

The major implication of our work is that the decay of intact proviruses in vitro is extremely rapid, perhaps as a result of enhanced expression. Gaining a better understanding of why intact proviruses decay faster in vitro might help the field identify strategies to purge the reservoir in vivo. When used wisely, in vitro models are a powerful tool to study the selective pressures shaping the viral landscape. Our finding that massively deleted sequences rarely succeed in integrating has several ramifications. It demonstrates that the total HIV DNA can differ substantially in character from the integrated HIV DNA under certain circumstances. The presence of unintegrated HIV DNA has the potential to obscure selection pressures and confound the interpretation of clinical studies, especially in the case of trials involving treatment interruptions.
KEYWORDS: HIV, latency, NGS, reservoir, sequencing, resting cells
ABSTRACT
Next-generation sequencing (NGS) represents a powerful tool to unravel the genetic make-up of the HIV reservoir, but limited data exist on its use in vitro. Moreover, most NGS studies do not separate integrated from unintegrated DNA, even though selection pressures on these two forms should be distinct. We reasoned we could use NGS to compare the infection of resting and activated CD4 T cells in vitro to address how the metabolic state affects reservoir formation and dynamics. To address these questions, we obtained HIV sequences 2, 4, and 8 days after NL4-3 infection of metabolically activated and quiescent CD4 T cells (cultured with 2 ng/ml interleukin-7). We compared the composition of integrated and total HIV DNA by isolating integrated HIV DNA using pulsed-field electrophoresis before performing sequencing. After a single-round infection, the majority of integrated HIV DNA was intact in both resting and activated T cells. The decay of integrated intact proviruses was rapid and similar in both quiescent and activated T cells. Defective forms accumulated relative to intact ones analogously to what is observed in vivo. Massively deleted viral sequences formed more frequently in resting cells, likely due to lower deoxynucleoside triphosphate (dNTP) levels and the presence of multiple restriction factors. To our surprise, the majority of these deleted sequences did not integrate into the human genome. The use of NGS to study reservoir dynamics in vitro provides a model that recapitulates important aspects of reservoir dynamics. Moreover, separating integrated from unintegrated HIV DNA is important in some clinical settings to properly study selection pressures.
IMPORTANCE The major implication of our work is that the decay of intact proviruses in vitro is extremely rapid, perhaps as a result of enhanced expression. Gaining a better understanding of why intact proviruses decay faster in vitro might help the field identify strategies to purge the reservoir in vivo. When used wisely, in vitro models are a powerful tool to study the selective pressures shaping the viral landscape. Our finding that massively deleted sequences rarely succeed in integrating has several ramifications. It demonstrates that the total HIV DNA can differ substantially in character from the integrated HIV DNA under certain circumstances. The presence of unintegrated HIV DNA has the potential to obscure selection pressures and confound the interpretation of clinical studies, especially in the case of trials involving treatment interruptions.
INTRODUCTION
Historically, T-cell activation has been considered an important aspect in the pathogenesis of HIV infection, since activated CD4 (aCD4) T cells appear more susceptible to infection and virion release (1–3) and are massively depleted during the first phase of viral decay after antiretroviral therapy (ART) initiation (4–6). On the other hand, resting CD4 (rCD4) T cells represent the major component of the HIV reservoir in individuals on long-term ART (7, 8). Some studies have suggested that the HIV reservoir of resting cells forms when aCD4 T cells revert to a resting state (9).
Studies comparing the kinetics of resting and activated T cells showed a dramatic difference in the tempo of infection (2, 10–15). While the pace of infection was much faster in aCD4 T cells, the final levels of integrated DNA after one round of infection were comparable. These studies also suggested the most important difference between the infection of these two cells was the superior ability of aCD4 T cells to release virions and initiate spreading infection (16). Multiple cellular factors have been suggested to restrict HIV infection in rCD4 T cells at multiple steps, including SAMHD1 for reverse transcription (17–19) and TRABD2A for virion release (20).
The advent of next-generation sequencing (NGS) has led to a new era in the study of HIV genetic make-up, with unprecedented opportunities in terms of both throughput and depth of analysis. While many groups, including our own, have been using NGS to study the proviral landscape of subjects on ART (21–26), there is limited literature on NGS as a tool to study in vitro models and no analysis of differences in the genetic landscape of integrated versus unintegrated HIV DNA. We realized that by studying the kinetics of integrated and unintegrated HIV DNA in vitro using NGS, we could gain a better understanding of selection pressures that would complement our longitudinal in vivo studies.
In the present study, we used an in vitro model of primary T-cell infection to study the HIV landscape in rCD4 and aCD4 T cells by NGS. Our data show that the frequency of intact integrated proviruses is similar in rCD4 and aCD4 T cells. However, massively deleted viral sequences are detected more often in resting cells, but they fail to integrate in the chromosomal DNA as efficiently as intact sequences. This results in similar levels of intact HIV DNA in both activated and resting T cells. We confirmed the importance of separating integrated versus unintegrated HIV DNA in individuals with HIV infection. Our results suggest it is essential to separate these two forms not only in in vitro models but also in clinical settings where unintegrated HIV DNA is expected to be present, because selection pressures on these two forms of DNA are distinct.
RESULTS
Comparison of total and intact HIV DNA dynamics in metabolically activated and resting CD4 T cells by long terminal repeat (LTR) quantitative PCR (qPCR) and a modified IPDA.
We previously compared the kinetics of HIV infection in aCD4 and rCD4 T cells using our primary T-cell model. With the advent of NGS, we wanted to monitor the kinetics of the intact and defective viral forms in metabolically active and resting cells. We isolated rCD4 T cells from peripheral blood mononuclear cells (PBMCs) of an uninfected donor. Half of the cells were infected with NL4-3 and then cultured for 8 days in the presence of saquinavir (SQV) and interleukin-7 (IL-7) (2 ng/ml). The other half was activated for 3 days using CD3/CD28 beads in the presence of IL-2 (50 UI/ml). After 3 days, cells were infected and then cultured for 8 days with SQV (to prevent new rounds of infection) and IL-2 (50 UI/ml). To study the kinetics of infection, we collected cells at multiple time points over 8 days and measured the levels of total HIV DNA. In line with previous studies (2, 11, 15), we confirmed that rCD4 T cells had a slower kinetics of infection than aCD4 T cells. For rCD4 T cells, the levels of total HIV DNA were the highest at day 2 (d2), while for aCD4 T cells the peak of infection was achieved earlier and then declined over time (Fig. 1), consistent with prior studies (12). We next estimated the number of intact viruses by intact proviral DNA assay (IPDA), sampling ∼10,000 cells per time point. In line with our PCR estimates, the number of intact viruses peaked earlier for aCD4 T cells (2.3 copies/cell at 6 h) and then declined over time (0.14 copies/cell at d8), while for rCD4 T cells the number of intact viruses declined from 1.1 copies/cell at d2 (peak infection) to 0.2 copies/cell at d8 (Fig. 1). Overall, IPDA combined with total HIV DNA measures suggested that a significant fraction of DNA in a single-round infection was intact, especially in aCD4 T cells, and that defective forms were more common in resting T cells.
FIG 1.

Dynamics of total and intact HIV DNA levels as determined by IPDA in rCD4 and aCD4 T cells after in vitro infection. Cells were either infected with NL4-3 or activated for 3 days with CD3/CD28 beads before infection. Cells were cultured with SQV and collected at 2, 4, 8, 24, 48, 96, and 192 h to measure total HIV DNA by qPCR and intact HIV DNA by a modified IPDA. Total and intact HIV DNA followed a similar pattern in terms of both peak infection and decay over time in culture.
Near-full-length viral sequencing of total HIV DNA reveals that the cellular metabolic status influences the formation of deleted viral sequences.
