

Abstract
Pathological cardiac hypertrophy is a classical hallmark of heart failure. At the molecular level, inhibition of histone deacetylase (HDAC) enzymes attenuate pathological cardiac hypertrophy in vitro and in vivo. Emodin is an anthraquinone that has been implicated in cardiac protection. However, it is not known if the cardio-protective actions for emodin are mediated through HDAC-dependent regulation of gene expression. Therefore, we hypothesized that emodin would attenuate pathological cardiac hypertrophy via inhibition of HDACs, and that these actions would be reflected in an emodin-rich food like rhubarb. In this study, we demonstrate that emodin and Turkish rhubarb containing emodin inhibit HDAC activity in vitro, with fast-on, slow-off kinetics. Moreover, we show that emodin increased histone acetylation in cardiomyocytes concomitant to global changes in gene expression; gene expression changes were similar to the well-established pan-HDAC inhibitor trichostatin A (TSA). We additionally present evidence that emodin inhibited phenylephrine (PE) and phorbol myristate acetate (PMA)-induced hypertrophy in neonatal rat ventricular myocytes (NRVMs). Lastly, we demonstrate that the cardioprotective actions of emodin are translated to an angiotensin II (Ang) mouse model of cardiac hypertrophy and fibrosis and are linked to HDAC inhibition. These data suggest that emodin blocked pathological cardiac hypertrophy, in part, by inhibiting HDAC-dependent gene expression changes.
Keywords: Emodin, histone deacetylase, HDAC, cardiac hypertrophy, food bioactives, heart failure
1. Introduction:
Pathological cardiac hypertrophy is a hallmark of heart failure that affects millions of people and costs billions of dollars each year [1]. In response to stress, muscle cells of the heart, or cardiomyocytes, enlarge which leads to cardiac dysmorphism and subsequent dysfunction, resulting in heart failure. Poor dietary habits such as the Western Diet are associated with cardiac hypertrophy [2]. Conversely, cardiac hypertrophy can be prevented through proper dietary management [3].
A heart-healthy diet is defined by leading experts as one consisting mainly of plant-based foods [4]. Indeed, reports from cellular studies [5] as well as human [6] and epidemiology analyses [7,8] suggest that plant-based foods are beneficial to overall heart health and deter heart disease. Plant-based foods like fruits, vegetables, whole grains and legumes contain essential macronutrients and micronutrients including fiber, vitamins and minerals. However, a building body of evidence suggests that benefits of these plant-based foods are independent of their essential nutrients [9]. This hints at the idea that other chemicals in these foods, i.e., phytochemicals, drive their efficacy. Indeed, phytochemicals have been shown to be cardioprotective; many of these early reports demonstrate protection via inhibition of oxidative stress, inflammation and shifts in intracellular signaling [10–13]. However, more recent evidence suggests that phytochemicals can regulate epigenetic modifications contributing to global changes in gene expression [14].
Epigenetic modifications differentially regulate gene expression independent of changes in DNA sequence. Lysine acetylation on histone tails is a reversible epigenetic modification that is regulated by two enzymes: histone deacetylases (HDACs) and histone acetyltransferases (HATs) [15]. HDACs remove acetyl groups from lysine residues on histones leading to nucleosome compaction and transcriptional repression. Eighteen identified mammalian HDACs have been separated into classes I (HDACs 1,2 3, 8), II (HDACs 4, 5, 6, 7, 9, 10), III (Sirt1–7) and IV (HDAC 11). Class II is further divided into sub-classes IIa and IIb. Of interest, inhibiting class I and II HDAC activity is efficacious in experimental models of HF [16,17] and several phytochemicals have been characterized as HDAC inhibitors [14]. However, it remains unclear if phytochemicals protect the heart through HDAC-dependent regulation of gene expression.
Emodin is an anthraquinone phytochemical found in plant-based foods like rhubarb, cabbage and beans [18–20]. Additionally, emodin-rich plants, including buckthorn and knotweed, have been used in traditional medicines for centuries against viral, bacterial and bowel abnormalities. In the heart, emodin has been reported to reduce mitochondrial oxidative stress [21] and attenuate inflammation [22]. Recently, we showed that emodin inhibited HDAC activity in a test tube [23]. In this report, we sought to elucidate the cardioprotective actions of emodin and emodin-rich rhubarb. We report that emodin inhibited HDAC activity and increased histone acetylation in cardiomyocytes concomitant to global changes in cardiac gene expression. Moreover, we report that emodin normalized cardiac gene expression changes similar to the well-known HDAC inhibitor TSA. Lastly, we demonstrate that emodin blocked pathological cardiac hypertrophy in vitro and in vivo, consistent with its role as an HDAC inhibitor.
2. Materials and Methods
2.1. Reagents
Emodin was purchased from SelleckChem (S2295) and Turkey rhubarb purchased through Prescribed for Life (Sb15-H091519). Phenylephrine (PE; 10 μM) was purchased through Tocris Bioscience, phorbol-12-myristate-13-acetate (PMA; 50 nM) and trichostatin A (TSA; 200 nM) were purchased through Sigma-Aldrich. Emodin, turkey rhubarb and TSA were prepared in dimethyl sulfoxide (DMSO, Pharmco-AAPER).
2.2. HDAC Activity Assays
Bovine heart tissue was procured from the University of Nevada, Wolf Pack Meats. Animal care and handling was approved by the University of Nevada, Reno, Institutional Animal Care and Use Committee. HDAC activity assays were completed as previously described [24]. Each substrate is based on ε-N-acylated lysine, derivatized on the carboxyl group with amino methylcoumarin (AMC) [25]. Heart tissue lysate was prepared in PBS (pH 7.4) containing 0.5% Triton X-100, 300 mM NaCl and protease/phosphatase inhibitor cocktail (ThermoFisher Scientific) using a Bullet Blender homogenizer (Next Advance). Tissue was clarified by centrifugation prior to determination of protein concentration using a BCA Protein Assay Kit (Pierce). Tissue (30 μg protein/well) was diluted in PBS for a total volume of 100 μl/well in a 96-well plate. For concentration-response determination, tissue was dosed with increasing semilog scale concentrations of emodin (SelleckChem) or Turkey Rhubarb (Prescribed for Life) for 2 hrs. For kinetic analysis, heart lysate was treated with 50 μM emodin or 100 mg/L turkey rhubarb at the prescribed time points. Class-specific HDAC substrates were added (5 μl of 1 mM DMSO stock solutions), and plates returned to the 37°C incubator for 2 hrs. HDAC substrates for HDAC activity experiments were as follows: ZLPA (Class I, GeneScript custom peptide), I1985 (Class IIa, Bachem, #4060676) and I-1875 (Class IIb, Bachem, #4033792). Developer/stop solution was added (50 μl per well of PBS with 1.5% Triton X-100, 3 μM TSA, and 0.75 mg/ml trypsin) and plates incubated at 37°C for 20 min. Subsequent to deacetylation, trypsin is used to release AMC, resulting in increased fluorescence. AMC fluorescence was measured via BioTek Synergy plate reader, with excitation and emission filters of 360 nm and 460 nm, respectively. Background signals from buffer blanks were subtracted, and GraphPad Prism used to calculate IC50 values for each compound. In addition, NRVMs were treated with PE (10 μM) and co-stimulated with either emodin (10 μM) or TSA (200 nM) for 48 hrs prior to cell lysis in PBS (0.5% Triton X-100, 300 mM NaCl and protease/phosphatase inhibitors). Protein concentrations were determined via BCA and HDAC activity assessed via class-selective HDAC substrates as described above. Fluorescence was measured via a BioTek Synergy plate reader (excitation filter at 360 nm and emission filter at 460 nm). Finally, male and female C57BL/6 mice were randomly assigned into groups to receive sham with vehicle (DMSO:PEG-300), angiotensin II (1.5 μg/kg/min) with vehicle or angiotensin II with emodin (30 mg/kg/day) for 14 days. On day 14, left ventricles were dissected and flash frozen, later to be lysed in PBS (0.5% Triton X-100, 300 mM NaCl and protease/phosphatase inhibitors). Protein concentrations were determined via BCA and HDAC activity assessed via class-selective HDAC substrates as described above. Fluorescence was measured via a BioTek Synergy plate reader (excitation filter at 360 nm and emission filter at 460 nm).
2.3. HPLC
An Agilent 1100 high performance liquid chromatography system, including a programmable solvent delivery pump, autosampler, and diode-array UV detector, was used for determination of emodin. Detection was set at 437 nm for emodin (6-methyl-1,3,8trihydroxyanthraquinone) analysis. Emodin separation was carried out using a Kinetex 5 um XBC18 100A 250 × 4.6mm column (Phenomenex, Torrance, CA). HPLC-grade methanol and water were used as reagents (Fisher Scientific). Isocratic separation was performed with a methanol/water (70:30, v/v) mobile phase at a flow rate of 0.8 mL/min. A stock emodin standard of 100 μg/mL was used to develop a seven-point calibration curve following serial dilution with methanol. Rhubarb extract was prepared in methanol at 100 mg/L. Each sample or standard was then sonicated for 30 minutes prior to triplicate injection via HPLC. A 30 μL aliquot of all samples or standards were injected directly into the HPLC system for quantitation. Emodin was identified in unknown samples by retention time matching between standards and unknowns, and the data was expressed in μg/mL.
2.4. Neonatal rat ventricular myocyte (NRVM) isolation and culture
NRVMs were prepared as previously described [26]. Briefly, hearts from 1–3 day-old SpragueDawley neonates were collected and digested in a solution containing trypsin (Gibco Life Technologies) and DNaseII from bovine (Sigma-Aldrich). Ventricular myocytes were isolated and then cultured in 100-mm dishes or 6-well plates that were coated with gelatin (0.2%, Sigma-Aldrich). Cardiomyocytes were placed in Minimum Eagles Medium (MEM, Genesee Scientific) with 10% calf serum, 2 mM L-glutamine and penicillin-streptomycin and incubated overnight. Media was replaced the next morning with Nurtidoma-SP (Roche Applied Science) and Dubelco’s Modified Eagles Medium (DMEM) prior to experimental treatments. Cells were co-spiked with either hypertrophic agonist, PE (10 μM) or PMA (50 nM), and emodin (10 μM), turkey rhubarb (100 mg/L) or TSA (200 nM) and incubated for 48 hours prior to being lysed or fixed for experiments described below.
