single cross-link within a protein or between two proteins necessarily requires two chemical modifications. As a result, the use of chemical cross-linking in the biopharmaceutical analysis also remain rare, although in some instances this technique had been shown to be useful vis-à-vis complementing information obtained via use of orthogonal techniques, such as HDX MS [69]. In such situations an argument can be made that once a specific method is validated using orthogonal (and commonly accepted) means, it can be used routinely even in the quality control settings.
Characterization of protein higher order structure with FPOP.
While classical chemical labeling and cross-linking techniques remain largely on the periphery of the efforts to characterize higher order structure of protein therapeutics, a new labeling approach introduced by Gross and Hambly in the previous decade [70] gave rise to an experimental tool that now enjoys unprecedented popularity both in industry and academia. This technique (termed FPOP, of Fast Photo-Oxidation of Proteins [71]) provides an elegant solution to the problem that makes the use of chemical labeling unpractical when applied to characterization of the higher order structure of protein therapeutics, namely distortions of the protein structure caused by multiple chemical modifications (vide supra). This problem is avoided in FPOP by making the duration of the chemical modification window narrow on the time scale of protein conformational transitions. As a result, the higher order structure of a protein can be probed in its native state without being affected by structural perturbations (despite placing multiple labels on single protein molecule). The labeling is irreversible (e.g., oxidation by OH· radicals), which allows a great deal of flexibility as far as the downstream processing of the labeled proteins. This provides a significant advantage over the HDX MS-based methods of the higher order structure analysis, where lability of amide hydrogen atoms places rather unforgiving restrictions on all protein processing/analysis steps from proteolysis to MS/MS. One significant disadvantage of FPOP compared to HDX MS is the significantly lower level of labels/reporters that are typically placed on a single protein molecule (a few labels per protein molecule as opposed to labeling every amide backbone in HDX MS), although the recent efforts to expand the repertoire of FPOP probes may address this problem at least to some extent [72, 73].
The protein labeling in FPOP is triggered by a very short pulse of an excimer laser; the wavelength is selected such as to maximize the photolytic production of radicals (e.g., OH· via photolysis of H2O2, which is present in the protein solution alongside the radical scavenger), and avoid interactions with all other components of the solution. The resultant radicals react with the protein by inducing oxidation, but the radical scavenger present in solution results in complete elimination of these reactive species almost immediately after the end of the laser pulse. The duration of the laser pulse (typically < 10−7 sec.) is short on the time scale of the protein conformational transitions; as a result, multiple oxidation events may occur within a single protein molecule without introducing artefacts (the laser pulse frequency and the protein solution flow rate are selected such as to avoid multiple irradiation of the same region of the flowing protein solution). It must be noted, however, that under some conditions the reactive species can persist in solution over much longer time intervals (> 10−2 sec.) [74], which would certainly be problematic vis-à-vis the reliability of FPOP measurements.
In recent years FPOP had been applied to solve multiple problems in the field of protein therapeutics ranging from assessing conformational integrity [75, 76] to mapping binding epitopes [52, 77, 78]. FPOP has a potential to tackle a range of other relevant problems, including assaying protein aggregation [79] and probing ligand-induced conformational changes [80]; consequently, the range of FPOP applications in the analysis of biopharmaceuticals is likely to continue to expand. Importantly, the information provided by FPOP is complementary to that derived from HDX MS measurements, which frequently prompts the deployment of integrated FPOP/HDX MS strategies [52, 78, 81]. An orthogonal approach to controlling the duration of the chemical labeling reactions may utilize a recently introduced cross-path reactive chromatography (XPRC) platform [82].
