

Human cytomegalovirus (HCMV) is the most common infectious cause of infant birth defects, resulting in permanent neurological disability for one newborn child every hour in the United States. After more than a half century of research and development, we remain without a clinically licensed vaccine or immunotherapeutic to reduce the burden of HCMV-associated disease. In this study, we sought to improve upon the glycoprotein B protein vaccine (gB/MF59), the most efficacious HCMV vaccine evaluated in a clinical trial, via targeted modifications to either the protein structure or vaccine formulation. Utilization of a novel vaccine platform, nucleoside-modified mRNA formulated in lipid nanoparticles, increased the durability and breadth of vaccine-elicited antibody responses. We propose that an mRNA-based gB vaccine may ultimately prove more efficacious than the gB/MF59 vaccine and should be further evaluated for its ability to elicit antiviral immune factors that can prevent HCMV-associated disease.
KEYWORDS: cytomegalovirus, glycoprotein B, vaccines
ABSTRACT
A vaccine to prevent maternal acquisition of human cytomegalovirus (HCMV) during pregnancy is a primary strategy to reduce the incidence of congenital disease. The MF59-adjuvanted glycoprotein B (gB) protein subunit vaccine (gB/MF59) is the most efficacious vaccine tested to date for this indication. We previously identified that gB/MF59 vaccination elicited poor neutralizing antibody responses and an immunodominant response against gB antigenic domain 3 (AD-3). Thus, we sought to test novel gB vaccines to improve functional antibody responses and reduce AD-3 immunodominance. Groups of juvenile New Zealand White rabbits were administered 3 sequential doses of the full-length gB protein with an MF59-like squalene-based adjuvant, the gB ectodomain protein (lacking AD-3) with squalene adjuvant, or lipid nanoparticle (LNP)-encapsulated nucleoside-modified mRNA encoding full-length gB. All vaccines were highly immunogenic with similar kinetics and comparable peak gB-binding and functional antibody responses. The AD-3-immunodominant IgG response following human gB/MF59 vaccination was closely mimicked in rabbits. Though gB ectodomain subunit vaccination eliminated targeting of epitopes in AD-3, it did not improve vaccine-elicited neutralizing or nonneutralizing antibody functions. gB nucleoside-modified mRNA-LNP-immunized rabbits exhibited an enhanced durability of vaccine-elicited antibody responses. Furthermore, the gB mRNA-LNP vaccine enhanced the breadth of IgG binding responses against discrete gB peptides. Finally, low-magnitude gB-specific T cell activity was observed in the full-length gB protein and mRNA-LNP groups, though not in ectodomain-vaccinated rabbits. Altogether, these data suggest that the use of gB nucleoside-modified mRNA-LNP vaccines is a viable strategy for improving on the partial efficacy of gB/MF59 vaccination and should be further evaluated in preclinical models.
IMPORTANCE Human cytomegalovirus (HCMV) is the most common infectious cause of infant birth defects, resulting in permanent neurological disability for one newborn child every hour in the United States. After more than a half century of research and development, we remain without a clinically licensed vaccine or immunotherapeutic to reduce the burden of HCMV-associated disease. In this study, we sought to improve upon the glycoprotein B protein vaccine (gB/MF59), the most efficacious HCMV vaccine evaluated in a clinical trial, via targeted modifications to either the protein structure or vaccine formulation. Utilization of a novel vaccine platform, nucleoside-modified mRNA formulated in lipid nanoparticles, increased the durability and breadth of vaccine-elicited antibody responses. We propose that an mRNA-based gB vaccine may ultimately prove more efficacious than the gB/MF59 vaccine and should be further evaluated for its ability to elicit antiviral immune factors that can prevent HCMV-associated disease.
INTRODUCTION
Human cytomegalovirus (HCMV) impacts 1 in 150 live-born infants, making this pathogen the most common cause of congenital infection worldwide (1, 2). Approximately 20% of infants infected with HCMV in utero will develop long-term sequelae, including microcephaly, intrauterine growth restriction, hearing/vision loss, or neurodevelopmental delay (3, 4). Furthermore, HCMV is the most prevalent infection among solid organ and hematopoietic stem cell transplant recipients, causing end-organ disease, such as gastroenteritis, pneumonitis, or hepatitis, and potentially predisposing these individuals to allograft rejection and/or failure (5, 6). However, we remain without a vaccine or immunotherapeutic intervention to reduce the burden of disease among newborn children and transplant recipients.
A variety of vaccine platforms and formulations have been evaluated in clinical trials for the prevention of both congenital (reviewed in reference 7) and transplant-associated (reviewed in reference 8) HCMV disease, of which the most efficacious has been the glycoprotein B (gB) subunit vaccine administered with MF59 squalene-based adjuvant (gB/MF59) (9). gB is the viral fusogen and is essential for entry into all cell types (10), including placental trophoblast progenitor cells (11). Furthermore, gB is highly expressed and an immune-dominant target following natural infection, making this protein an attractive target for vaccination. gB/MF59 subunit vaccination demonstrated moderate (∼50%) efficacy in blocking HCMV infection and host seroconversion in populations of HCMV-seronegative postpartum women (12) and adolescent women (13). Furthermore, in transplant recipients, this vaccine protected against HCMV viremia and reduced the clinical need for antiviral treatment (14).
Analysis of samples obtained from both postpartum and transplant-recipient gB vaccinees revealed two key observations regarding the target and function of gB-elicited antibody responses that inform our understanding of the partial vaccine efficacy. First, we identified that vaccination elicited an extraordinarily robust response against antigenic domain 3 (AD-3), a nonneutralizing epitope in the C-terminal region of the protein that is presumed to be cytosolic (intraluminal) (15). Second, we noted that gB-specific antibodies elicited in postpartum women and transplant recipients were predominantly nonneutralizing, suggesting that the mechanism of partial protection against viral acquisition was not the induction of neutralizing antibodies (15, 16). However, the protective nonneutralizing function remains unclear: we identified that gB/MF59 vaccinees had high-magnitude viral phagocytosis activity, though the magnitude was not associated with infection status (15). These results led us to hypothesize that we might improve upon the gB/MF59 vaccine through the rational design of novel immunogens that minimize responses against the nonneutralizing AD-3 epitope.
Here we present the findings of an investigation into the immunogenicity of two novel gB vaccines aiming to reduce exposure to AD-3: a truncated gB protein subunit vaccine (lacking the AD-3 epitope) administered with the MF59-like squalene-based adjuvant AddaVax (the gB ectodomain [gB ecto] vaccine) and a gB nucleoside-modified mRNA vaccine packaged in lipid nanoparticles (LNPs) (the gB mRNA-LNP [gB mRNA] vaccine). The primary goal of this investigation was to assess the magnitude and function of antibody responses elicited by rationally designed, next-generation gB vaccines head-to-head against the partially efficacious gB/MF59 subunit vaccine in a small-animal model. While animal challenge models are available for CMV (guinea pig cytomegalovirus [GPCMV], rhesus monkey cytomegalovirus [RhCMV]), we chose to initially assess the quality of the antibody responses elicited by novel HCMV immunogens in direct comparison to those elicited by the clinically tested vaccine. New Zealand White rabbits were selected because we previously identified that neutralizing and nonneutralizing antibodies are elicited in rabbits by vaccination and that Fc receptor-independent and -dependent effector functions can be measured in vitro (17). Three groups of rabbits were vaccinated with either (i) full-length gB plus AddaVax (the immunogen from the gB/MF59 vaccine trial) (gB FL), (ii) the gB ectodomain plus AddaVax (gB ecto), or (iii) gB mRNA-LNP (gB mRNA). We anticipated that the gB ectodomain and gB mRNA-LNP vaccines would have a reduced targeting of AD-3 and an enhanced neutralizing and/or nonneutralizing function, resulting in a superior vaccine that might be deployed to prevent both congenital and transplant-associated HCMV disease.