2.5. Immunoblotting
Cells were lysed in PBS containing 300 mM NaCl, 0.5% Triton-X and HALT™ protease/phosphatase inhibitors. Cell lysate was then sonicated and centrifuged (16,000g for 5min) prior to BCA for protein quantification. Samples were resolved with SDS-PAGE and transferred to a nitrocellulose membrane prior to overnight incubation with primary antibodies for acetyl histone H3 lysine residues 9/14 (H3K9/14; Cell Signaling Technology, 9677), H3K18 (Cell Signaling Technology, 13998s) and H3K27 (Cell Signaling Technology, 8173s) as well as Total histone H3 (Cell Signaling Technology, 4499), phosphorylated ERK (Cell Signaling Technology, 4370), total ERK (Santa Cruz Biotechnology; Sc‐1647) and atrial natriuretic factor (ANP, Santa Cruz Biotechnology, Sc-515701). The next day, horseradish peroxidase‐conjugated secondary antibodies (Southern Biotech) were used prior to exposing with SuperSignal West Pico Chemiluminescence System (Thermo Fisher Scientific) on a ChemiDoc XRS+ Imager (BioRad).
2.6. Immunostaining
NRVMs were plated in 6-well dishes and treated as described above. Plates were fixed with 4% paraformaldehyde at room temperature for 20 minutes and prepared for immunostaining as previously described [27]. After fixation, cells were permeabilized with PBS containing bovine serum albumin (3%, Fisher Bioreagents, BP1605) and Nonidet NP-40 (0.1%, Sigma-Aldrich IGEPAL CA-630) prior to being incubated for two hours with a primary antibody containing ANF (1:1000, Phoenix Pharmaceuticals, H-005–24) and α-actinin (1:750 Sigma A-7811). Cells were then incubated with a secondary antibody cocktail (donkey anti-mouse FITC, Jackson ImmunoResearch; goat anti-rabbit Cy3, Jackson ImmunoResearch) for one hour and briefly washed and incubated with Hoechst (10 μM, Invitrogen H3570) in PBS. Cells were washed with PBS and imaged via the EVOS FL Cell Imaging System (Thermo Fisher Scientific) at 20×. Twenty images of cells were taken per well. Cells were then analyzed for cell area and ANF expression via Image J (NIH Software).
2.7. Cell viability
NRVMs, spiked with either DMSO (veh), emodin (Emod, 10μM), phenylephrine (PE, 10μM) or PE + Emod, were exposed to Invitrogen alamarBlue™ HS Cell Viability Reagent (Thermo Fisher Scientific) for cell viability analyses. Following manufacturer instructions, NRVMs were incubated with Invitrogen alamarBlue™ HS Cell Viability Reagent (10:1, media with NRVMs:Invitrogen alamarBlue™ HS Cell Viability Reagent) in gelatin-coated 96-well plates for 1 hour. Invitrogen alamarBlue™ HS Cell Viability Reagent is resazurin based, which upon entering an NRVM will be reduced to the highly fluorescent resorufin. After 1 hour incubation, fluorescence was measured via BioTek Synergy plate reader, with excitation and emission filters of 530 nm and 590 nm, respectively. Results were normalized to DMSO control and expressed as % cell viability.
2.8. RNA-sequencing
To analyze transcriptome-wide changes, RNA was isolated from NRVMs using QIAzol (Qiagen). RNA integrity was assessed via RNA Agilent Bioanalyzer; all samples achieved a RIN value >8. 500 ng of RNA was used for cDNA library preparation with the Ribo-Zero Human/Mouse/Rat TruSeq Stranded Total RNA kit from Illumina. Set-B Adapters (Illumina) were used for the cDNA library prep. Validation of library prep was performed with the Agilent Bioanalyzer and sequencing performed in the Genomics Center at the University of Nevada Reno via the NextSeq 500.
To assure the sequencing performance and library quality, we applied the RNA-SeQC [28] tool to assess the data quality of each sequencing dataset. We used the Sailfish pipeline [29] to quantify the mRNA expression from the raw sequencing data, using the Ensembl [30] rat gene annotation (Rnor_6.0). Transcript per million reads (TPM) was used as the unit of human gene expression level. We used the edgeR algorithm [31] to compare the groupwise gene expression pattern. The TMM algorithm implemented in the edgeR package was applied for reads count normalization and effective library size estimation. Groupwise differential expression was estimated by the likelihood ratio test implicated in the edgeR package. The genes were false discovery rate < 5% were deemed differentially expressed. Supplementary Table 1 illustrates genes examined via RNA-sequencing.
2.9. Real-time qPCR
RNA was isolated as described above and RNA quantity determined via NanoDrop Spectrometry ND1000. 500 ng of RNA was reverse transcribed to cDNA via Verso cDNA Synthesis Kit (ThermoFisher Scientific). Quantitative real-time polymerase chain reaction (qPCR) was used to determine mRNA expression for select genes. In short, cDNA underwent qPCR with Apex qPCR GREEN Master Mix (Genesee Scientific, 42–120) and the following IDT Primers were used: rat atrial natriuretic peptide/factor (ANP, forward- GCC GGT AGA AGA RGA GGT CAT, reverse- GCT TCC TCA GTC TGC TCA CTC A); rat b-type natriuretic peptide (BNP, forward- GGT GCT GCC CCA GAT GAT T, reverse- CTG GAG ACT GGC TAG GAC TTC); rat skeletal muscle alpha actin (Kcnc3, forward-, reverse-); and 18s ribosomal RNA (forward- GCC GCT AGA GGT GAA ATT CTT A, reverse- CTT TCG CTC TGG TCC GTC TT). Fluorescence was detected in real-time using the BioRad CF96X qPCR instrument.
2.10. Experimental animals
Nine-week old C57BL/6 mice were housed at the University of Nevada, Reno. All mice were maintained on standard chow and housed under standard 12h light/dark cycle. Animal care and use was approved by the Institutional Animal Use and Care Committee at the University of Nevada, Reno. At ten-weeks of age, C57BL/6 male and female mice were randomized to receive vehicle control (Sham) or angiotensin II (Ang) for 14 days. 14-day micro-osmotic pumps (Alzet, model 1002) were subcutaneously implanted in mice under isoflurane anesthesia; pumps contained angiotensin II (Bachem) at 1.5 μg/kg/min or vehicle control. Mice in both groups were further randomized to receive: 1) sham dosed with vehicle (1:1 DMSO:PEG-300 (Acros Organics)), 2) Ang osmotic pump dosed with vehicle or 3) Ang osmotic pump dosed with emodin (30 mg/kg/day). Mice received intraperitoneal (IP) injection of vehicle or emodin every day for 14 days. Three days prior to the end of the study, systolic blood pressure measurements were taken via tail cuff using the Coda High Throughput System (Kent Scientific); the first two days were used for acclimation and third day for data collection. At the end of the study, whole hearts and left ventricles (LVs) were weighed and compared to tibia length for morphology analyses. LVs were also dissected for histology and HDAC activity experiments.
2.11. Histology
Left ventricles (LVs) were fixed in 4% paraformaldehyde for 24 hours prior to 70% ethanol housing. LVs were then processed using the Leica ASP300S and paraffin embedded using Leica EG1160. PicroSirius Red for collagen staining was performed on LVs cross-sectioned at 5 μm as previously described [32]. Stained LVs were imaged using the Keyence BZ-X700, and collagen staining was quantified using ImageJ software.
2.12. Statistical Methods
A minimum of three experiments with an n = 3 per experimental treatment group was performed and data quantified. One-way ANOVA with Tukey’s post-hoc was performed unless otherwise specified using GraphPad7 (GraphPad Software, La Jolla, CA). p < 0.05 was considered significant.
3. Results:
3.1. Emodin and emodin-rich rhubarb inhibited HDAC activity in a dose-dependent, fast-on/slow-off manner
Emodin is a phytochemical commonly found in rhubarb [18,20]. Our lab previously identified emodin as a class I, IIa and IIb HDAC inhibitor in vitro (Fig. 1A and [23]). As such, we postulated that emodin-rich rhubarb would phenocopy HDAC inhibition. Indeed, rhubarb inhibited class I and II HDAC activity in a dose-dependent manner (IC50=100 mg/L, Fig. 1A). Similar to emodin, rhubarb inhibited recombinant class I, IIa and IIb HDACs (Supplemental Figure 1), only HDACs 2 and 6 differed between emodin and rhubarb. These experiments demonstrated that both emodin and rhubarb inhibited HDAC activity at a single point in time. To determine HDAC inhibition kinetics, we incubated cardiac tissue with emodin (50 μM) or rhubarb (100 mg/L) for 0.5, 1, 2, 4, 8, 12 or 24 hours prior to analyzing class I, IIa and IIb HDAC activity. Emodin rapidly inhibited HDAC activity (0.5 hours), with prolonged HDAC inhibition out through 24-hours (Fig. 1B). Rhubarb HDAC inhibition kinetics phenocopied emodin (Fig. 1B). As the phytochemical profile of a plant can be affected by soil content, climate and other environmental factors, we examined emodin content via high-performance liquid chromatography (HPLC). We confirmed that emodin is a key component of a turkey rhubarb extract (P4L, Fig. 2). Surprisingly, 100mg/L of turkey rhubarb contained 2.67 μg/ml of emodin which approximated to 10 μM, further supporting the postulate that emodin-rich rhubarb inhibited HDAC activity. These results collectively suggest that emodin is a fast-on, slow-off pan-HDAC inhibitor that is likely responsible for rhubarb-dependent HDAC inhibition.
Figure 1. Emodin and rhubarb inhibit HDAC activity in bovine cardiac tissue with fast-on, slow-off kinetics.