Conclusions and outlook
Despite being a relatively recent addition to the experimental toolbox used to characterize biopharmaceutical products’ higher order structure, MS is now commonly viewed as a reliable platform capable of addressing the most challenging questions and dealing with very complex systems. MS-based methods of assessing higher order structure are now routinely used in characterization of systems as complex as mAbs, and have become increasingly common in regulatory filings [47]. It will be interesting to see if the great success enjoyed by MS as a tool for characterizing the higher order structure of protein therapeutics in vitro could be translated to in vivo studies. Indeed, conformational changes suffered by protein therapeutics post-administration should have significant impact on both efficacy and safety, but reliable experimental tools capable of detecting and characterizing such events in vivo remain wanting. Should MS prove capable of filling this gap, the impact on the studies of pharmacokinetics and pharmacodynamics of protein therapeutics would be enormous indeed (recent application of the FPOP technology to in vivo studies in primitive model organisms [83] and human cells [84] look particularly promising in this regard). A related field where mass spectrometry can make significant contributions in the future is process analytical technology (PAT) [85–87] in the productions of protein therapeutics. This field has been traditionally dominated by less technologically sophisticated, but rugged methods of analysis due to the need to carry out measurements on-line (and ideally in-line) in real time [88]. However, mass spectrometry is now being actively tested as an alternative, which is capable of providing information on the conformational integrity of protein therapeutics during their production [89].
Above and beyond protein therapeutics, MS-based methods for characterization of higher order structure are likely to accelerate the development of other types of biopharmaceutical products, such as gene therapy products. The potential of native MS to characterize entities as complex as viral capsids and intact viruses has already been demonstrated [90, 91], and there is no doubt that the role of mass spectrometry in the field of gene therapy will become as significant as it is today in the realm of protein therapeutics.
Highlights.
Higher order structure of protein therapeutics is an important quality attribute, which determines both potency and safety of these medicines
A number of the state-of-the-art tools experimental biophysics (e.g., NMR and X-ray crystallography) are poorly suited for routine analysis and characterization of biopharmaceutical products
Mass spectrometry enables characterization of higher order structure of biopharmaceuticals as complex as monoclonal antibodies at an unprecedented level of detail, and has already become an integral part of the analytical toolbox in biopharma
Acknowledgements
The work presented in this manuscript was made possible by the generous support from the National Institutes of Health (R01 GM132673), National Science Foundation (CHE-1709552), U.S. Pharmacopeia, Amgen, Biogen, Shire and Pfizer. All MS data shown here were acquired at the Mass Spectrometry Core facility at UMass-Amherst.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors declare no conflict of interest
References
- [1].Poppe L, Jordan JB, Rogers G, Schnier PD, On the Analytical Superiority of 1D NMR for Fingerprinting the Higher Order Structure of Protein Therapeutics Compared to Multidimensional NMR Methods, Anal. Chem 87(11) (2015) 5539–5545. [DOI] [PubMed] [Google Scholar]