RESULTS
gB-specific IgG magnitude and durability.
Rabbits were vaccinated with one of three different gB vaccines: full-length gB (gB FL), a gB ectodomain (gB ecto), or nucleoside-modified gB mRNA encapsulated in a lipid nanoparticle (gB mRNA) (a comparison of the immunogen structures is shown in Fig. 1; the vaccine schedule is provided in Fig. 2). We first assessed the ability of the IgG elicited by all three vaccines to bind soluble full-length gB (Fig. 3A) and gB ectodomain (Fig. 3B) proteins by a binding antibody multiplex assay (BAMA), as well as to bind cell-associated gB on the surface of gB-transfected cells by flow cytometry (Fig. 3C). All vaccines were highly immunogenic, with the peak immunogenicity (10 weeks) binding magnitude being similar to that for both soluble and cell-associated gB. However, gB mRNA-immunized rabbits had enhanced binding to both soluble and cell-associated gB at the time of animal necropsy (20 weeks), indicating the superior durability of the mRNA vaccine-elicited antibody responses compared to the gB FL-elicited antibody responses. This distinction was most pronounced for binding to cell-associated gB (median percentage of phycoerythrin [PE]-positive cells at 20 weeks, 29.1% for gB mRNA-vaccinated rabbits versus 18.8% for gB FL-vaccinated rabbits; P = 0.01, Kruskal-Wallis test with the post hoc Mann-Whitney U test). Additionally, we evaluated the avidity of vaccine-elicited IgG responses by a plate-based, urea wash enzyme-linked immunosorbent assay (ELISA) (Fig. 3D). We noted a slightly reduced median relative avidity index (RAI; measured against soluble gB FL protein) in gB mRNA-immunized rabbits, though the difference was not statistically significant.
FIG 1.

Comparison of immunogen structure. The full open reading frame (ORF) of HCMV gB is shown, from the N terminus (N-Term) on the left to the C terminus (C-Term) on the right. The major antigenic regions indicated include AD-1 (orange), AD-2 site 1 (red), AD-2 site 2 (yellow), AD-3 (purple), AD-4 (domain 2) (green), and AD-5 (domain 1) (blue). Numbers indicate the approximate number of amino acid residues dividing each region of interest (the variable is dependent on the precise strain). The structures of the gB immunogens employed are indicated by black bars for gB FL (blue), gB ecto (green), and gB mRNA (purple). Data for the ribbon diagram were adapted from Burke et al. (64) and Hebner et al. (65).
FIG 2.
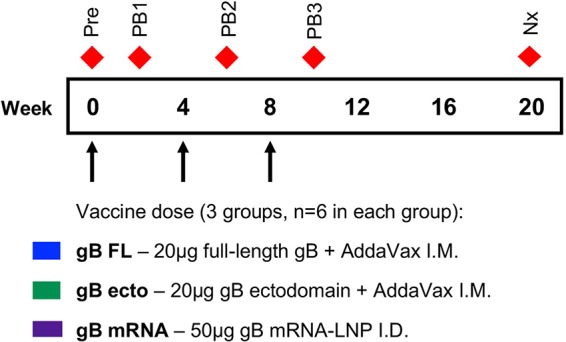
Vaccination and sampling timeline. At 0, 4, and 8 weeks, juvenile New Zealand White rabbits were administered 50-μg doses of either full-length gB protein plus AddaVax intramuscularly (blue) or the gB ectodomain protein plus AddaVax intramuscularly (green) or lipid nanoparticle-packaged gB mRNA intradermally (purple). Blood was sampled at the following time points (indicated by red diamonds): preimmune (0 weeks; Pre), post-boost 1 (2 weeks; PB1), post-boost 2 (6 weeks; PB2), post-boost 3 (10 weeks; PB3), and necropsy (20 weeks; Nx).
FIG 3.

gB mRNA-LNP vaccination elicits more durable binding antibody responses against soluble and cell-associated gB than gB FL and gB ecto vaccination. (A, B) IgG binding to full-length gB (A) and gB ectodomain (B) proteins was assessed by BAMA. (C) IgG binding to gB-transfected cells was measured by flow cytometry. (D) gB-binding avidity against full-length gB was assessed using urea-wash ELISA. All proteins were strain matched (Towne). IgG responses for full-length gB vaccinees are shown in blue, those for gB ectodomain are shown in green, and those for gB mRNA-LNP are shown in purple. Binding responses were assessed for the preimmune, 10-week (post-boost 3, peak immunogenicity), and 20-week (necropsy) time points. Data points represent the results for individual animals, with the lines designating the median. The dotted black line indicates the mean preimmune response plus 2 standard deviations. *, P < 0.05, Kruskal-Wallis with the post hoc Mann-Whitney U test. (E) The 50% end dilution (ED50) was calculated according to the method of Reed and Muench (62) using the plasma dilution that resulted in a 50% reduction in ELISA-binding optical density. Following log10 transformation, binding was modeled using a linear model and projected out to 50 weeks following initial vaccination (only the projection out to 33 weeks is shown). The R2 model for linear fit is displayed. The results for full-length gB vaccinees are shown in blue, those for gB ectodomain vaccinees are shown in green, and those for gB mRNA-LNP vaccinees are shown in purple.
To further evaluate the vaccine-elicited antibody response durability, we assessed binding against the gB FL immunogen by plate-based ELISA. We noted a log-linear decay of vaccine-elicited antibody responses from peak immunogenicity (10-week time point) until necropsy (20-week time point) and performed modeling thereof (Fig. 3E). A linear fit was noted for the log-transformed values for all 3 groups (for gB FL-vaccinated rabbits, R2 = 0.42; for gB ecto-vaccinated rabbits, R2 = 0.44; for gB mRNA-vaccinated rabbits, R2 = 0.38; for all comparisons, P < 0.001) (Fig. 3E). Log-linear models of antibody response decay were employed to project the response magnitude at time points following animal necropsy (Table 1). Antibody responses following vaccination with gB FL or gB ecto were projected to be undetectable at 50 weeks following initial vaccination, whereas gB mRNA-elicited responses were projected to decay at a lower rate and to remain detectable at 50 weeks (Table 1). Notably, the 95% prediction intervals of these projections overlap, suggesting only a trend toward this increased durability.
TABLE 1.
Projected durability of vaccine-elicited gB binding past week 20
| Wk | Projected mean (ED50b; 95% prediction interval) |
||
|---|---|---|---|
| gB FL | gB ecto | gB mRNA | |
| 20a | 110 (20–617) | 135 (32–562) | 174 (60–490) |
| 30 | 22 (3–186) | 32 (6–186) | 68 (19–245) |
| 40 | 4 (1–72) | 8 (1–76) | 27 (5–145) |
| 50 | 1 (1–31) | 2 (1–33) | 11 (1–89) |
Data for week 20 represent the calculated mean and confidence interval at the time of animal necropsy.
ED50, 50% effective dose.
Linear gB epitope binding.