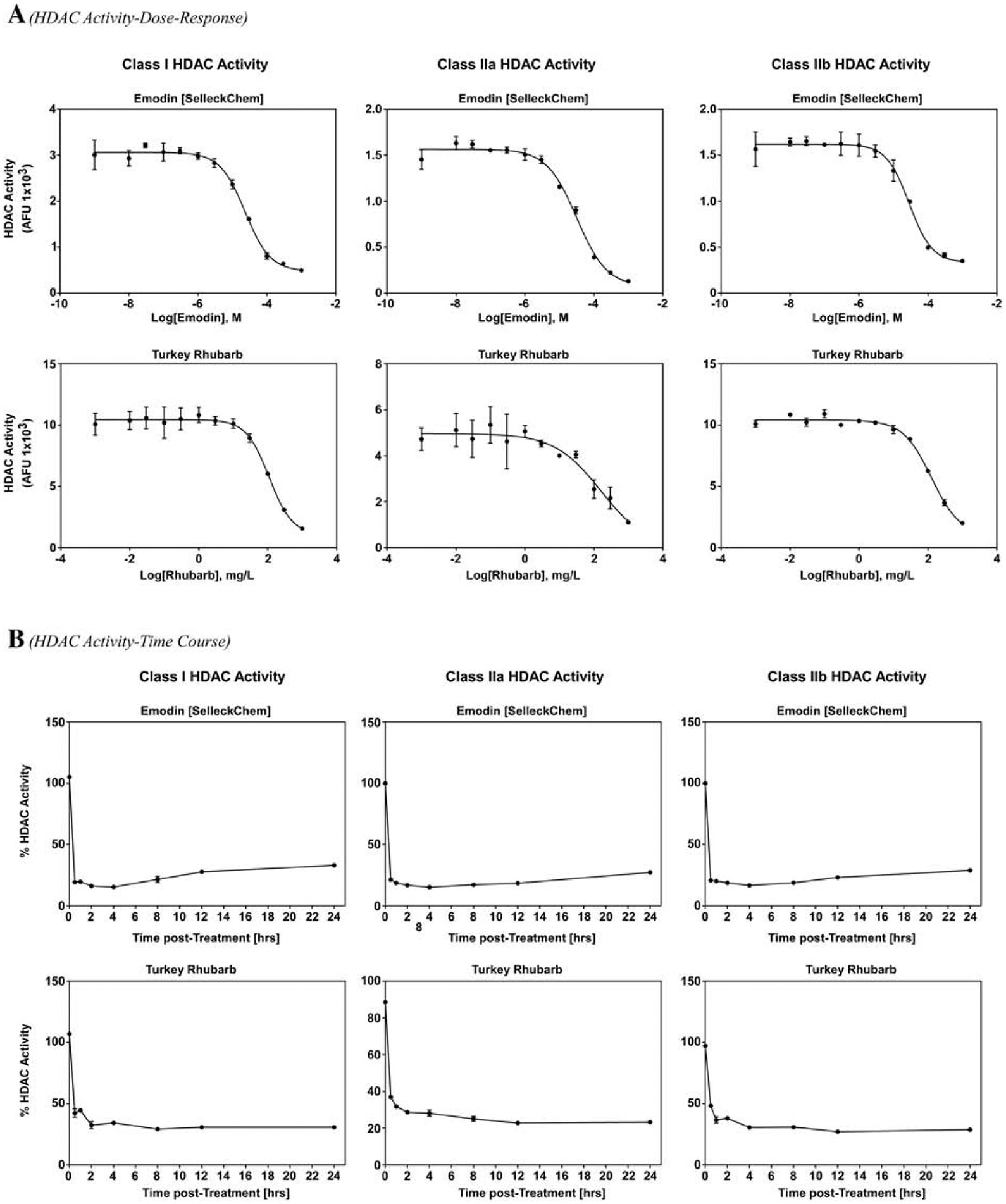
A) Bovine cardiac tissue was treated for 2 hours with increasing doses of emodin (Top Panel) or turkey rhubarb (bottom panel) prior to incubation with cell permeable fluorogenic HDAC substrates for 2 hours and developer solution for 20 minutes. B) Bovine cardiac lysate was treated with emodin (10 μM; Top Panel) or turkey rhubarb (100 mg/L) over time (24 hrs max) prior to incubation with the cell permeable fluorogenic HDAC substrates followed by developer solution. Fluorescence was assessed via BioTek Synergy plate reader with excitation/emission set at 360/460 nm.
Figure 2. Turkey rhubarb contains appreciable emodin as determined by HPLC.
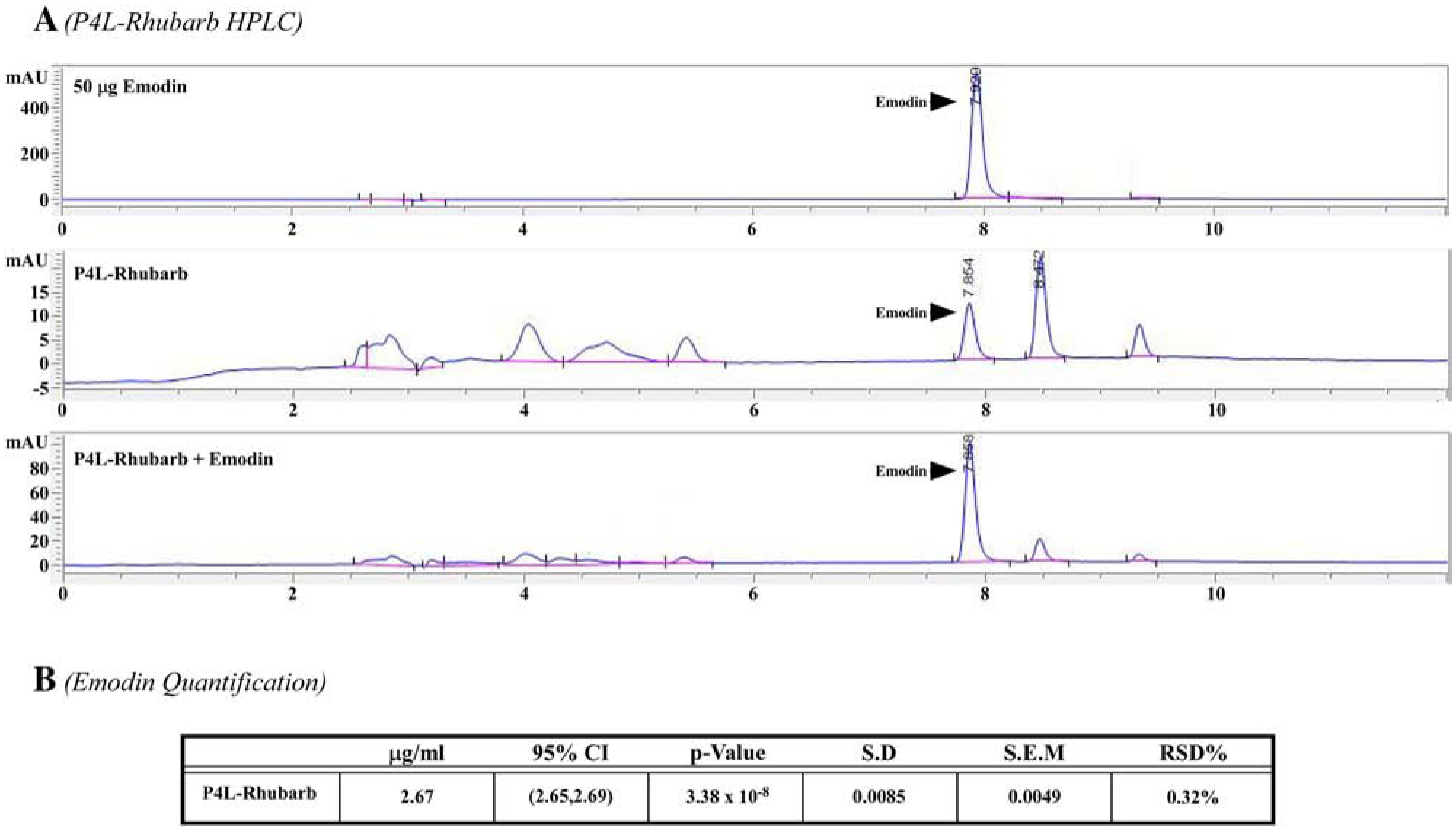
High Performance Liquid Chromatography (HPLC) was used to determine emodin concentrations within turkey rhubarb, 50 μg emodin standard was used to determine emodin peaks and compare emodin concentrations within turkey rhubarb. Turkey rhubarb spiked with emodin was used to verify the emodin peak. B) Emodin concentration was quantified.
3.2. Emodin attenuated HDAC activity concomitant with increased histone acetylation in cardiac myocytes
In vitro analysis above demonstrated that emodin inhibited HDAC activity, however, these findings do not demonstrate inhibitory actions for this compound within cells or tissue. Therefore, we sought to elucidate the actions of emodin on HDAC activity and histone acetylation in neonatal rat ventricular myocytes (NRVMs). We postulated that emodin would inhibit HDAC activity in NRVMs. To test this postulate, NRVMs were co-spiked with phenylephrine (PE, 10 μM) in the absence or presence of vehicle control, emodin (10 μM) or the well-established pan-HDAC inhibitor Trichostatin A (TSA, 200 nM). Cells were lysed after 48 hours for protein to assess HDAC activity. We report that emodin significantly inhibited class I, IIa and IIb HDAC activity, similar to TSA (Fig. 3A). As HDACs catalytically reduce histone acetylation and we’ve shown that emodin inhibits HDAC activity, we postulated that emodin would increase histone acetylation in cardiomyocytes. To test this, we treated NRVMs with PE in the absence or presence of emodin as described above. As hypothesized, emodin increased histone H3 acetylation on lysine residues 9/14 (Ac-H3K9/14), 18 (Ac-H3K18) and 27 (Ac-H3K27) in NRVMs (Fig. 3B and 3C). Finally, a cell viability assay to verify that HDAC inhibition and cardiac hypertrophy described below were not secondary to cell death. NRVMs were dosed with either vehicle (DMSO), emodin, PE or PE + emodin at concentrations used in experiments above. No significant differences were observed between treatments (Fig. 3D), suggesting that emodin at 10 μM is not cardiotoxic.
Figure 3. Emodin inhibited HDAC activity in cardiac myocytes concomitant to increased histone acetylation.
