

Abstract
Pancreatic ductal adenocarcinoma (PDAC) cells have an exceptional ability to invade nerves through pronounced crosstalk between nerves and cancer cells; however, the mechanism of PDAC cell invasion remains to be elucidated. Here, we demonstrate the therapeutic potential of telomerase-specific oncolytic adenoviruses, OBP-301 and tumor suppressor p53-armed OBP-702, against human PDAC cells. Highly invasive PDAC cells exhibited higher levels of phosphorylated extracellular signal-regulated kinases 1 and 2 (ERK1/2) expression independent of KRAS expression; ERK1/2 inhibitor or small interfering RNA (siRNA) treatment significantly reduced the migration and invasion of PDAC cells, suggesting that the ERK signaling pathway is associated with the invasiveness of PDAC cells. OBP-702 infection suppressed ERK signaling and inhibited PDAC cell migration and invasion more efficiently than OBP-301. OBP-702 also effectively inhibited PDAC cell invasion even when invasiveness was enhanced by administration of motility stimulators, such as nerve and neurosecretory factors. Moreover, noninvasive whole-body imaging analyses showed that OBP-702 significantly suppressed tumor growth in an orthotopic PDAC xenograft model, although both viruses were equally effective against subcutaneous tumors, suggesting that OBP-702 can influence the orthotopic tumor microenvironment. Our data suggest that oncolytic virus-mediated disruption of ERK signaling is a promising antitumor strategy for attenuating the invasiveness of PDAC cells.
Keywords: pancreatic cancer, adenovirus, telomerase, p53, ERK, migration, invasion, nerve, tumor microenvironment
Graphical Abstract
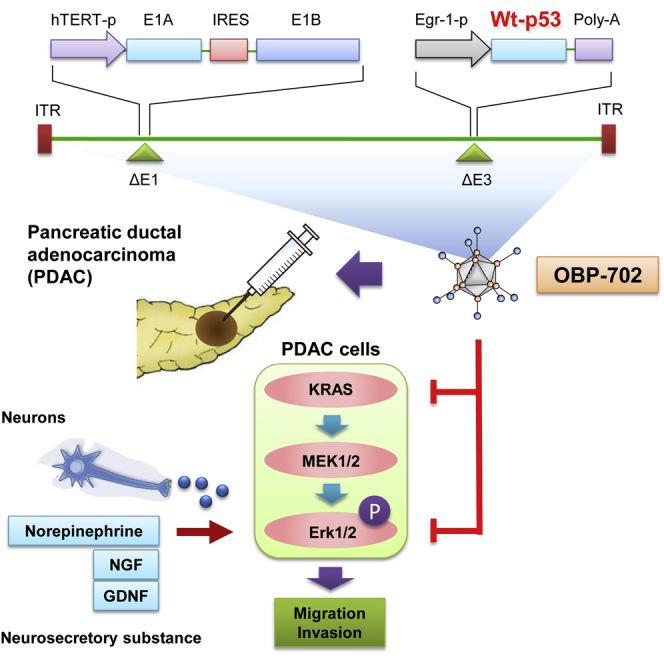
Telomerase-specific oncolytic adenovirus OBP-702 armed with the wild-type p53 tumor suppressor suppressed ERK signaling and inhibited pancreatic ductal adenocarcinoma (PDAC) cell migration and invasion, even when enhanced by nerve-derived motility stimulators. OBP-702 also significantly suppressed tumor growth in an orthotopic PDAC xenograft model by influencing the tumor microenvironment.
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is the most lethal type of cancer. The disease has a poor prognosis, with a 5-year survival rate of only 8%, despite recent advances in the treatment of PDAC.1 The poor prognosis of PDAC is primarily due to early onset of local recurrence and distant metastasis. Perineural invasion (PNI) is one of the main causes of recurrence and metastasis after curative surgery.2,3 The presence of cancer cells within the epineurial, perineurial, or endoneurial spaces of the neural sheath, as defined by Liebig et al.,4 occurs in PDAC at a higher incidence (80%–100%) than any other type of cancer.2 The underlying mechanism of PNI reportedly involves mutual neurotrophic crosstalk between PDAC cells and surrounding nerves.5 Nerves in the tumor microenvironment stimulate the growth and invasion of PDAC cells via the secretion of neurotransmitters (catecholamines and acetylcholine) and neurotrophic growth factors (nerve growth factor [NGF] and glial cell line-derived neurotrophic factor [GDNF]).6 Although nerve infiltration in the tumor microenvironment plays a crucial role in the progression of PDAC, there is currently no attractive strategy for limiting the invasiveness of PDAC cells.
The primary treatment option for locally advanced or metastatic PDAC is chemotherapy.7 Gemcitabine has been used as the standard first-line treatment; however, the 1-year survival rate of PDAC patients after treatment with gemcitabine is only 17%–23%.8 Recently, interest in the chemotherapeutic use of FOLFILINOX,9 nab-paclitaxel,10 and the molecular-targeted agent erlotinib11 has grown; however, these therapies do not markedly improve the clinical outcome of PDAC patients. Oncolytic virotherapy recently emerged as a novel antitumor therapy against PDAC.12, 13, 14 The telomerase-specific virus OBP-301 (telomelysin), in which the human telomerase reverse transcriptase (hTERT) promoter drives expression of the E1A and E1B genes for tumor-specific virus replication, exhibits broad-spectrum antitumor effects against many types of cancer, including PDAC.15, 16, 17 We also generated a modified OBP-301 variant (OBP-702) that induces the tumor suppressor gene p53 by inserting the Egr1 promoter-driven p53 expression cassette into the E3 region of OBP-301.18 OBP-702 exhibited greater antitumor efficacy than OBP-301 through activation of the p53-mediated signaling pathway independent of p53 status in targeted tumor cells,18, 19, 20 suggesting that OBP-702 has therapeutic potential against various p53-inactivated cancers, including PDAC.21
In the present study, we hypothesized that the mitogen-activated protein kinase (MAPK) signaling pathway is associated with invasiveness of PDAC cells. We evaluated the therapeutic potential of the telomerase-specific oncolytic adenovirus OBP-301 and p53-activating virus OBP-702 against the malignant behavior of PDAC cells. Moreover, in vivo preclinical experiments using an orthotopic PDAC xenograft tumor model were performed to assess the virus-mediated antitumor activity.
Results
In Vitro Cytopathic Effect of OBP-301 and OBP-702 against p53 Mutant PDAC Cells
To determine the therapeutic potential of telomerase-specific oncolytic adenoviruses for treating PDAC, we investigated the in vitro cytopathic effect of OBP-301 and OBP-702 against four human PDAC cell lines (Capan-1, MIA PaCa-2, BxPC-3, and Panc-1) with p53 mutations using an sodium 3’-[1-(phenylaminocarbonyl)-3,4-tetrazolium]-bis (4-methoxy6-nitro) benzene sulfonic acid hydrate (XTT) assay of cell viability on day 3 after viral infection. Infection with OBP-301 at high doses (multiplicity of infection [MOI] of 50 and 100) significantly suppressed cell viability, whereas infection with OBP-702 at either low (MOI of 5 and 10) or high (MOI of 50 and 100) doses significantly suppressed the viability of all human PDAC cell lines examined (Figure 1A), demonstrating more profound antitumor efficacy of OBP-702 than OBP-301 for treating PDAC. OBP-702 is generated by inserting the p53 expression cassette into the E3 region of OBP-301.18 To rule out the possibility that E3 modification induces the profound antitumor effect of OBP-702, we analyzed the cytopathic effect of OBP-401, in which the non-toxic green fluorescent protein (GFP) expression cassette is inserted into the E3 region of OBP-301.22 The cytopathic effect of GFP-expressing OBP-401 was almost similar with that of OBP-301 in Capan-1, BxPC-3, and Panc-1 cells, although MIA PaCa-2 cells slightly showed higher sensitivity to OBP-401 than OBP-301 (Figure S1). These results suggest that OBP-702 induces more profound antitumor effect than OBP-301 in PDAC cells, probably because of p53 activation rather than E3 modification.