Employing a peptide microarray library that spans the entire gB open reading frame (ORF) (Towne strain), we previously observed an AD-3 epitope immunodominance in human gB/MF59 vaccinees, with 78% of the gB peptide-binding IgG response being directed against this singular region (18). Using the same tool, we determined that rabbits administered the gB FL vaccine had a nearly identical AD-3 epitope immunodominance (a 74% total gB peptide-binding IgG response) (Fig. 4A; Table 2). AD-3 linear peptide binding was dramatically reduced in the gB ecto (<1%) and gB mRNA (46%) groups, though it still remained the dominant response elicited by gB mRNA (Table 2). In the absence of AD-3, the furin cleavage site was the dominant response for the gB ecto vaccine (Table 2). Furthermore, gB mRNA-vaccinated rabbits had a slightly reduced total peptide binding compared to gB FL-vaccinated rabbits (Fig. 4B; the median peptide-binding mean fluorescence intensity [MFI] sum was 96,629 for gB FL-vaccinated rabbits and 48,051 for gB mRNA-vaccinated rabbits; P was not significant by the Kruskal-Wallis test with the post hoc Mann-Whitney U test), though both the gB FL and gB mRNA groups had greater total peptide binding than the gB ecto group (for both comparisons, P < 0.05, Kruskal-Wallis test with the post hoc Mann-Whitney U test). Importantly, there was an enhanced breadth of peptide-binding responses in gB mRNA-vaccinated rabbits compared to that in both the gB FL and gB ecto groups (Fig. 4D; median number of discrete peptides bound, gB FL-vaccinated rabbits = 44.5, gB ecto-vaccinated rabbits = 28.5, gB mRNA-vaccinated rabbits = 85; for both comparisons, P < 0.05, Kruskal-Wallis test with the post hoc Mann-Whitney U test). Of note, the pattern of linear peptide binding and breadth for mRNA-immunized rabbits appeared to be analogous to that elicited by natural HCMV infection (18).
FIG 4.

gB mRNA-LNP vaccination reduced AD-3 immunodominance, yet it enhanced the breadth of the linear peptide-binding IgG response. (A) The magnitude of binding of rabbit antibodies to a 15-mer peptide library spanning the entire Towne gB ORF (180 unique peptides) was assessed at week 10 (post-boost 3, peak immunogenicity). Each row indicates the results for a single rabbit. Peptides corresponding to distinct gB antigenic domains are indicated along the x axis. (B, C) The sum of total peptide-binding MFI to both the full-length gB protein (B) and those peptides corresponding to the AD-3 epitope (C). (D, E) Number of unique peptides with a binding response of >100 MFI for the full-length gB protein (C) and the AD-3 epitope (E). The responses for full-length gB vaccinees are shown in blue, those for gB ectodomain vaccinees are shown in green, and those for gB mRNA-LNP vaccinees are shown in purple. Data points represent the results for individual animals, with the lines designating the median. *, P < 0.05, Kruskal-Wallis test with the post hoc Mann-Whitney U test.
TABLE 2.
Median peptide-binding MFI
| gB region | Median MFI (% total binding) |
||
|---|---|---|---|
| gB FL | gB ecto | gB mRNA | |
| AD-1 | 1,560 (1.6) | 1,642 (4.4) | 3,603 (7.5) |
| AD-2 site 1 | 210 (0.2) | 1,919 (5.2) | 1,217 (2.5) |
| AD-2 site 2 | 750 (0.8) | 1,611 (4.3) | 2,643 (5.5) |
| AD-3 | 71,239 (73.7) | 890 (2.4) | 24,137 (50.2) |
| AD-4 | 3,733 (3.9) | 5,064 (13.6) | 3,135 (6.5) |
| AD-5 | 8,680 (9.0) | 4,172 (11.2) | 5,971 (12.4) |
| Cleavage | 7,526 (7.8) | 17,008 (45.7) | 2,838 (5.9) |
| Other | 2,931 (3.0) | 4,909 (13.2) | 4,507 (9.4) |
| Total | 96,629 | 37,215 | 48,051 |
Vaccine-elicited IgG binding to neutralizing epitopes and HCMV neutralization.
We next examined IgG binding to regions of gB known to be targeted by neutralizing antibodies: domain 1 (AD-4), domain 1 (AD-4) plus domain 2 (AD-5), AD-1, and AD-2 (Fig. 5A to D). Notably, we did not identify in any of the 3 vaccine groups vaccine-elicited antibodies against AD-2 (Fig. 5D), an epitope known to be the target of potently neutralizing gB-specific antibodies associated with reduced viremia in transplant recipients (19) and protection against congenital transmission (20). Furthermore, low responses against AD-1 were noted (Fig. 5C). The kinetics of vaccine-elicited IgG binding against domain 1 as well as domains 1 and 2 mirrored those of binding to soluble gB protein (Fig. 3), However, at 20 weeks, both gB ecto-vaccinated rabbits and gB mRNA-vaccinated rabbits had enhanced binding against domain 1 compared to gB FL-vaccinated rabbits (median domain 1 MFI at 20 weeks, gB FL-vaccinated rabbits = 571, gB ecto-vaccinated rabbits = 2,343, gB mRNA-vaccinated rabbits = 2,730; for both comparisons, P < 0.05, Kruskal-Wallis test with the post hoc Mann-Whitney U test) (Fig. 5A).
FIG 5.

Enhanced durability of HCMV-neutralizing antibodies following gB mRNA-LNP vaccination. (A to D) Vaccine-elicited binding to glycoprotein B neutralizing epitope domain 1 (A), domains 1 and 2 (B), AD-1 (C), and AD-2 site 1 (D) was assessed by BAMA. (E to J) Neutralization of either autologous virus Towne (E, F) or heterologous virus AD169r (G to J) in the absence of purified rabbit complement (−C) (E, G, I) and the presence of purified rabbit complement (+C) (F, H, J) on MRC-5 fibroblast cells (E to H) and ARPE-19 epithelial cells (I, J). The results for full-length gB vaccinees are shown in blue, those for gB ectodomain vaccinees are shown in green, and those for gB mRNA-LNP vaccinees are shown in purple. Antibody responses were assessed for the preimmune, 10-week (post-boost 3, peak immunogenicity), and 20-week (necropsy) time points. Data points represent individual animals, with the lines designating the median. The dotted black lines indicate the mean preimmune response plus 2 standard deviations. *, P < 0.05, Kruskal-Wallis test with the post hoc Mann-Whitney U test.
Furthermore, we assessed the magnitude of vaccine-elicited HCMV neutralization against autologous (vaccine strain) Towne virus in fibroblast cells in the presence and absence of rabbit complement but identified little difference between vaccination groups (Fig. 5E and F). We also measured the neutralization of heterologous (cross strain) AD169 revertant virus (AD169r) in fibroblast and epithelial cell lines (Fig. 5G to J). All three vaccines had similar peak heterologous neutralization at 10 weeks (median AD169r 50% inhibitory dose [ID50] in fibroblasts at 10 weeks with purified rabbit complement, gB FL-vaccinated rabbits = 158, gB ecto-vaccinated rabbits = 184, gB mRNA-vaccinated rabbits = 174). gB mRNA-vaccinated rabbits had 2-fold higher virus neutralization at 20 weeks than the other groups, indicating a superior response durability, though this distinction was not statistically significant (median AD169r ID50 in fibroblast at 20 weeks with purified rabbit complement, gB FL-vaccinated rabbits = 49, gB ecto-vaccinated rabbits = 62, gB mRNA-vaccinated rabbits = 120; P = 0.12, Kruskal-Wallis test with the post hoc Mann-Whitney U test). Notably, the neutralizing responses measured in both fibroblasts and epithelial cells were similar in magnitude, as might be expected for gB-specific antibodies.
Vaccine-elicited engagement of Fcγ receptors and nonneutralizing effector functions.
Subsequently, we investigated the ability of vaccine-elicited antibodies to engage Fcγ receptors (FcγR; specifically, FcγRI, FcγRIIa, FcγRIIb, and FcγRIIIa), which is a prerequisite for Fc-mediated effector functions (Fig. 6A to D). Intriguingly, a single dose of the gB mRNA vaccine (but not the gB FL or gB ecto vaccine) resulted in the rapid development of antigen-specific IgG that could engage with Fc receptors (Table 3), which might have been due to the robust induction of T follicular helper cells, which facilitate B cell maturation and class switching (21). This distinction was most notable for FcγRIIa and FcγRIIb (median FcγRIIa MFI at 2 weeks, 0 for gB FL-vaccinated rabbits versus 128 for gB mRNA-vaccinated rabbits; P = 0.02, Kruskal-Wallis test with the post hoc Mann-Whitney U test). At peak immunogenicity (10 weeks), the gB-specific antibody engagement of Fcγ receptors was similar between the vaccine groups. However, at the time of necropsy, we generally noted enhanced median binding to FcγRs among gB mRNA-vaccinated rabbits versus gB FL-vaccinated rabbits, though this comparison was significant only for FcγRIIIa (median FcγRIIIa MFI at 20 weeks, 3,929 for gB FL-vaccinated rabbits versus 10,933 for gB mRNA-vaccinated rabbits; P = 0.02, Kruskal-Wallis test with the post hoc Mann-Whitney U test).