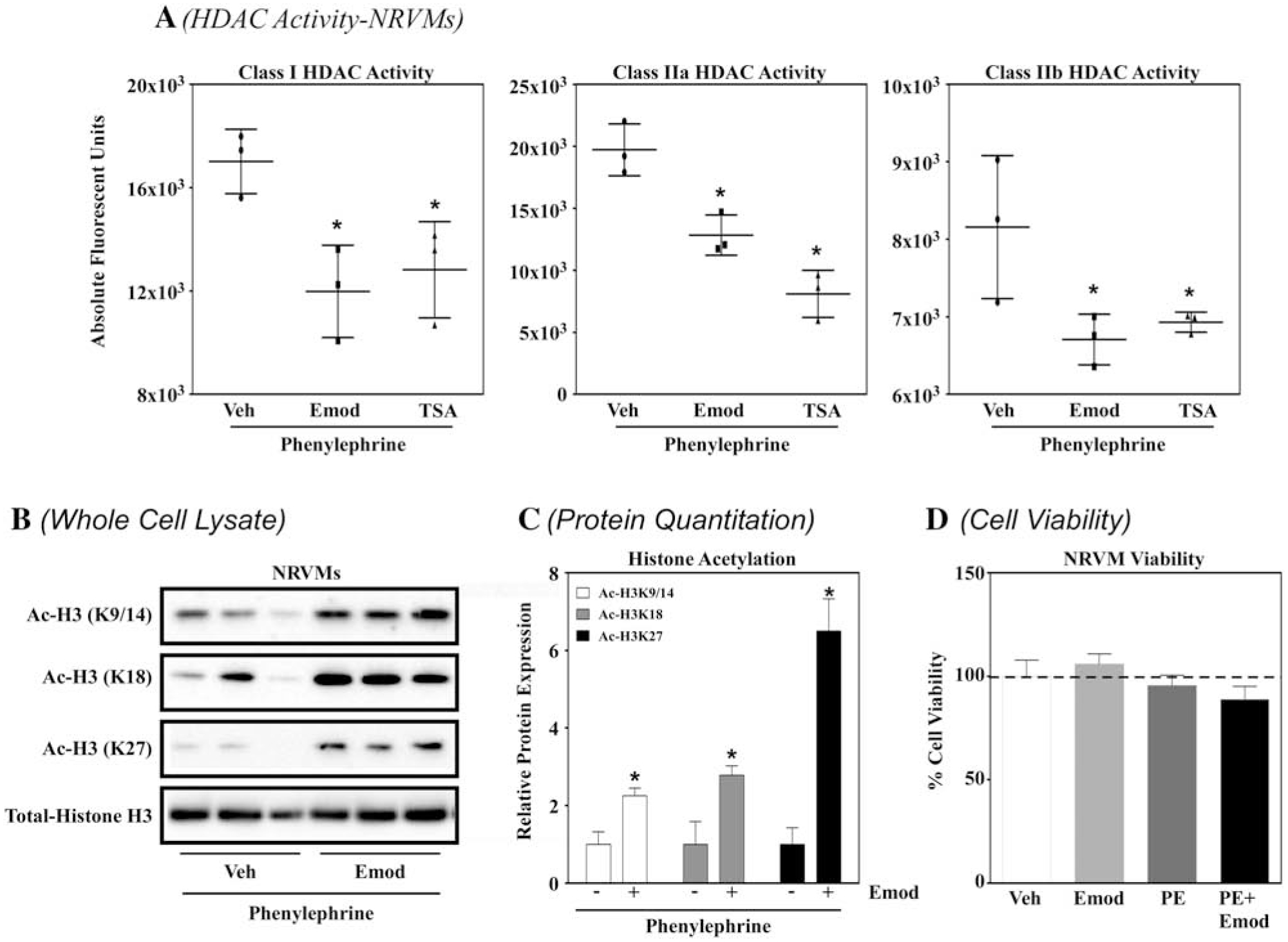
A) Neonatal rat ventricular myocytes (NRVMs) were incubated with phenylephrine (10 μM; PE) with or without emodin (10 μM; Emod) or trichostatin A (200 nM; TSA) for 48 hours prior to protein lysis. Cells were lysed and incubated against the cell permeable fluorogenic HDAC substrates for 2 hours and developer solution for 20 minutes. Fluorescence was assessed via BioTek Synergy plate reader with excitation/emission set at 360/460 nm. B) NRVMs were incubated with phenylephrine (10 μM; PE) with or without emodin (10 μM; Emod) or trichostatin A (200 nM; TSA) for 48 hours prior to protein lysis. Cell lysate was then incubated against antibodies for acetylated histone 3 at lysine (K) residues K9/14 (Ac-H3K9/14), K18 (Ac-H3K18), K27 (Ac-H3K27) as well as against total histone H3 (TotalHistone H3) prior to analysis via immunoblot. C) Acetyl Histone H3 proteins were normalized to total H3 and quantitation performed via Image J software. D) NRVMs were treated with DMSO or emodin (10 μM; Emod) with or without phenylephrine (10 μM; PE) for 48 hours prior to incubation with Invitrogen alamarBlue™ HS Cell Viability Reagent (10:1, media with NRVMs:Invitrogen alamarBlue™ HS Cell Viability Reagent) for 1 hour. Fluorescence was measured via BioTek Synergy plate reader, with excitation and emission filters of 530 nm and 590 nm, respectively. All statistical analyses were run in GraphPad Prism Software. One-way ANOVA with Tukey’s Post-hoc analysis was used for HDAC activity assays, while student’s t-test with Welch’s Correction used for immunoblot analysis.
3.3. Emodin and rhubarb blocked receptor- and intracellular-mediated cardiomyocyte hypertrophy
HDAC inhibitors have been shown to block cardiomyocyte hypertrophy [16]. Thus, we postulated that emodin and emodin-rich rhubarb (P4L) would attenuate receptor- and intracellular signaling-mediated cardiac hypertrophy in NRVMs. Cells were co-spiked with either emodin (10 μM) or rhubarb (P4L, 100 mg/L) and either PE (10 μM) or PMA (50 nM) and incubated for 48 hours prior to being fixed and stained with antibodies against the sarcomere protein α-actinin (green), the hypertrophic marker atrial natriuretic factor (ANF, red) and DAPI (nuclear stain, blue). Cardiomyocytes co-spiked with emodin and either agonist were significantly smaller than those spiked without emodin (Fig. 4A, 4B, 4D and 4E). Rhubarb (P4L, 100 mg/L) similarly attenuated agonist-induced cardiac hypertrophy (Fig. 4A, 4B, 4D and 4E). Moreover, as emodin-treated NRVMs co-spiked with or without phenylephrine showed no significant differences in cell viability (Fig. 3D), cardiotoxicity did not drive these observed differences in NRVM size. Therefore, emodin blocked cardiomyocyte hypertrophy that was induced either by receptor- or intracellular-mediated agonists, suggesting that emodin elicits cardioprotective actions within the cell.
Figure 4. Emodin and rhubarb inhibit cardiomyocyte hypertrophy.
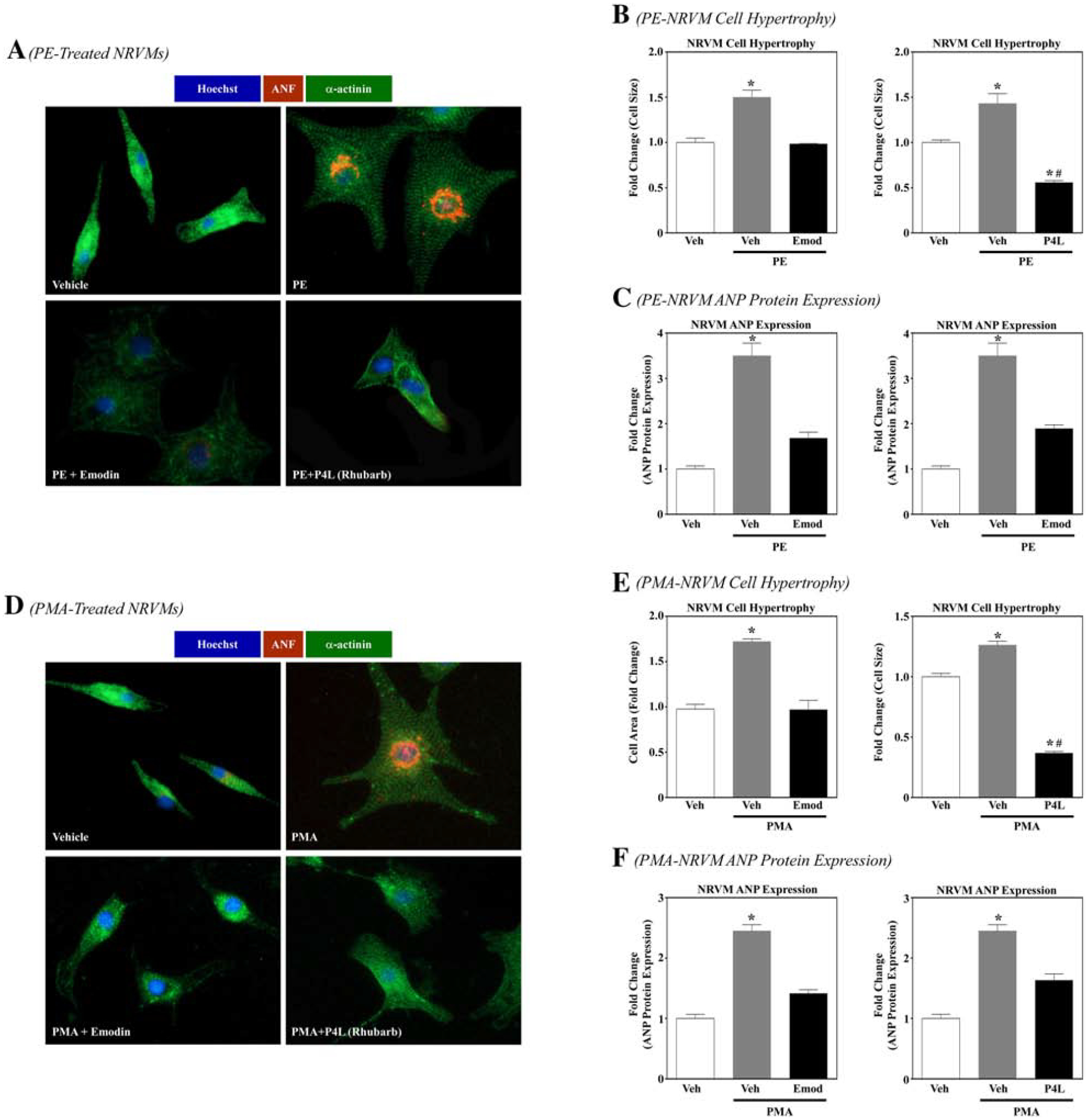
A) Neonatal rat ventricular myocytes (NRVMs) were stimulated to hypertrophy with phenylephrine (10 μM; PE) in the absence or presence of Emodin (10 μM) or rhubarb (100 mg/L; P4L) for 48 hrs. Cells were fixed and immunostained with antibodies directed against α-actinin or atrial natruetic factor (ANF). Cell nuclei were stained with Hoechst. Cells were visualized with EVOS microscopy. Ten pictures were taken per well and cell size (area) and ANF expression (pixels) were calculated using Image J software. GraphPad Prism was used to examine statistical significance. One-way ANOVA with Tukey’s post-hoc analysis was used. Significance was set at p<0.05. B) Cell area and C) ANF expression was quantified. D) NRVMs were stimulated to hypertrophy with phorbol myristate acetate (50 nM; PMA) in the absence or presence of Emodin (10 μM) or rhubarb (100 mg/L; P4L) for 48 hrs. Cells were fixed and immunostained with antibodies directed against α-actinin or atrial natriuretic peptide (ANP). Cell nuclei were stained with Hoechst. Cells were visualized with the Invitrogen EVOS FL microscope. Ten pictures were taken per well and cell size (area) and ANP expression (pixels) were calculated using Image J software. GraphPad Prism was used to examine statistical significance. One-way ANOVA with Tukey’s post-hoc analysis was used. Significance was set at p<0.05. E) Cell area and F) ANP expression was quantified.