Figure 1.

Induction of Autophagy- and Apoptosis-Related Death of Human Pancreatic Ductal Adenocarcinoma (PDAC) Cells Infected with OBP-301 or OBP-702
(A) The cytopathic effect of OBP-301 and OBP-702 against four PDAC cell lines (Capan-1, MIA PaCa-2, BxPC-3, Panc-1). Cell viability was determined 72 h after infection with OBP-301 or OBP-702 at the indicated MOI using an XTT assay. Cell viability was calculated relative to that of mock-infected cells, the viability of which was set at 1.0. Data are expressed as mean ± SD (n = 5). ∗p < 0.05 (versus 0 MOI). (B and C) Expression of the apoptosis markers PARP and cleaved PARP (C-PARP), autophagy marker p62, viral EIA, and p53 proteins in PDAC cells infected with OBP-301 or OBP-702 at the indicated MOI for 72 h. β-Actin was assayed as a loading control.
BxPC-3 and Panc-1 cells were more sensitive to OBP-702 than Capan-1 and MIA PaCa-2 cells (Figure 1A). To explore the mechanism of high sensitivity to OBP-702 in PDAC cells, we analyzed the expression level of coxsackie and adenovirus receptor (CAR) in four human PDAC cell lines by flow cytometry. BxPC-3 and Panc-1 cells showed higher CAR expression than Capan-1 and MIA PaCa-2 cells (Figure S2). These results suggest that BxPC-3 and Panc-1 cells are sensitive to OBP-702 due to high CAR expression.
To explore the mechanism of the oncolytic adenovirus-induced cytopathic effect against PDAC cells, we conducted western blot analyses of apoptosis and autophagy (Figures 1B and 1C). OBP-301 infection at a high dose resulted in a slight increase in the expression of the apoptosis-specific marker cleaved poly(ADP-ribose) polymerase (C-PARP) in Capan-1, MIA PaCa-2, and BxPC-3 cells, but not in Panc-1 cells. Decreased expression of the autophagy-specific marker p62 was observed in all PDAC cells treated with OBP-301 at high doses, suggesting that OBP-301 primarily induces autophagy rather than apoptosis. By contrast, OBP-702 infection at low and high doses resulted in increased C-PARP expression and decreased p62 expression (Figure 1C), suggesting that both apoptosis and autophagy are involved in the OBP-702-induced antitumor effect. Moreover, p53 expression increased in cells treated with OBP-702 but decreased in cells treated with OBP-301, although both OBP-301 and OBP-702 effectively induced adenoviral E1A expression. These results indicate that the telomerase-specific oncolytic adenoviruses OBP-301 and OBP-702 exhibit cytopathic effects that involve autophagy and apoptosis in p53 mutant PDAC cells.
Invasive Phenotype of PDAC Cells Is Associated with Activation of the MAPK Signaling Pathway
Next, we sought to characterize the migration and invasion of PDAC cells using Boyden chamber-based assays. Both Capan-1 and MIA PaCa-2 cells exhibited low migration and invasion capabilities, whereas BxPC-3 and Panc-1 cells exhibited a high capacity to migrate and invade (Figure 2A), suggesting that BxPC-3 and Panc-1 cells migrate and invade tissues more aggressively than Capan-1 and MIA PaCa-2 cells. By contrast, analyses of cell proliferation showed that MIA PaCa-2 and Panc-1 cells proliferate more rapidly than Capan-1 and BxPC-3 cells (Figure S1). These results suggest that although BxPC-3 and Panc-1 cells exhibit an invasive phenotype, it is not correlated with rapid cell proliferation.
Figure 2.

Characterization of the Migration and Invasion Properties of PDAC Cells
(A) The number of migrating or invading cells in five randomly selected fields was determined under a light microscope. Representative photomicrographs of migrating and invading cells stained with crystal violet. Original magnification ×100. Data are expressed as mean ± SD (n = 5). (B) Expression of KRAS, phospho-ERK1/2 (p-ERK1/2), and ERK1/2 proteins in PDAC cells. β-Actin was assayed as a loading control.
Because constitutive activation of the KRAS-MAPK signaling pathway is associated with the malignant phenotype of PDAC cells,23,24 we evaluated the expression of KRAS and extracellular signal-regulated kinase (ERK) proteins. Capan-1, MIA PaCa-2, and Panc-1 cells are KRAS mutant type, and BxPC-3 cells are KRAS wild type. Western blot analyses demonstrated that high and moderate KRAS expression were observed in highly invasive Panc-1 and BxPC-3 cells and poorly invasive Capan-1 cells, whereas poorly invasive MIA PaCa-2 cells exhibited low KRAS expression, suggesting no relationship between invasive phenotype and KRAS expression. In contrast, the expression of phosphorylated ERK1/2 was high in highly invasive BxPC-3 and Panc-1 cells, but not in poorly invasive Capan-1 and MIA PaCa-2 cells (Figure 2B). These results suggest that activation of the ERK signaling pathway is associated with the migration and invasion capabilities of PDAC cells independent of KRAS status.
Oncolytic Viruses Inhibit the Migration and Invasion of PDAC Cells by Suppressing the KRAS-MAPK Signaling Pathway
The therapeutic potential of oncolytic adenoviruses for inhibiting PDAC cell migration and invasion was analyzed using highly invasive BxPC-3 and Panc-1 cells. Although infection with OBP-301 and OBP-702 for 24 h did not decrease the viability of either BxPC-3 or Panc-1 cells (Figure S4A), the migration and invasion capabilities of BxPC-3 and Panc-1 cells were significantly suppressed by infection with OBP-301 and OBP-702 for 24 h in a dose-dependent manner. Although the effect of OBP-702 was much stronger than that of OBP-301 in BxPC-3 cells, the viruses exhibited similar effects against Panc-1 cells (Figures 3A and S4B). These results suggest that both OBP-301 and OBP-702 suppress the migration and invasion capabilities of PDAC cells without affecting their cell viabilities.
Figure 3.

OBP-301 and OBP-702 Inhibit the Migration and Invasion of Highly Invasive PDAC Cells by Suppressing KRAS-ERK Signaling
(A) Migration and invasion assay using highly invasive PDAC cells infected with OBP-301 or OBP-702 at the indicated MOI for 24 h. ∗p < 0.05 (versus 0 MOI). (B) Expression of KRAS, phospho-ERK1/2 (p-ERK1/2), and ERK1/2 proteins in highly invasive PDAC cells infected with OBP-301 or OBP-702 at the indicated MOI for 24 h. (C) Migration and invasion assay and western blot analysis of KRAS expression in highly invasive PDAC cells transfected with 10 nM KRAS siRNA (si-KRAS) for 72 h. (D) Migration and invasion assay and western blot analysis of p-ERK1/2 and ERK1/2 expression in highly invasive PDAC cells transfected with 10 nM ERK1/2 siRNA (si-ERK1/2) for 72 h. Control siRNA (si-con) was used as a control. β-Actin was assayed as a loading control. The number of migrating and invading cells after treatment was calculated relative to that of non-treated cells, which was set at 1.0. Data are expressed as mean ± SD (n = 5). ∗p < 0.05 (versus si-con).