FIG 6.

gB mRNA-LNP vaccination elicits long-lived gB-specific IgG-Fcγ receptor engagement, though a reduced durability of the virion phagocytosis response. (A to D) Vaccine-elicited IgG engagement of FcγRI (A), FcγRIIa (B), FcγRIIb (C), and FcγRIIIa (D) was assessed by BAMA. (E) Phagocytosis of fluorophore-coupled whole HCMV (TB40/E) virions was measured by flow cytometry. (F) The upregulation of CD107a on the surface of NK cells in the presence of HCMV-infected cells was assessed by flow cytometry as an approximation of ADCC activity. The results for full-length gB vaccinees are shown in blue, those for gB ectodomain vaccinees are shown in green, and those for gB mRNA-LNP vaccinees are shown in purple. Antibody responses were assessed at the preimmune, 10-week (post-boost 3, peak immunogenicity), and 20-week (necropsy) time points. Data points represent individual animals, with the lines designating the median. Dotted black lines indicate the mean preimmune response plus 2 standard deviations. *, P < 0.05, Kruskal-Wallis test with the post hoc Mann-Whitney U test.
TABLE 3.
Rapid induction of gB-specific IgG that engage Fcγ receptor following a single mRNA vaccine dose
| Fc receptor | Median MFI |
||
|---|---|---|---|
| gB FL | gB ecto | gB mRNA | |
| FcγRI | 0 | 19 | 528 |
| FcγRIIa | 0 | 0 | 129a |
| FcγRIIb | 0 | 0 | 207a |
| FcγRIIIa | 95 | 98 | 1,770 |
, P < 0.05 (Kruskal-Wallis test with the post hoc Mann-Whitney U test) for comparison of gB mRNA to both gB FL and gB ecto.
We next investigated the magnitude of nonneutralizing, Fc effector functions mediated by vaccine-elicited antibodies. First, we measured the antibody-dependent cellular phagocytosis (ADCP) of whole HCMV virions (TB40/E strain) (Fig. 6E). We identified similar peak phagocytosis activity (10 weeks), but intriguingly, we noted enhanced phagocytosis in the gB FL vaccinees at 20 weeks, indicating more robust phagocytosis durability in this group (median percentage of phagocytosing cells, 10.1% for gB FL vaccinees versus 7.1% for gB mRNA vaccinees; P < 0.01, Kruskal-Wallis test with the post hoc Mann-Whitney U test). Next, we assessed natural killer (NK) cell degranulation activity (CD107a upregulation) when vaccine-elicited antibodies were incubated with HCMV-infected cells (Fig. 6F), which we previously identified to approximate antibody-dependent cellular cytotoxicity (ADCC) for rabbits (17). However, we were unable to measure NK cell degranulation activity for the majority of animals, suggesting that antibodies mediating this nonneutralizing effector function are not a dominant response elicited by the gB FL, gB ecto, or gB mRNA vaccines.
gB non-ectodomain-directed antibodies can mediate whole-HCMV-virion phagocytosis.
Since gB FL-vaccinated rabbits had both (i) an extraordinarily robust antibody response against AD-3 and (ii) superior whole-virion phagocytosis activity, we sought to investigate whether AD-3-directed antibodies could mediate phagocytosis. We depleted gB ectodomain-specific IgG from vaccinated rabbit sera, and depletion was confirmed by ELISA against the same protein (Fig. 7A). Furthermore, we established that AD-3-specific antibodies remained in the gB ectodomain-depleted sera of gB FL vaccinees by measuring the antibody responses to an immunodominant AD-3 peptide (Fig. 7B). Interestingly, ectodomain-depleted rabbit sera were able to mediate low-level HCMV virion phagocytosis (Fig. 7C) (median percentage of phagocytosing cells for gB FL, preimmune sera = 0.96%, mock-depleted sera = 2.91%, gB ecto-depleted sera = 1.93%), suggesting that nonectodomain epitopes (e.g., AD-3 or membrane-proximal external region) can be bound by circulating IgG that can subsequently mediate nonneutralizing effector functions. Furthermore, the magnitude of vaccine-elicited binding to AD-3 linear peptides of nondepleted plasma correlated strongly with the phagocytosis mediated by gB ectodomain-depleted serum antibodies (Fig. 7D; r = 0.75, P < 0.001, Spearman rank correlation).
FIG 7.

Phagocytosis-mediating antibodies directed against gB AD-3. (A, B) Binding antibodies targeting the gB ectodomain are not detectable in gB ectodomain-depleted sera (A), though antibodies targeting an immunodominant linear peptide within the AD-3 region (QDKGQKPNLLDRLRH) persist (B). Mock, mock-depleted sera using HIV-1 gp120; gB ecto, gB ectodomain-depleted sera; AUC, area under the curve. (C) Phagocytosis of whole HCMV (TB40/E) virions remained measurable by flow cytometry for samples depleted of ectodomain-targeting antibodies (gB ecto-dep.; i.e., with AD-3-specific antibodies remaining). (D) Spearman correlation of AD-3 peptide-binding IgG magnitude with phagocytosis activity mediated by gB ectodomain-depleted serum antibodies. The results for full-length gB vaccinees are shown in blue, those for gB ectodomain vaccinees are shown in green, and those for gB mRNA-LNP vaccinees are shown in purple. Data points represent individual animals, with the lines designating the median. Dotted black lines indicate the mean preimmune response plus 2 standard deviations.
gB-specific T cell responses.
Lastly, we investigated the magnitude of antigen-specific T cells by intracellular cytokine staining. Purified spleen mononuclear cells were either not stimulated, incubated with mitogen concanavalin A (ConA), or incubated with pooled gB peptides (the gating strategy is shown in Fig. 8A to C). Subsequently, the concentration of gamma interferon (IFN-γ)-positive (IFN-γ+) live T cells was identified for each group/treatment (Fig. 8D). For animals from each vaccine group, ConA nonspecifically stimulated a population of T cells to produce IFN-γ+ (for all comparisons, P < 0.05, Friedman test with the post hoc Wilcoxon matched-pairs signed-rank test). When the percentage of unstimulated IFN-γ+ T cells was subtracted from that of gB-stimulated IFN-γ+ T cells (Fig. 8E), we observed a modest gB-specific T cell response that was the most pronounced in gB FL- and gB mRNA-vaccinated rabbits (median percentage of gB-specific IFN-γ+ T cells, gB FL-vaccinated rabbits = 0.22%, gB ecto-vaccinated rabbits = 0%, gB mRNA-vaccinated rabbits = 0.38%; P was not significant by the Kruskal-Wallis test). In addition to spleen cells, we attempted to identify antigen-specific T cells in peripheral blood as well as mesenteric lymph nodes, but no IFN-γ+ cells were observed using this method upon either ConA or gB peptide stimulation.
FIG 8.