Activation of intracellular signaling cascades such as the mitogen-activated protein kinase (MAPK) pathway as well as re-activation of fetal genes are common features involved in pathological cardiac hypertrophy. Moreover, HDAC inhibition has been shown to attenuate MAPK activation [33] and the fetal gene program [16,17]. We thus hypothesized that emodin would suppress MAPK activation and attenuate the fetal gene ANF in NRVMs. To test this hypothesis, PE treated NRVMs were co-spiked with emodin as described above. Cells were then lysed for immunoblotting experiments or fixed and stained for ANF protein expression. Similar to cardiac hypertrophy, emodin significantly attenuated the MAPK, extracellular signal-regulated kinase (ERK) phosphorylation (Supp. Fig. 2A and 2B). In addition, emodin attenuated ANF (part of the fetal gene program) protein expression (Fig. 4A, 4C, 4D, 4F, Supp. Fig. 2A and 2C). Similar to emodin, rhubarb (P4L) also inhibited ANF protein expression (Fig. 4A, 4C, 4D, 4F).
3.4. Emodin reversed stress-induced changes in the cardiomyocyte transcriptome similar to TSA
HDAC inhibitors such as the well-established inhibitor TSA regulate differential gene expression in cardiomyocytes [34]. Moreover, HDAC inhibitors attenuate the fetal gene program. We thus postulated that emodin and TSA would similarly reverse PE-induced differential gene expression in NRVMs. To test this postulate, cells were treated with vehicle or PE. PE-treated NRVMs were co-spiked with either vehicle control, emodin (10 μM) or TSA (200 nM), incubated for 48 hours and then lysed for RNA-sequencing. We report that emodin normalized 54 genes that were upregulated with PE, 30 of which overlapped with TSA (Fig. 5A and 5B). Furthermore, 18 genes were normalized with emodin that were downregulated in PE-treated NRVMs, 12 genes overlapped with TSA (Fig. 5A and 5B). As mentioned above, pathological cardiac hypertrophy is linked to re-activation of the fetal gene program and HDAC inhibitors attenuate this re-activation [16,17]. In keeping with these reports, emodin reversed agonist-induced mRNA expression of the fetal genes atrial natriuretic peptide (ANP) and brain natriuretic peptide (BNP) (Fig. 5C), as examined by qPCR. Finally, our heat map showed that emodin also upregulated genes that were suppressed by PE. Indeed, qPCR demonstrated that emodin increased potassium voltage-gated channel subfamily C member 3 (Kcnc3) mRNA (Fig. 5C). Combined, these data demonstrate that emodin reverses stress-induced differential gene expression in cardiomyocytes, similar to the pan-HDAC inhibitor TSA.
Figure 5. Emodin and TSA normalized PE-mediated cardiac gene expression changes.
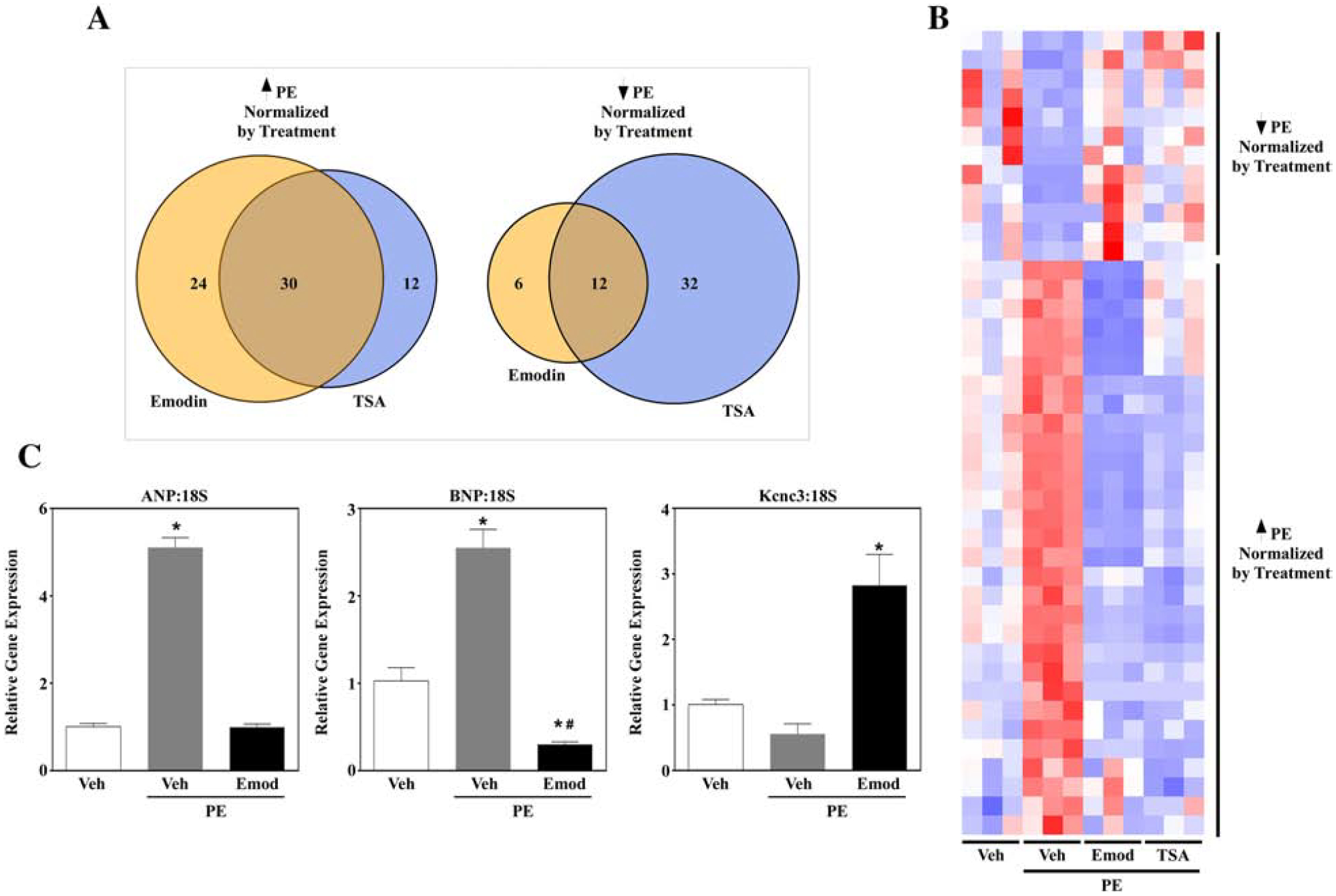
Neonatal rat ventricular myocytes (NRVMs) were stimulated to hypertrophy with phenylephrine (10 μM; PE) in the absence or presence of Emodin (10 μM) or Trichostatin A (200 nM; TSA) for 48 hrs. RNA was isolated via Trizol prior to RNA-sequencing analysis. A) Venn diagrams demonstrate gene expression overlap for emodin and TSA. TSA and emodin normalized PE-induced cardiac gene (A; Left Panel) as well as PE-suppressed (A; Right Panel) gene expression. B) A heat map representing overlap for TSA and emodin normalization of PE-mediated genes. C) Pathological hypertrophy is linked to re-activation of fetal genes. NRVMs stimulated as described above were lysed for RNA. Quantitative PCR was used to examine fetal gene profiles for atrial natriuretic peptide (ANP), brain natriuretic peptide (BNP) and potassium voltage-gated channel subfamily C member 3 (Kcnc3). GraphPad Prism was used to examine statistical significance via one-way ANOVA with Tukey’s post-hoc. Significance was set at p<0.05.
3.5. Emodin attenuated pathological cardiac hypertrophy and fibrosis in angiotensin II-infused mice
To elucidate the role of emodin in regulating cardiac hypertrophy in vivo, we treated angiotensin II (Ang)-infused male and female C57BL/6 mice with or without 30 mg/kg/day of emodin (Emod). After 14-days with Ang treatment in the absence or presence of Emod, heart weight (HW) and left ventricle (LV) weight was examined and date normalized to tibia length (TL). As anticipated, Ang significantly increased systolic blood pressure (Table 1), HW and LV weight in male and female mice compared to vehicle control mice (Fig. 6A and 6C). However, HW and LV weight were significantly attenuated with Emod treatment for male or female mice, Emod did not differ from Veh control (Fig. 6A and 6C). Interestingly, differences in cardiac hypertrophy were not due to attenuation of systolic blood pressure, as no significant difference was observed between Ang and Ang+Emod male and female mice (Table 1). HDAC inhibitors have also been shown to block angiotensin II-induced cardiac fibrosis [32]. Consistent with this, Emod significantly attenuated Ang-induced cardiac fibrosis in male and female mice (Fig. 6B and 6D). Of note, treatment with Emod significantly inhibited class I and IIa HDAC activity compared to Ang-treated male and female mice (Supplemental Fig. 3). Combined, these data suggest that emodin protects the heart from pathological cardiac hypertrophy and fibrosis in vivo, in part, by inhibiting HDAC activity.
Table 1.
Body weight and blood pressure of animals in the emodin prevention trial
| Emodin Prevention Trial | ||||||
|---|---|---|---|---|---|---|
| Males | Females | |||||
| Vehicle (n=10) | ANG II (n=13) | ANG II + Emodin (n=9) | Vehicle (n=8) | ANG II (n=8) | ANG II +Emodin (n=8) | |
| Study Start BW (g) | 26.38 ± 2.44 | 26.40 ± 2.47 | 26.55 ± 1.75 | 19.08 ± 0.83 | 18.83 ± 1.05 | 18.90 ± 1.09 |
| Study End BW (g) | 26.89 ± 2.44 | 27.43 ± 2.39 | 24.07 ± 1.62 | 21.16 ± 0.87 | 21.25 ± 0.56 | 20.09 ± 1.45 |
| Systolic Blood Pressure (mmHg) | 124.95 ± 16.33 | 141.17 ± 15.80* | 134.85 ± 21.11* | 123.98 ± 9.36 | 149.32 ± 14.77* | 137.59 ± 8.99* |
ANG II; Angiotensin II; Data were analyzed by One-way ANOVA, with Tukey’s post-hoc. Significance was defined as p<0.05.
Figure 6. Emodin attenuated angiotensin II-induced pathological hypertrophy and fibrosis in male and female mice.