To explore the mechanism of oncolytic adenovirus-mediated suppression of the invasive phenotype of PDAC cells, we conducted western blot analyses of KRAS and ERK protein expression. OBP-301 and OBP-702 induced a similar dose-dependent decrease in the expression of KRAS protein in BxPC-3 and Panc-1 cells (Figure 3B). By contrast, the expression of phosphorylated ERK1/2 decreased in OBP-702-infected PDAC cells, but not OBP-301-infected cells (Figure 3B). These results suggest that OBP-702 suppresses ERK expression more efficiently than OBP-301.
To clarify the role of KRAS-MAPK signaling in the invasive phenotype of PDAC cells, we conducted migration and invasion assays using KRAS siRNA and ERK1/2 siRNA. Treatment with KRAS siRNA or ERK1/2 siRNA significantly suppressed the migration and invasion capabilities of BxPC-3 and Panc-1 cells compared with control siRNA treatment (Figures 3C and 3D). Interestingly, the effect of ERK1/2 siRNA was much stronger than that of KRAS siRNA in BxPC-3 cells, but the effects of both siRNAs were similar in Panc-1 cells. Moreover, treatment with the ERK1/2 inhibitor SCH772984 had a similar effect as treatment with ERK1/2 siRNA in BxPC-3 and Panc-1 cells (Figure S3). These results suggest that suppression of ERK signaling plays a critical role in OBP-702-mediated suppression of the invasive phenotype of PDAC cells.
Oncolytic Viruses Suppress the Migration and Invasion Capabilities of PDAC Cells via Stimulation with Dorsal Root Ganglion and Neurosecretory Factors
In the pancreatic microenvironment, nerves play a crucial role in mediating the invasive phenotype of PDAC cells.6 The role of nerves in enhancing the invasive phenotype of PDAC cells was investigated using dorsal root ganglia (DRGs) obtained from athymic nude mice. Neurite outgrowth was confirmed in DRGs cultured in a Matrigel-coated dish for 7 days (Figure S4A), indicating that harvested DRGs are suitable for use as nerve tissues. Indeed, the addition of DRGs significantly increased the migration and invasion capabilities of BxPC-3 and Panc-1 cells (Figure S4B). To investigate the therapeutic potential of oncolytic adenoviruses in suppressing the nerve-stimulated invasive phenotype of PDAC cells, we conducted migration and invasion assays using highly invasive BxPC-3 and Panc-1 cells co-cultured with DRGs. Both OBP-301 and OBP-702 significantly suppressed the migration and invasion capabilities of BxPC-3 and Panc-1 cells in the presence of DRGs (Figure S4C). These results suggest that OBP-301 and OBP-702 suppress the nerve-stimulated invasive potential of PDAC cells.
Several neurosecretory factors are involved in the nerve-stimulated invasion of PDAC cells.6 To evaluate which neurosecretory factors enhance the invasive phenotype of PDAC cells similar to DRGs, we examined three neurosecretory factors: norepinephrine (NE), NGF, and GDNF. Administration of NE significantly increased the migration capability of BxPC-3 cells, whereas administration of NGF and GDNF significantly enhanced the migration capability of Panc-1 cells (Figures 4A and S5A). Western blot analyses demonstrated that all three neurosecretory factors increased the expression of phosphorylated ERK in BxPC-3 and Panc-1 cells (Figure 4B).
Figure 4.

OBP-301 and OBP-702 Inhibit the Migration and Invasion of Highly Invasive PDAC Cells Stimulated with Neurosecretory Factors
(A) Migration assay using highly invasive PDAC cells stimulated with norepinephrine (NE), nerve growth factor (NGF), or glial cell line-derived neurotrophic factor (GDNF) at the indicated doses. ∗p < 0.05 (versus control). (B) Expression of phospho-ERK1/2 (p-ERK1/2) and ERK1/2 proteins in highly invasive PDAC cells stimulated with NE (100 μM), NGF (100 ng/mL), or GDNF (100 ng/mL) for 24 h. β-Actin was assayed as a loading control. (C) Migration and invasion assay using highly invasive PDAC cells stimulated with NE (100 μM), NGF (100 ng/mL), or GDNF (100 ng/mL) for 24 h in the presence of OBP-301 (100 MOI) or OBP-702 (100 MOI). The number of migrating and invading cells after treatment was calculated relative to that of non-treated cells, which was set at 1.0. Data are expressed as mean ± SD (n = 5). ∗p < 0.05 (versus NE, NGF, or GDNF).
To investigate the therapeutic potential of oncolytic adenoviruses for suppressing the neurosecretory factor-enhanced invasive phenotype of PDAC cells, we conducted migration and invasion assays using highly invasive BxPC-3 and Panc-1 cells co-cultured with neurosecretory factors. OBP-301 and OBP-702 significantly suppressed the migration and invasion capabilities of BxPC-3 and Panc-1 cells in the presence of the neurosecretory factors (Figure 4C). Although the effect of OBP-702 was much stronger than that of OBP-301 in BxPC-3 cells, the viruses exhibited similar effects against Panc-1 cells. Moreover, similar to OBP-301 and OBP-702, the ERK1/2 inhibitor SCH772984 significantly suppressed the migration and invasion capabilities of BxPC-3 and Panc-1 cells after stimulation with the neurosecretory factors (Figure S5C). These results suggest that both OBP-301 and OBP-702 suppress the invasive potential of PDAC cells stimulated with abundant neurosecretory factors.
Oncolytic Viruses Suppress Tumor Growth in a Subcutaneous BxPC-3 Xenograft Tumor Model
To evaluate the antitumor effect of oncolytic adenoviruses in PDAC tumors, we used a subcutaneous BxPC-3 xenograft tumor model. Intratumoral injection of OBP-301 and OBP-702 significantly suppressed tumor growth compared with mock treatment, whereas there was no significant difference in the volumes of OBP-301- and OBP-702-treated tumors (Figures 5A–5C). Histopathologic analyses of tumor tissues on day 37 revealed large necrotic areas in OBP-702-treated tumors, as compared with OBP-301- or mock-treated tumors (Figure 5D). Immunohistologic analysis of expression of the cell proliferation marker Ki-67 revealed a significantly lower percentage of Ki-67-positive cancer cells in OBP-702-treated tumors compared with OBP-301- or mock-treated tumors (Figures 5D and 5E). These results suggest that both OBP-301 and OBP-702 suppress the growth of PDAC tumors, and OBP-702 inhibits the proliferation of PDAC cells in tumors more strongly than OBP-301.
Figure 5.

OBP-301 and OBP-702 Inhibit Tumor Growth in a Subcutaneous BxPC-3 Tumor Model
(A) BxPC-3 cells (5 × 106 cells/site) were inoculated into the right flanks of athymic nude mice. OBP-301 (108 PFUs), OBP-702 (108 PFUs), or PBS (mock) was injected into the tumors on days 0, 2, and 4. Data are expressed as mean tumor volume ± SD (mock group: n = 8; OBP-301 and OBP-702 groups: n = 9). (B) Macroscopic appearance of representative mice on day 37 after first treatment. (C) Macroscopic appearance of all isolated tumors on day 37 after first treatment. (D) Histologic analysis of BxPC-3 tumors. Paraffin-embedded sections of BxPC-3 tumors were stained with hematoxylin and eosin (H&E) or anti-Ki-67 antibody. Left two images and right two images represent H&E and Ki-67 staining, respectively, with low (×100) and high (×400) original magnification. High-magnification images are the area outlined by a black square in the low-magnification images. (E) Percentage of Ki-67-positive cells in each group. The percentage of Ki-67-positive cells was calculated from three random fields of tumor section in each group under microscopy. ∗p < 0.05.