- [2].Arbogast LW, Delaglio F, Schiel JE, Marino JP, Multivariate Analysis of Two-Dimensional 1H, 13C Methyl NMR Spectra of Monoclonal Antibody Therapeutics To Facilitate Assessment of Higher Order Structure, Anal. Chem 89(21) (2017) 11839–11845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Houde DJ, Berkowitz SA, Biophysical Characterization of Proteins in Developing Biopharmaceuticals, Elsevier Science Bv, Amsterdam, 2015. [Google Scholar]
- [4].Fukuhara K, Tsuji T, Toi K, Takao T, Shimonishi Y, Verification by mass spectrometry of the primary structure of human interleukin-2, J. Biol. Chem 260(19) (1985) 10487–94. [PubMed] [Google Scholar]
- [5].Sasaki H, Bothner B, Dell A, Fukuda M, Carbohydrate structure of erythropoietin expressed in Chinese hamster ovary cells by a human erythropoietin cDNA, J. Biol. Chem 262(25) (1987) 12059–76. [PubMed] [Google Scholar]
- [6].Raschdorf F, Dahinden R, Maerki W, Richter WJ, Merryweather JP, Location of disulphide bonds in human insulin-like growth factors (IGFs) synthesized by recombinant DNA technology, Biomed. Environ. Mass Spectrom 16(1–12) (1988) 3–8. [DOI] [PubMed] [Google Scholar]
- [7].Kaltashov IA, Bobst CE, Abzalimov RR, Mass spectrometry-based methods to study protein architecture and dynamics, Protein Sci. 22(5) (2013) 530–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Kaltashov IA, Eyles SJ, Studies of biomolecular conformations and conformational dynamics by mass spectrometry, Mass Spectrom. Rev 21(1) (2002) 37–71. [DOI] [PubMed] [Google Scholar]
- [9].Nguyen DN, Becker GW, Riggin RM, Protein mass spectrometry: applications to analytical biotechnology, J. Chromatogr. A 705(1) (1995) 21–45. [DOI] [PubMed] [Google Scholar]
- [10].Bobst CE, Abzalimov RR, Houde D, Kloczewiak M, Mhatre R, Berkowitz SA, Kaltashov IA, Detection and characterization of altered conformations of protein pharmaceuticals using complementary mass spectrometry-based approaches, Anal. Chem 80(19) (2008) 7473–7481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Houde D, Arndt J, Domeier W, Berkowitz S, Engen JR, Characterization of IgG1 conformation and conformational dynamics by hydrogen/deuterium exchange mass spectrometry, Anal. Chem 81(7) (2009) 2644–2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Alexandrov ML, Gall LN, Krasnov NV, Nikolaev VI, Pavlenko VA, Shkurov VA, Ion extraction from solutions at atmospheric pressure - a method of mass spectrometric analysis of bioorganic substances., Dokl. Acad. Nauk SSSR 277(2) (1984) 379–383. [Google Scholar]
- [13].Yamashita M, Fenn JB, Electrospray ion source. Another variation on the free-jet theme, J. Phys. Chem 88(20) (1984) 4451–4459. [Google Scholar]
- [14].Smith RD, Lightwahl KJ, Winger BE, Loo JA, Preservation of noncovalent associations in electrospray ionization mass spectrometry - Multiply charged polypeptide and protein dimers, Org. Mass Spectrom 27(7) (1992) 811–821. [Google Scholar]
- [15].Chowdhury SK, Katta V, Chait BT, Probing conformational changes in proteins by mass spectrometry., J. Am. Chem. Soc 112 (1990) 9012–9013. [Google Scholar]
- [16].Konermann L, Douglas DJ, Unfolding of proteins monitored by electrospray ionization mass spectrometry: a comparison of positive and negative ion modes., J. Am. Soc. Mass Spectrom 9(12) (1998) 1248–1254. [DOI] [PubMed] [Google Scholar]
- [17].Kaltashov IA, Mohimen A, Estimates of protein surface areas in solution by electrospray ionization mass spectrometry, Anal. Chem 77(16) (2005) 5370–5379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kaltashov IA, Abzalimov RR, Do ionic charges in ESI MS provide useful information on macromolecular structure?, J. Am. Soc. Mass Spectrom 19(9) (2008) 1239–1246. [DOI] [PubMed] [Google Scholar]