We were interested to know if the IPDA estimates of intact DNA would predict intact sequences estimated by sequencing. To address this question, we measured the decay of intact viral forms in total HIV DNA in rCD4 versus aCD4 T cells by NGS. We performed HIV near-full-length sequencing at limiting dilution, obtaining a total of 993 sequences. We used samples collected at days 2, 4, and 8 after infection.
We found that the number of intact viruses declined over time in both aCD4 and rCD4 T cells, similar to our IPDA prediction (Fig. 2). The absolute number of intact viral sequences ranged from 0.7 copies/cell at d2 to 0.1 copies/cell at d8 in aCD4 T cells and from 1.2 at d2 to 0.2 copies/cell at d8 in rCD4 T cells (Fig. 2). We estimated the decay of intact viral forms to be faster than that of defective ones, with similar patterns in rCD4 and aCD4 T cells. In fact, the half-life of intact viral sequences was 2.5 days in aCD4 T cells and 2.4 days in rCD4 T cells. Taken together, these data suggest that deleted viral DNA sequences form more often in resting cells. Moreover, the faster decline of intact viral sequences found in both resting and activated T cells suggests higher selection pressure against intact over defective forms in culture, possibly due to higher viral expression leading to higher cytotoxicity. Another possible explanation is that defective viral forms lack critical recognition sequences for innate sensing and might be less efficiently targeted for degradation than intact ones. Importantly, the predominant massively deleted viruses we sequenced in rCD4 T cells generally were not detected by the primers used in the IPDA assay. While IPDA provided strong estimates of decay of the intact sequences, it did not capture the dynamics of deleted forms accurately.
FIG 2.

Dynamics of intact and defective DNA forms as determined by NGS in the total fraction of HIV DNA isolated from rCD4 and aCD4 T cells. Cells were collected at d2, d4, and d8 after a single-round infection with NL4-3, and viral sequences were amplified by NGS. The number of intact viral sequences declined in both aCD4 and rCD4 T cells over time in culture, with a faster decay than that of defective ones. Moreover, massively deleted sequences formed more often in resting cells. We did not perform NGS on DNA collected at earlier time points due to limited sample availability.
Near-full-length sequencing of integrated HIV DNA shows important differences in the dynamics of intact versus defective proviruses.
To study whether similar patterns occurred in the integrated fraction of HIV DNA, we used pulsed-field electrophoresis (BluePippin; Sage Science) to select the high-molecular-weight fraction of DNA (>20 kb) from the same samples shown in Fig. 2. This fraction is expected to be enriched for integrated HIV DNA, as previously shown by others (27). NGS revealed a similar decline in the intact HIV copies/cell over time in both activated and resting T cells. This number declined from 0.3 at d2 to 0.05 copies/cell at d8 in aCD4 T cells and from 0.4 to 0.1 copies/cell in rCD4 T cells (Fig. 3). The calculated half-life of integrated intact proviruses was very similar in both cell types (2.2 days in aCD4 and 2.4 days in rCD4 T cells), as predicted by in vivo modeling studies (28, 29). On the other hand, defective proviruses only minimally declined (Fig. 3). This could be because an intact provirus induces more cytotoxicity than a defective one, leading to the preferential death of the infected cell harboring it. In conclusion, our results suggest a striking difference in the genetic make-up of the total compared to the integrated fraction of HIV DNA in rCD4 T cells.
FIG 3.

Dynamics of intact and defective proviruses isolated from genomic DNA in rCD4 and aCD4 T cells after removing unintegrated HIV DNA by pulsed-field electrophoresis. Cells were collected at d2, d4, and d8 after a single-round infection with NL4-3, and integrated HIV DNA was isolated by pulsed-field electrophoresis (BluePippin). Proviral sequences were amplified by NGS. The absolute number of intact proviruses declined in both aCD4 and rCD4 T cells, while defective proviruses changed minimally over time.
Size distribution of the viral sequences isolated from total and integrated HIV DNA suggests that massively deleted viral forms occur more readily in rCD4 T cells but fail to integrate efficiently.
To our surprise, despite the fact that deleted reverse transcripts formed more frequently in rCD4 T cells (Fig. 2), these deleted forms were rarely detected in the host chromosomal DNA in a single-round infection.
We wanted to know if the character of the deleted viruses was different in rCD4 versus aCD4 T cells. In Fig. 4, we combined all our sequences for total versus integrated HIV DNA in resting as well as activated T cells and analyzed them by size. Viral sequences were grouped in 1,000-bp bins to build histograms for both rCD4 and aCD4 T cells, differentiating the total from the integrated HIV fraction (Fig. 4 and Table 1).
FIG 4.
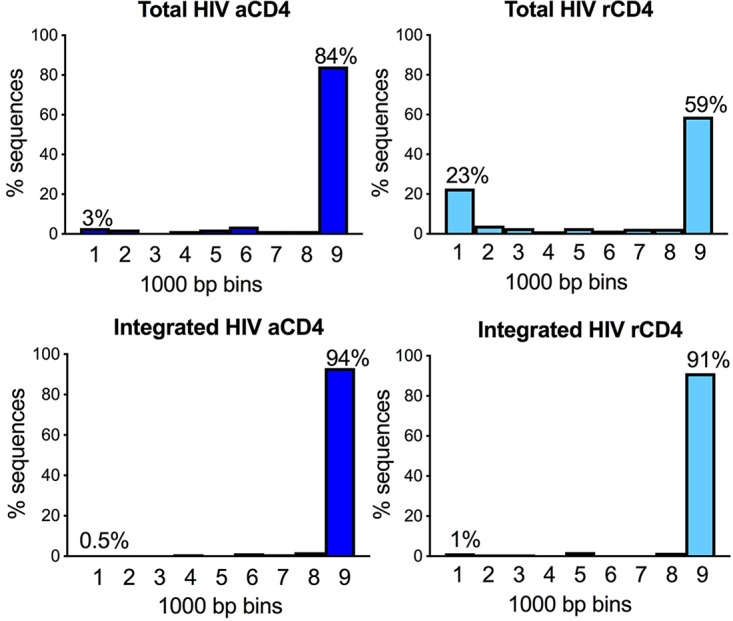
Size distribution of HIV DNA sequences isolated from total and integrated HIV of rCD4 and aCD4 T cells. Sequences were grouped in 1,000-bp bins to build histograms. The percentage of sequences with <1,000 bp was significantly higher in the total DNA fraction of rCD4 T cells (23%) than in integrated HIV DNA (1%, P < 0.0001 by chi-square test) as well as DNA isolated from aCD4 T cells (0.5 to 3%, P < 0.0001 by chi-square test).
TABLE 1.
Distribution of the viral sequences according to their sizea
| Viral size (bp) | Frequency [no. (%)] of: |
|||
|---|---|---|---|---|
| aCD4 total HIV | aCD4 integrated HIV | rCD4 total HIV | rCD4 integrated HIV | |
| 0–1,000 | 4 (3) | 1 (0.5) | 71 (22.8) | 5 (1.5) |
| 1,001–2,000 | 3 (2.2) | 1 (0.5) | 13 (4.2) | 3 (0.9) |
| 2,001–3,000 | 0 | 0 | 9 (2.9) | 3 (0.9) |
| 3,001–4,000 | 2 (1.5) | 2 (1) | 4 (1.3) | 2 (0.6) |
| 4,001–5,000 | 3 (2.2) | 0 | 9 (2.9) | 7 (2) |
| 5,001–6,000 | 5 (3.7) | 3 (1.5) | 5 (1.6) | 2 (0.6) |
| 6,001–7,000 | 2 (1.5) | 2 (1) | 8 (2.6) | 2 (0.6) |
| 7,001–8,000 | 2 (1.5) | 4 (2) | 8 (2.6) | 6 (1.7) |
| 8,001–9,000 | 113 (84.4) | 191 (93.5) | 185 (59.3) | 313 (91.3) |
| Total | 134 | 204 | 312 | 343 |
We compared the size distribution of viral sequences from rCD4 versus aCD4 T cells in the integrated and total fractions of HIV DNA. Sequences were grouped in 1,000-bp bins, and the frequency of viral sequences in each bin was used to build the histograms presented in Fig. 4. The frequency of viral sequences shorter than 1,000 bp was significantly higher in total HIV DNA from resting cells than in total HIV DNA from activated cells (P < 0.0001 by chi-square test). Moreover, it was significantly higher than the frequency of massively deleted viral sequences observed in integrated HIV DNA (P < 0.0001 by chi-square test).