OBP-702 Suppresses Tumor Growth in an Orthotopic BxPC-3-Luc Xenograft Tumor Model
To assess the therapeutic efficacy of OBP-301 and OBP-702 against PDAC tumors within the pancreatic microenvironment, we used an orthotopic BxPC-3-Luc (luciferase) xenograft tumor model, in which Luc activity is associated with the viability of PDAC cells in tumors. Analyses using a noninvasive in vivo imaging system (IVIS) demonstrated that OBP-702 significantly suppressed the viability of PDAC cells in tumors, whereas OBP-301 did not suppress the viability of PDAC cells in tumors when compared with mock treatment (Figures 6A and 6B). These results suggest that OBP-702 has more potential to suppress the viability of PDAC cells within the pancreatic tumor microenvironment than OBP-301.
Figure 6.

OBP-702 Inhibits Tumor Growth in an Orthotopic BxPC-3-Luc Xenograft Tumor Model
BxPC-3-Luc cells (3 × 106 cells/site) were inoculated into the pancreatic tails of athymic nude mice. OBP-301 (108 PFUs/tumor), OBP-702 (108 PFUs/tumor), or PBS (mock) was injected into the tumors on days 0, 2, and 4. Luminescence in tumor tissues was analyzed using an IVIS system at 0, 7, 14, 21, and 28 days after first treatment. (A) Representative photographs of mock-, OBP-301-, or OBP-702-treated tumor-bearing mice. (B) Data are expressed as mean ± SD (mock group: n = 12; OBP-301 and OBP-702 groups: n = 10). ∗p < 0.05.
Discussion
The prognosis of patients with PDAC is poor due to the high invasiveness, local recurrence, and distant metastasis characteristics of this disease.2 PNI, one of the most common hallmarks of PDAC, is associated with disease recurrence and pain. Therefore, therapies that target the invasiveness of PDAC cells are needed in order to improve the clinical outcome of these patients. In this study, we demonstrated that the telomerase-specific oncolytic adenoviruses OBP-301 and OBP-702 suppress the growth and invasion of PDAC cells independent of KRAS status via the induction of autophagy and apoptosis and suppression of ERK signaling. Moreover, the p53-activating OBP-702-mediated antitumor effect was stronger than the p53-nonexpressing OBP-301-mediated effect against PDAC tumors within the pancreatic tumor microenvironment, which includes nerve tissues and abundant neurosecretory factors. Thus, a p53-armed oncolytic virotherapy would be a promising strategy for inhibiting the growth and invasion of PDAC cells via p53 activation and suppression of ERK signaling.
The p53 tumor suppressor gene is one of the most frequently mutated genes in PDAC cells,25,26 which suggests that gene therapy targeting p53 would be effective against p53-inactivated PDAC.20 However, whether p53 restoration plays a crucial role in the growth and invasion of PDAC cells remains unclear. We recently reported that OBP-702-mediated p53 overexpression induces apoptosis- and autophagy-related death in a variety of malignant tumor cell types.18,19 Consistent with these findings, we confirmed that OBP-702 exhibits antitumor effects in p53 mutant PDAC cells via induction of autophagy and apoptosis. Rosenfeldt et al.27 reported that autophagy suppresses PDAC progression in KRAS mutant and p53-deficient mice. By contrast, Todoric et al.28 demonstrated that autophagy prevents the development of PDAC in KRAS mutant acinar cells. Because p53 is a strong inducer of autophagy,29,30 p53-mediated autophagy induction could play a tumor-suppressive role in the development and progression of p53-inactivated PDAC. Thus, OBP-702 would have therapeutic potential for eliminating p53 mutant PDAC cells by inducing p53-mediated autophagy.
Activation of MAPK signaling is a key factor in the development, growth, and progression of PDAC tumors.23,24 Our data revealed that highly invasive BxPC-3 and Panc-1 cells exhibit ERK activation, supporting the hypothesis that ERK plays a critical role in the invasive phenotype of PDAC cells. Previous reports demonstrated that Panc-1 cells are KRAS mutant with ERK activation,31 whereas BxPC-3 cells are KRAS wild type.32 Recently, Chen et al.33 revealed that BxPC-3 cells have an in-frame BRAF deletion in association with ERK activation. A recent report by the Cancer Genome Atlas Research Network also indicated the presence of an in-frame BRAF deletion in KRAS-wild-type PDAC tumors.34 Therefore, ERK activation in highly invasive BxPC-3 and Panc-1 cells may be associated with KRAS and BRAF mutations. However, poorly invasive Capan-1 and MIA PaCa-2 cells do not exhibit ERK activation despite harboring KRAS mutations.32 Shibata et al.35 suggested that in addition to KRAS mutations, mutations in p53, along with several reprogramming factors, are necessary to activate pancreatic ERK signaling in mice. Although ERK activation plays a key role in the invasive phenotype of PDAC cells, these cells exhibit distinct diversity in terms of their genetic background with respect to ERK activation.
An abundance of neurosecretory factors is a common feature of the pancreatic microenvironment,2, 3, 4, 5, 6 and this can contribute to local recurrence, distant metastasis, and poor prognosis in PDAC patients. Our data demonstrated that several neurosecretory factors (NE, NGF, GDNF) promote the migration and invasion of highly invasive BxPC-3 and Panc-1 cells via ERK activation. Regarding the role of ERK activation in the invasion of BxPC-3 and Panc-1 cells, Yang et al.36 recently revealed that ERK activation induces the downregulation of Raf kinase inhibitory protein (RKIP), which contributes to the invasive and metastatic phenotype of BxPC-3 and Panc-1 cells. RKIP is a downstream target of tumor suppressor p53, by which RKIP induction inhibits the activation of ERK signaling.37 Because p53-activating OBP-702, but not p53-nonexpressing OBP-301, suppressed the activation of ERK in highly invasive BxPC-3 and Panc-1 cells, p53-mediated RKIP activation may play a critical role in suppressing ERK activation in highly invasive PDAC cells in concert with neurosecretory factors.
Subcutaneous and orthotopic xenograft tumor models using human PDAC cell lines are frequently employed for in vivo evaluation of therapeutic potential.38 We confirmed the in vivo antitumor effect of OBP-301 and OBP-702 using a subcutaneous BxPC-3 xenograft tumor model. Previous reports have suggested that epidermal growth factor receptor (EGFR) plays a critical role in the development of PDAC tumors.39,40 As we recently confirmed that OBP-301 suppresses EGFR expression via the accumulation of adenoviral E1A, resulting in autophagy-related death of malignant tumor cells,41 the antitumor effect of OBP-301 and OBP-702 in subcutaneous PDAC tumors may be associated at least in part with EGFR suppression. By contrast, experiments using an orthotopic BxPC-3 xenograft tumor model demonstrated the antitumor effect of OBP-702, but not OBP-301. Orthotopic BxPC-3 xenograft tumors have been shown to exhibit high metastatic potential, with invasive characteristics similar to those of clinical PDAC tumors.42 Hayes et al.43 recently reported that ERK suppression reduces the growth of PDAC tumors independent of KRAS status. Because BxPC-3 cells have an in-frame BRAF deletion in association with ERK activation,33 the OBP-702-mediated antitumor effect against orthotopic PDAC tumors may be associated with ERK suppression. Further experiments would be warranted to explore the molecular mechanism of the OBP-702-mediated antitumor effect against PDAC tumors.
