

Abstract
Purpose
We hypothesized that four criteria could help identify malignant mesotheliomas (MMs) most likely linked to germline mutations of BAP1 or of other genes: family history of MM, BAP1-associated cancers, or multiple malignancies; or age younger than 50 years.
Patients and Methods
Over the course of 7 years, 79 patients with MM met the four criteria; 22 of the 79 (28%) reported possible asbestos exposure. They were screened for germline BAP1 mutations by Sanger sequencing and by targeted next-generation sequencing (tNGS) for germline mutations in 55 additional cancer-linked genes. Deleterious mutations detected by tNGS were validated by Sanger sequencing.
Results
Of the 79 patients, 43 (16 probands and 27 relatives) had deleterious germline BAP1 mutations. The median age at diagnosis was 54 years and median survival was 5 years. Among the remaining 36 patients with no BAP1 mutation, median age at diagnosis was 45 years, median survival was 9 years, and 12 had deleterious mutations of additional genes linked to cancer. When compared with patients with MMs in the SEER cohort, median age at diagnosis (72 years), median survival for all MM stages (8 months), and stage I (11 months) were significantly different from the 79 patients with MM in the current study (P < .0001).
Conclusion
We provide criteria that help identify a subset of patients with MM who had significantly improved survival. Most of these patients were not aware of asbestos exposure and carried either pathogenic germline mutations of BAP1 or of additional genes linked to cancer, some of which may have targeted-therapy options. These patients and their relatives are susceptible to development of additional cancers; therefore, genetic counseling and cancer screening should be considered.
INTRODUCTION
Malignant mesothelioma (MM) is an aggressive cancer with a median survival of approximately 12 months from diagnosis. Conventional wisdom dictates that MM occurs in professions exposed to high levels of asbestos for many years. MM is commonly diagnosed when individuals are 70 to 80 years old and approximately 30 to 60 years from initial exposure. Since the 1980s, asbestos use has been entirely banned in western Europe and restricted in the United States. Cohorts of professionals exposed to high levels of asbestos are reduced disappearing because of old age. Presently, MM is increasingly being diagnosed in young individuals, and in women, with no known history of exposure.1,2
Through a 14-year study of an epidemic of MM in Cappadocia, Turkey, where more than 50% of the population exposed to erionite fibers died of MM,3,4 we hypothesized, and then proved, that susceptibility to MM was transmitted in a Mendelian fashion. We formulated the hypothesis that the cause of the epidemic was gene–environment interaction.3-5 While investigating this hypothesis, we discovered that heterozygous germline BAP1-inactivating mutations (BAP1+/-) caused a very high incidence of MM in some US families apparently not exposed to asbestos.6 We found that BAP1 mutations and susceptibility to MM were transmitted through the course of multiple generations.7 We reported that MM developed in BAP1+/- mice (homozygous BAP1-/- knockout mice mutations are embryonically lethal) after exposure to low doses of asbestos, which rarely cause MM in wild-type mice.8 We and others found that human germline BAP1 mutations were also associated with uveal melanoma (UVM), cutaneous melanoma (CM), clear-cell renal cell carcinoma (ccRCC), and breast and other cancers.9-19 We named this condition the BAP1 cancer syndrome.9,10 In parallel studies, we and others discovered that acquired somatic BAP1 mutations and deletions were present in more than 60% of patients with MM,20-24 90% of metastatic UVM,25 15% of ccRCC,26 and in other cancers.27 BAP1 regulates DNA repair by homologous recombination,28,29 Ca2+-dependent cell death,29 and cellular metabolism.30,31 To our knowledge, it is unknown if, in addition to BAP1, germline mutations in other genes predispose to MM.
Here, we studied patients with MM who had a family history of MM and/or other cancers and/or early-onset MM. We found that inherited germline mutations are more frequent in this subgroup of patients, and that their presence influenced survival and helped identify relatives at risk for MM. We discuss opportunities for prevention and early detection in carriers of germline mutations that predispose to MM, and possible therapeutic implications.
METHODS
Study Oversight and Study Population
After we determined that carriers of BAP1+/- mutations developed MM,6 some patients with MM contacted us directly, through their physicians, or through the Mesothelioma Applied Research Foundation to have germline testing for BAP1+/-. We offered free testing to patients with pleural and peritoneal MM who met one or more of the following criteria that, based on our experience,6,7 would make them and/or their relatives more likely to carry BAP1+/- in the germline: (1) first- or second-degree relatives with MM; (2) proband or one first- or second-degree relative diagnosed with UVM, CM, and ccRCC—malignancies frequent in carriers of BAP1+/-; (3) history of multiple cancers (any cancer) in the majority of first- and second-degree relatives; and (4) early MM onset (age < 50 years; the incidence of MM before age 50 years is very rare and suggestive of genetic predisposition or environmental exposure since childhood).1,32
Written informed consent was received from all patients. Collection and use of patient information and samples were in accordance with the Declaration of Helsinki (1995) and the World Medical Association (2013 revision), approved by University of Hawaii (institutional review board [IRB] no.CHS14406), New York University (IRB no. i8896), and Hyogo College of Medicine (IRB no. RINHI244).
Over 7 years, 79 patients with MM who met the inclusion criteria were tested for BAP1+/- and were screened for mutations in 55 additional genes (including tumor suppressor genes, oncogenes, DNA repair genes, and genes somatically mutated in MM) by targeted next-generation sequencing (tNGS).