- [19].Bobst CE, Thomas JJ, Salinas P, Savickas P, Kaltashov IA, Impact of oxidation on protein therapeutics: Conformational dynamics of intact and oxidized acid-β-glucocerebrosidase at near-physiological pH, Protein Sci 19(12) (2010) 2366–2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Abzalimov RR, Bobst CE, Salinas PA, Savickas P, Thomas JJ, Kaltashov IA, Studies of pH-dependent self-association of a recombinant form of arylsulfatase A with electrospray ionization mass spectrometry and size-exclusion chromatography, Anal. Chem 85(3) (2013) 1591–1596. [DOI] [PubMed] [Google Scholar]
- [21].Nguyen SN, Bobst CE, Kaltashov IA, Mass spectrometry-guided optimization and characterization of a biologically active transferrin-lysozyme model drug conjugate, Mol. Pharm 10 (2013) 1988–2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Wang G, de Jong RN, van den Bremer ET, Beurskens FJ, Labrijn AF, Ugurlar D, Gros P, Schuurman J, Parren PW, Heck AJ, Molecular Basis of Assembly and Activation of Complement Component C1 in Complex with Immunoglobulin G1 and Antigen, Mol. Cell 63(1) (2016) 135–45. [DOI] [PubMed] [Google Scholar]
- [23].Strasser J, de Jong RN, Beurskens FJ, Wang G, Heck AJR, Schuurman J, Parren P, Hinterdorfer P, Preiner J, Unraveling the Macromolecular Pathways of IgG Oligomerization and Complement Activation on Antigenic Surfaces, Nano Lett. 19(7) (2019) 4787–4796. [DOI] [PubMed] [Google Scholar]
- [24].Jurchen JC, Williams ER, Origin of asymmetric charge partitioning in the dissociation of gas-phase protein homodimers, J. Am. Chem. Soc 125(9) (2003) 2817–2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Felitsyn N, Kitova EN, Klassen JS, Thermal decomposition of a gaseous multiprotein complex studied by blackbody infrared radiative dissociation. Investigating the origin of the asymmetric dissociation behavior, Anal. Chem 73(19) (2001) 4647–4661. [DOI] [PubMed] [Google Scholar]
- [26].Abzalimov RR, Frimpong AK, Kaltashov IA, Gas-phase processes and measurements of macromolecular properties in solution: On the possibility of false positive and false negative signals of protein unfolding, Int. J. Mass Spectrom 253(3) (2006) 207–216. [Google Scholar]
- [27].Muneeruddin K, Thomas JJ, Salinas PA, Kaltashov IA, Characterization of small protein aggregates and oligomers using size exclusion chromatography with on-line detection by native electrospray ionization mass spectrometry, Anal. Chem 86(21) (2014) 10692–10699. [DOI] [PubMed] [Google Scholar]
- [28].Kaltashov IA, Pawlowski JW, Yang W, Muneeruddin K, Yao H, Bobst CE, Lipatnikov AN, LC/MS at the whole protein level: studies of biomolecular structure and interactionsusing native LC/MS and cross-path reactive chromatography (XP-RC) MS, Methods (San Diego, Calif.) 144 (2018) 14–26. [DOI] [PubMed] [Google Scholar]
- [29].Abzalimov RR, Kaltashov IA, Electrospray ionization mass spectrometry of highly heterogeneous protein systems: Protein ion charge state assignment via incomplete charge reduction, Anal. Chem 82(18) (2010) 7523–7526. [DOI] [PubMed] [Google Scholar]
- [30].Wang G, Johnson AJ, Kaltashov IA, Evaluation of electrospray ionization mass spectrometry as a tool for characterization of small soluble protein aggregates, Anal. Chem 84(3) (2012) 1718–1724. [DOI] [PubMed] [Google Scholar]
- [31].Muneeruddin K, Bobst CE, Frenkel R, Houde D, Turyan I, Sosic Z, Kaltashov IA, Characterization of a PEGylated protein therapeutic by ion exchange chromatography with on-line detection by native ESI MS and MS/MS, Analyst 142(2) (2017) 336–344. [DOI] [PubMed] [Google Scholar]
- [32].Joubert MK, Luo Q, Nashed-Samuel Y, Wypych J, Narhi LO, Classification and characterization of therapeutic antibody aggregates, J. Biol. Chem 286(28) (2011) 25118–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Fukuda J, Iwura T, Yanagihara S, Kano K, Factors to Govern Soluble and Insoluble Aggregate-formation in Monoclonal Antibodies, Anal. Sci 31(12) (2015) 1233–1240. [DOI] [PubMed] [Google Scholar]