Precision medicine therapies based on genetic alterations have recently emerged for the treatment of various types of cancer, including PDAC.44,45 Recent genomic sequencing studies revealed that most PDAC cells harbor genetic alterations in four major genes: KRAS, TP53, CDKN2A, and SMAD4.46 Yachida et al.47 suggested that among these four driver gene mutations, a smaller number of driver gene alterations could serve as a prognostic factor for PDAC patients with longer survival. Hingorani et al.48 demonstrated that concomitant expression of the KRASG12D and TP53R172H mutated genes induced PDAC cell invasion and metastasis in a genetically engineered mice model, suggesting that KRAS and TP53 represent potent therapeutic targets among the four driver genes. Although KRAS protein plays a critical role in cancers involving KRAS mutations,49 efforts to develop drugs targeting mutant KRAS protein in a tumor-specific manner have not been successful. Our tumor-specific oncolytic viruses OBP-301 and OBP-702 efficiently suppressed KRAS expression in PDAC cells. In addition, recent reports demonstrated that p53 restoration suppresses the invasion and metastasis of lung cancer cells in KRAS and TP53 mutant mice.50,51 Indeed, p53-activating OBP-702 suppressed the invasion of highly invasive PDAC cells more strongly than p53-nonexpressing OBP-301. Thus, OBP-702 could be a useful agent for targeting KRAS and TP53 gene alterations in PDAC cells as a novel precision medicine tool.
In conclusion, we demonstrated that the p53-armed oncolytic adenovirus OBP-702 exerts an inhibitory effect on the growth and invasion of PDAC cells via the induction of p53 expression, autophagy, and apoptosis. OBP-702 effectively inhibited the neurosecretory factor-enhanced migration and invasion of PDAC cells via suppression of ERK signaling. Moreover, the OBP-702-induced antitumor activity was effective in both subcutaneous and orthotopic PDAC xenograft tumor models. These data suggest that OBP-702-mediated p53 reactivation is a promising antitumor strategy for use against p53-inactivated PDAC tumors. We have recently reported that combination therapy with OBP-301 and chemotherapy or radiotherapy is more effective than monotherapy of OBP-301 in various types of cancer, such as esophageal cancer,52 gastric cancer,53 and osteosarcoma.54 Therefore, the preclinical studies to confirm the therapeutic potential of OBP-702 are currently underway for PDAC cells in combination with chemotherapy, radiotherapy, and immune checkpoint inhibitors. Further clinical study is also warranted to confirm the safety and feasibility of using p53-armed telomerase-specific oncolytic adenovirus OBP-702 in patients with invasive PDAC.
Materials and Methods
Cell Lines
The human PDAC cell lines Capan-1, MIA PaCa-2, BxPC-3, and Panc-1 were obtained from the American Culture Type Collection. BxPC-3 cells stably transfected with the firefly Luc expression vector (BxPC-3-Luc) were obtained from the Japanese Collection of Research Bioresources Cell Bank (JCRB, Osaka, Japan). Cells were cultured for no longer than 5 months following resuscitation. Authentication was not performed by the authors. MIA PaCa-2 and Panc-1 cells were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum (FBS). BxPC-3 and BxPC-3-Luc cells were maintained in RPMI-1640 supplemented with 10% FBS. Capan-1 cells were maintained in Iscove’s modified Dulbecco’s medium supplemented with 20% FBS. All media were supplemented with 100 U/mL penicillin and 100 mg/mL streptomycin. Cells were maintained at 37°C in a humidified atmosphere with 5% CO2.
Recombinant Adenoviruses
The telomerase-specific replication-competent adenovirus OBP-301 (telomelysin), in which the promoter element of the hTERT gene drives the expression of E1A and E1B genes, was constructed and characterized previously.15, 16, 17 OBP-702 was constructed by modifying OBP-301 to express the exogenous p53 gene by inserting a human wild-type p53 gene expression cassette driven by the Egr-1 promoter into the E3 region of OBP-301.18 OBP-401 was constructed by modifying OBP-301 to express the GFP by inserting a GFP gene expression cassette into the E3 region of OBP-301.22 Recombinant adenoviruses were purified using cesium chloride step gradients, and virus titers were determined using a plaque-forming assay with 293 cells. Viruses were stored at −80°C.
Reagents
NE, NGF, and GDNF were purchased from Sigma-Aldrich (St. Louis, MO, USA). Control siRNA, KRAS siRNA, ERK1 siRNA, and ERK2 siRNA were purchased from Applied Biosystems (Foster City, CA, USA).
Cell Viability Assay
Cells were seeded in 96-well plates at a density of 103 cells/well (MIA PaCa-2, BxPC-3, Panc-1) or 5 × 103 cells/well (Capan-1) 24 h before virus infection. Cells were infected with OBP-301, OBP-702, or OBP-401 at a MOI of 0, 1, 5, 10, 50, or 100 plaque-forming units (PFUs)/cell. Cell viability was determined on day 3 after virus infection using a Cell Proliferation Kit II (Roche, Indianapolis, IN, USA) according to the manufacturer’s protocol.
Western Blot Analysis
Cells were seeded in a 100-mm dish at a density of 105 cells/dish 24 h before virus infection. Cells were infected with OBP-301 or OBP-702 at the indicated MOI for 72 h. Whole-cell lysates were prepared at the indicated time points. Proteins were electrophoresed on 6%–15% SDS polyacrylamide gels and then transferred onto polyvinylidene difluoride membranes (Hybond-P; GE Healthcare, Buckinghamshire, UK). Primary antibodies used were as follows: mouse anti-p62 monoclonal antibody (mAb) (Medical & Biological Laboratories, Nagoya, Japan), mouse anti-p53 mAb (Santa Cruz Biotechnology, Santa Cruz, CA, USA), mouse anti-Ad5 E1A mAb (BD Pharmingen, Franklin Lakes, NJ, USA), rabbit anti-KRAS mAb (Abcam, Cambridge, MA, USA), rabbit anti-PARP polyclonal antibody (pAb), rabbit anti-ERK1/2 mAb, rabbit anti-phospho-ERK1/2 mAb (Cell Signaling Technology, Danvers, MA, USA), and mouse anti-β-actin mAb (Sigma-Aldrich). Secondary antibodies used were as follows: horseradish peroxidase-conjugated antibodies against rabbit IgG or mouse IgG (GE Healthcare). Immunoreactive bands on the blots were visualized using enhanced chemiluminescence substrates (ECL Plus; GE Healthcare).