Germline DNA was extracted from saliva or peripheral blood.20 Clinical information was collected through the medical records and patient interviews. Personal and family histories of cancers and asbestos exposure were self-reported and obtained using a standardized questionnaire approved by the IRBs and complemented, when possible, with patient interviews (Appendix Fig A1, online only). Patients were observed up to 20 years.
BAP1 Sequencing and Immunohistochemistry
BAP1 was amplified by polymerase chain reaction and sequenced in its entirety.20 BAP1 staining was performed as described.20
tNGS and Data Analysis
tNGS was performed using an Illumina Truseq Custom Amplicon (Illumina, San Diego, CA), and the Agilent Haloplex Custom kit (Agilent Technologies, Santa Clara, CA) as described previously.21 tNGS sequencing data were submitted to the DDBJ Japanese genotype-phenotype archive for genetic and phenotypic human data (http://www.ncbi.nlm.nih.gov/pubmed/25477381) under accession number JGAS00000000108. Pathogenic variants were extracted using Combined Annotation Dependent Depletion (CADD) score (version 1.3; http://cadd.gs.washington.edu/), which is among the recommended strategies for selection of deleterious mutations according to the guidelines of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.33 CADD can quantitatively prioritize functional, deleterious, and disease-causing variants across a wide range of functional categories, effect sizes, and genetic architectures, and is used to prioritize causal variation in research and clinical settings.
A CADD score of 15 is the recommended cutoff to identify deleterious mutations. A CADD score of 20 indicates that a variant is among the top 1% of deleterious variants in the human genome.34 Final selection of potentially pathogenic variants was verified by the assessment of quality scores and visual inspection of the data using StrandNGS (Agilent Technologies, Santa Clara, CA). The Database for Annotation, Visualization and Integrated Discovery, which provides a comprehensive set of functional gene annotation tools, was used for functional and biologic annotation analysis (version 6.8; https://david.ncifcrf.gov/).
RESULTS
BAP1 Testing
A total of 52 unrelated patients with MM met the recruitment criteria and their DNA was sequenced for the presence of BAP1 mutations. Deleterious mutations were detected, in the BAP1 gene, in 16 patients (30.7%), and 36 carried germline wild-type BAP1 (BAP1WT). A total of 153 first- and second-degree relatives of 12 of the 16 BAP1+/- patients volunteered for BAP1 testing (n = 90 women [58.8%]; n = 63 men [41.2%]). Among them, 66 (43.1%) carried BAP1+/-: 23 men (34.8%) and 43 women (65.2%). As expected, their BAP1 mutations were identical to those found in their related probands. Among the 153 relatives, MM developed in 27 of 66 carriers of germline BAP1+/-, therefore, these relatives were included in our study, resulting in a total of 79 MM patients examined (Tables 1-3). There were no significant survival or other differences among the MMs in relatives and probands. The remaining 39 family members with BAP1+/- were too young for MM to have developed and were alive at the end of the study. MM did not develop in any of the 87 relatives with BAP1WT.
Table 1.
Patient Characteristics

Table 3.
Deleterious Germline Variants in Cancer Susceptibility Genes Identified in Patients With Familial and Early-Onset Malignant Mesothelioma

Among BAP1+/- carriers, the oldest patient in whom MM developed was age 75 years; the youngest was age 29 years (Table 1). Pathology reports documenting the tumor histologic subtype or tumor biopsy–specimen slides for review were available for 37 of 79 patients. All BAP1+/- MMs and almost all BAP1WT MMs were of the epithelial type (Table 2). Fifty-seven of 79 patients (72%) did not report any asbestos exposure. Unstained MM tissue sections available for seven patients carrying BAP1+/- were analyzed by immunohistochemistry, and no nuclear BAP1 staining was observed (Appendix Fig A2, online only), providing evidence of biallelic BAP1 inactivation.1,20,35
Table 2.
Clinical Characteristics of Familial and Early-Onset Mesotheliomas
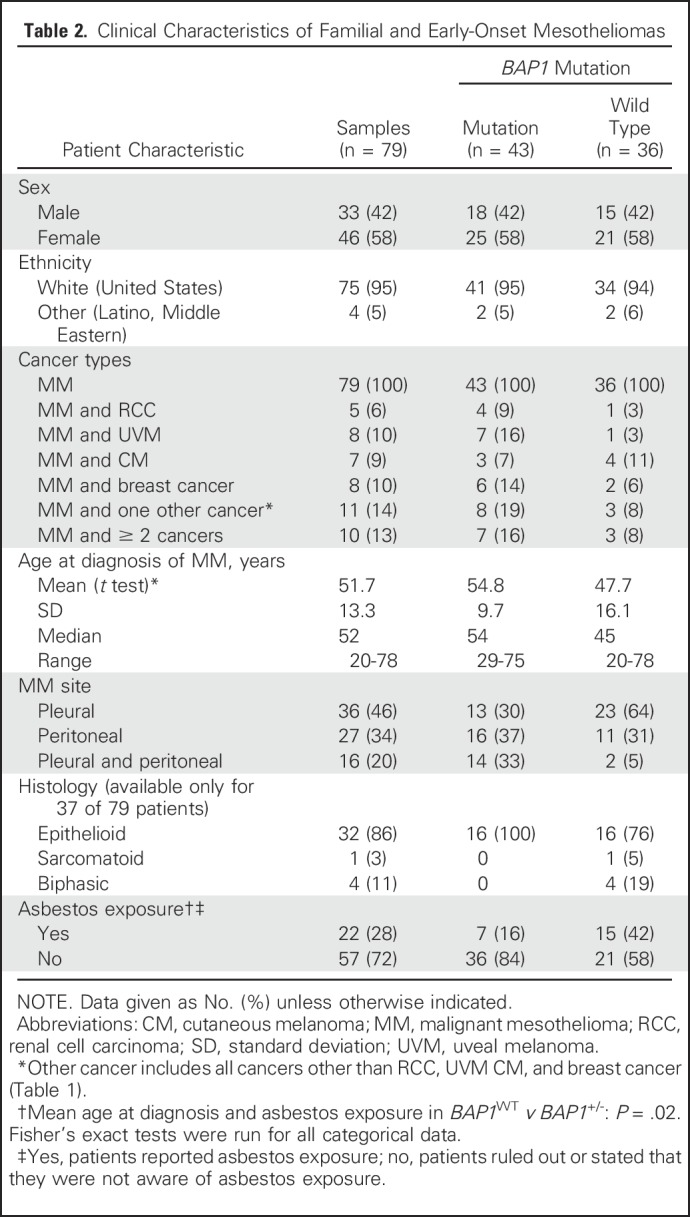