- [34].Mirza UA, Cohen SL, Chait BT, Heat-induced conformational changes in proteins studied by electrospray ionization mass spectrometry, Anal. Chem 65(1) (1993) 1–6. [DOI] [PubMed] [Google Scholar]
- [35].Wang G, Abzalimov RR, Kaltashov IA, Direct monitoring of heat-stressed biopolymers with temperature-controlled electrospray ionization mass spectrometry, Anal. Chem 83(8) (2011) 2870–2876. [DOI] [PubMed] [Google Scholar]
- [36].Shoemaker GK, Soya N, Palcic MM, Klassen JS, Temperature-dependent cooperativity in donor-acceptor substrate binding to the human blood group glycosyltransferases, Glycobiology 18(8) (2008) 587–92. [DOI] [PubMed] [Google Scholar]
- [37].Charrow J, Enzyme replacement therapy for Gaucher disease, Expert Opin. Biol. Ther 9(1) (2009) 121–31. [DOI] [PubMed] [Google Scholar]
- [38].Wang G, Bondarenko PV, Kaltashov IA, Multi-step conformational transitions in heat-treated protein therapeutics can be monitored in real time with temperature-controlled electrospray ionization mass spectrometry, Analyst 143 (2018) 670–677. [DOI] [PubMed] [Google Scholar]
- [39].Bohrer BC, Mererbloom SI, Koeniger SL, Hilderbrand AE, Clemmer DE, Biomolecule Analysis by Ion Mobility Spectrometry, Ann. Rev. Anal. Chem 1 (2008) 293–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Campuzano ID, Lippens JL, Ion mobility in the pharmaceutical industry: an established biophysical technique or still niche?, Curr. Opin. Chem. Biol 42 (2018) 147–159. [DOI] [PubMed] [Google Scholar]
- [41].D’Atri V, Causon T, Hernandez-Alba O, Mutabazi A, Veuthey JL, Cianferani S, Guillarme D, Adding a new separation dimension to MS and LC-MS: What is the utility of ion mobility spectrometry?, Journal of separation science 41(1) (2018) 20–67. [DOI] [PubMed] [Google Scholar]
- [42].Zhao Y, Abzalimov RR, Kaltashov IA, Interactions of Intact Unfractionated Heparin with Its Client Proteins Can Be Probed Directly Using Native Electrospray Ionization Mass Spectrometry, Anal. Chem 88(3) (2016) 1711–1718. [DOI] [PubMed] [Google Scholar]
- [43].Minsky BB, Abzalimov RR, Niu C, Zhao Y, Kirsch Z, Dubin PL, Savinov SN, Kaltashov IA, Mass Spectrometry Reveals a Multifaceted Role of Glycosaminoglycan Chains in Factor Xa Inactivation by Antithrombin, Biochemistry 57(32) (2018) 4880–4890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Brown CJ, Woodall DW, El-Baba TJ, Clemmer DE, Characterizing Thermal Transitions of IgG with Mass Spectrometry, J. Am. Soc. Mass Spectrom 30(11) (2019) 2438–2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Englander SW, Hydrogen exchange and mass spectrometry: A historical perspective, J. Am. Soc. Mass Spectrom 17(11) (2006) 1481–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Wei H, Mo J, Tao L, Russell RJ, Tymiak AA, Chen G, Iacob RE, Engen JR, Hydrogen/deuterium exchange mass spectrometry for probing higher order structure of protein therapeutics: methodology and applications, Drug Discov. Today 19(1) (2014) 95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Rogstad S, Faustino A, Ruth A, Keire D, Boyne M, Park J, A Retrospective Evaluation of the Use of Mass Spectrometry in FDA Biologics License Applications, J. Am. Soc. Mass Spectrom 28(5) (2017) 786–794. [DOI] [PubMed] [Google Scholar]
- [48].Konermann L, Stocks BB, Pan Y, Tong X, Mass spectrometry combined with oxidative labeling for exploring protein structure and folding, Mass Spectrom. Rev 29(4) (2010) 651–667. [DOI] [PubMed] [Google Scholar]
- [49].Han JC, Han GY, A procedure for quantitative determination of Tris(2-carboxyethyl)phosphine, an odorless reducing agent more stable and effective than dithiothreitol, Anal. Biochem 220(1) (1994) 5–10. [DOI] [PubMed] [Google Scholar]