Total HIV DNA amplified from rCD4 T cells contained a significantly larger fraction of short sequences (less than 1,000 bp) than total HIV DNA from aCD4 T cells (P < 0.0001 by chi-square test). In fact, 23% of the analyzed sequences were less than 1,000 bp, unlike the 3% observed in the total DNA fraction of aCD4 T cells. This was expected since reverse transcription occurs more slowly in resting cells (11). Unexpectedly, the proportion of massively deleted proviruses among the purified high-molecular-weight DNA was similar in rCD4 and aCD4 T cells, with ∼0.5 to 1% of sequences smaller than 1,000 bp. Overall, these data suggest massively deleted viral forms are generated more frequently in metabolically quiescent cells, but most of these deleted forms do not appear to successfully integrate (Fig. 3 and 4 and Table 1). Why preintegration complexes with deleted proviruses are less likely to successfully integrate remains unclear, but several possible mechanisms are proposed in Discussion.
Separating integrated from unintegrated HIV DNA might have important implications in clinical studies.
Our in vitro results suggested the importance of isolating integrated HIV DNA prior to NGS to avoid data misinterpretation. We were interested in evaluating if this was true in a clinical setting. We purified DNA from PBMC aliquots of a chronically infected subject off ART (Fig. 5A). PBMCs of this ART-naive individual were collected on the day before ART initiation. We separated the high- versus low-molecular-weight DNA by BluePippin technology as described in Materials and Methods and amplified HIV sequences from both fractions. The high-molecular-weight DNA is expected to be enriched for integrated HIV, while the low-molecular-weight fraction contains unintegrated HIV. We amplified 84 sequences from the integrated fraction and 34 from the unintegrated fraction. We also obtained 123 HIV sequences from the pool of total unfractionated DNA (Fig. 5A). We measured by qPCR the levels of HIV LTR DNA to estimate the absolute number of viruses in each fraction. In contrast to what we observed in vitro, we found that low-molecular-weight DNA harbored more intact viruses (50%) than the high-molecular-weight fraction (30%). In the total HIV DNA fraction, we found 34% of viruses were intact. As a control, we isolated integrated HIV DNA from PBMCs of a subject on long-term ART (∼10 years) and found no evidence of unintegrated HIV DNA (Fig. 5B). The high-molecular-weight DNA was used to amplify 96 proviruses. We found that the fraction of intact proviruses (7/96, 7%) was similar to the one observed in viral sequences obtained from the pool of total HIV DNA (6/84 sequences, 7%). These results suggest that in settings where ongoing replication is expected, such as structured treatment interruption (STI) trials, recent ART initiation, or chronic infection, isolation of integrated HIV DNA should precede NGS to avoid the amplification of unintegrated viral genomes, which can complicate data interpretation.
FIG 5.

Number of intact HIV sequences in the high- and low-molecular-weight fraction of DNA in individuals with HIV infection on and off ART. (A) BluePippin was used to separate high (>20 kb; HMW; 84 sequences)- and low (<12 kb; LMW; 34 sequences)-molecular-weight DNA from PBMCs of one ART-naive individual with untreated HIV infection. We then performed NGS as well as HIV LTR qPCR, using unfractionated DNA as a control (total DNA; 123 sequences). Intact HIV represented 50% of the sequences obtained from unintegrated HIV (7,874 copies/million CD4s), while it made up 30% of the DNA obtained from integrated HIV, corresponding to 4,198 copies/million CD4 T cells. (B) NGS was used to compare the frequency of intact proviruses retrieved from the HMW versus unfractionated DNA of one individual on long-term ART. We found no evidence of unintegrated DNA, since the fraction and absolute number of intact proviruses (7%) was similar in the two samples. A consequence of these results is that when unintegrated DNA is present, as occurs with ongoing replication, the percentage of defective viral sequences is decreased. In our proof-of-concept example, 66% of viral sequences were defective in total HIV DNA when ongoing replication occurred. However, after 3 years on ART, 97% of the HIV sequences from the same subject were defective as a result of strong selection pressures on intact proviruses (see also Fig. 6 and reference 22).
Moreover, our results suggest that major selection pressures must occur in vivo to account for the predominance of deleted proviruses observed in subjects on ART (Fig. 6).
FIG 6.

HIV sequences undergo strong pre- and postintegration selection. Here, we provide a visual summary of possible mechanisms contributing to the preintegration bottleneck for massively deleted viral sequences in rCD4 T cells as well as the postintegration selection of intact proviruses. (A) Negative selection for defective viral sequences occurs prior to integration. As a result of multiple factors, including low dNTPs and higher levels of restriction factors, HIV reverse transcription is slow in resting cells and might result in the generation of more deleted viral sequences than activated cells. Preintegration complexes (PICs) containing intact DNA sequences appear to translocate the nuclear pore more efficiently (B) than deleted ones (A). This is reasonable, since deleted sequences generally lack structural elements, such as the central polypurine tract (cPPT), that might contribute to nuclear import. Additionally, reverse transcriptase pausing might provide a longer temporal window for innate restriction factors, including Mx2, to have an effect. This, in turn, could lead to the decreased efficiency of nuclear import. (B) Negative selection for intact forms after integration. Cells harboring intact proviruses are more likely to be cleared as a result of both viral and immune cytotoxicity. (Courtesy of Mary Leonard; reproduced with permission.)
DISCUSSION
In this study, we used NGS to address how the cellular metabolic state affects reservoir formation and dynamics. As we predicted, we found that reverse transcripts with massive deletions formed more frequently in resting cells, but to our surprise, these massively deleted reverse transcripts were only rarely detected in the host chromosome. This, in turn, suggests a strong selection against massively deleted viruses in the steps preceding integration. As a result of these selection pressures, the majority of integrated HIV DNA after in vitro infection is intact in both resting and activated T cells. Moreover, our study revealed another selection pressure occurring after the step of integration, since the integrated intact HIV DNA decayed more rapidly than the integrated defective DNA. As discussed below, these findings have important implications for utilizing in vitro models as a tool to study reservoir dynamics in vivo.
Our approach of separating metabolically active from resting T cells as well as separating integrated from unintegrated HIV DNA was essential for elucidating these important differential selection pressures. Notably, no obvious distinctions between intact and total HIV DNA could be identified when total HIV DNA measurements were compared to intact HIV estimates by IPDA (Fig. 1), consistent with other reports (21, 25) and with the fact that a large fraction of total DNA is intact in a single-round infection. On the other hand, the use of NGS gave us the opportunity to unveil important differences in the fate of intact versus defective viral sequences. In fact, we found that intact HIV decayed rapidly while defective viral forms were more stable. This proved true when we examined total DNA (containing both integrated and unintegrated HIV; Fig. 2) or high-molecular-weight DNA (containing integrated and chromosomal DNA; Fig. 3). Moreover, when we separated integrated from unintegrated DNA, we identified a bottleneck between the step of reverse transcription and integration preferentially affecting the massively deleted reverse transcripts (Fig. 3 and 4).
Short defective transcripts form more frequently in rCD4 T cells.