Full-length gB and gB mRNA-LNP vaccines elicit antigen-specific T cells in spleen of the majority of vaccinated rabbits. Rabbit spleen cells were either not stimulated (NS) or incubated with concanavalin A (ConA) or with a pool of gB peptides (gB) and then stained for KEN-5 (a rabbit pan-T cell marker) and IFN-γ before flow cytometric analysis. (A) The cell population was selected using forward and side scatter. (B) Live rabbit T cells were identified using LIVE/DEAD Aqua stain (Invitrogen) as well as a KEN-5-specific antibody (AF647). (C) IFN-γ+ T cell subpopulation identified by intracellular cytokine staining (AF488). (D) The percentages of live T cells that stained positive for IFN-γ are plotted. (E) For each animal, the difference between the percentage of gB-stimulated and unstimulated cells is plotted. The results for full-length gB vaccinees are shown in blue, those for gB ectodomain vaccinees are shown in green, and those for gB mRNA-LNP vaccinees are shown in purple. Data points represent individual animals, with the lines designating the median. *, P < 0.05, Friedman test with the post hoc Wilcoxon matched-pairs signed-rank test. FSC-A, forward scatter area; SSC-A, side scatter area.
DISCUSSION
Glycoprotein B, a homotrimeric viral fusogen that is essential for entry into all cell types, has long been a leading HCMV vaccine candidate (9). Yet over the past decade, following the discovery that the most potent HCMV-neutralizing antibodies in human sera target the gH/gL/UL128-131A pentameric complex (PC) (22, 23), the focus has expanded from gB vaccine development. We propose that gB remains relevant, given that the gB/MF59 protein subunit vaccine achieved moderate vaccine efficacy in preventing primary HCMV infection and seroconversion (12, 13), a feat unparalleled in the HCMV vaccine field (24). Furthermore, the gB/MF59 vaccine reduced viremia and demonstrated a protective benefit in transplant recipients (14). Importantly, these partial successes were achieved without the elicitation of robust neutralizing antibody responses (15, 16). It is certainly possible that both gB and the pentameric complex may be critical antigenic components to an efficacious HCMV vaccine. Indeed, an HCMV gB plus PC mRNA vaccine recently underwent a successful phase 1 trial by Moderna (the mRNA-1647 vaccine, ClinicalTrials.gov registration number NCT03382445). Therefore, the optimization of both the gB and PC vaccine components remains of paramount importance. In this investigation, we sought to improve upon the gB antigen and vaccine/delivery platform, comparing the novel vaccine immunogenicity head-to-head against the gB/MF59 vaccine immunogenicity in a preclinical model.
HCMV preclinical vaccine development is hindered by CMV species specificity (25, 26). Though validated preclinical challenge models exist for guinea pigs (GPCMV) (27–29) and rhesus monkeys (RhCMV) (25, 30), herein we chose to test the immunogenicity of HCMV vaccines in an animal model for two reasons: (i) direct comparison to the clinically tested gB/MF59 vaccine and (ii) the ability to employ existing HCMV-specific tools/assays to dissect out binding specificity. Rabbits in particular were selected, given our previously validated ability to measure neutralizing and nonneutralizing in vitro functionality (17). In this study, therefore, we were able to epitope map and define the function of antibodies elicited by these three experimental vaccines and subsequently compare our results to previously reported immune correlates of protection (31). HCMV-neutralizing IgG has previously been associated with reduced viral systemic dissemination (20, 30, 32). Specifically, antibodies targeting gB AD-2 are correlated with a reduced incidence of viremia and congenital disease (19, 20), though AD-2-specific antibodies were not elicited to any appreciable extent by the vaccines tested in this investigation (Fig. 5H). Furthermore, nonneutralizing antibodies targeting gB and other surface glycoproteins are well described to have a protective role in preventing HCMV acquisition (15, 16), reducing viremia (16, 33), and blocking tissue-invasive replication (33, 34), though the precise mechanism remains unknown. Lastly, HCMV-specific CD4+ and CD8+ T cells have been widely implicated in reducing HCMV acquisition (35, 36), viral replication (25, 37–45), and the incidence of congenital/transplant-associated disease (46, 47). Therefore, while we remain without a well-validated immune correlate of protection, we can harness the responses measured in vitro for hypothesis generation and to carry a candidate vaccine forward into preclinical challenge models.
Given the uncertainty regarding precise epitope specificities or immune effector functions protective against HCMV-associated disease, we regarded enhanced durability and peptide-binding breadth to be desirable attributes for vaccine development and the primary outcomes of interest in this study. Overall, we identified that the experimental vaccines (gB FL, gB ecto, and gB mRNA) elicited binding at comparable magnitudes and comparable functional antibody responses at peak immunogenicity. The most distinction between the vaccine-elicited immunogenicity was observed only at the latest time point (20 weeks, which was 12 weeks after the final vaccine dose), signifying the variable durability of the elicited antibody responses. Nucleoside-modified mRNA-LNP vaccines have been well described to produce sustained antigen presentation with robust T follicular helper cell and germinal center B cell stimulation, resulting in extraordinarily durable antibody responses after even a single vaccine dose in small and large animals (21, 48–50). Indeed, we identified that the durability of the gB mRNA vaccine-elicited responses exceeded that of the gB FL vaccine-elicited responses for nearly all measured antibody binding/functional responses (Table 4; comparisons were statistically significant for soluble and cell-associated gB binding, binding to gB domain 1 and domains 1 and 2, heterologous virus neutralization, and engagement of FcγRIIIa). Though the improvement in antibody response durability following mRNA vaccination was not dramatic, it is nevertheless notable, given that gB/MF59 is an extraordinarily immunogenic vaccine (51). Furthermore, we observed an enhanced breadth of the peptide-binding immune response in gB mRNA-immunized rabbits, resulting in the targeting of unconventional/subdominant epitopes that are not seen in protein subunit vaccinees (Fig. 4; Table 2). mRNA-vaccinated rabbits appear to have a gB peptide-binding fingerprint that closely resembles that in seropositive individuals elicited by natural host infection (15). Importantly, while HCMV gB mRNA vaccines have been tested previously in a preclinical model (52), this is the first such investigation to test a gB mRNA-based vaccine alongside the partially effective gB/MF59 protein subunit vaccination and to directly compare the epitope specificity, durability, and neutralizing/nonneutralizing function of the antibodies elicited by these two vaccine platforms.
TABLE 4.
Summary of vaccine-elicited immune responses at week 20a
| Category | Median response magnitude | Value for the following vaccine: |
||
|---|---|---|---|---|
| gB FL | gB ecto | gB mRNA | ||
| gB binding | gB FL binding (log10 MFI) | 2.85 | 3.37 | 3.53 * |
| gB ecto binding MFI (log10 MFI) | 2.77 | 3.19 | 3.34 * | |
| Cell-associated gB binding (% of cells) | 18.81 | 18.58 | 29.05 * | |
| RAI (gB FL target) | 0.93 | 0.90 | 0.78 | |
| Peptide bindingb | Total binding sum (log10 MFI) | 5.25 | 4.74 | 5.04 |
| AD-3 binding sum (log10 MFI) | 5.12 | 2.27 | 4.77 | |
| No. of peptides bound (>100 MFI) | 44.5 | 28.5 | 85 * | |
| Neutralizing epitope binding and neutralization | Domain 1 binding (log10 MFI) | 2.76 | 3.37 | 3.44 * |
| Domain 1 + 2 binding (log10 MFI) | 2.96 | 3.49 | 3.03 | |
| AD-1 binding (log10 MFI) | NMR | NMR | NMR | |
| AD-2 binding (log10 MFI) | NMR | NMR | NMR | |
| AD169r Fibro Neut + C (log10 ID50) | 1.69 | 1.79 | 2.08 | |
| AD169r Epi Neut + C (log10 ID50) | 1.91 | 1.83 | 2.29 | |
| Fcγ receptor binding and function | FcγRI binding (log10 MFI) | 3.22 | 3.41 | 3.59 |
| FcγRIIa binding (log10 MFI) | 3.14 | 3.24 | 3.29 | |
| FcγRIIb binding (log10 MFI) | 1.71 | 2.27 | 3.07 | |
| FcγRIIIa binding (log10 MFI) | 3.59 | 3.69 | 4.04 * | |
| TB40/E virion phagocytosis (% of cells) | 10.06* | 7.04 | 7.06 | |
| NK cell degranulation (% of cells) | NMR | NMR | NMR | |
| T cells | Splenic gB-specific T cells (% of live T cells) | 0.22 | NMR | 0.38 |
Data in boldface indicate the maximal response. Fibro Neut, neutralization in fibroblasts; Epi Neut, neutralization in epithelial cells; +C, in the presence of complement; NMR, no measurable response above the baseline levels; *, P < 0.05, Kruskal-Wallis test with the post hoc Mann-Whitney U test (compared to the results for gB FL).