Migration and Invasion Assay
Migration assay is used to investigate the mobility of tumor cells toward a chemo-attractant through non-coated membrane. Invasion assay is used to investigate the mobility of tumor cells with invasive capability through extracellular matrix-coated membrane. Cell migration and invasion assays were conducted using 24-well Boyden chambers equipped with 8-μm pore size filter membranes and 8-μm pore size filter membranes coated with Matrigel, respectively (BD Biosciences). For the assays, 10% FBS-containing medium was placed in the lower chambers to be used as a chemoattractant. To characterize PDAC cell migration and invasion, we placed 105 PDAC cells in a 500-μL volume of serum-free medium in the upper chambers and incubated them at 37°C for 24 h. Next, to assess the effect of oncolytic adenoviruses and neurosecretory factors, we placed 5 × 104 cells (BxPC-3) or 2.5 × 104 cells (Panc-1) in the upper chambers for the migration assay, and 105 cells (BxPC-3) or 5 × 104 cells (Panc-1) were placed in the upper chambers for the invasion assay. Twenty-four hours after treatment with oncolytic adenoviruses and neurosecretory factors, migrating or invading cells on the bottom surface of the membrane were stained with crystal violet (Sigma-Aldrich) and counted under a microscope (original magnification ×100) in five randomly selected fields.
In Vivo Subcutaneous BxPC-3 Xenograft Tumor Model
Animal experimental protocols were approved by the Ethics Review Committee for Animal Experimentation of Okayama University School of Medicine (No. OKU-2015364). BxPC-3 xenograft tumors were produced on the right flank of 6-week-old female BALB/c-nu/nu mice (CLEA Japan, Tokyo, Japan) by subcutaneous injection of 5 × 106 cells in 100 μL of PBS. When tumors had grown to a diameter of 5–7 mm, OBP-301, OBP-702, or PBS was injected intratumorally at a MOI of 108 PFUs/50 μL. Oncolytic adenovirus was injected on days 0, 2, and 4. The perpendicular diameter of each tumor was measured every 3 or 4 days, and tumor volume was calculated using the following formula: tumor volume (mm3) = a × b2 × 0.5, where a represents the longest diameter, b represents the shortest diameter, and 0.5 is the constant for calculating the volume of an ellipsoid.
Histopathologic Analysis
Subcutaneous tumors were fixed in 10% neutralized formalin and embedded in paraffin blocks. Sections were stained with hematoxylin and eosin to assess the tumor region. Proliferating tumor cells within tumor tissues were detected by immunostaining with rabbit anti-Ki67 mAb (Abcam) using standard techniques. Photographs of immunostained sections were obtained under light microscopy. The total number of cells and total number of Ki67-immunoreactive cells were calculated in five randomly selected fields in each tumor using ImageJ software.
In Vivo Orthotopic BxPC-3-Luc Xenograft Tumor Model
Animal experimental protocols were approved by the Ethics Review Committee for Animal Experimentation of Okayama University School of Medicine (No. OKU-2011062). BxPC-3-Luc cells suspended in Matrigel at a concentration of 3 × 106 cells per 20 μL were inoculated into the pancreatic tail of 6-week-old female BALB/c-nu/nu mice during laparotomy with a small left-flank abdominal incision. Three weeks later, mice were injected intratumorally with OBP-301 or OBP-702 at a MOI of 1.0 × 108 PFUs/20 μL, or mice were injected with PBS during laparotomy on days 0, 7, and 14. Tumor progression was monitored by intraperitoneal injection of the substrate luciferin (VivoGlo Luciferin; Promega, Madison, WI, USA) at a dose of 150 mg/kg body weight. Images were collected in the supine position every few minutes from 10 to 30 min after luciferin injection using a Xenogen IVIS Lumina Imaging System (Caliper Life Sciences, Cheshire, UK), and photons emitted from the abdominal region were quantified using Xenogen Living Image Software (Caliper Life Sciences).
Statistical Analysis
Data are expressed as the mean ± SD. Significant differences were assessed using the Student’s t test. Statistical significance was defined as a p value of less than 0.05.
Author Contributions
H.T. and T. Fujiwara developed the concept and designed research; T.K., H.T., T.I., H.A., T. Fushimi, and R.S. performed research and acquired data; T.K., H.T., T.I., S. Kuroda, S. Kikuchi, R.Y., Y. Umeda, and F. T. analyzed and interpreted data; Y. Urata and H. M. supplied materials; and T.K., H.T., and T. Fujiwara wrote and reviewed the manuscript.
Conflicts Of Interest
Y. Urata is the President and CEO of Oncolys BioPharma, Inc., the manufacturer of OBP-301 (telomelysin). H.T. and T. Fujiwara are consultants for Oncolys BioPharma. The other authors declare no competing interests.
Acknowledgments
We thank Tomoko Sueishi and Tae Yamanishi for their technical support in the production of viruses and the histopathologic tissue preparation. This study was supported in part by grants from the Japan Agency for Medical Research and Development (17929761 to T. Fujiwara) and grants from the Ministry of Education, Science, and Culture, Japan (25293283 and 16H05416 to T. Fujiwara; 25462057 and 16K10596 to H.T.).
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omto.2020.03.016.
Supplemental Information
References
- 1.Siegel R.L., Miller K.D., Jemal A. Cancer statistics, 2018. CA Cancer J. Clin. 2018;68:7–30. doi: 10.3322/caac.21442. [DOI] [PubMed] [Google Scholar]
- 2.Bapat A.A., Hostetter G., Von Hoff D.D., Han H. Perineural invasion and associated pain in pancreatic cancer. Nat. Rev. Cancer. 2011;11:695–707. doi: 10.1038/nrc3131. [DOI] [PubMed] [Google Scholar]
- 3.Demir I.E., Ceyhan G.O., Liebl F., D’Haese J.G., Maak M., Friess H. Neural invasion in pancreatic cancer: the past, present and future. Cancers (Basel) 2010;2:1513–1527. doi: 10.3390/cancers2031513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liebig C., Ayala G., Wilks J.A., Berger D.H., Albo D. Perineural invasion in cancer: a review of the literature. Cancer. 2009;115:3379–3391. doi: 10.1002/cncr.24396. [DOI] [PubMed] [Google Scholar]
- 5.Ceyhan G.O., Demir I.E., Altintas B., Rauch U., Thiel G., Müller M.W., Giese N.A., Friess H., Schäfer K.H. Neural invasion in pancreatic cancer: a mutual tropism between neurons and cancer cells. Biochem. Biophys. Res. Commun. 2008;374:442–447. doi: 10.1016/j.bbrc.2008.07.035. [DOI] [PubMed] [Google Scholar]
- 6.Jobling P., Pundavela J., Oliveira S.M., Roselli S., Walker M.M., Hondermarck H. Nerve-Cancer Cell Cross-talk: A Novel Promoter of Tumor Progression. Cancer Res. 2015;75:1777–1781. doi: 10.1158/0008-5472.CAN-14-3180. [DOI] [PubMed] [Google Scholar]