- [50].Bobst CE, Zhang M, Kaltashov IA, Existence of a noncanonical state of iron-bound transferrin at endosomal pH revealed by hydrogen exchange and mass spectrometry, J. Mol. Biol 388(5) (2009) 954–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Kaltashov IA, Bobst CE, Abzalimov RR, Berkowitz SA, Houde D, Conformation and dynamics of biopharmaceuticals: transition of mass spectrometry-based tools from academe to industry, J. Am. Soc. Mass Spectrom 21(3) (2010) 323–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Li J, Wei H, Krystek SR, Bond D, Brender TM, Cohen D, Feiner J, Hamacher N, Harshman J, Huang RYC, Julien SH, Lin Z, Moore K, Mueller L, Noriega C, Sejwal P, Sheppard P, Stevens B, Chen G, Tymiak AA, Gross ML, Schneeweis LA, Mapping the Energetic Epitope of an Antibody/Interleukin-23 Interaction with Hydrogen/Deuterium Exchange, Fast Photochemical Oxidation of Proteins Mass Spectrometry, and Alanine Shave Mutagenesis, Anal. Chem 89(4) (2017) 2250–2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Baerga-Ortiz A, Hughes CA, Mandell JG, Komives EA, Epitope mapping of a monoclonal antibody against human thrombin by H/D-exchange mass spectrometry reveals selection of a diverse sequence in a highly conserved protein, Protein Sci. 11(6) (2002) 1300–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Jensen PF, Larraillet V, Schlothauer T, Kettenberger H, Hilger M, Rand KD, Investigating the interaction between the neonatal Fc receptor and monoclonal antibody variants by hydrogen/deuterium exchange mass spectrometry, Mol. Cell. Proteomics 14(1) (2015) 148–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Rand KD, Zehl M, Jorgensen TJ, Measuring the hydrogen/deuterium exchange of proteins at high spatial resolution by mass spectrometry: overcoming gas-phase hydrogen/deuterium scrambling, Acc. Chem. Res 47(10) (2014) 3018–27. [DOI] [PubMed] [Google Scholar]
- [56].Abzalimov RR, Bobst CE, Kaltashov IA, A new approach to measuring protein backbone protection with high spatial resolution using H/D exchange and electron capture dissociation, Anal. Chem 85(19) (2013) 9173–9180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Abzalimov RR, Kaltashov IA, Controlling hydrogen scrambling in multiply charged protein ions during collisional activation: Implications for top-down hydrogen/deuterium exchange MS utilizing collisional activation in the gas phase, Anal. Chem 82(3) (2010) 942–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Hoerner JK, Xiao H, Dobo A, Kaltashov IA, Is there hydrogen scrambling in the gas phase? Energetic and structural determinants of proton mobility within protein ions, J. Am. Chem. Soc 126(24) (2004) 7709–7717. [DOI] [PubMed] [Google Scholar]
- [59].Kaltashov IA, Bobst CE, Abzalimov RR, H/D exchange and mass spectrometry in the studies of protein conformation and dynamics: Is there a need for a top-down approach?, Anal. Chem 81(19) (2009) 7892–7899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Pan J, Han J, Borchers CH, Konermann L, Hydrogen/deuterium exchange mass spectrometry with top-down electron capture dissociation for characterizing structural transitions of a 17 kDa protein, J. Am. Chem. Soc 131 (2009) 12801–12808. [DOI] [PubMed] [Google Scholar]
- [61].Abzalimov RR, Kaplan DA, Easterling ML, Kaltashov IA, Protein conformations can be probed in top-down HDX MS experiments utilizing electron transfer dissociation of protein ions without hydrogen scrambling, J. Am. Soc. Mass Spectrom 20(8) (2009) 1514–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Brodie NI, Huguet R, Zhang T, Viner R, Zabrouskov V, Pan J, Petrotchenko EV, Borchers CH, Top-Down Hydrogen-Deuterium Exchange Analysis of Protein Structures Using Ultraviolet Photodissociation, Anal. Chem 90(5) (2018) 3079–3082. [DOI] [PubMed] [Google Scholar]