Our work suggests that deleted reverse transcripts form more frequently in rCD4 T cells than in aCD4 T cells. However, these deleted viral intermediates do not integrate as efficiently as intact ones. While our study does not address the mechanisms behind these observations, we hypothesize that in rCD4 T cells the cellular environment itself (with higher levels of restriction factors [reviewed in reference 30]) and lower levels of deoxynucleoside triphosphates (dNTPs) create favorable conditions for the generation of these short preintegration viral forms. Early in vitro studies reported restriction at the step of reverse transcription in rCD4 T cells (1–3). However, subsequent studies showed that reverse transcription occurs in rCD4 T cells, although with slower kinetics (2, 10–15), and it can be boosted by the knockdown of cellular restriction factors as well as the addition of dNTPs (11, 17, 31, 32). Previous work from our group showed that dNTPs increase the integration rate in rCD4 T cells without inducing T-cell activation (11). We also hypothesize that pausing of the reverse transcriptase (RT) on the HIV template contributes to the formation of deleted preintegration sequences, increasing the chance that RT could dissociate and then reassociate in a different region of the template, potentially leading to the formation of deleted viral sequences (33, 34).
There is a preintegration bottleneck for massively deleted viral forms.
In our in vitro model, we were surprised to notice that short defective viruses did not integrate as efficiently as intact ones. Reverse transcription occurs at a lower rate in rCD4 T cells, likely due to the presence of multiple restriction factors (30, 35) as well as lower levels of dNTPs. This, in turn, might result in the generation of more deleted reverse transcripts than those of activated T cells (Fig. 2). We speculate that massively deleted preintegration complexes (PIC) enter the nucleus less efficiently (Fig. 6). One of the reasons might be the lack of the central DNA flap, which has been proposed to mediate HIV nuclear import (36). Moreover, while some PICs might protect the RT complex more efficiently from host factors, resulting in minimal pausing and successful integration, other PICs might provide less protection for the RT complex (Fig. 6). As a consequence, restriction factors might more easily access the RT complex, increasing pausing and deletion frequency (30). Pausing can provide a wider temporal window for immune sensing to regulate viral nuclear import. Restriction factors, including Mx2, are likely induced after infection, limiting the nuclear entry of the viral genome (30, 37–39). We are aware that the interaction between HIV and host factors is complex and can occur either in the cytoplasm or in the nucleus. For this reason, while technically challenging, we envision future fractionation studies helping to identify the major contributors to this preintegration bottleneck.
Our in vitro model mirrors important aspects of decay in vivo, although clearance is significantly accelerated.
We found that the vast majority of integrated HIV DNA was represented by intact proviruses in both rCD4 and aCD4 T cells. As previously mentioned, limited NGS data exist to study in vitro models. At first inspection, our results appear to disagree with previous literature (25), but this discrepancy can be explained by the range of activation states as well as the presence of DNA intermediates in the cultures. Imamichi et al. (21) reported ∼20% of deleted viral sequences in vitro (similar to our experience with phytohemagglutinin [PHA]; not shown), while Bruner et al. found ∼40% of deleted forms in a single-round infection (25). This difference might be due to a different degree of cellular activation in the sample. Regardless, in both cases the authors did not separate integrated from unintegrated HIV DNA (21, 25). Neglecting to separate these two fractions might mask important aspects of reservoir selection that influence its formation and dynamics.
Our in vitro and in vivo data suggest selection against intact HIV is much stronger than that against defective HIV DNA.
Our in vitro model is consistent with longitudinal data for subjects on ART showing that intact proviruses are cleared faster than defective ones. However, decay is dramatically faster in vitro than in vivo. This accelerated decay could be due to an altered cytokine milieu that may reduce T-cell longevity in vitro. Alternatively, the absence of CD8 T cells from our in vitro model may lead to enhanced HIV expression, as is proposed to occur in CD8 depletion studies (40, 41). Our data suggest that the postintegration selection for defective forms in vivo must be very strong, since the defective forms appear to integrate inefficiently, making up less than 10% of integrated DNA in a single-round infection. Similar to in vivo forms, the deleted forms appear to be more stable than intact ones. We were surprised to observe a similar decay of intact proviruses in resting and activated CD4 T cells, since activated cells express higher levels of toxic proteins and have a superior ability to release virus (16). This might be due to the fact that even low-level HIV expression, as occurs in resting cells, could be sufficient to trigger viral clearance, perhaps by innate sensing. Moreover, these findings are consistent with in vivo modeling studies (29) that have postulated similar decay in metabolically active and quiescent cells.
The predominance of intact proviruses in vitro has important implications for ex vivo superinfection models.
Our data reinforce the idea that major selective pressures shape the reservoir in vivo, since the vast majority of proviruses are defective in subjects on ART while in a single-round infection they are mostly intact. Models utilizing CD4 T-cell superinfection to study CD8-mediated viral clearance might not reflect in vivo conditions, since the majority of viral sequences in superinfected cells would be intact and therefore more easily cleared by effector cells (42, 43). This is different from what we observe in vivo in individuals on long-term ART, where only a small fraction of proviruses is intact.
Is the separation of integrated from unintegrated HIV DNA clinically relevant?
Overall, our findings suggest the importance of separating integrated from unintegrated HIV DNA. While it is generally thought that subjects on ART only have minimal levels of unintegrated HIV DNA, nonetheless this might not be the case in several clinical settings. As an example, in STI studies (44), the impact of an intervention could be masked by ongoing replication and/or clonal expansion. Without isolating the integrated HIV fraction, there is a risk to miss whether a change in the size and composition of the “true” reservoir occurred. A similar argument can be raised in studies utilizing NGS to define the pre-ART composition of the reservoir or the change in the viral landscape in the first months after ART initiation (45). The use of BluePippin was proposed by the Richman laboratory as an approach to measure integrated HIV DNA (27). We further advanced the use of this technique by providing proof of concept of the striking difference that exists between sequences obtained from total versus integrated HIV DNA in vitro. Moreover, we bolstered these findings in a clinical setting by showing that the composition of the proviral landscape is different in the low-molecular-weight DNA (containing unintegrated HIV DNA) from that of the high-molecular-weight DNA (containing integrated HIV and chromosomal DNA) in a subject off ART (Fig. 5).
Our study has several limitations. First, due to the limited number of defective sequences, we were not able to study selective pressures exerted on different categories of defective viruses. Second, we only performed NGS on rCD4 T cells maintained in culture with low levels of IL-7. Our rationale was based on literature showing that IL-7-treated T cells exhibit minimal phenotype changes, even at higher dose (46), and have longer survival in culture (47). Third, in our in vitro experiments, we only allowed one round of infection. This is different from what is observed in vivo, where even early ART initiation occurs after many rounds of replication. Future experiments designed to capture the viral landscape after multiple rounds of infection might provide additional insights into reservoir dynamics. Lastly, one potential limitation is that we used sequence samples from only one in vitro inoculation. However, we made the deliberate choice to sequence deeply over a thousand HIV amplicons at limiting dilution rather than performing many replicate experiments. Notably, in our in vitro system, the frequency of defective viral sequences was extremely low. If we had chosen to obtain fewer sequences in replicates, we likely would not have detected any defective virus in the integrated HIV DNA fraction.
In conclusion, our in vitro model of rCD4 T-cell infection recapitulates important aspects of reservoir dynamics. While reservoir decay is accelerated in vitro, our model could be a powerful tool for preclinical testing. Moreover, our results suggest the clinical importance of separating unintegrated HIV DNA from integrated HIV DNA to study differential selection pressures.
MATERIALS AND METHODS
Apheresis.
Both HIV-negative and HIV-positive subjects enrolled in the study underwent apheresis at the University of Pennsylvania according to protocol number 704904, approved by the Institutional Review Board (IRB). Each subject signed informed written consent prior to enrollment. The pre-ART sample used in this study was provided by S. Migueles (National Institutes of Health), who follows his institutional protocol with IRB approval. For the individual off ART, plasma viral load at the time of cell collection was 10,443 copies/ml and CD4 T-cell count was 312 cells/μl. For the individual on ART, the CD4 T-cell count was 768 cells/μl and plasma viral load was 35 copies/ml.