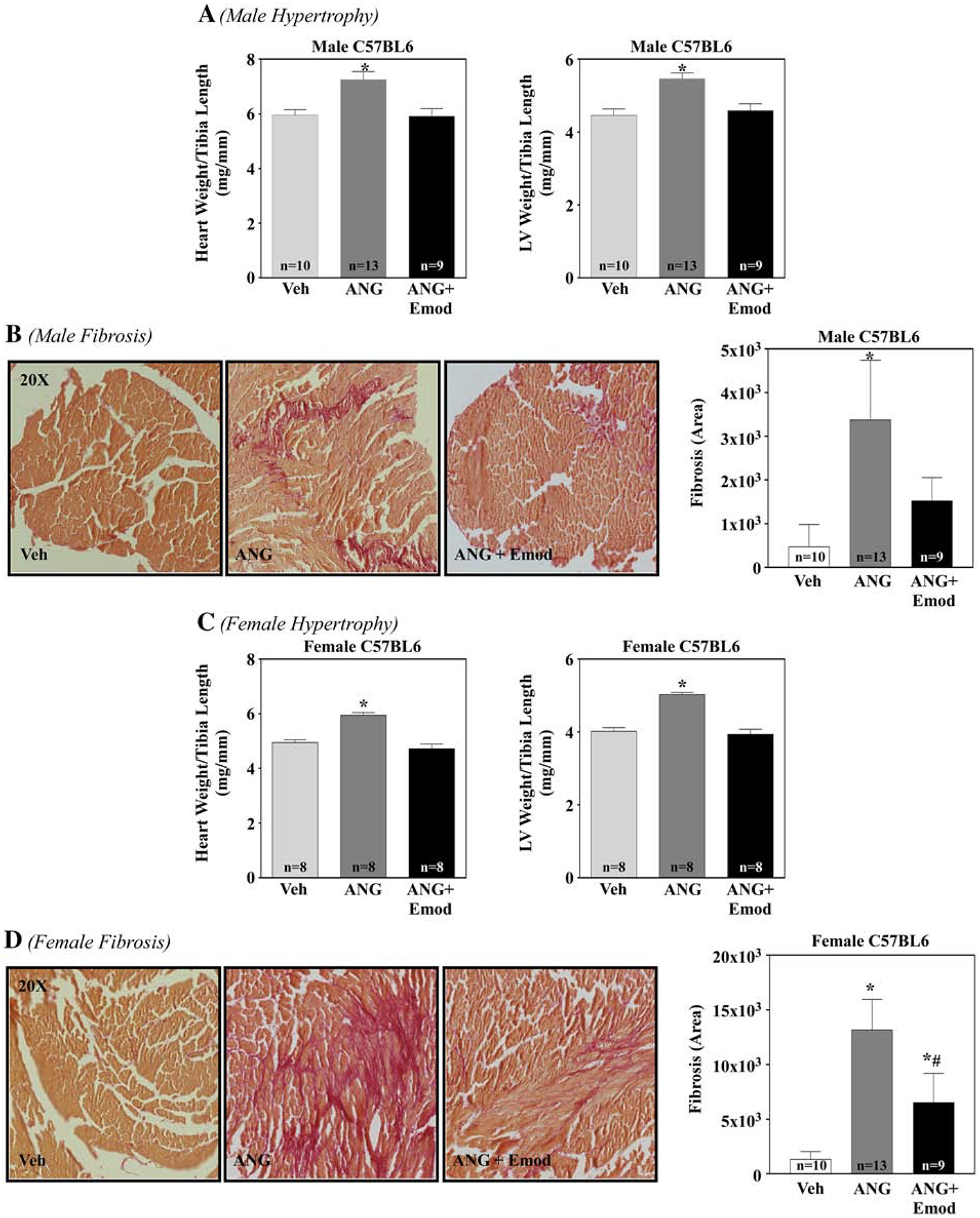
C57BL/6 male and female mice were surgically implanted with a sham (Veh) or micro-osmotic pump containing angiotensin II (Ang; 1.5 μg/kg/min) and dosed with or without emodin (Emod, 30 mg/kg/day) for 14 days. 14 days post Ang, whole hearts and left ventricles were dissected and assessed for hypertrophy and fibrosis. Whole heart weight to tibia length (HW/TL, mg/mm) and left ventricle weight to tibia length (LV/TL, mg/mm) of sham (Veh), Ang and Ang+Emod treated C57BL/6 male (A) and female (C) mice were examined at study end (14 days). Left ventriclular collagen of sham (Veh), Ang and Ang+Emod treated C57BL/6 mice was assessed via PicroSirius Red staining for male (B) and female (D) mice. ImageJ software was used to determine fibrotic area. GraphPad Prism was used to examine statistical significance. One-way ANOVA with Tukey’s post-hoc analysis was used. Significance was set at p<0.05.
4. Discussion:
In this study, we showed that emodin attenuated pathological cardiac hypertrophy in vitro and in vivo, with in vivo findings further demonstrating that emodin attenuated cardiac fibrosis. A rhubarb extract rich in emodin, as confirmed via HPLC (Fig. 2), similarly attenuated agonist-induced cardiomyocyte hypertrophy. These cardioprotective events correlated with increased histone acetylation and attenuated HDAC activity in NRVMs treated with emodin. Additionally, cardiac lysate spiked with either emodin or rhubarb inhibited HDAC activity in a dose-dependent, fast-on, slow-off kinetic manner. Emodin also inhibited HDAC activity in the hearts of mice exposed to angiotensin II. Finally, PE induced differential changes in the cardiomyocyte transcriptome; emodin reversed these changes similar to the well-established pan-HDAC inhibitor, TSA. Combined, our data support our postulate that dietary food bioactive HDAC inhibitors like emodin attenuate cardiac hypertrophy via transcriptome-wide changes in gene expression.
Class I and II HDAC inhibition is efficacious in primary cell culture and animal models of pathological cardiac hypertrophy. Antos and colleagues [16] first reported the pan-HDAC inhibitor, TSA dose-dependently attenuated cardiomyocyte hypertrophy; this was concomitant to increased histone acetylation and inhibition of the fetal gene program, which is a set of genes(e.g., ANF and BNP) that are re-activated in hypertrophic models in vitro and in vivo as well as in human HF [35–37]. Later reports further demonstrated that TSA increased histone acetylation and attenuated cardiac hypertrophy and fetal gene program re-activation in mice exposed to pressure-overload-induced hypertrophy [17]. Consistent with these reports, our data showed that emodin inhibited cardiac myocyte hypertrophy concomitant to increased histone acetylation and inhibition of HDAC activity and the fetal gene program. HDACs regulate the removal of acetyl marks from nucleosomal histones and as such control DNA accessibility leading to global changes in gene expression [38]. Not surprisingly, TSA has been shown to alter the transcriptome in NRVMs, normalizing gene expression changes in response to pathological stress [16,39]. Consistent with these findings, emodin reversed stress-induced changes in the cardiomyocyte transcriptome similar to TSA, supporting the postulate that emodin inhibits cardiac enlargement via epigenetic regulation of HDAC activity. It should be noted however, that complete overlap was not observed between emodin and TSA, suggesting that emodin potentially regulates gene expression through other diet-gene mechanisms. Of interest, some of these overlapping genes were involved in pathological cardiac hypertrophy and muscle contraction including the myosin light and heavy chains and cardiac troponin as well as inflammatory mediators like interleukin 6. Despite the non-complete overlap in gene expression, these are the first reports, to our knowledge, demonstrating transcriptome wide changes in cardiac myocytes in response to the dietary HDAC inhibitor emodin.
In addition to pan-HDAC inhibitors, class I selective HDAC inhibitors have been shown to increase histone acetylation and inhibit cardiomyocyte hypertrophy in NRVMs [40]. Moreover, class I selective HDAC inhibition was shown to block transverse aortic constriction-induced cardiac hypertrophy [40] as well as angiotensin II-induced fibrosis in mice [32]. Unlike class I selective inhibitors, inhibition of the class II HDAC, HDAC6 was shown to improve cardiac contractile function in a mouse model of hypertension, independent of changes to cardiac hypertrophy [41]. Combined, these reports would suggest that targeting class I and II HDACs contribute to cardioprotection via improvements in cardiac enlargement and contractile function. In this study, we report that emodin and emodin-rich rhubarb inhibited class I and II HDAC activity, suggesting that cardioprotection noted for fruit and vegetable intake is likely mediated through a myriad of epigenetic and non-epigenetic mechanisms that contribute to normalization of the transcriptome and improvements in muscle function.
Early reports involving non-epigenetic regulation showed that emodin ameliorated oxidative stress and inflammation in the heart [42–44]. For example, emodin attenuated inflammation and apoptosis in cardiac myocytes in response to ischemia/reperfusion (I/R) [42]. Here, Ye and colleagues [42] showed that emodin dose-dependently reduced the nuclear factor kappa B (NF-κB) inflammasome pathway. Many intracellular signaling cascades play fundamental roles in regulating pathological cardiac hypertrophy and contribute to cardiac dysfunction, including NF-κB and the MAPKs [45–48]. For example, nuclear ERK activation has been shown sufficient to drive cardiac enlargement and fibrosis in mice [47,48]. Of significance, HDAC inhibitors, including TSA, have been shown to down-regulate NF-κB [49] and MAPK activation [33]. In particular, class I inhibition of HDACs was shown to attenuate ERK activation in cardiac myocytes; this partially contributed to inhibition of pathological cardiac hypertrophy [33]. Consistent with this report, we showed that emodin inhibited ERK phosphorylation in NRVMs. Combined, these data would suggest that emodin elicits cardioprotection via regulation of intracellular signaling cascades that are dependent on HDAC activity (e.g. ERK and NF-κB).
Emodin is found in many plants, with high concentrations noted in rhubarb [18,19]. We were unable to find studies that examined rhubarb consumption or emodin bioavailability from rhubarb consumption, yet would speculate low circulating levels of the compound after a meal. While it is unlikely that rhubarb is consumed frequently or in large amounts, it should be noted that our dose of emodin approximated 10 μM in 100 mg/L of rhubarb, our dose used in these studies. This dose of rhubarb would be considered low in a meal. In addition, we reported fast-on and slow-off HDAC inhibition with rhubarb or emodin. From these kinetic data, we would speculate that rhubarb need not be ingested frequently for HDAC inhibition. Lastly, we would argue that an emodin dietary supplement could also be considered for HDAC inhibition. Despite these speculations, however, emodin bioavailability remains low [18,50], suggesting the need for research examining its interactions with the microbiome on heart health. Others have taken a more direct approach by examining ways to improve emodin bioavailability e.g. with nanoparticle encapsulation [51].