Peptide binding using week 10 sera.
A secondary focus of this investigation was to gain a better understanding of the implications of the gB/MF59-elicited immune-dominant response directed against gB AD-3 (15). We previously hypothesized that the absence of neutralizing antibodies in gB/MF59 vaccinees may be attributable to the dominant responses against the AD-3 decoy epitope, which diverted antibody targeting away from more functional epitopes (24). We directly tested this hypothesis with our gB ecto group by excluding AD-3 from the immunogen. Intriguingly, we failed to see any consistent increase in the magnitude of functional neutralizing/nonneutralizing antibodies in gB ecto-vaccinated rabbits, suggesting that inclusion of the AD-3 epitope in the vaccine immunogen does not hinder the development of more functional antibodies. Is it therefore possible that AD-3-directed antibodies have any functional or protective role that might account for the 50% vaccine efficacy observed in gB/MF59 vaccinees? In this study, we noted that AD-3-specific antibodies can mediate nonneutralizing antibody effector functions, including whole-virion phagocytosis (Fig. 6C), and that the magnitude of overall phagocytosis activity is well correlated with the magnitude of AD-3-specific antibody binding (Fig. 7D). This finding raises the question of how AD-3 (which is presumed to be intraluminal/cytosolic) is expressed on the surface of the virion to be accessible for phagocytosis-mediating antibodies. Furthermore, our observations emphasize the possibility that AD-3-directed antibodies may have played a distinct role in the 50% vaccine efficacy achieved following gB/MF59 immunization (12).
A limitation to this study is the dissimilarity in vaccine dose and route of delivery between comparison groups. The protein subunit vaccines (gB FL, gB ecto) were administered intramuscularly (i.m.) at a dose of 20 μg, mimicking the protocol for gB/MF59-immunized humans in the partially efficacious clinical trials (12, 13, 24). In this study, we aimed to compare the antibody responses elicited by gB vaccines administered at the most effective dose and via the most effective route. Therefore, a 50-μg dose of the gB nucleoside-modified mRNA-LNP was employed since we have previously shown that mRNA-elicited responses are not dose dependent beyond this quantity of immunogen (48). Furthermore, intradermal (i.d.) immunization was employed for the mRNA group, as we and others have previously noted the superiority of this route for an mRNA-LNP vaccine formulation (50, 53, 54). We therefore cannot rule out the possibility that our results are dose or route dependent, that is, that a higher dose or a different immunization route of gB FL protein might not have achieved a peptide-binding breadth and a durability of responses similar to those achieved in gB mRNA-LNP-vaccinated rabbits. Furthermore, while rabbits provide an excellent model to study vaccine immunogenicity, this investigation was restricted by the rabbit immunologic toolbox. We were able to measure Fcγ receptor engagement in this study, though we lacked the ability to identify the mechanism behind variable Fcγ receptor engagement (e.g., IgG subclass or Fc glycosylation). Furthermore, while we were able to identify gB-specific T cells in spleen, we were unable to (i) identify antigen-specific T cells in peripheral blood or (ii) parse out T cell subsets (CD4+ cells, CD8+ cells, etc.). Nevertheless, the results described are sufficient justification for subsequent testing of the gB mRNA-LNP vaccine in nonhuman primate preclinical challenge models and/or human clinical trials.
This comparison of the immune responses elicited by next-generation HCMV vaccines head-to-head against those elicited by gB/MF59 (gB FL) immunization informs future rational HCMV vaccine development efforts. We hypothesize that gB will constitute an essential component of any efficacious HCMV vaccine, and, therefore, gB vaccine formulations must be optimized to elicit functional and protective antibody responses. Herein, we have demonstrated that gB nucleoside-modified mRNA-LNP immunization improves the durability and peptide-binding breadth of the gB-specific antibody repertoire in comparison to those achieved with the partially efficacious gB/MF59 vaccine. Additionally, we note that gB ectodomain immunization did not elicit antibody responses that were functionally superior to those elicited by gB FL, suggesting that the immunodominant AD-3 response does not interfere with the development of functional antibodies. While we await well-validated immune correlates of protection to guide rational HCMV vaccine development efforts, we propose that the durable and epitopically broad responses elicited by the gB mRNA-LNP formulation make this vaccine candidate highly appealing for subsequent evaluation. Therefore, testing of this vaccine platform in nonhuman primate (RhCMV gB) or guinea pig (GPCMV gB) challenge models as well as human immunogenicity trials is warranted to rigorously interrogate its ability to elicit immune factors protective against congenital HCMV infection and transplant-associated disease.
MATERIALS AND METHODS
gB ectodomain immunogen production.
The sequence encoding the ectodomain segment (amino acid residues 1 to 696) of the Towne strain (GenBank accession number FJ616285.1) HCMV glycoprotein B (gB) was tagged at the 3′ end with a polyhistidine tag, and the furin cleavage site at residue 457 was mutated from RTKR to STKS. The nucleotide sequence was codon optimized for mammalian cells and then cloned into the pcDNA3.1(+) mammalian expression vector (Invitrogen) via the BamHI site at the 5′ end and the EcoRI site at the 3′ end. Subsequently, the plasmid was transiently transfected into Expi293i cells using ExpiFectamine 293 transfection reagents (Thermo Fisher Scientific) according to the manufacturer’s instructions. The culture supernatant was harvested after 5 days of incubation at 37°C in 8% CO2 and then purified using nickel-nitrilotriacetic acid resin (Thermo Fisher Scientific). The purity, identity, and correct molecular weight were confirmed by Western blotting using monoclonal antibodies specific for gB AD-2, gB domain 1, and gB domain 2 (described in reference 15), followed by alkaline phosphatase AP-conjugated anti-human IgG (Sigma-Aldrich). Finally, the protein was tested for the presence of endotoxin using a Pierce Limulus amoebocyte lysate chromogenic endotoxin quantitation kit (Thermo Fisher Scientific). gB protein ectodomain aliquots were stored at −80°C at a concentration of ∼1 μg/μl and then thawed at <60 min prior to injection.
gB mRNA production and formulation into lipid nanoparticles.
The modified mRNA encoding HCMV gB (Towne strain, GenBank accession number FJ616285.1) was produced as previously described (55) using T7 RNA polymerase (MEGAscript; Ambion) on a codon-optimized (56) linearized plasmid (the sequence is available upon request). The mRNA was transcribed to contain a 101-nucleotide-long poly(A) tail. To generate modified nucleoside-containing mRNA, m1Ψ-5′-triphosphate (TriLink) was used instead of UTP. The mRNA was then capped using an m7G capping kit with 2′-O-methyltransferase (ScriptCap; CellScript). The mRNA was purified by fast protein liquid chromatography (FPLC; Akta purifier; GE Healthcare) as described previously (57), analyzed by electrophoresis using denaturing or native agarose gels, and stored at −20°C. The FPLC-purified m1Ψ-containing HCMV gB mRNA and poly(C) RNA (Sigma) were encapsulated in LNPs using a self-assembly process in which an aqueous solution of mRNA at pH 4.0 was rapidly mixed with a solution of lipids dissolved in ethanol (58). The LNPs used in this study were similar in composition to those described previously (58, 59), which contain an ionizable cationic lipid (proprietary to Acuitas), phosphatidylcholine, cholesterol, and polyethylene glycol-lipid (50:10:38.5:1.5, mol/mol), and were encapsulated at an RNA-to-total-lipid ratio of ∼0.05 (wt/wt). They had a diameter of ∼80 nm, as measured by dynamic light scattering using a Zetasizer Nano ZS (Malvern Instruments Ltd.) instrument. The mRNA-LNP formulations were stored at −80°C at a concentration of mRNA of ∼1 μg/μl and then thawed at <60 min prior to injection.