In vitro experiments.
rCD4 T cells were negatively selected from PBMCs of uninfected donors using the EasySep human CD4 T-cell isolation kit (Stemcell Technologies). Approximately 20 million cells were either immediately infected or activated for 3 days prior to infection using CD3/CD28 beads (0.5 bead per cell; Miltenyi) in RPMI supplemented with IL-2 (50 UI/ml), 30% fetal bovine serum (FBS), and penicillin-streptomycin. Both rCD4 and aCD4 T cells were infected with NL4-3 by centrifuging viral supernatant on cells at 1,200 × g for 2 h at 25°C (10). After spinoculation, cells were placed in the incubator for 2 h to allow fusion, washed twice with medium, and then treated with DNase for 30 min to remove any residual HIV plasmid DNA. Cells then were resuspended in fresh RPMI supplemented with 30% FBS and penicillin-streptomycin. Saquinavir (SQV; 1 μM) was added to allow only a single round of infection. rCD4 T cells were supplemented with IL-7 (2 ng/ml), which has been shown at low concentrations to increase cell survival with minimal effects on proliferation (48). aCD4 T cells were cultured in the presence of IL-2 (50 UI/ml). Cell viability was monitored over time using the trypan blue exclusion method. rCD4 and aCD4 T cells were collected at 2, 4, 8, 24, 48, 96, and 192 h after infection and pellets frozen until further use.
DNA isolation, size selection, and quantification of HIV DNA.
DNA was isolated from rCD4 and aCD4 T cells using the Gentra Puregene cell kit (Qiagen). About half of each sample was used for quantification of total HIV DNA and for NGS (total HIV fraction). This fraction contained both integrated and unintegrated HIV DNA. The remaining half was used to isolate high-molecular-weight DNA (20 to 80 kb) using BluePippin (Sage Science). The BluePippin platform uses pulsed-field electrophoresis for DNA size selection (27). Table 2 shows that we were able to robustly and consistently remove low-molecular-weight DNA (containing unintegrated HIV) from the high-molecular-weight fraction (which is enriched for integrated HIV and chromosomal DNA). Up to 5 μg of total DNA was diluted in a volume of 30 μl. Ten microliters of loading solution (Sage Science) was added to the DNA, and the total volume of 40 μl was loaded on a BluePippin 0.75% agarose dye-free cassette according to the manufacturer’s protocol. One lane of the cassette was reserved for the marker (Sage Science). We used a high-pass protocol, setting the cutoff for DNA collection at 20 kb. The DNA fraction we collected was used to measure HIV DNA levels and for proviral sequencing (integrated HIV fraction). For both the total and integrated HIV samples, we confirmed the expected DNA size by fragment analysis (AATI fragment analyzer; not shown). HIV DNA was measured using primers against the HIV LTR (22). For the experiments that involved separating integrated from unintegrated DNA in individuals with HIV infection, we isolated DNA from PBMCs and then collected the integrated HIV DNA fraction as described above. To isolate unintegrated HIV DNA from PBMCs of the subject off ART, we performed multiple consecutive runs to collect DNA for NGS. We used ∼40 μg of total DNA and loaded it on either a BluePippin 0.75% agarose dye-free cassette (S1 marker) or 1.5% agarose dye-free cassette (R2 marker) to collect the DNA with size ranging between 200 bp and 12 kb.
TABLE 2.
BluePippin technology robustly and efficiently isolates high-molecular-weight DNAa
| Sample | Pre-BluePippin |
Post-BluePippin |
Efficiency (%) | ||||
|---|---|---|---|---|---|---|---|
| Albumin/well (no. of copies) | HIV LTR/well (no. of copies) | HIV/cell | Albumin/well (no. of copies) | HIV LTR/well (no. of copies) | HIV/cell | ||
| 1 | 19,510 | 39,489 | 2.0 | 3,122 | 4.1 | 0.001 | 99.990 |
| 2 | 16,154 | 135,658 | 8.4 | 10,295 | 90.3 | 0.009 | 99.933 |
| 3 | 22,132 | 266,301 | 12.0 | 4,479 | 12.9 | 0.003 | 99.995 |
| 4 | 19,839 | 374,192 | 18.9 | 6,708 | 9.5 | 0.001 | 99.997 |
NL4-3 was digested with two restriction enzymes (FspI and NcoI) to generate a linearized plasmid (10,565 nucleotides). The plasmid DNA was isolated using BluePippin (marker S1, 10- to 18-kb protocol, collection between 9.8 and 11.5 kb) and then diluted with uninfected genomic DNA at incremental ratios of HIV to genomic DNA (samples 1 to 4). Half of the sample was used to isolate high-molecular-weight DNA by BluePippin (U1 marker, high-pass protocol, collection cutoff of 20 kb) before HIV LTR qPCR (see Materials and Methods for more details). This fraction was expected to contain genomic DNA but not linear HIV. The remaining half (genomic and linear HIV DNA) was used for downstream HIV qPCR without further manipulation. We performed qPCR for albumin to estimate the number of cells/well and HIV copies/cell. As shown, BluePippin removed the vast majority of linear HIV DNA from high-molecular-weight genomic DNA (>99.9% in every sample).
HIV DNA was quantified by total HIV against the LTR (22). First-step PCRs were cycled using the Nexus master cycler (Eppendorf), and qPCRs were cycled on a 7500 FAST real-time instrument (ThermoFisher). PCR conditions for the first round were 95°C for 2 min; 95°C for 15 s, 64°C for 45 s, and 72°C for 1 min for 12 cycles; and then 72°C for 10 min. Fifteen microliters of the first-round PCRs was run on the qPCR instrument using the same primers. PCR conditions were 95°C for 15 s and then 95°C for 10 s and 60°C for 20 s for 40 cycles (22).
Viral amplification and sequencing.
Near-full-length HIV sequencing was performed using a two-step nested PCR approach as previously described (22). Briefly, both primer sets were designed within the LTRs and staggered to avoid localized LTR amplification as well as LTR-related PCR artifacts. We used a long-range and high-fidelity polymerase enzyme for both reactions (Platinum SuperFi PCR master mix; ThermoFisher). In the first PCR, DNA was diluted so that ≤30% of wells were positive for HIV DNA. Nested PCRs were visualized by gel electrophoresis, and reaction mixtures containing more than one band were excluded from further analysis. PCR amplicons were purified using the DNA Clean & Concentrator kit (Zymogen). Amplicons were prepared using the Nextera library preparation kit (Illumina) for sequencing on a MiniSeq system using a mid-output flow cell (Illumina).
Sequence assembly and removal of double proviruses.
The bioinformatic pipeline used for viral assembly has been described previously (22), the only difference being the use of NL4-3 instead of HXB2 as the reference sequence for in vitro experiments. Intact viral sequences were defined as those containing a nearly complete psi packaging site, including SL2 and at least 2 other stem loops, and 9 complete open reading frames for all HIV genes. For sequences retrieved from individuals with HIV infection, we allowed for truncated Nef and Tat genes, as commonly identified in infectious strains of HIV (49). Additional requirements for intact viral sequences were the presence of major donor site 1 (D1) or a GT dinucleotide cryptic donor site located four nucleotides downstream (50) and the presence of major donor site 4 (D4). We also required the presence of splice acceptor site A5, A7, and either A4a, A4b, or A4c, as well as an intact Rev-responsive element (RRE) sequence (50). A more detailed description of the workflow is provided in reference 22. To identify hypermutated HIV sequences, all viruses were aligned using MAFFT with the E-INSi algorithm and a 1.8 gap penalty, and an intact HIV sequence was selected as the reference. The aligned HIV sequences were checked against the reference for hypermutation using the LANL Hypermut 2 program. The viral sequence with the lowest chance of being a hypermutated one (as determined by the Hypermut program) was selected as the reference, and once again Hypermut 2 was run on the alignment. Viral sequences determined to be hypermutated with a P value of <0.05 were counted as hypermutated ones. As expected (51), for in vitro infected cells, we did not identify any hypermutated virus.