In this study we focused on emodin within rhubarb and neglected the actions for the other phytochemicals present. These additional phytochemicals likely played a role in the discrepancy observed against the recombinant HDACs, in which emodin inhibited all HDACs but HDAC 4 while rhubarb inhibited all HDACs except for HDACs 2, 4 and 6 (Supplemental Fig. 1). It should be noted, however, that emodin and rhubarb similarly attenuated stress-induced cardiac hypertrophy in NRVMs, suggesting that emodin is the primary compound in rhubarb that epigenetically alters the transcriptome contributing to anti-hypertrophic actions in the heart.
Lastly, reports have reviewed the therapeutic potential of HDAC inhibition in pre-clinical models of HF [52,53]; however, no study or trial has been published (to our knowledge) that cites HDAC inhibition as the primary or secondary target of any pharmacological agent in the human heart. Several FDA-approved HDAC inhibitors, such as Vorinostat (i.e. SAHA), are currently on the market to treat human T-cell lymphoma, with several more in clinical trials for various cancers (clinicaltrials.gov). It should be noted however that Xie and colleagues [54] showed efficacy for Vorinostat in I/R-induced cardiac dysfunction; this study was important as it showed that HDAC inhibitors could reverse stress-induced cardiac damage in a large animal model (i.e. rabbit) and therefore has established efficacy for moving SAHA into clinical trials for HF patients. However, clinical trials are expensive and the duration from pre-clinical experiments to FDA-approval is lengthy. Dietary compounds provide intriguing preventative or therapeutic options for their current lack-of-oversight from the FDA, per the Dietary Supplement Health and Education Act of 1994 (DSHEA), and can reach the market without human study in a timely manner. It will be interesting to see if emodin attenuates pathological cardiac hypertrophy via HDAC inhibition in an animal model of HF. These experiments are currently underway.
Supplementary Material
Recombinant HDACs were incubated with A) emodin (10 μM) or B) turkey rhubarb (100 mg/Ll P4L) for 2 hours prior to the addition of cell permeable HDAC substrates. Developer solution was added for 20 minutes fluorescence assessed via BioTek Synergy plate reader with excitation/emission set at 360/460 nm. GraphPad Prism software was used to visualize data and determine significance (p<0.05). One-way ANOVA with Bonferroni’s Post-hoc was used to determine significance.
A) Neonatal rat ventricular myocytes (NRVMs) were incubated with emodin (10 μM; Emod) for 48 hours prior to protein lysis. Cells were lysed and incubated against antibodies for atrial natriuretic peptide (ANP), phosphorylated ERK1/2 (phospho-ERK1/2) and total-ERK1/2 prior to analysis via immunoblot. B) Phospho-ERK1/2 and C) ANP were normalized to total-ERK1/2 and quantitation performed via Image J software. All statistical analyses were run in GraphPad Prism Software. One-way ANOVA with Tukey’s Post-hoc analysis was used to determine significance set at p<0.05.
Classe I, IIa and IIb HDAC activity was examined in the left ventricles (LV) of AngII-treated male (A) and female (B) C57BL/6 mice treated with or without emodin (Emod) for 14 days. LV lysate was incubated with substrates selective of either class I, IIa or IIb HDACs for 2 hours prior to developer solution and fluorescence assessment via the BioTek Synergy plate reader (excitation/emission set at 360/460 nm). GraphPad Prism software was used to visualize data and determine significance (p<0.05). Student’s t-test with Welch’s Correction was used to determine significance.
Funding:
This work is supported by the USDA NIFA (Hatch-NEV00727, Hatch-NEV00767), the Dennis Meiss & Janet Ralston Fund for Nutri-epigenetic Research, the National Institute for General Medical Sciences (NIGMS) of the NIH (P20 GM130459) and the National Heart, Lung, and Blood Institute of the NIH (R15 HL143496) and NSF EPSCOR Track II (OIA-1826801) to B.S.F. Core facilities used for Research were supported by NIGMS of the NIH (P20GM103554).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Benjamin EJ, Virani SS, Callaway CW, Chang AR, Cheng S, Chiuve SE, et al. Heart Disease and Stroke Statistics—2018 Update: A Report From the American Heart Association. Circulation 2018;137:CIR.0000000000000558. doi: 10.1161/CIR.0000000000000558. [DOI] [PubMed] [Google Scholar]
- [2].Lim SS, Vos T, Flaxman AD, Danaei G, Shibuya K, Adair-Rohani H, et al. A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990–2010: A systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012;380:2224–60. doi: 10.1016/S0140-6736(12)61766-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Butler T. Dietary management of heart failure: room for improvement? Br J Nutr 2016;115:1202–17. doi: 10.1017/S000711451500553X. [DOI] [PubMed] [Google Scholar]
- [4].US Department Health and Human Services, Dietary Guidelines Advisory Committee. Scientific Report of the 2015 Dietary Guidelines Advisory Committee. Washingt USDA US Dep Heal Hum Serv 2015;53:1689–99. doi: 10.1017/CBO9781107415324.004. [DOI] [Google Scholar]
- [5].Upadhyay S, Dixit M. Role of polyphenols and other phytochemicals on molecular signaling. Oxid Med Cell Longev 2015;2015:1–15. doi: 10.1155/2015/504253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Levitan EB, Lewis CE, Tinker LF, Eaton CB, Ahmed A, Manson JE, et al. Mediterranean and DASH diet scores and mortality in women with heart failure: The Women’s Health Initiative. Circ Heart Fail 2013;6:1116–23. doi: 10.1161/CIRCHEARTFAILURE.113.000495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Fardet A, Boirie Y. Associations between food and beverage groups and major dietrelated chronic diseases: An exhaustive review of pooled/meta-analyses and systematic reviews. Nutr Rev 2014;72:741–62. doi: 10.1111/nure.12153. [DOI] [PubMed] [Google Scholar]
- [8].McEvoy CT, Temple N, Woodside JV. Vegetarian diets, low-meat diets and health: a review. Public Health Nutr 2012;15:2287–94. doi: 10.1017/S1368980012000936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Schwingshackl L, Boeing H, Stelmach-Mardas M, Gottschald M, Dietrich S, Hoffmann G, et al. Dietary Supplements and Risk of Cause-Specific Death, Cardiovascular Disease, and Cancer: A Systematic Review and Meta-Analysis of Primary Prevention Trials. Adv Nutr An Int Rev J 2017;8:27–39. doi: 10.3945/an.116.013516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Islam MA, Alam F, Solayman M, Khalil MI, Kamal MA, Gan SH. Dietary Phytochemicals: Natural Swords Combating Inflammation and Oxidation-Mediated Degenerative Diseases. Oxid Med Cell Longev 2016;2016:1–25. doi: 10.1155/2016/5137431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Egert S, Bosy-Westphal A, Seiberl J, Kürbitz C, Settler U, Plachta-Danielzik S, et al. Quercetin reduces systolic blood pressure and plasma oxidised low-density lipoprotein concentrations in overweight subjects with a high-cardiovascular disease risk phenotype: A double-blinded, placebo-controlled cross-over study. Br J Nutr 2009;102:1065–74. doi: 10.1017/S0007114509359127. [DOI] [PubMed] [Google Scholar]
- [12].Seifried HE, Anderson DE, Fisher EI, Milner JA. A review of the interaction among dietary antioxidants and reactive oxygen species. J Nutr Biochem 2007;18:567–79. doi: 10.1016/j.jnutbio.2006.10.007. [DOI] [PubMed] [Google Scholar]
- [13].Perez-Gregorio R, Simal-Gandara J. A Critical Review of Bioactive Food Components, and of their Functional Mechanisms, Biological Effects and Health Outcomes. Curr Pharm Des 2017;23:2731–41. doi: 10.2174/1381612823666170317122913. [DOI] [PubMed] [Google Scholar]
- [14].Evans L, Ferguson B. Food Bioactive HDAC Inhibitors in the Epigenetic Regulation of Heart Failure. Nutrients 2018;10:1120. doi: 10.3390/nu10081120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Yang X-JJ, Seto E. HATs and HDACs: from structure, function and regulation to novel strategies for therapy and prevention. Oncogene 2007;26:5310–8. doi: 10.1038/sj.onc.1210599. [DOI] [PubMed] [Google Scholar]
- [16].Antos CL, McKinsey TA, Dreitz M, Hollingsworth LM, Zhang CL, Schreiber K, et al. Dose-dependent blockade to cardiomyocyte hypertrophy by histone deacetylase inhibitors. J Biol Chem 2003;278:28930–7. doi: 10.1074/jbc.M303113200. [DOI] [PubMed] [Google Scholar]
- [17].Kong Y, Tannous P, Lu G, Berenji K, Rothermel BA, Olson EN, et al. Suppression of class I and II histone deacetylases blunts pressure-overload cardiac hypertrophy. Circulation 2006;113:2579–88. doi: 10.1161/CIRCULATIONAHA.106.625467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Dong X, Fu J, Yin X, Cao S, Li X, Lin L, et al. Emodin: A Review of its Pharmacology, Toxicity and Pharmacokinetics. Phyther Res 2016;30:1207–18. doi: 10.1002/ptr.5631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Mueller SO, Schmitt M, Dekant W, Stopper H, Schlatter J, Schreier P, et al. Occurrence of emodin, chrysophanol and physcion in vegetables, herbs and liquors. Genotoxicity and anti-genotoxicity of the anthraquinones and of the whole plants. Food Chem Toxicol 1999;37:481–91. doi: 10.1016/S0278-6915(99)00027-7. [DOI] [PubMed] [Google Scholar]
- [20].Li Y, Liu H, Ji X, Li J. Optimized separation of pharmacologically active anthraquinones in Rhubarb by capillary electrochromatography. Electrophoresis, vol. 21, 2000, p. 3109–15. doi:. [DOI] [PubMed] [Google Scholar]
- [21].Du Y, Ko KM. Effects of emodin treatment on mitochondrial ATP generation capacity and antioxidant components as well as susceptibility to ischemia-reperfusion injury in rat hearts: Single versus multiple doses and gender difference. Life Sci 2005;77:2770–82. doi: 10.1016/j.lfs.2005.03.027. [DOI] [PubMed] [Google Scholar]
- [22].Wu Y, Tu X, Lin G, Xia H, Huang H, Wan J, et al. Emodin-mediated protection from acute myocardial infarction via inhibition of inflammation and apoptosis in local ischemic myocardium. Life Sci 2007;81:1332–8. doi: 10.1016/j.lfs.2007.08.040. [DOI] [PubMed] [Google Scholar]