- [63].Pan J, Zhang S, Parker CE, Borchers CH, Subzero temperature chromatography and top-down mass spectrometry for protein higher-order structure characterization: method validation and application to therapeutic antibodies, J Am Chem Soc 136(37) (2014) 13065–71. [DOI] [PubMed] [Google Scholar]
- [64].Pan J, Zhang S, Borchers CH, Comparative higher-order structure analysis of antibody biosimilars using combined bottom-up and top-down hydrogen-deuterium exchange mass spectrometry, Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics 1864(12) (2016) 1801–1808. [DOI] [PubMed] [Google Scholar]
- [65].Pan J, Zhang S, Chou A, Borchers CH, Higher-order structural interrogation of antibodies using middle-down hydrogen/deuterium exchange mass spectrometry, Chem. Sci 7(2) (2016) 1480–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Limpikirati P, Liu T, Vachet RW, Covalent labeling-mass spectrometry with non-specific reagents for studying protein structure and interactions, Methods (San Diego, Calif.) 144 (2018) 79–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Chu F, Thornton DT, Nguyen HT, Chemical cross-linking in the structural analysis of protein assemblies, Methods (San Diego, Calif.) 144 (2018) 53–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Madsen JA, Yin Y, Qiao J, Gill V, Renganathan K, Fu W-Y, Smith S, Anderson J, Covalent Labeling Denaturation Mass Spectrometry for Sensitive Localized Higher Order Structure Comparisons, Anal. Chem 88(4) (2016) 2478–2488. [DOI] [PubMed] [Google Scholar]
- [69].Zhang MM, Beno BR, Huang RY, Adhikari J, Deyanova EG, Li J, Chen G, Gross ML, An Integrated Approach for Determining a Protein-Protein Binding Interface in Solution and an Evaluation of Hydrogen-Deuterium Exchange Kinetics for Adjudicating Candidate Docking Models, Anal. Chem 91(24) (2019) 15709–15717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Hambly DM, Gross ML, Laser flash photolysis of hydrogen peroxide to oxidize protein solvent-accessible residues on the microsecond timescale, J. Am. Soc. Mass Spectrom 16(12) (2005) 2057–63. [DOI] [PubMed] [Google Scholar]
- [71].Li KS, Shi L, Gross ML, Mass Spectrometry-Based Fast Photochemical Oxidation of Proteins (FPOP) for Higher Order Structure Characterization, Acc. Chem. Res 51(3) (2018) 736–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Cheng M, Zhang B, Cui W, Gross ML, Laser-Initiated Radical Trifluoromethylation of Peptides and Proteins: Application to Mass-Spectrometry-Based Protein Footprinting, Angew. Chem. Int. Ed. Engl 56(45) (2017) 14007–14010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Zhang MM, Rempel DL, Gross ML, A Fast Photochemical Oxidation of Proteins (FPOP) platform for free-radical reactions: the carbonate radical anion with peptides and proteins, Free Radic. Biol. Med 131 (2019) 126–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Vahidi S, Konermann L, Probing the Time Scale of FPOP (Fast Photochemical Oxidation of Proteins): Radical Reactions Extend Over Tens of Milliseconds, J. Am. Soc. Mass Spectrom 27(7) (2016) 1156–64. [DOI] [PubMed] [Google Scholar]
- [75].Jones LM, Zhang H, Cui W, Kumar S, Sperry JB, Carroll JA, Gross ML, Complementary MS methods assist conformational characterization of antibodies with altered S-S bonding networks, J. Am. Soc. Mass Spectrom 24(6) (2013) 835–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Cornwell O, Bond NJ, Radford SE, Ashcroft AE, Long-Range Conformational Changes in Monoclonal Antibodies Revealed Using FPOP-LC-MS/MS, Anal. Chem 91(23) (2019) 15163–15170. [DOI] [PubMed] [Google Scholar]