Modified IPDA.
IPDA was performed using a modified version of the method recently published by Siliciano’s laboratory (52). For the amplification of HIV targets, we used the primers, probes, and cycling conditions published in reference 52 and reported in Table 3. Briefly, ∼10,000 cells were assayed in duplicate in a 20-μl volume using ddPCR Supermix for probes (no dUTP) on the Bio-Rad QX200 platform. As a control for sheared DNA, we simultaneously set up a reaction on a separate aliquot using two sets of primers annealing to the human CD4 gene (Table 3). The two amplicons generated are spaced at the same distance as the HIV amplicons.
TABLE 3.
Primers and probes used for the modified IPDA assay utilized in the study
| Primera | Sequence |
|---|---|
| Ψ F* | CAGGACTCGGCTTGCTGAAG |
| Ψ R* | CAGGACTCGGCTTGCTGAAG |
| Ψ Probe (FAM-MGB)* | TTTTGGCGTACTCACCAGT |
| Env F* | AGTGGTGCAGAGAGAAAAAAGAGC |
| Env R* | GTCTGGCCTGTACCGTCAGC |
| Env Probe (VIC-MGB)* | CCTTGGGTTCTTGGGA |
| CD4 F pair 1 | GAGACTGTGCTAGACTCCTCTCT |
| CD4 R pair 1 | CACTCCCTCTCTGATCTTGCTATTT |
| CD4 probe pair 1 (FAM-MGB) | CAGCTCAGTGTCCAGAG |
| CD4 F pair 2 | CTGGGTGACAGAGTGGATCTAAAC |
| CD4 R pair 2 | GAGGTGATGGTGGAAGAAGGTATG |
| CD4 probe pair 2 (VIC-MGB) | ATTTACAGTCAAGCCTCAAAG |
Asterisks indicate data published in reference 52.
Statistical analysis and graphing.
Statistical processes were performed using R and Microsoft Excel software. GraphPad Prism software was used for graphing. Viral decay was calculated using linear regression on log-transformed data. Confidence intervals were computed using previously described models of HIV qPCR and digital PCR uncertainty (53–56).
Data availability.
All relevant data used in the manuscript are available upon request. The proviral sequences obtained from the HIV-positive individuals in the study are available in GenBank under accession numbers MK383384 to MK385589 and in reference 22.
ACKNOWLEDGMENTS
CD4 cellular gene control primer sequences were from David N. Levy. We thank Mary Leonard for the artwork in Fig. 6.
This work was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under award numbers R01AI12001, R21AI116216, and UM1AI126617 (U. O’Doherty) and R03AI136710 (R. Zurakowski) with cofunding support from the National Institute on Drug Abuse, the National Institute of Mental Health, and the National Institute of Neurological Disorders and Stroke.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
M.R.P., M.P.B., and D.J.V. performed the NGS experiments. M.R.P. set up the cell cultures and performed the modified IPDA experiments. R.Z. provided statistical support for data analysis. M.R.P. and U.O. designed the experiments and wrote the manuscript. All authors read and approved the final version of the manuscript.
REFERENCES
- 1.Stevenson M, Stanwick TL, Dempsey MP, Lamonica CA. 1990. HIV-1 replication is controlled at the level of T cell activation and proviral integration. EMBO J 9:1551–1560. doi: 10.1002/j.1460-2075.1990.tb08274.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zack JA, Arrigo SJ, Weitsman SR, Go AS, Haislip A, Chen I. 1990. HIV-1 entry into quiescent primary lymphocytes: molecular analysis reveals a labile, latent viral structure. Cell 61:213–222. doi: 10.1016/0092-8674(90)90802-L. [DOI] [PubMed] [Google Scholar]
- 3.Bukrinsky MI, Stanwick TL, Dempsey MP, Stevenson M. 1991. Quiescent T lymphocytes as an inducible virus reservoir in HIV-1 infection. Science 254:423–427. doi: 10.1126/science.1925601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Perelson AS, Neumann AU, Markowitz M, Leonard JM, Ho DD. 1996. HIV-1 dynamics in vivo: virion clearance rate, infected cell life-span, and viral generation time. Science 271:1582–1586. doi: 10.1126/science.271.5255.1582. [DOI] [PubMed] [Google Scholar]
- 5.Wei X, Ghosh SK, Taylor ME, Johnson VA, Emini EA, Deutsch P, Lifson JD, Bonhoeffer S, Nowak MA, Hahn BH, Saag MS, Shaw GM. 1995. Viral dynamics in human immunodeficiency virus type 1 infection. Nature 373:117–122. doi: 10.1038/373117a0. [DOI] [PubMed] [Google Scholar]
- 6.Ho DD, Neumann AU, Perelson AS, Chen W, Leonard JM, Markowitz M. 1995. Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature 373:123–126. doi: 10.1038/373123a0. [DOI] [PubMed] [Google Scholar]
- 7.Chun TW, Stuyver L, Mizell SB, Ehler LA, Mican JA, Baseler M, Lloyd AL, Nowak MA, Fauci AS. 1997. Presence of an inducible HIV-1 latent reservoir during highly active antiretroviral therapy. Proc Natl Acad Sci U S A 94:13193–13197. doi: 10.1073/pnas.94.24.13193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Finzi D, Hermankova M, Pierson T, Carruth LM, Buck C, Chaisson RE, Quinn TC, Chadwick K, Margolick J, Brookmeyer R, Gallant J, Markowitz M, Ho DD, Richman DD, Siliciano RF. 1997. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science 278:1295–1300. doi: 10.1126/science.278.5341.1295. [DOI] [PubMed] [Google Scholar]
- 9.Shan L, Deng K, Gao H, Xing S, Capoferri AA, Durand CM, Rabi SA, Laird GM, Kim M, Hosmane NN, Yang HC, Zhang H, Margolick JB, Li L, Cai W, Ke R, Flavell RA, Siliciano JD, Siliciano RF. 2017. Transcriptional reprogramming during effector-to-memory transition renders CD4+ T cells permissive for latent HIV-1 infection. Immunity 47:766–775. doi: 10.1016/j.immuni.2017.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Swiggard WJ, Baytop C, Yu JJ, Dai J, Li C, Schretzenmair R, Theodosopoulos T, O'Doherty U. 2005. Human immunodeficiency virus type 1 can establish latent infection in resting CD4+ T cells in the absence of activating stimuli. J Virol 79:14179–14188. doi: 10.1128/JVI.79.22.14179-14188.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Plesa G, Dai J, Baytop C, Riley JL, June CH, O'Doherty U. 2007. Addition of deoxynucleosides enhances human immunodeficiency virus type 1 integration and 2LTR formation in resting CD4+ T cells. J Virol 81:13938–13942. doi: 10.1128/JVI.01745-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Swiggard WJ, O'Doherty U, McGain D, Jeyakumar D, Malim MH. 2004. Long HIV type 1 reverse transcripts can accumulate stably within resting CD4+ T cells while short ones are degraded. AIDS Res Hum Retroviruses 20:285–295. doi: 10.1089/088922204322996527. [DOI] [PubMed] [Google Scholar]