- 7.Sheahan A.V., Biankin A.V., Parish C.R., Khachigian L.M. Targeted therapies in the management of locally advanced and metastatic pancreatic cancer: a systematic review. Oncotarget. 2018;9:21613–21627. doi: 10.18632/oncotarget.25085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Burris H.A., 3rd, Moore M.J., Andersen J., Green M.R., Rothenberg M.L., Modiano M.R., Cripps M.C., Portenoy R.K., Storniolo A.M., Tarassoff P. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J. Clin. Oncol. 1997;15:2403–2413. doi: 10.1200/JCO.1997.15.6.2403. [DOI] [PubMed] [Google Scholar]
- 9.Conroy T., Desseigne F., Ychou M., Bouché O., Guimbaud R., Bécouarn Y., Adenis A., Raoul J.L., Gourgou-Bourgade S., de la Fouchardière C., Groupe Tumeurs Digestives of Unicancer. PRODIGE Intergroup FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011;364:1817–1825. doi: 10.1056/NEJMoa1011923. [DOI] [PubMed] [Google Scholar]
- 10.Von Hoff D.D., Ervin T., Arena F.P., Chiorean E.G., Infante J., Moore M., Seay T., Tjulandin S.A., Ma W.W., Saleh M.N. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 2013;369:1691–1703. doi: 10.1056/NEJMoa1304369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moore M.J., Goldstein D., Hamm J., Figer A., Hecht J.R., Gallinger S., Au H.J., Murawa P., Walde D., Wolff R.A., National Cancer Institute of Canada Clinical Trials Group Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J. Clin. Oncol. 2007;25:1960–1966. doi: 10.1200/JCO.2006.07.9525. [DOI] [PubMed] [Google Scholar]
- 12.Rahal A., Musher B. Oncolytic viral therapy for pancreatic cancer. J. Surg. Oncol. 2017;116:94–103. doi: 10.1002/jso.24626. [DOI] [PubMed] [Google Scholar]
- 13.Ady J.W., Heffner J., Klein E., Fong Y. Oncolytic viral therapy for pancreatic cancer: current research and future directions. Oncolytic Virother. 2014;3:35–46. doi: 10.2147/OV.S53858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nattress C.B., Halldén G. Advances in oncolytic adenovirus therapy for pancreatic cancer. Cancer Lett. 2018;434:56–69. doi: 10.1016/j.canlet.2018.07.006. [DOI] [PubMed] [Google Scholar]
- 15.Kawashima T., Kagawa S., Kobayashi N., Shirakiya Y., Umeoka T., Teraishi F., Taki M., Kyo S., Tanaka N., Fujiwara T. Telomerase-specific replication-selective virotherapy for human cancer. Clin. Cancer Res. 2004;10:285–292. doi: 10.1158/1078-0432.ccr-1075-3. [DOI] [PubMed] [Google Scholar]
- 16.Fujiwara T., Urata Y., Tanaka N. Telomerase-specific oncolytic virotherapy for human cancer with the hTERT promoter. Curr. Cancer Drug Targets. 2007;7:191–201. doi: 10.2174/156800907780058835. [DOI] [PubMed] [Google Scholar]
- 17.Hashimoto Y., Watanabe Y., Shirakiya Y., Uno F., Kagawa S., Kawamura H., Nagai K., Tanaka N., Kumon H., Urata Y., Fujiwara T. Establishment of biological and pharmacokinetic assays of telomerase-specific replication-selective adenovirus. Cancer Sci. 2008;99:385–390. doi: 10.1111/j.1349-7006.2007.00665.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yamasaki Y., Tazawa H., Hashimoto Y., Kojima T., Kuroda S., Yano S., Yoshida R., Uno F., Mizuguchi H., Ohtsuru A. A novel apoptotic mechanism of genetically engineered adenovirus-mediated tumour-specific p53 overexpression through E1A-dependent p21 and MDM2 suppression. Eur. J. Cancer. 2012;48:2282–2291. doi: 10.1016/j.ejca.2011.12.020. [DOI] [PubMed] [Google Scholar]
- 19.Hasei J., Sasaki T., Tazawa H., Osaki S., Yamakawa Y., Kunisada T., Yoshida A., Hashimoto Y., Onishi T., Uno F. Dual programmed cell death pathways induced by p53 transactivation overcome resistance to oncolytic adenovirus in human osteosarcoma cells. Mol. Cancer Ther. 2013;12:314–325. doi: 10.1158/1535-7163.MCT-12-0869. [DOI] [PubMed] [Google Scholar]
- 20.Tazawa H., Kagawa S., Fujiwara T. Advances in adenovirus-mediated p53 cancer gene therapy. Expert Opin. Biol. Ther. 2013;13:1569–1583. doi: 10.1517/14712598.2013.845662. [DOI] [PubMed] [Google Scholar]
- 21.Iacobuzio-Donahue C.A. Genetic evolution of pancreatic cancer: lessons learnt from the pancreatic cancer genome sequencing project. Gut. 2012;61:1085–1094. doi: 10.1136/gut.2010.236026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kishimoto H., Kojima T., Watanabe Y., Kagawa S., Fujiwara T., Uno F., Teraishi F., Kyo S., Mizuguchi H., Hashimoto Y. In vivo imaging of lymph node metastasis with telomerase-specific replication-selective adenovirus. Nat. Med. 2006;12:1213–1219. doi: 10.1038/nm1404. [DOI] [PubMed] [Google Scholar]
- 23.Furukawa T. Impacts of activation of the mitogen-activated protein kinase pathway in pancreatic cancer. Front. Oncol. 2015;5:23. doi: 10.3389/fonc.2015.00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Neuzillet C., Hammel P., Tijeras-Raballand A., Couvelard A., Raymond E. Targeting the Ras-ERK pathway in pancreatic adenocarcinoma. Cancer Metastasis Rev. 2013;32:147–162. doi: 10.1007/s10555-012-9396-2. [DOI] [PubMed] [Google Scholar]
- 25.Vincent A., Herman J., Schulick R., Hruban R.H., Goggins M. Pancreatic cancer. Lancet. 2011;378:607–620. doi: 10.1016/S0140-6736(10)62307-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ryan D.P., Hong T.S., Bardeesy N. Pancreatic adenocarcinoma. N. Engl. J. Med. 2014;371:1039–1049. doi: 10.1056/NEJMra1404198. [DOI] [PubMed] [Google Scholar]
- 27.Rosenfeldt M.T., O’Prey J., Morton J.P., Nixon C., MacKay G., Mrowinska A., Au A., Rai T.S., Zheng L., Ridgway R. p53 status determines the role of autophagy in pancreatic tumour development. Nature. 2013;504:296–300. doi: 10.1038/nature12865. [DOI] [PubMed] [Google Scholar]
- 28.Todoric J., Antonucci L., Di Caro G., Li N., Wu X., Lytle N.K., Dhar D., Banerjee S., Fagman J.B., Browne C.D. Stress-activated NRF2-MDM2 cascade controls neoplastic progression in pancreas. Cancer Cell. 2017;32:824–839.e8. doi: 10.1016/j.ccell.2017.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tang J., Di J., Cao H., Bai J., Zheng J. p53-mediated autophagic regulation: A prospective strategy for cancer therapy. Cancer Lett. 2015;363:101–107. doi: 10.1016/j.canlet.2015.04.014. [DOI] [PubMed] [Google Scholar]
- 30.Mrakovcic M., Fröhlich L.F. p53-Mediated Molecular Control of Autophagy in Tumor Cells. Biomolecules. 2018;8:e14. doi: 10.3390/biom8020014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Giehl K., Skripczynski B., Mansard A., Menke A., Gierschik P. Growth factor-dependent activation of the Ras-Raf-MEK-MAPK pathway in the human pancreatic carcinoma cell line PANC-1 carrying activated K-ras: implications for cell proliferation and cell migration. Oncogene. 2000;19:2930–2942. doi: 10.1038/sj.onc.1203612. [DOI] [PubMed] [Google Scholar]