- [23].Godoy LD, Lucas JE, Bender AJ, Romanick SS, Ferguson BS. Targeting the epigenome: Screening bioactive compounds that regulate histone deacetylase activity. Mol Nutr Food Res 2016:1600744. doi: 10.1002/mnfr.201600744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Lemon DD, Horn TR, Cavasin MA, Jeong MY, Haubold KW, Long CS, et al. Cardiac HDAC6 Catalytic Activity is Induced in Response to Chronic Hypertension. J Mol Cell Cardiol 2012;51:41–50. doi: 10.1016/j.yjmcc.2011.04.005.Cardiac. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Heltweg B, Dequiedt F, Marshall BL, Brauch C, Yoshida M, Nishino N, et al. Subtype selective substrates for histone deacetylases. J Med Chem 2004;47:5235–43. doi: 10.1021/jm0497592. [DOI] [PubMed] [Google Scholar]
- [26].Palmer JN, Hartogensis WE, Patten M, Fortuin FD, Long CS. Interleukin-1β induces cardiac myocyte growth but inhibits cardiac fibroblast proliferation in culture. J Clin Invest 1995;95:2555–64. doi: 10.1172/JCI117956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Reid BG, Stratton MS, Bowers S, Cavasin MA, Demos-Davies KM, Susano I, et al. Discovery of novel small molecule inhibitors of cardiac hypertrophy using high throughput, high content imaging. J Mol Cell Cardiol 2016;97:106–13. doi: 10.1016/j.yjmcc.2016.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Deluca DS, Levin JZ, Sivachenko A, Fennell T, Nazaire MD, Williams C, et al. RNA-SeQC: RNA-seq metrics for quality control and process optimization. Bioinformatics 2012. doi: 10.1093/bioinformatics/bts196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Patro R, Mount SM, Kingsford C. Sailfish enables alignment-free isoform quantification from RNA-seq reads using lightweight algorithms. Nat Biotechnol 2014. doi: 10.1038/nbt.2862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Cunningham F, Amode MR, Barrell D, Beal K, Billis K, Brent S, et al. Ensembl 2015. Nucleic Acids Res 2015. doi: 10.1093/nar/gku1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Robinson MD, McCarthy DJ, Smyth GK. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2009. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Williams SM, Golden-Mason L, Ferguson BS, Schuetze KB, Cavasin MA, Demos-Davies K, et al. Class I HDACs regulate angiotensin II-dependent cardiac fibrosis via fibroblasts and circulating fibrocytes. J Mol Cell Cardiol 2014;67:112–25. doi: 10.1016/j.yjmcc.2013.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Ferguson BS, Harrison BC, Jeong MY, Reid BG, Wempe MF, Wagner FF, et al. Signaldependent repression of DUSP5 by class I HDACs controls nuclear ERK activity and cardiomyocyte hypertrophy. Proc Natl Acad Sci U S A 2013;110:9806–11. doi: 10.1073/pnas.1301509110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Blakeslee WW, Lin YH, Stratton MS, Tatman PD, Hu T, Ferguson BS, et al. Class I HDACs control a JIP1-dependent pathway for kinesin-microtubule binding in cardiomyocytes. J Mol Cell Cardiol 2017;112:74–82. doi: 10.1016/j.yjmcc.2017.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Yoshimura M, Yasue H, Okumura K, Ogawa H, Jougasaki M, Mukoyama M, et al. Different secretion patterns of atrial natriuretic peptide and brain natriuretic peptide in patients with congestive heart failure. Circulation 1993;87:464–9. doi: 10.1161/01.CIR.87.2.464. [DOI] [PubMed] [Google Scholar]
- [36].Brown LA, Nunez DJR, Wilkins MR. Differential regulation of natriuretic peptide receptor messenger RNAs during the development of cardiac hypertrophy in the rat. J Clin Invest 1993;92:2702–12. doi: 10.1172/JCI116887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Nakagawa O, Itoh H, Harada M, Komatsu Y, Yoshimasa T, Nakao K. Gene regulation of brain natriuretic peptide in cardiocyte hypertrophy by alpha1-adrenergic stimulation. Clin Exp Pharmacol Physiol Suppl 1995;22:S183–5. [DOI] [PubMed] [Google Scholar]
- [38].Ito K, Adcock IM. Histone Acetylation and Histone Deacetylation. Mol Biotechnol 2003;20:099–106. doi: 10.1385/mb:20:1:099. [DOI] [PubMed] [Google Scholar]
- [39].Kaneda R, Ueno S, Yamashita Y, Choi YL, Koinuma K, Takada S, et al. Genome-wide screening for target regions of histone deacetylases in cardiomyocytes. Circ Res 2005;97:210–8. doi: 10.1161/01.RES.0000176028.18423.07. [DOI] [PubMed] [Google Scholar]
- [40].Gallo P, Latronico MVG, Gallo P, Grimaldi S, Borgia F, Todaro M, et al. Inhibition of class I histone deacetylase with an apicidin derivative prevents cardiac hypertrophy and failure. Cardiovasc Res 2008;80:416–24. doi: 10.1093/cvr/cvn215. [DOI] [PubMed] [Google Scholar]
- [41].Demos-Davies KM, Ferguson BS, Cavasin MA, Mahaffey JH, Williams SM, Spiltoir JI, et al. HDAC6 contributes to pathological responses of heart and skeletal muscle to chronic angiotensin-II signaling. AJP Hear Circ Physiol 2014;307:H252–8. doi: 10.1152/ajpheart.00149.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Ye B, Chen X, Dai S, Han J, Liang X, Lin S, et al. Emodin alleviates myocardial ischemia/reperfusion injury by inhibiting gasdermin D-mediated pyroptosis in cardiomyocytes. Drug Des Devel Ther 2019;Volume 13:975–90. doi: 10.2147/dddt.s195412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Zhang X, Qin Q, Dai H, Cai S, Zhou C, Guan J. Emodin protects h9c2 cells from hypoxiainduced injury by up-regulating mir-138 expression. Brazilian J Med Biol Res 2019;52:e7994. doi: 10.1590/1414-431x20187994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Xu K, Zhou T, Huang Y, Chi Q, Shi J, Zhu P, et al. Anthraquinone emodin inhibits tumor necrosis factor alpha-induced calcification of human aortic valve interstitial cells via the NF-κB pathway. Front Pharmacol 2018;9:1328. doi: 10.3389/fphar.2018.01328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Hall G, Hasday JD, Rogers TB. Regulating the regulator: NF-κB signaling in heart. J Mol Cell Cardiol 2006;41:580–91. doi: 10.1016/j.yjmcc.2006.07.006. [DOI] [PubMed] [Google Scholar]
- [46].Wang Y. Mitogen-activated protein kinases in heart development and diseases. Circulation 2007;116:1413–23. doi: 10.1161/CIRCULATIONAHA.106.679589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Ruppert C, Deiss K, Herrmann S, Vidal M, Oezkur M, Gorski A, et al. Interference with ERKThr188 phosphorylation impairs pathological but not physiological cardiac hypertrophy. Proc Natl Acad Sci 2013;110:7440–5. doi: 10.1073/pnas.1221999110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Lorenz K, Schmitt JP, Schmitteckert EM, Lohse MJ. A new type of ERK1/2 autophosphorylation causes cardiac hypertrophy. Nat Med 2009;15:75–83. doi: 10.1038/nm.1893. [DOI] [PubMed] [Google Scholar]
- [49].Place RF, Noonan EJ, Giardina C. HDAC inhibition prevents NF-κB activation by suppressing proteasome activity: Down-regulation of proteasome subunit expression stabilizes IκBα. Biochem Pharmacol 2005;70:394–406. doi: 10.1016/j.bcp.2005.04.030. [DOI] [PubMed] [Google Scholar]
- [50].Liu W, Feng Q, Li Y, Ye L, Hu M, Liu Z. Coupling of UDP-glucuronosyltransferases and multidrug resistance-associated proteins is responsible for the intestinal disposition and poor bioavailability of emodin. Toxicol Appl Pharmacol 2012;265:316–24. doi: 10.1016/j.taap.2012.08.032. [DOI] [PubMed] [Google Scholar]
- [51].Wang S, Chen T, Chen R, Hu Y, Chen M, Wang Y. Emodin loaded solid lipid nanoparticles: Preparation, characterization and antitumor activity studies. Int J Pharm 2012;430:238–46. doi: 10.1016/j.ijpharm.2012.03.027. [DOI] [PubMed] [Google Scholar]
- [52].McKinsey TA. Therapeutic Potential for HDAC Inhibitors in the Heart. Annu Rev Pharmacol Toxicol 2012;52:303–19. doi: 10.1146/annurev-pharmtox-010611-134712. [DOI] [PubMed] [Google Scholar]
- [53].McKinsey TA, Olson EN. Toward transcriptional therapies for the failing heart: Chemical screens to modulate genes. J Clin Invest 2005;115:538–46. doi: 10.1172/JCI24144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Xie M, Kong Y, Tan W, May H, Battiprolu PK, Pedrozo Z, et al. Histone deacetylase inhibition blunts ischemia/reperfusion injury by inducing cardiomyocyte autophagy. Circulation 2014;129:1139–51. doi: 10.1161/CIRCULATIONAHA.113.002416. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Recombinant HDACs were incubated with A) emodin (10 μM) or B) turkey rhubarb (100 mg/Ll P4L) for 2 hours prior to the addition of cell permeable HDAC substrates. Developer solution was added for 20 minutes fluorescence assessed via BioTek Synergy plate reader with excitation/emission set at 360/460 nm. GraphPad Prism software was used to visualize data and determine significance (p<0.05). One-way ANOVA with Bonferroni’s Post-hoc was used to determine significance.
A) Neonatal rat ventricular myocytes (NRVMs) were incubated with emodin (10 μM; Emod) for 48 hours prior to protein lysis. Cells were lysed and incubated against antibodies for atrial natriuretic peptide (ANP), phosphorylated ERK1/2 (phospho-ERK1/2) and total-ERK1/2 prior to analysis via immunoblot. B) Phospho-ERK1/2 and C) ANP were normalized to total-ERK1/2 and quantitation performed via Image J software. All statistical analyses were run in GraphPad Prism Software. One-way ANOVA with Tukey’s Post-hoc analysis was used to determine significance set at p<0.05.
Classe I, IIa and IIb HDAC activity was examined in the left ventricles (LV) of AngII-treated male (A) and female (B) C57BL/6 mice treated with or without emodin (Emod) for 14 days. LV lysate was incubated with substrates selective of either class I, IIa or IIb HDACs for 2 hours prior to developer solution and fluorescence assessment via the BioTek Synergy plate reader (excitation/emission set at 360/460 nm). GraphPad Prism software was used to visualize data and determine significance (p<0.05). Student’s t-test with Welch’s Correction was used to determine significance.