- [77].Li KS, Chen G, Mo J, Huang RY, Deyanova EG, Beno BR, O’Neil SR, Tymiak AA, Gross ML, Orthogonal Mass Spectrometry-Based Footprinting for Epitope Mapping and Structural Characterization: The IL-6 Receptor upon Binding of Protein Therapeutics, Anal. Chem 89(14) (2017) 7742–7749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Zhang Y, Wecksler AT, Molina P, Deperalta G, Gross ML, Mapping the Binding Interface of VEGF and a Monoclonal Antibody Fab-1 Fragment with Fast Photochemical Oxidation of Proteins (FPOP) and Mass Spectrometry, J. Am. Soc. Mass Spectrom 28(5) (2017) 850–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Li KS, Rempel DL, Gross ML, Conformational-Sensitive Fast Photochemical Oxidation of Proteins and Mass Spectrometry Characterize Amyloid Beta 1–42 Aggregation, J. Am. Chem. Soc 138(37) (2016) 12090–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Liu XR, Rempel DL, Gross ML, Composite Conformational Changes of Signaling Proteins upon Ligand Binding Revealed by a Single Approach: Calcium-Calmodulin Study, Anal. Chem 91(19) (2019) 12560–12567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Shi L, Liu T, Gross ML, Huang Y, Recognition of Human IgG1 by Fcgamma Receptors: Structural Insights from Hydrogen-Deuterium Exchange and Fast Photochemical Oxidation of Proteins Coupled with Mass Spectrometry, Biochemistry 58(8) (2019) 1074–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Pawlowski JW, Carrick I, Kaltashov IA, Integration of On-Column Chemical Reactions in Protein Characterization by Liquid Chromatography/Mass Spectrometry: Cross-Path Reactive Chromatography, Anal. Chem 90(2) (2018) 1348–1355. [DOI] [PubMed] [Google Scholar]
- [83].Espino JA, Jones LM, Illuminating Biological Interactions with in Vivo Protein Footprinting, Anal. Chem 91(10) (2019) 6577–6584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Johnson DT, Punshon-Smith B, Espino JA, Gershenson A, Jones LM, Implementing In-Cell Fast Photochemical Oxidation of Proteins in a Platform Incubator with a Movable XY Stage, Anal. Chem 92(2) (2020) 1691–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Rathore AS, Bhambure R, Ghare V, Process analytical technology (PAT) for biopharmaceutical products, Anal. Bioanal. Chem 398(1) (2010) 137–54. [DOI] [PubMed] [Google Scholar]
- [86].Read EK, Shah RB, Riley BS, Park JT, Brorson KA, Rathore AS, Process analytical technology (PAT) for biopharmaceutical products: Part II. Concepts and applications, Biotechnol. Bioeng 105(2) (2010) 285–95. [DOI] [PubMed] [Google Scholar]
- [87].Read EK, Park JT, Shah RB, Riley BS, Brorson KA, Rathore AS, Process analytical technology (PAT) for biopharmaceutical products: Part I. concepts and applications, Biotechnol. Bioeng 105(2) (2010) 276–84. [DOI] [PubMed] [Google Scholar]
- [88].Guerra A, von Stosch M, Glassey J, Toward biotherapeutic product real-time quality monitoring, Crit. Rev. Biotechnol 39(3) (2019) 289–305. [DOI] [PubMed] [Google Scholar]
- [89].Furuki K, Toyo’oka T, Yamaguchi H, A novel rapid analysis using mass spectrometry to evaluate downstream refolding of recombinant human insulin-like growth factor-1 (mecasermin), Rapid Commun. Mass Spectrom 31(15) (2017) 1267–1278. [DOI] [PubMed] [Google Scholar]
- [90].van de Waterbeemd M, Snijder J, Tsvetkova IB, Dragnea BG, Cornelissen JJ, Heck AJ, Examining the Heterogeneous Genome Content of Multipartite Viruses BMV and CCMV by Native Mass Spectrometry, J. Am. Soc. Mass Spectrom 27(6) (2016) 1000–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Keifer DZ, Motwani T, Teschke CM, Jarrold MF, Acquiring Structural Information on Virus Particles with Charge Detection Mass Spectrometry, J. Am. Soc. Mass Spectrom 27(6) (2016) 1028–36. [DOI] [PMC free article] [PubMed] [Google Scholar]