- 32.Deer E.L., González-Hernández J., Coursen J.D., Shea J.E., Ngatia J., Scaife C.L., Firpo M.A., Mulvihill S.J. Phenotype and genotype of pancreatic cancer cell lines. Pancreas. 2010;39:425–435. doi: 10.1097/MPA.0b013e3181c15963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen S.H., Zhang Y., Van Horn R.D., Yin T., Buchanan S., Yadav V., Mochalkin I., Wong S.S., Yue Y.G., Huber L. Oncogenic BRAF Deletions That Function as Homodimers and Are Sensitive to Inhibition by RAF Dimer Inhibitor LY3009120. Cancer Discov. 2016;6:300–315. doi: 10.1158/2159-8290.CD-15-0896. [DOI] [PubMed] [Google Scholar]
- 34.Cancer Genome Atlas Research Network Integrated genomic characterization of pancreatic ductal adenocarcinoma. Cancer Cell. 2017;32:185–203.e13. doi: 10.1016/j.ccell.2017.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shibata H., Komura S., Yamada Y., Sankoda N., Tanaka A., Ukai T., Kabata M., Sakurai S., Kuze B., Woltjen K. In vivo reprogramming drives Kras-induced cancer development. Nat. Commun. 2018;9:2081. doi: 10.1038/s41467-018-04449-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang K., Li Y., Lian G., Lin H., Shang C., Zeng L., Chen S., Li J., Huang C., Huang K., Chen Y. KRAS promotes tumor metastasis and chemoresistance by repressing RKIP via the MAPK-ERK pathway in pancreatic cancer. Int. J. Cancer. 2018;142:2323–2334. doi: 10.1002/ijc.31248. [DOI] [PubMed] [Google Scholar]
- 37.Lee S.J., Lee S.H., Yoon M.H., Park B.J. A new p53 target gene, RKIP, is essential for DNA damage-induced cellular senescence and suppression of ERK activation. Neoplasia. 2013;15:727–737. doi: 10.1593/neo.121862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Behrens D., Walther W., Fichtner I. Pancreatic cancer models for translational research. Pharmacol. Ther. 2017;173:146–158. doi: 10.1016/j.pharmthera.2017.02.013. [DOI] [PubMed] [Google Scholar]
- 39.Ardito C.M., Grüner B.M., Takeuchi K.K., Lubeseder-Martellato C., Teichmann N., Mazur P.K., Delgiorno K.E., Carpenter E.S., Halbrook C.J., Hall J.C. EGF receptor is required for KRAS-induced pancreatic tumorigenesis. Cancer Cell. 2012;22:304–317. doi: 10.1016/j.ccr.2012.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Navas C., Hernández-Porras I., Schuhmacher A.J., Sibilia M., Guerra C., Barbacid M. EGF receptor signaling is essential for k-ras oncogene-driven pancreatic ductal adenocarcinoma. Cancer Cell. 2012;22:318–330. doi: 10.1016/j.ccr.2012.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tazawa H., Yano S., Yoshida R., Yamasaki Y., Sasaki T., Hashimoto Y., Kuroda S., Ouchi M., Onishi T., Uno F. Genetically engineered oncolytic adenovirus induces autophagic cell death through an E2F1-microRNA-7-epidermal growth factor receptor axis. Int. J. Cancer. 2012;131:2939–2950. doi: 10.1002/ijc.27589. [DOI] [PubMed] [Google Scholar]
- 42.Metildi C.A., Kaushal S., Hoffman R.M., Bouvet M. In vivo serial selection of human pancreatic cancer cells in orthotopic mouse models produces high metastatic variants irrespective of Kras status. J. Surg. Res. 2013;184:290–298. doi: 10.1016/j.jss.2013.03.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hayes T.K., Neel N.F., Hu C., Gautam P., Chenard M., Long B., Aziz M., Kassner M., Bryant K.L., Pierobon M. Long-Term ERK Inhibition in KRAS-Mutant Pancreatic Cancer Is Associated with MYC Degradation and Senescence-like Growth Suppression. Cancer Cell. 2016;29:75–89. doi: 10.1016/j.ccell.2015.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Knudsen E.S., O’Reilly E.M., Brody J.R., Witkiewicz A.K. Genetic Diversity of Pancreatic Ductal Adenocarcinoma and Opportunities for Precision Medicine. Gastroenterology. 2016;150:48–63. doi: 10.1053/j.gastro.2015.08.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dreyer S.B., Chang D.K., Bailey P., Biankin A.V. Pancreatic Cancer Genomes: Implications for Clinical Management and Therapeutic Development. Clin. Cancer Res. 2017;23:1638–1646. doi: 10.1158/1078-0432.CCR-16-2411. [DOI] [PubMed] [Google Scholar]
- 46.Biankin A.V., Waddell N., Kassahn K.S., Gingras M.C., Muthuswamy L.B., Johns A.L., Miller D.K., Wilson P.J., Patch A.M., Wu J., Australian Pancreatic Cancer Genome Initiative Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature. 2012;491:399–405. doi: 10.1038/nature11547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yachida S., White C.M., Naito Y., Zhong Y., Brosnan J.A., Macgregor-Das A.M., Morgan R.A., Saunders T., Laheru D.A., Herman J.M. Clinical significance of the genetic landscape of pancreatic cancer and implications for identification of potential long-term survivors. Clin. Cancer Res. 2012;18:6339–6347. doi: 10.1158/1078-0432.CCR-12-1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hingorani S.R., Wang L., Multani A.S., Combs C., Deramaudt T.B., Hruban R.H., Rustgi A.K., Chang S., Tuveson D.A. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7:469–483. doi: 10.1016/j.ccr.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 49.McCormick F. KRAS as a Therapeutic Target. Clin. Cancer Res. 2015;21:1797–1801. doi: 10.1158/1078-0432.CCR-14-2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Junttila M.R., Karnezis A.N., Garcia D., Madriles F., Kortlever R.M., Rostker F., Brown Swigart L., Pham D.M., Seo Y., Evan G.I., Martins C.P. Selective activation of p53-mediated tumour suppression in high-grade tumours. Nature. 2010;468:567–571. doi: 10.1038/nature09526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Feldser D.M., Kostova K.K., Winslow M.M., Taylor S.E., Cashman C., Whittaker C.A., Sanchez-Rivera F.J., Resnick R., Bronson R., Hemann M.T., Jacks T. Stage-specific sensitivity to p53 restoration during lung cancer progression. Nature. 2010;468:572–575. doi: 10.1038/nature09535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kuroda S., Fujiwara T., Shirakawa Y., Yamasaki Y., Yano S., Uno F., Tazawa H., Hashimoto Y., Watanabe Y., Noma K. Telomerase-dependent oncolytic adenovirus sensitizes human cancer cells to ionizing radiation via inhibition of DNA repair machinery. Cancer Res. 2010;70:9339–9348. doi: 10.1158/0008-5472.CAN-10-2333. [DOI] [PubMed] [Google Scholar]
- 53.Yano S., Tazawa H., Hashimoto Y., Shirakawa Y., Kuroda S., Nishizaki M., Kishimoto H., Uno F., Nagasaka T., Urata Y. A genetically engineered oncolytic adenovirus decoys and lethally traps quiescent cancer stem-like cells in S/G2/M phases. Clin. Cancer Res. 2013;19:6495–6505. doi: 10.1158/1078-0432.CCR-13-0742. [DOI] [PubMed] [Google Scholar]
- 54.Osaki S., Tazawa H., Hasei J., Yamakawa Y., Omori T., Sugiu K., Komatsubara T., Fujiwara T., Sasaki T., Kunisada T. Ablation of MCL1 expression by virally induced microRNA-29 reverses chemoresistance in human osteosarcomas. Sci. Rep. 2016;6:28953. doi: 10.1038/srep28953. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.