Among 33 patients with MM who were younger than 50 years, 13 carried the BAP1+/- mutation, nine had germline mutations in other genes (discussed later in this section), and 11 had no germline mutations among the genes tested. Among the 13 patients with BAP1+/- who were younger than 50 years, 12 had an extensive family history of cancer types associated with the BAP1 cancer syndrome (ie, MM, UVM, ccRCC); one had a strong family history of cancers not associated with the BAP1 cancer syndrome. In contrast, only one of 20 patients with BAP1WT who were younger than 50 years had a first-degree relative with MM (P < .01).
Survival
Survival data were available for 77 of the 79 patients with MM: 43 of 43 patients with germline BAP1+/- mutations and 34 of 36 with BAP1WT (Fig 1). Median survival was 5 years among the 43 patients with BAP1+/-, and 15% of these 43 patients were alive 10 years after diagnosis of MM, compared with a median survival of 9 years among the 34 patients with BAP1WT, of whom 41% survived ≥ 10 years after diagnosis. The survival curve in each of the two MM subgroups of our cohort, BAP1+/- and BAP1WT, was compared with that of the SEER cohort36 (Fig 1A). Median survival in the SEER cohort (all stages) was 8 months from diagnosis. When patients in SEER with stage I MM were selected, median survival was 11 months. Therefore, the MM cohort we studied, including patients with BAP1+/- or BAP1WT mutations had a significantly better survival compared with the MM SEER dataset (P < .001; this applies to all comparisons except BAP1+/- v BAP1WT, where P < .16; Fig 1A).
Fig 1.

Kaplan-Meier MM survival probability versus years with number at risk. (A) 1. Familial and early-onset BAP1WT MM (median survival, 9 years; 10-year survival, 41%); 2. Familial and early-onset BAP1+/- MM (median survival, 5 years; 10-year survival, 15%); 3. SEER, stage I (median survival, 11 months; 10-year survival, 9.2%); 4. SEER, all stages (median survival, 8 months; 10-year survival, 3.3%). (B) Familial MM cohort by onset: 1. age < 50 years (median survival, 10 years; 10-year survival, 41%); 2. age ≥ 50 years (median survival, 4 years; 10-year survival, 15%). Rows below figure indicate the number of patients alive in each cohort per year. All patients were treated in the United States. Because patients in our cohort and in the SEER cohort were treated at different institutions, we do not have information on the exact treatment each of them received. BAP1+/-, heterozygous BAP1-inactivating mutations; BAP1WT, wild-type BAP1; MM, malignant mesothelioma.
Irrespective of BAP1 status, 33 patients who developed MM before they reached age 50 years had a median survival of 10 years, compared with a median survival of 4 years among the 44 patients who developed MM at a later age (P < .0006; Fig 1B). There was no significant survival difference based on MM site, single versus multiple malignancies or patient’s sex (Appendix Fig A3, online only).
In addition to Kaplan-Meier log-rank tests, we ran a multivariate Cox proportional hazards regression analysis. The variables, entered simultaneously, were BAP1 status (BAP1+/- v BAP1WT; P = .16), age (< 50 years v ≥ 50 years; P = .004), site (pleural, peritoneal, or both; P = .19), and sex (male v female, P = .87). A notable change from the Kaplan-Meier analysis was that the difference in survival between patients with MM and other cancers and patients with MM only reached significance (P = .02). Additional studies in a larger cohort are needed to verify this finding.
tNGS
Among 36 patients with MM with BAP1WT, 34 were further screened by tNGS for germline mutations in 56 genes (including BAP1 and other tumor suppressor genes, oncogenes, DNA repair genes, and genes previously found somatically mutated in MM; Appendix Table A1, online only). In parallel, we tested 11 germline DNA samples from the MM subcohort with BAP1+/- mutations. To exclude polymorphisms, we focused on mutations with an allele frequency less than 0.005 in the Exome Aggregation Consortium (ExAC) database, which comprises 60,706 unrelated individual sequences (exac.broadinstitute.org). Next, we used the CADD score to identify pathogenic mutations among those identified as rare with ExAC. First, we evaluated all the BAP1 mutations detected, which included truncating, frameshift, splice site, and nonsynonymous mutations. All BAP1 mutations found by tNGS (which were consistent with previous Sanger sequencing data) had a CADD score greater than 20 (Table 3), supporting previous work showing that they are pathogenic1,6,7,9,10.