- 13.Vatakis DN, Bristol G, Wilkinson TA, Chow SA, Zack JA. 2007. Immediate activation fails to rescue efficient human immunodeficiency virus replication in quiescent CD4+ T cells. J Virol 81:3574–3582. doi: 10.1128/JVI.02569-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Agosto LM, Yu JJ, Dai J, Kaletsky R, Monie D, O'Doherty U. 2007. HIV-1 integrates into resting CD4+ T cells even at low inoculums as demonstrated with an improved assay for HIV-1 integration. Virology 368:60–72. doi: 10.1016/j.virol.2007.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Spina CA, Guatelli JC, Richman DD. 1995. Establishment of a stable, inducible form of human immunodeficiency virus type 1 DNA in quiescent CD4 lymphocytes in vitro. J Virol 69:2977–2988. doi: 10.1128/JVI.69.5.2977-2988.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pace MJ, Graf EH, Agosto LM, Mexas AM, Male F, Brady T, Bushman FD, O'Doherty U. 2012. Directly infected resting CD4+ T cells can produce HIV Gag without spreading infection in a model of HIV latency. PLoS Pathog 8:e1002818. doi: 10.1371/journal.ppat.1002818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baldauf H-M, Pan X, Erikson E, Schmidt S, Daddacha W, Burggraf M, Schenkova K, Ambiel I, Wabnitz G, Gramberg T, Panitz S, Flory E, Landau NR, Sertel S, Rutsch F, Lasitschka F, Kim B, König R, Fackler OT, Keppler OT. 2012. SAMHD1 restricts HIV-1 infection in resting CD4(+) T cells. Nat Med 18:1682–1687. doi: 10.1038/nm.2964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.White TE, Brandariz-Nuñez A, Valle-Casuso JC, Amie S, Nguyen LA, Kim B, Tuzova M, Diaz-Griffero F. 2013. The retroviral restriction ability of SAMHD1, but not its deoxynucleotide triphosphohydrolase activity, is regulated by phosphorylation. Cell Host Microbe 13:441–451. doi: 10.1016/j.chom.2013.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ayinde D, Casartelli N, Schwartz O. 2012. Restricting HIV the SAMHD1 way: through nucleotide starvation. Nat Rev Microbiol 10:675–680. doi: 10.1038/nrmicro2862. [DOI] [PubMed] [Google Scholar]
- 20.Liang G, Zhao L, Qiao Y, Geng W, Zhang X, Liu M, Dong J, Ding H, Sun H, Shang H. 2019. Membrane metalloprotease TRABD2A restricts HIV-1 progeny production in resting CD4+ T cells by degrading viral polyprotein Gag. Nat Immunol 20:711–723. doi: 10.1038/s41590-019-0385-2. [DOI] [PubMed] [Google Scholar]
- 21.Imamichi H, Dewar RL, Adelsberger JW, Rehm CA, O’Doherty U, Paxinos EE, Fauci AS, Lane HC. 2016. Defective HIV-1 proviruses produce novel protein-coding RNA species in HIV-infected patients on combination antiretroviral therapy. Proc Natl Acad Sci U S A 113:201609057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pinzone MR, VanBelzen DJ, Weissman S, Bertuccio MP, Cannon LM, Venanzi-Rullo E, Migueles S, Jones RB, Mota T, Joseph SB, Groen K, Pasternak AO, Hwang WT, Sherman B, Vourekas A, Nunnari G, O’Doherty U. 2019. Longitudinal HIV sequencing reveals reservoir expression leading to decay which is obscured by clonal expansion. Nat Commun 10:1–12. doi: 10.1038/s41467-019-08431-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee GQ, Orlova-Fink N, Einkauf K, Chowdhury FZ, Sun X, Harrington S, Kuo H-H, Hua S, Chen H-R, Ouyang Z, Reddy K, Dong K, Ndung'u T, Walker BD, Rosenberg ES, Yu XG, Lichterfeld M. 2017. Clonal expansion of genome-intact HIV-1 in functionally-polarized Th1 CD4 T cells. J Clin Investig 127:2689–2696. doi: 10.1172/JCI93289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hiener B, Horsburgh BA, Eden J-S, Barton K, Schlub TE, Lee E, von Stockenstrom S, Odevall L, Milush JM, Liegler T, Sinclair E, Hoh R, Boritz EA, Douek D, Fromentin R, Chomont N, Deeks SG, Hecht FM, Palmer S. 2017. Identification of genetically intact HIV-1 proviruses in specific CD4 + T cells from effectively treated participants. Cell Rep 21:813–822. doi: 10.1016/j.celrep.2017.09.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bruner KM, Murray AJ, Pollack RA, Soliman MG, Laskey SB, Capoferri AA, Lai J, Strain MC, Lada SM, Hoh R, Ho Y-C, Richman DD, Deeks SG, Siliciano JD, Siliciano RF. 2016. Defective proviruses rapidly accumulate during acute HIV-1 infection. Nat Med 22:1043–1049. doi: 10.1038/nm.4156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ho Y-C, Shan L, Hosmane NN, Wang J, Laskey SB, Rosenbloom DIS, Lai J, Blankson JN, Siliciano JD, Siliciano RF. 2013. Replication-competent noninduced proviruses in the latent reservoir increase barrier to HIV-1 cure. Cell 155:540–551. doi: 10.1016/j.cell.2013.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lada SM, Huang K, VanBelzen DJ, Montaner LJ, O'Doherty U, Richman DD. 2018. Quantitation of integrated HIV provirus by pulsed-field gel electrophoresis and droplet digital PCR. J Clin Microbiol 56:1–10. doi: 10.1128/JCM.01158-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Andrade A, Guedj J, Rosenkranz SL, Lu D, Mellors J, Kuritzkes DR, Perelson AS, Ribeiro RM, ACTG A5249s Protocol Team. 2015. Early HIV RNA decay during raltegravir-containing regimens exhibits two distinct subphases (1a and 1b). AIDS 29:2419–2426. doi: 10.1097/QAD.0000000000000843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cardozo EF, Andrade A, Mellors JW, Kuritzkes DR, Perelson AS, Ribeiro RM. 2017. Treatment with integrase inhibitor suggests a new interpretation of HIV RNA decay curves that reveals a subset of cells with slow integration. PLoS Pathog 13:e1006478-18. doi: 10.1371/journal.ppat.1006478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.D Urbano V, De Crignis E, Re MC. 2018. Host restriction factors and human immunodeficiency virus (HIV-1): a dynamic interplay involving all phases of the viral life cycle. Curr HIV Res 16:184–207. doi: 10.2174/1570162X16666180817115830. [DOI] [PubMed] [Google Scholar]
- 31.Descours B, Cribier A, Chable-Bessia C, Ayinde D, Rice G, Crow Y, Yatim A, Schwartz O, Laguette N, Benkirane M, Schawartz O, Laguette N, Benkirane M. 2012. SAMHD1 restricts HIV-1 reverse transcription in quiescent CD4+ T-cells. Retrovirology 9:87. doi: 10.1186/1742-4690-9-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baldauf HM, Stegmann L, Schwarz SM, Ambiel I, Trotard M, Martin M, Burggraf M, Lenzi GM, Lejk H, Pan X, Fregoso OI, Lim ES, Abraham L, Nguyen LA, Rutsch F, König R, Kim B, Emerman M, Fackler OT, Keppler OT. 2017. Vpx overcomes a SAMHD1-independent block to HIV reverse transcription that is specific to resting CD4 T cells. Proc Natl Acad Sci U S A 114:2729–2734. doi: 10.1073/pnas.1613635114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Piekna-Przybylska D, Bambara RA. 2011. Requirements for efficient minus strand strong-stop DNA transfer in human immunodeficiency virus 1. RNA Biol 8:230–236. doi: 10.4161/rna.8.2.14802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Operario DJ, Balakrishnan M, Bambara RA, Kim B. 2006. Reduced dNTP interaction of human immunodeficiency virus type 1 reverse transcriptase promotes strand transfer. J Biol Chem 281:32113–32121. doi: 10.1074/jbc.M604665200. [DOI] [PubMed] [Google Scholar]