Animal care and sample collection.
Juvenile New Zealand White rabbits (approximately 10 weeks of age) were purchased from Robinson Services Inc. (Mocksville, NC) and housed at Duke University. For blood collections, animals were sedated with 1-mg/kg-of-body-weight subcutaneous acepromazine and topical 1% lidocaine applied to the ears. EDTA-anticoagulated blood was collected via auricular venipuncture. Plasma was separated from whole blood by centrifugation, and peripheral blood mononuclear cells (PBMCs) were isolated by density gradient centrifugation using Lympholyte cell separation medium (Cedarlane Laboratories). The animals were euthanized using 0.5 ml of subcutaneously injected xylazine (100 mg/ml) plus ketamine (500 mg/ml), mixed together in a 1:5 ratio, followed by 0.5 ml of intracardiac pentobarbital sodium plus phenytoin sodium (Euthasol). Lymphocytes were isolated from spleen and mesenteric lymph nodes by manual tissue disruption and crushing through a 100-μm-mesh-size cell strainer, followed by density gradient centrifugation with Lympholyte cell separation medium.
Animal vaccination.
Three groups of female juvenile New Zealand White rabbits (n = 6) were given different vaccines: (i) 20 μg intramuscular full-length gB protein (a generous gift of Sanofi Pasteur) combined 1:1 (vol/vol) with the MF59-like squalene-based adjuvant AddaVax (Invivogen), (ii) 20 μg intramuscular gB ectodomain protein combined 1:1 (vol/vol) with AddaVax, or (iii) 50 μg intradermal nucleoside-modified gB mRNA packaged in lipid nanoparticles. The 20-μg dose for the protein subunit vaccines was selected to mimic the dose used in the human clinical trial protocol (12–14). The 50-μg dose for mRNA was selected given our previous findings that the mRNA vaccine-elicited response magnitude is not dose dependent above this threshold (48). Vaccine doses were administered monthly for three consecutive months. For intramuscular (i.m.) protein subunit vaccine administration, the rabbits were sedated with 1-mg/kg subcutaneous acepromazine and then injected with the vaccine dose in the rear thigh (alternating sides between monthly doses). For intradermal (i.d.) mRNA-LNP vaccine administration, the rabbits were sedated with 1-mg/kg subcutaneous acepromazine as well as 2% inhaled isoflurane, and the saddle of the rabbit was shaved. Chilled, sterile phosphate-buffered saline (PBS) was added to each vaccine dose to a total volume of 300 μl, the diluted vaccine was divided into 6 equal fractions of 50 μl each, and then the rabbit saddle was injected intradermally with 3 injections on each side of the spine (injection sites varied between monthly doses). Different routes of vaccination were selected for the subunit and the mRNA vaccine platforms because we sought to compare these gB vaccine formulations administered by the most effective route and have previously identified that the mRNA vaccines work most efficiently after intradermal administration (50).
Cell culture.
Human retinal pigment epithelial (ARPE-19) cells (ATCC) were maintained for a maximum of 35 passages in Dulbecco’s modified Eagle medium (DMEM)–Ham’s F-12 medium (DMEM-F12) supplemented with 10% fetal calf serum (FCS), 2 mM l-glutamine, 1 mM sodium pyruvate, 50 U/ml penicillin, 50 μg/ml streptomycin and gentamicin, and 1% epithelial growth cell supplement (ScienCell). Human lung (MRC-5) fibroblasts (ATCC) were maintained for a maximum of 20 passages in DMEM containing 20% FCS, 50 U/ml penicillin, and 50 μg/ml streptomycin. Human epithelial kidney (HEK293T) cells (ATCC) were maintained for a maximum of 35 passages in DMEM containing 10% FCS, 25 mM HEPES buffer, 50 U/ml penicillin, and 50 μg/ml streptomycin. Human monocyte (THP-1) cells (ATCC) were maintained for a maximum of 35 passages in RPMI 1640 medium containing 10% FCS. All cell lines were tested for the presence of mycoplasma biannually.
Virus growth.
AD169 revertant virus (AD169r; a gift from Merck) (60) and BADrUL131-Y4 virus (61) stocks were propagated on ARPE-19 cells in T75 culture flasks. Towne virus (ATCC) was propagated on MRC-5 cells in T75 culture flasks. The supernatant, containing cell-free virus, was collected when 90% of the cells showed cytopathic effects and was then cleared of cell debris by low-speed centrifugation before passage through a 0.45-μm-pore-size filter. Viral infections of ARPE-19 cells were carried out in a similar medium, but the medium contained only 5% FCS and lacked cell growth supplement.
Binding antibody multiplex assay (BAMA).
Antibody responses against the gB full-length protein, the gB ectodomain, and gB epitopes (the creation of which is described in references 14 and 20) were assessed by a multiplex ELISA. In brief, carboxylated fluorescent beads (Luminex) were covalently coupled to purified HCMV antigens and subsequently incubated with maternal plasma in assay diluent (phosphate-buffered saline, 5% normal goat serum [NGS], 0.05% Tween 20, 1% Blotto milk, 0.5% polyvinyl alcohol, 0.8% polyvinylpyrrolidone). The antigen panel included full-length gB (courtesy of Sanofi Pasteur), the gB ectodomain protein, gB domain 1, gB domain 2, gB domains 1 and 2, gB AD-1 (MyBioSource), and biotinylated linear gB AD-2 (biotin-NETIYNTTLKYGD). HCMV glycoprotein-specific antibodies were detected with phycoerythrin-conjugated goat anti-human IgG (2 μg/ml; Southern Biotech). The beads were washed and acquired on a Bio-Plex 200 instrument (Bio-Rad), and results were expressed as the mean fluorescence intensity. Testing was performed at a panel of prevaccination time points to determine the nonspecific baseline levels of binding. Minimal background activity was observed, so the threshold for positivity for each antigen was set at the mean value for negative-control serum binding to each antigen plus 3 standard deviations. Blank beads were used in all assays to account for nonspecific binding. All assays included tracking of the HCMV immunoglobulin (Cytogam; CSL Behring) standard by the use of Levy-Jennings charts. The preset assay criteria for sample reporting were a coefficient of variation per duplicate values of ≤20% for each sample and ≥100 beads counted per sample. All samples were analyzed at the same dilution for each antigen: full-length gB, gB ectodomain, gB domain 1, gB domain 2, and gB domains 1 and 2 were assessed at a 1:500 dilution; gB AD-1 and gB AD-2 were assessed at a 1:50 dilution. These dilutions were predetermined to be within the linear range of the assay, based on testing serial dilutions of a small subset of plasma samples.
Fcγ receptor engagement.
The binding of vaccine-elicited serum antibodies to Fcγ receptors was characterized using a multiplex Fcγ receptor BAMA, employing the reagents and quality control methods described above. In brief, HCMV gB (full length) was covalently coupled to streptavidin (Rockland)-coupled fluorescent beads (Luminex). Serum samples were diluted 1:500, then incubated in duplicate in a 96-well microplate with gB-streptavidin-coupled beads, and then washed and incubated with one of the following biotinylated Fcγ receptor tetramers: FcγRIa, FcγRIIa (clone H131), FcγRIIb, and FcγRIIIa (clone V158) (Fcγ receptor proteins were courtesy of Kevin Saunders). Fcγ receptor engagement was detected using mouse anti-human IgG-PE (MyBioSource), followed by a final wash. Data were acquired on a BioPlex-200 instrument (Luminex).
gB-transfected cell binding.