Therefore, we applied the same approach and cutoff values (ExAC < 0.005; CADD > 20) to identify pathogenic mutations detected using this 56-gene panel. We found that 12 of 34 patients with MM with BAP1WT (35%) contained one germline mutation in 11 of the 55 additional genes tested (two patients had two different deleterious MLH1 variants). Also, five of 11 DNA samples from patients with MM with BAP1+/- mutations contained one additional germline mutation, each in five different genes; all variants had a CADD score greater than 20 (Tables 1 and 4; Fig 2). All mutations were validated by polymerase chain reaction and Sanger sequencing.
Table 4.
Cancer Susceptibility Genes Identified as Mutated in Patients With Familial and Early-Onset Malignant Mesothelioma
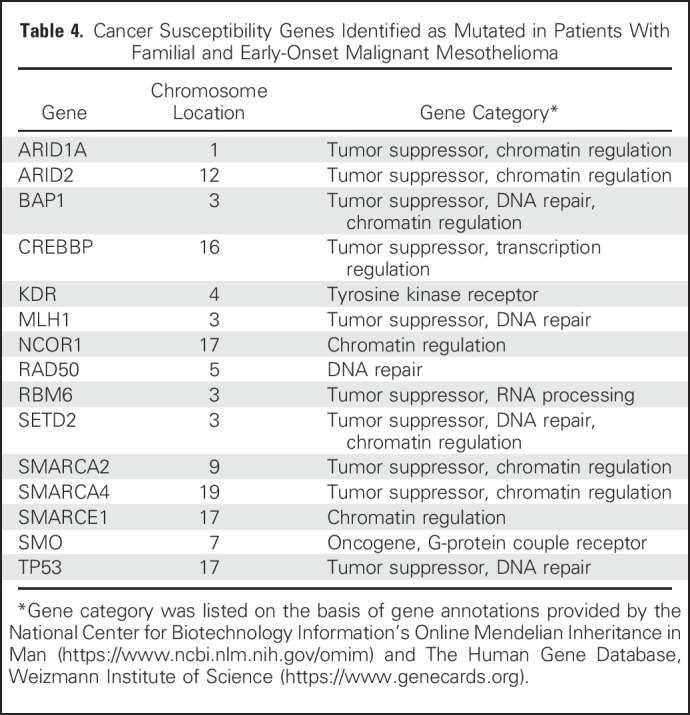
Fig 2.

Deleterious germline variants identified in patients with familial malignant mesothelioma (MM). Pie charts show the number of gene variants identified by targeted next-generation sequencing in (A) the familial MM cohort and, separately, in the (B) BAP1WT and (C) BAP1+/- family cohorts. Selected variants with a frequency < 0.005 in the Exome Aggregation Consortium database have a Combined Annotation Dependent Depletion score > 20. BAP+/-, BAP1-inactivating mutations; BAP1WT, wild-type BAP1.
DISCUSSION
Based on our experience studying families carrying germline BAP1+/-,6,7,9 we used a strict set of criteria to select those MMs that we hypothesized were likely caused by germline mutations of BAP1 or of other genes. We studied 79 patients with MM (52 probands and 27 relatives; Table 1), 43 carried germline BAP1+/- (16 probands and 27 relatives); 36 of 52 probands were BAP1WT.
Most patients with MM who carried BAP1+/- mutations had first- or second-degree relatives with MM, UVM, and ccRCC (Table 1). None of the 36 patients with MM among probands with BAP1WT had a family history of UVM or ccRCC; however, 12 of 36 had deleterious germline mutations of additional genes that, when mutated, cause cancer syndromes, such as MLH1 (Lynch syndrome),37 TP53 (Li-Fraumeni syndrome),38 and/or mutations in genes that regulate DNA repair,39 or were mutations of genes previously found somatically mutated in MM21,40(Tables 1 and 4; Fig 2).
In our cohort, selected for the clinical red flags suggesting heritability, survival was significantly better than in those with “classic” sporadic MM, even when compared with patients with stage I disease in SEER36 (Fig 1). Within our cohort, patients with BAP1 mutations had a worse survival than those with no BAP1 mutations. Sex and pleural or peritoneal location did not influence survival (Appendix Fig A3). Early age at cancer onset is often related to a genetic mutation41-43; in our cohort, the subset of patients with early age at MM onset had the best survival rate (Fig 1B). Moreover, among BAP1+/-carriers in whom multiple tumors developed, the improved prognosis seemed to apply to all tumor types.
The high percentage (72%) of patients with MM who reported no asbestos exposure in our cohort is not entirely surprising, given their relatively young age, in most cases, and because of the presence of pathogenic germline mutations. One could speculate that MM developing in the absence of asbestos exposure may have a different biology and improved prognosis.
Improved survival has also been observed in carriers of other germline mutations that predispose to various cancer syndromes. For example, patients with colorectal cancer who are affected by familial adenomatous polyposis or by Lynch syndrome have a better prognosis than do patients with sporadic colorectal cancer.44 Similarly, patients with gastric cancer who carry CDH1 mutations have a better survival rate compared with sporadic gastric cancers.45 Our current hypothesis is that the improved prognosis is caused by changes in the tumor microenvironment, in turn caused by the presence of heterozygous germline mutations in all cells. We are investigating this hypothesis.