- 35.Rausell A, Muñoz M, Martinez R, Roger T, Telenti A, Ciuffi A. 2016. Innate immune defects in HIV permissive cell lines. Retrovirology 13:43. doi: 10.1186/s12977-016-0275-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Arhel NJ, Souquere-Besse S, Munier S, Souque P, Guadagnini S, Rutherford S, Prévost MC, Allen TD, Charneau P. 2007. HIV-1 DNA flap formation promotes uncoating of the pre-integration complex at the nuclear pore. EMBO J 26:3025–3037. doi: 10.1038/sj.emboj.7601740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dicks MDJ, Betancor G, Jimenez-Guardeño JM, Pessel-Vivares L, Apolonia L, Goujon C, Malim MH. 2018. Multiple components of the nuclear pore complex interact with the amino-terminus of MX2 to facilitate HIV-1 restriction. PLoS Pathog 14:e1007408–e1007424. doi: 10.1371/journal.ppat.1007408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kane M, Yadav SS, Bitzegeio J, Kutluay SB, Zang T, Wilson SJ, Schoggins JW, Rice CM, Yamashita M, Hatziioannou T, Bieniasz PD. 2013. Mx2 is an interferon induced inhibitor of HIV-1 infection. Nature 502:563–566. doi: 10.1038/nature12653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goujon C, Moncorgé O, Bauby H, Doyle T, Ward CC, Schaller T, Hué S, Barclay WS, Schulz R, Malim MH. 2013. Human MX2 is an interferon-induced post-entry inhibitor of HIV-1 infection. Nature 502:559–562. doi: 10.1038/nature12542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cartwright EK, Spicer L, Smith SA, Lee D, Fast R, Paganini S, Lawson BO, Nega M, Easley K, Schmitz JE, Bosinger SE, Paiardini M, Chahroudi A, Vanderford TH, Estes JD, Lifson JD, Derdeyn CA, Silvestri G. 2016. CD8+ lymphocytes are required for maintaining viral suppression in SIV-infected macaques treated with short-term antiretroviral therapy. Immunity 45:656–668. doi: 10.1016/j.immuni.2016.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Klatt NR, Shudo E, Ortiz AM, Engram JC, Paiardini M, Lawson B, Miller MD, Else J, Pandrea I, Estes JD, Apetrei C, Schmitz JE, Ribeiro RM, Perelson AS, Silvestri G. 2010. CD8+ lymphocytes control viral replication in SIVmac239-infected rhesus macaques without decreasing the lifespan of productively infected cells. PLoS Pathog 6:e1000747. doi: 10.1371/journal.ppat.1000747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Graf EH, Pace MJ, Peterson BA, Lynch LJ, Chukwulebe SB, Mexas AM, Shaheen F, Martin JN, Deeks SG, Connors M, Migueles SA, O'Doherty U. 2013. Gag-positive reservoir cells are susceptible to HIV-specific cytotoxic T lymphocyte mediated clearance in vitro and can be detected in vivo. PLoS One 8:e71879. doi: 10.1371/journal.pone.0071879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang S-H, Ren Y, Thomas AS, Chan D, Mueller S, Ward AR, Patel S, Bollard CM, Cruz CR, Karandish S, Truong R, Macedo AB, Bosque A, Kovacs C, Benko E, Piechocka-Trocha A, Wong H, Jeng E, Nixon DF, Ho Y-C, Siliciano RF, Walker BD, Jones RB. 2018. Latent HIV reservoirs exhibit inherent resistance to elimination by CD8+ T cells. J Clin Investig 128:876–889. doi: 10.1172/JCI97555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sharaf R, Lee GQ, Sun X, Etemad B, Aboukhater LM, Hu Z, Brumme ZL, Aga E, Bosch RJ, Wen Y, Namazi G, Gao C, Acosta EP, Gandhi RT, Jacobson JM, Skiest D, Margolis DM, Mitsuyasu R, Volberding P, Connick E, Kuritzkes DR, Lederman MM, Yu XG, Lichterfeld M, Li JZ. 2018. HIV-1 proviral landscapes distinguish posttreatment controllers from noncontrollers. J Clin Investig 128:4074–4085. doi: 10.1172/JCI120549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee GQ, Reddy K, Einkauf KB, Gounder K, Chevalier JM, Dong KL, Walker BD, Yu XG, Ndung’u T, Lichterfeld M. 2019. HIV-1 DNA sequence diversity and evolution during acute subtype C infection. Nat Commun 10:2737. doi: 10.1038/s41467-019-10659-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Scripture-Adams DD, Brooks DG, Korin YD, Zack JA. 2002. Interleukin-7 induces expression of latent human immunodeficiency virus type 1 with minimal effects on T-cell phenotype. J Virol 76:13077–13082. doi: 10.1128/jvi.76.24.13077-13082.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vassena L, Proschan M, Fauci AS, Lusso P. 2007. Interleukin 7 reduces the levels of spontaneous apoptosis in CD4+ and CD8+ T cells from HIV-1-infected individuals. Proc Natl Acad Sci U S A 104:2355–2360. doi: 10.1073/pnas.0610775104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Trinité B, Chan CN, Lee CS, Levy DN. 2016. HIV-1 Vpr- and reverse transcription-induced apoptosis in resting peripheral blood CD4 T cells and protection by common gamma-chain cytokines. J Virol 90:904–916. doi: 10.1128/JVI.01770-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Clark E, Nava B, Caputi M. 2017. Tat is a multifunctional viral protein that modulates cellular gene expression and functions. Oncotarget 8:27569–27581. doi: 10.18632/oncotarget.15174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Purcell DF, Martin MA. 1993. Alternative splicing of human immunodeficiency virus type 1 mRNA modulates viral protein expression, replication, and infectivity. J Virol 67:6365–6378. doi: 10.1128/JVI.67.11.6365-6378.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sheehy AM, Gaddis NC, Choi JD, Malim MH. 2002. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 418:646–650. doi: 10.1038/nature00939. [DOI] [PubMed] [Google Scholar]
- 52.Bruner KM, Wang Z, Simonetti FR, Bender AM, Kwon KJ, Sengupta S, Fray EJ, Beg SA, Antar AAR, Jenike KM, Bertagnolli LN, Capoferri AA, Kufera JT, Timmons A, Nobles C, Gregg J, Wada N, Ho YC, Zhang H, Margolick JB, Blankson JN, Deeks SG, Bushman FD, Siliciano JD, Laird GM, Siliciano RF. 2019. A quantitative approach for measuring the reservoir of latent HIV-1 proviruses. Nature 566:120–125. doi: 10.1038/s41586-019-0898-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Strain MC, Lada SM, Luong T, Rought SE, Gianella S, Terry VH, Spina CA, Woelk CH, Richman DD. 2013. Highly precise measurement of HIV DNA by droplet digital PCR. PLoS One 8:e55943–e55949. doi: 10.1371/journal.pone.0055943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cannon LM, Vargas-Garcia CA, Jagarapu A, Piovoso MJ, Zurakowski R. 2018. HIV 2-LTR experiment design optimization. PLoS One 13:e0206700–e0206720. doi: 10.1371/journal.pone.0206700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Buzon MJ, Sun H, Li C, Shaw A, Seiss K, Ouyang Z, Martin-Gayo E, Leng J, Henrich TJ, Li JZ, Pereyra F, Zurakowski R, Walker BD, Rosenberg ES, Yu XG, Lichterfeld M. 2014. HIV-1 persistence in CD4+ T cells with stem cell-like properties. Nat Med 20:139–142. doi: 10.1038/nm.3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Luo R, Cardozo EF, Piovoso MJ, Wu H, Buzon MJ, Martinez-Picado J, Zurakowski R. 2013. Modelling HIV-1 2-LTR dynamics following raltegravir intensification. J R Soc Interface 10:20130186. doi: 10.1098/rsif.2013.0186. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All relevant data used in the manuscript are available upon request. The proviral sequences obtained from the HIV-positive individuals in the study are available in GenBank under accession numbers MK383384 to MK385589 and in reference 22.