BAP1+/- segregated in family members who were affected by cancer in this study and in previous studies.6,7,9,11-19 Among the 153 relatives tested, MM developed in 27 of 66 carriers of germline BAP1+/- (those in whom tumors did not develop were still quite young), whereas MM did not develop in any of the 87 relatives with BAP1WT. Moreover, in tumor cells of BAP1+/- carriers, BAP1 nuclear staining was absent (Appendix Fig A2), indicating loss of heterozygosity.20 Also, MM can develop in BAP1+/- mice even when not exposed to asbestos or to other carcinogens.46 These findings support causation. Some patients with BAP1+/- mutations (five of 11 tested) carried germline mutations of additional genes. It is possible that these mutations, together with BAP1+/-, contribute to and influence the tumor phenotype; in some families, there is a prevalence of MM; in others, a prevalence of UVM; and in others, a prevalence of ccRCC.1
There are some limitations to our study. Only 22 of 79 patients reported asbestos exposure, limiting the statistical power to assess any hypothesis in relation to asbestos exposure and presence of germline mutations predisposing to MM. Moreover, because the exposure was self-reported, it was subject to recall biases. It is possible that some patients carried additional germline mutations in genes not included in our 56-gene panel. Although using larger panels to identify genetic mutations increases the yield of positive findings, the results are more difficult to interpret and may cause unnecessary anxiety in patients and their relatives.47 Therefore, we designed and used a targeted gene panel including well-known tumor suppressors, genes somatically mutated in MM, and genes associated with cancer syndromes, several of which are DNA repair genes (Table 3). Among the mutations detected, we considered only those with an allele frequency less than 0.005 in the ExAC database—thus too rare to be considered polymorphisms. To further increase specificity, we used a stricter criterion (CADD score > 20) than the recommended cutoff of a CADD score greater than 15 to identify deleterious mutations.
Although we used strict criteria, it remains possible that not all these mutations contributed to tumor development; segregation of these mutations in family members with cancer, and/or loss of heterozygosity in tumor tissue, as observed for BAP1, will be required to definitively prove causality. The use of strict criteria and a tNGS strategy limited to genes that, when mutated, are anticipated to be deleterious, reduced the risk of false-positive results. However, this same strict approach increased the risk of false-negative results: deleterious mutations would not have been identified if their CADD score was less than 20 or if genes containing pathogenic mutations were not included in our gene panel. Thus, we may have underestimated the fraction of MMs linked to genetic mutations in our cohort.
Two abstracts that are relevant to our study have been presented after our initial submission. Hassan et al48 reported that 12% of 239 patients with MM, studied at the US National Cancer Institute, carried a pathogenic germline mutation—BAP1 was the most commonly affected gene (7%)—and that women, especially, had a second cancer diagnosis or had relatives with MM, melanoma, or breast cancer. They observed a significantly improved survival rate among patients with pleural MMs who were carriers of germline mutations. Panou et al49 reported that 12% of 198 patients with MM studied at the University of Chicago carried pathogenic germline mutations—BAP1 was the most common (3%)—especially those with peritoneal MM, minimal asbestos exposure, young age, and a second cancer diagnosis.
Together, these studies provide compelling evidence that there is a subset of MMs that developed in carriers of pathogenic germline mutations. Therefore, we recommend that patients with the clinical red flags denoting heritability for MM (ie, familial history of MM or other cancers and young age) should undergo genetic testing by tNGS using a gene panel similar to the one we used or a larger gene panel covering DNA repair genes and tumor suppressor genes, because these were the genes we and our colleagues39,48,49 found most commonly mutated in the germline of patients with MM. Ideally, when economically feasible, all patients with MM should be tested. A proportion of these germline mutations may be actionable, and patients can be enrolled in targeted clinical trials. Moreover, patients with MM who carry germline mutations have a significantly improved prognosis. This knowledge is relevant to the patients, their relatives, and the physicians who have to plan their care. These patients are susceptible to development of multiple cancers, and thus must be screened, at least by a thorough history and physical examination for early detection of additional malignancies, especially UVM, CM, ccRCC, and breast cancers. Detection of these malignancies could lead to treatment with curative radical excision at an earlier stage. Furthermore, carriers of germline BAP1 and TP53 mutations, and of other DNA repair genes, are much more susceptible to secondary malignancies after radiation therapy; thus, whenever possible, ultrasound and whole-body magnetic resonance imaging should be used in place of computed tomography scans.50 Finally, family members found to have inherited the same deleterious mutations will benefit from cancer screening that can be life saving.1
Genetic testing must be carried with proper support from genetic counselors, as thoroughly discussed in the context of pancreatic cancer, a malignancy with a similarly dismal prognosis as MM, and thus a malignancy that stands out for possible therapeutic benefits when actionable mutations are detected.51
ACKNOWLEDGMENT
We thank all patients with malignant mesothelioma who kindly agreed to participate in the study.
Appendix
Fig A1.

Study flow chart. (*) This age was chosen because the incidence of malignant mesothelioma (MM) before age 50 years is rare and suggestive of genetic predisposition or environmental exposure since childhood. ccRCC, clear-cell renal cell carcinoma; CM, cutaneous melanoma; MARF, Mesothelioma Applied Research Foundation; NYU, New York University; tNGS, targeted next-generation sequencing; UVM, uveal melanoma.
Fig A2.

Representative immunostain. Note absence of nuclear staining in tumor cells, which is evidence of biallelic BAP1 inactivation, whereas tumor-infiltrating mononuclear phagocytes show nuclear staining because they retain one BAP1 wild-type allele.
Fig A3.

Kaplan-Meier familial mesothelioma survival probability versus years, with number at risk. (A) MM site: 1. pleura (median survival, 6 years; 10-year survival, 25%); 2. peritoneum (median survival, 6 years; 10-year survival, 24%); 3. both (median survival, 6 years; 10-year survival, 27%). (B) Primary relatives: 1. MM only (median survival, 6 years; 10-year survival, 25%); 2. MM plus other cancers (median survival, 7 years; 10-year survival, 28%). (C) Sex: female (median survival, 7 years; 10-year survival, 25%);male (median survival, 5 years; 10-year survival, 29%). MM, malignant mesothelioma; Pe, peritoneal; Pl, pleural.
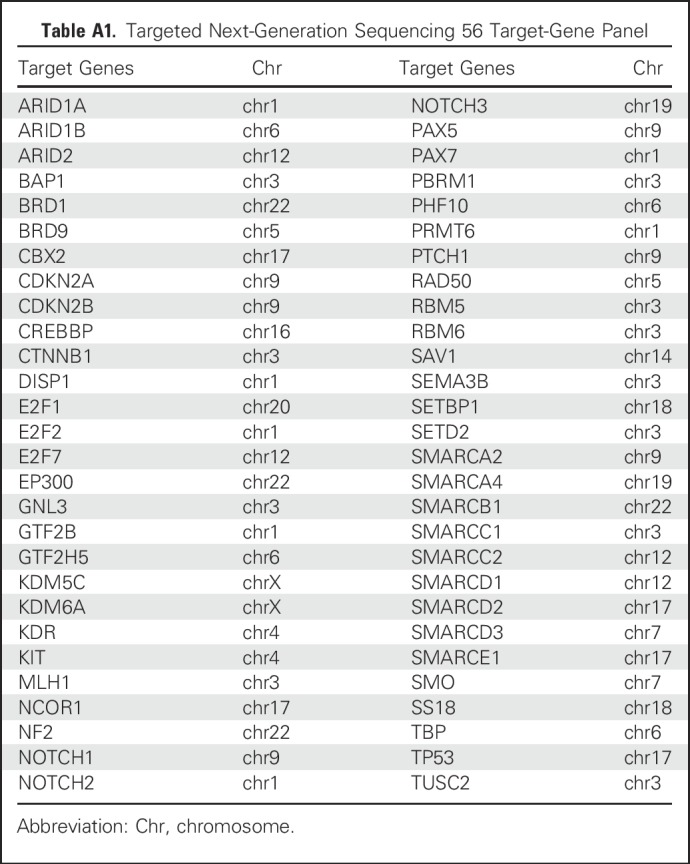
Table A1.
Targeted Next-Generation Sequencing 56 Target-Gene Panel
Footnotes
This work was supported by Department of Defense Grant No. CA150220 to H.Y. and M.C.; National Cancer Institute (NCI) Grant No. R01 CA198138 (M.C.); the University of Hawaii Foundation (M.C.), which received unrestricted donations to support cancer and mesothelioma research from the Melohn family endowment; from Honeywell International (M.C.); The Riviera United 4-a Cure grant (M.C. and H.Y.); the Early Detection Research Network NCI Grant No. 5U01CA111295-08 (H.I.P., H.Y.); donations from Belluck and Fox (H.I.P.); Grant-in-Aid for Scientific Research Grants KAKENHI 26460689 (Y.Y.), 25460710 (M.E.), 24590715 and 15K08658 (T.T.); and a Grant-in-Aid for Researchers from Hyogo College of Medicine, 2017 (Y.Y.).
AUTHOR CONTRIBUTIONS
Conception and design: Haining Yang, Michele Carbone
Financial support: Michele Carbone, Yoshie Yoshikawa, Haining Yang
Collection and assembly of data: Sandra Pastorino, Harvey I. Pass, Masaki Nasu, Yasutaka Takinishi, Michael Minaai, Chandra Goparaju, Mika Tanji, Michele Carbone
Data analysis and interpretation: Sandra Pastorino, Yoshie Yoshikawa, Harvey I. Pass, Mitsuru Emi, Masaki Nasu, Ian Pagano, Ryuji Yamamoto, Tomoko Hashimoto-Tamaoki, Masaki Ohmuraya, Keisuke Goto, Kavita Y. Sarin, Angela Bononi, Andrea Napolitano, Giovanni Gaudino, Mary Hesdorffer, Haining Yang, Michele Carbone
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
A Subset of Mesotheliomas With Improved Survival Occurring in Carriers of BAP1 and Other Germline Mutations
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/site/ifc.
Sandra Pastorino
No relationship to disclose
Yoshie Yoshikawa
No relationship to disclose
Harvey I. Pass
Consulting or Advisory Role: Astra Zeneca
Speakers' Bureau: Genentech (I), Genomic Health (I)
Mitsuru Emi
No relationship to disclose
Masaki Nasu
No relationship to disclose
Ian Pagano
No relationship to disclose
Yasutaka Takinishi
No relationship to disclose
Ryuji Yamamoto
No relationship to disclose
Michael Minaai
No relationship to disclose
Tomoko Hashimoto-Tamaoki
Stock or Other Ownership: Takeda Pharmaceutical, Eisai
Patents, Royalties, Other Intellectual Property: Hyogo College of Medicine (Inst)
Masaki Ohmuraya
No relationship to disclose
Keisuke Goto
No relationship to disclose
Chandra Goparaju
No relationship to disclose
Kavita Y. Sarin
Stock or Other Ownership: NFlection, Myotherix, Seylanmed
Consulting or Advisory Role: NFlection
Research Funding: NFlection
Mika Tanji
No relationship to disclose
Angela Bononi
No relationship to disclose
Andrea Napolitano
Travel, Accommodations, Expenses: Eli Lilly, Eisai, Pierre Fabre
Giovanni Gaudino
No relationship to disclose
Mary Hesdorffer
No relationship to disclose
Haining Yang
No relationship to disclose
Michele Carbone
Patents, Royalties, Other Intellectual Property: Pending patent applications on BAP1 and mesothelioma diagnosis; provides medical/legal consulting related to MM
REFERENCES
- 1.Carbone M, Kanodia S, Chao A, et al. : Consensus report of the 2015 Weinman International Conference on Mesothelioma. J Thorac Oncol 11:1246-1262, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Carbone M, Bedrossian CW: The pathogenesis of mesothelioma. Semin Diagn Pathol 23:56-60, 2006 [DOI] [PubMed] [Google Scholar]
- 3.Carbone M, Emri S, Dogan AU, et al. : A mesothelioma epidemic in Cappadocia: Scientific developments and unexpected social outcomes. Nat Rev Cancer 7:147-154, 2007 [DOI] [PubMed] [Google Scholar]
- 4.Emri SA: The Cappadocia mesothelioma epidemic: Its influence in Turkey and abroad. Ann Transl Med 5:239, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roushdy-Hammady I, Siegel J, Emri S, et al. : Genetic-susceptibility factor and malignant mesothelioma in the Cappadocian region of Turkey. Lancet 357:444-445, 2001 [DOI] [PubMed] [Google Scholar]
- 6.Testa JR, Cheung M, Pei J, et al. : Germline BAP1 mutations predispose to malignant mesothelioma. Nat Genet 43:1022-1025, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carbone M, Flores EG, Emi M, et al. : Combined genetic and genealogic studies uncover a large BAP1 cancer syndrome kindred tracing back nine generations to a common ancestor from the 1700s. PLoS Genet 11:e1005633, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Napolitano A, Pellegrini L, Dey A, et al. : Minimal asbestos exposure in germline BAP1 heterozygous mice is associated with deregulated inflammatory response and increased risk of mesothelioma. Oncogene 35:1996-2002, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carbone M, Ferris LK, Baumann F, et al. : BAP1 cancer syndrome: Malignant mesothelioma, uveal and cutaneous melanoma, and MBAITs. J Transl Med 10:179, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carbone M, Yang H, Pass HI, et al. : BAP1 and cancer. Nat Rev Cancer 13:153-159, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haugh AM, Njauw CN, Bubley JA, et al. : Genotypic and phenotypic features of BAP1 cancer syndrome: A report of 8 new families and review of cases in the literature. JAMA Dermatol 153:999-1006, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wadt KA, Aoude LG, Johansson P, et al. : A recurrent germline BAP1 mutation and extension of the BAP1 tumor predisposition spectrum to include basal cell carcinoma. Clin Genet 88:267-272, 2015 [DOI] [PubMed] [Google Scholar]
- 13.Abdel-Rahman MH, Pilarski R, Cebulla CM, et al. : Germline BAP1 mutation predisposes to uveal melanoma, lung adenocarcinoma, meningioma, and other cancers. J Med Genet 48:856-859, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Farley MN, Schmidt LS, Mester JL, et al. : A novel germline mutation in BAP1 predisposes to familial clear-cell renal cell carcinoma. Mol Cancer Res 11:1061-1071, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Popova T, Hebert L, Jacquemin V, et al. : Germline BAP1 mutations predispose to renal cell carcinomas. Am J Hum Genet 92:974-980, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wiesner T, Obenauf AC, Murali R, et al. : Germline mutations in BAP1 predispose to melanocytic tumors. Nat Genet 43:1018-1021, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.de la Fouchardière A, Cabaret O, Savin L, et al. : Germline BAP1 mutations predispose also to multiple basal cell carcinomas. Clin Genet 88:273-277, 2015 [DOI] [PubMed] [Google Scholar]
- 18.Klebe S, Driml J, Nasu M, et al. : BAP1 hereditary cancer predisposition syndrome: A case report and review of literature. Biomark Res 3:14, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kittaneh M, Berkelhammer C: Detecting germline BAP1 mutations in patients with peritoneal mesothelioma: Benefits to patient and family members. J Transl Med 16:194, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nasu M, Emi M, Pastorino S, et al. : High incidence of somatic BAP1 alterations in sporadic malignant mesothelioma. J Thorac Oncol 10:565-576, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yoshikawa Y, Emi M, Hashimoto-Tamaoki T, et al. : High-density array-CGH with targeted NGS unmask multiple noncontiguous minute deletions on chromosome 3p21 in mesothelioma. Proc Natl Acad Sci USA 113:13432-13437, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bueno R, Stawiski EW, Goldstein LD, et al. : Comprehensive genomic analysis of malignant pleural mesothelioma identifies recurrent mutations, gene fusions and splicing alterations. Nat Genet 48:407-416, 2016 [DOI] [PubMed] [Google Scholar]
- 23.Lo Iacono M, Monica V, Righi L, et al. : Targeted next-generation sequencing of cancer genes in advanced stage malignant pleural mesothelioma: A retrospective study. J Thorac Oncol 10:492-499, 2015 [DOI] [PubMed] [Google Scholar]
- 24.Guo G, Chmielecki J, Goparaju C, et al. : Whole-exome sequencing reveals frequent genetic alterations in BAP1, NF2, CDKN2A, and CUL1 in malignant pleural mesothelioma. Cancer Res 75:264-269, 2015 [DOI] [PubMed] [Google Scholar]
- 25.Harbour JW, Onken MD, Roberson ED, et al. : Frequent mutation of BAP1 in metastasizing uveal melanomas. Science 330:1410-1413, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Peña-Llopis S, Vega-Rubín-de-Celis S, Liao A, et al. : BAP1 loss defines a new class of renal cell carcinoma. Nat Genet 44:751-759, 2012. [Erratum in Nat Genet. 44:072, 2012] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lin M, Zhang L, Hildebrandt MAT, et al. : Common, germline genetic variations in the novel tumor suppressor BAP1 and risk of developing different types of cancer. Oncotarget 8:74936-74946, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yu H, Pak H, Hammond-Martel I, et al. : Tumor suppressor and deubiquitinase BAP1 promotes DNA double-strand break repair. Proc Natl Acad Sci USA 111:285-290, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bononi A, Giorgi C, Patergnani S, et al. : BAP1 regulates IP3R3-mediated Ca2+ flux to mitochondria suppressing cell transformation. Nature 546:549-553, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bononi A, Yang H, Giorgi C, et al. : Germline BAP1 mutations induce a Warburg effect. Cell Death Differ 24:1694-1704, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Amelio I: Genes versus Environment: cytoplasmic BAP1 determines the toxic response to environmental stressors in mesothelioma. Cell Death Dis 8:e2907, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baumann F, Flores E, Napolitano A, et al. : Mesothelioma patients with germline BAP1 mutations have 7-fold improved long-term survival. Carcinogenesis 36:76-81, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Richards S, Aziz N, Bale S, et al. : Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17:405-424, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kircher M, Witten DM, Jain P, et al. : A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 46:310-315, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Carbone M, Shimizu D, Napolitano A, et al. : Positive nuclear BAP1 immunostaining helps differentiate non-small cell lung carcinomas from malignant mesothelioma. Oncotarget 7:59314-59321, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.National Cancer Institute : SEER Cancer Statistics Review, 1975-2014. https://seer.cancer.gov/csr/1975_2014/
- 37.Ryan NAJ, Morris J, Green K, et al. : Association of mismatch repair mutation with age at cancer onset in Lynch syndrome: Implications for stratified surveillance strategies. JAMA Oncol 3:1702-1706, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Varley JM: Germline TP53 mutations and Li-Fraumeni syndrome. Hum Mutat 21:313-320, 2003 [DOI] [PubMed] [Google Scholar]
- 39.Betti M, Casalone E, Ferrante D, et al. : Germline mutations in DNA repair genes predispose asbestos-exposed patients to malignant pleural mesothelioma. Cancer Lett 405:38-45, 2017 [DOI] [PubMed] [Google Scholar]
- 40.Yoshikawa Y, Sato A, Tsujimura T, et al. : Biallelic germline and somatic mutations in malignant mesothelioma: Multiple mutations in transcription regulators including mSWI/SNF genes. Int J Cancer 136:560-571, 2015 [DOI] [PubMed] [Google Scholar]
- 41.Pearlman R, Frankel WL, Swanson B, et al. : Prevalence and spectrum of germline cancer susceptibility gene mutations among patients with early-onset colorectal cancer. JAMA Oncol 3:464-471, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dite GS, Jenkins MA, Southey MC, et al. : Familial risks, early-onset breast cancer, and BRCA1 and BRCA2 germline mutations. J Natl Cancer Inst 95:448-457, 2003 [DOI] [PubMed] [Google Scholar]
- 43.Paulo P, Maia S, Pinto C, et al. : Targeted next generation sequencing identifies functionally deleterious germline mutations in novel genes in early-onset/familial prostate cancer. PLoS Genet 14:e1007355, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bertario L, Russo A, Sala P, et al. : Survival of patients with hereditary colorectal cancer: Comparison of HNPCC and colorectal cancer in FAP patients with sporadic colorectal cancer. Int J Cancer 80:183-187, 1999 [DOI] [PubMed] [Google Scholar]
- 45.Fang WL, Chang SC, Lan YT, et al. : Molecular and survival differences between familial and sporadic gastric cancers. BioMed Res Int 2013:396272, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kadariya Y, Cheung M, Xu J, et al. : Bap1 Is a bona fide tumor suppressor: Genetic evidence from mouse models carrying heterozygous germline Bap1 mutations. Cancer Res 76:2836-2844, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tung N, Domchek SM, Stadler Z, et al. : Counselling framework for moderate-penetrance cancer-susceptibility mutations. Nat Rev Clin Oncol 13:581-588, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hassan R, Morrow B, Walsh T, et al. : Inherited predisposition to malignant mesothelioma (MM) due to mutations in DNA repair genes. J Clin Oncol 36, 2018. (15_suppl; abstr 8504) [Google Scholar]
- 49.Panou V, Gadiraju M, Wolin A, et al. : Frequency of germline mutations in cancer susceptibility genes in malignant mesothelioma. J Clin Oncol 36, 2018. (15_suppl; abstr 8504) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Heymann S, Delaloge S, Rahal A, et al. : Radio-induced malignancies after breast cancer postoperative radiotherapy in patients with Li-Fraumeni syndrome. Radiat Oncol 5:104, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shindo K, Yu J, Suenaga M, et al. : Deleterious germline mutations in patients with apparently sporadic pancreatic adenocarcinoma. J Clin Oncol 35:3382-3390, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]