Binding of vaccine-elicited antibodies to trimeric gB expressed on cell membranes was assessed by flow cytometry as described previously (15). Briefly, HEK293T cells were grown overnight to ∼50% confluence and then cotransfected, using the Effectine transfection reagent (Qiagen), with a green fluorescent protein (GFP)-expressing plasmid (a gift of Maria Blasi, Duke University) and a second plasmid encoding the full-length Towne strain gB (SinoBiological). Transfected cells were incubated for 2 days at 37°C in 5% CO2, washed with Dulbecco’s PBS (DPBS; Gibco), and then removed from the flask using an enzyme-free cell dissociation buffer (Thermo Fisher Scientific). Cells were washed in wash buffer (DPBS plus 1% FBS), and then 100,000 live cells were added to each well of a 96-well V-bottom plate (Corning). After centrifugation (1,200 × g, 5 min), the cells were resuspended in 1:6,250-diluted serum samples and incubated for 2 h at 37°C in 5% CO2. Next, the cells were washed and stained with LIVE/DEAD Aqua dead cell stain (Thermo Fisher Scientific) diluted 1:1,000 for 20 min at room temperature. Afterwards, the cells were washed, then resuspended in PE-conjugated goat anti-human IgG Fc (eBioscience) diluted 1:200 in wash buffer, and then incubated for 25 min at 4°C. Following two additional wash steps, the cells were resuspended and fixed in DPBS–1% formalin. Events were acquired on an LSR Fortessa machine (BD Biosciences) using the high-throughput sampler (HTS). The percentage of PE-positive cells was calculated from the live, GFP-positive cell population and reported for each sample. The background binding of each plasma sample was corrected for by using cells transfected with the GFP-expressing plasmid alone.
ELISA and avidity.
ELISA plates (384-well plates; Corning) were coated overnight at 4°C with 30 ng full-length gB per well and then blocked with assay diluent (1× PBS containing 4% whey, 15% normal goat serum, and 0.5% Tween 20). Threefold dilutions of sera were then added to the plate. Finally, bound IgG was detected with a horseradish peroxidase (HRP)-conjugated polyclonal goat anti-monkey IgG (Rockland), and developed using the SureBlue Reserve tetramethylbenzidine (TMB) peroxidase substrate (KPL). The 50% end dilution (ED50) was calculated as the plasma dilution that resulted in a 50% reduction in the binding optical density, as determined by the method of Reed and Muench (62). To assess antibody binding avidity, following addition of the sera, duplicate wells that were treated for 5 min with either 7 M urea or 1× PBS serum dilutions that resulted in an optical density (OD) value of between 0.6 and 1.2 in the absence of urea treatment (dilution range = 1:30 to 1:1,000) were used to determine the relative avidity index (RAI). Indexes were calculated as the ratio of the OD of the urea-treated wells to the OD of the PBS-treated wells.
Glycoprotein B peptide microarray.
Binding to gB linear peptides was assessed as described previously (15). In brief, 15-mer peptides covering the entire gB open reading frame (Towne strain) and neighboring peptides overlapping by 10 residues (a total of 188 peptides) were synthesized and printed to a PepStar multiwell array (JPT Peptide) in triplicate. Microarray binding was performed manually using individual slides immobilized in an ArraySlide 24-4 chamber (JPT Peptide). First, the arrays were blocked with blocking buffer (PBS containing 1% milk Blotto, 5% NGS, and 0.05% Tween 20) and then incubated, first, with serum diluted 1:250 in blocking buffer and, second, with anti-human IgG conjugated to Alexa Fluor 647 (AF467; Jackson ImmunoResearch) diluted in blocking buffer (0.75 μg/ml). The arrays were washed in wash buffer (1× Tris-buffered saline buffer plus 0.1% Tween 20) between steps using an automated plate washer (BioTek model ELX50). To measure the fluorescence, the arrays were scanned at a wavelength of 635 nm using an InnoScan 710 device (Innopsys) at a photomultiplier voltage setting of 580 and 100% laser power. Images were analyzed using Mapix software (Innopsys) and reviewed manually for accurate automated peptide identification. The intensity of binding of the sera to each peptide was corrected with the surrounding background fluorescence. The median fluorescent intensity of each of the 3 replicates is reported.
Neutralization.
The neutralization titers of patient sera were measured by a high-throughput immunofluorescence assay as previously described (15). Briefly, MRC-5 cells were seeded into 96-well flat-bottom plates and incubated for 2 days at 37°C in 5% CO2 to achieve 100% confluence. Rabbit serum was incubated at 55°C for 1 h to heat inactivate the proteins that might cause nonspecific cellular cytotoxicity (e.g., complement). Once the cells were confluent, 3-fold dilutions (1:10 to 1:30,000) of heat-inactivated rabbit serum in infection media were incubated with Towne (ATCC) or AD169r (Merck Laboratories) virus stock at a multiplicity of infection (MOI) of 1.0 in a total volume of 50 μl for 45 min at 37°C. To assess neutralization in the presence of complement, plasma/virus was diluted in infection medium containing purified rabbit complement (Cedarlane Laboratories) at a final dilution of 1:4. Immune complexes were added in duplicate to wells containing MRC-5 cells and then subsequently incubated for 18 h at 37°C. Infected cells were then fixed for 10 min with 3.7% paraformaldehyde, permeabilized for 10 min with Triton X-100, and subsequently processed for immunofluorescence with mouse anti-HCMV IE-1 monoclonal antibody (MAB810; Millipore), followed by goat anti-mouse IgG-Alexa Fluor 488 (AF488; Millipore) and DAPI (4′,6-diamidino-2-phenylindole) nuclear stain. Total cells and AF488-positive infected cells per well were counted on a Cellomics ArrayScan reader (Thermo Fisher Scientific). Neutralization titers (ID50) were calculated according to the method of Reed and Muench (62), using the plasma dilution that resulted in a 50% reduction in the percentage of infected cells compared to the number of cells in control wells infected with virus only.
Whole-HCMV-virion phagocytosis.
The ability of vaccine-elicited antibodies to facilitate phagocytosis of whole HCMV virions was assessed as previously (15). Briefly, 107 PFU of concentrated, sucrose gradient-purified HCMV TB40/E-mCherry virus was buffer exchanged with 1× PBS and sodium bicarbonate (final concentration, 0.1 M), and then AF647–N-hydroxysuccinimide ester (Invitrogen) was added for direct viral conjugation at room temperature for 1 h with constant agitation. The reaction was quenched with 1 M Tris-HCl, pH 8.0, and then the labeled virus was diluted 25 times in wash buffer (PBS plus 0.1% fetal bovine serum [FBS]). Serum samples were diluted 1:10 in wash buffer, and then 10 μl of diluted serum was combined with 10 μl of diluted, fluorophore-conjugated virus in a round-bottom, 96-well plate (Corning) and allowed to incubate at 37°C for 2 h. Following this incubation step, 25,000 THP-1 cells, suspended in 200 μl primary growth medium, were added to each well. The plates were centrifuged at 1,200 × g and 4°C for 1 h in a spinoculation step and then incubated at 37°C for an additional hour. The cells were resuspended, transferred to a 96-well V-bottom plate, and then washed twice prior to fixation in 100 μl DPBS–1% formalin. Events were acquired on an LSR Fortessa machine (BD Biosciences) using the HTS. The percentage of AF647-positive cells was calculated from the full THP-1 cell population and reported for each sample. A cutoff for sample positivity was defined as the mean value for the prevaccination sera (n = 18) plus 2 standard deviations.
NK cell CD107a degranulation assay.
Cell-surface expression of CD107a was used as a marker for natural killer (NK) cell degranulation, which we have previously shown to have good agreement with antibody-dependent cellular cytotoxicity activity for rabbit serum (17). MRC-5 cells were infected with the BadrUL131-Y4 strain at an MOI of 1.0 for 48 h at 37°C at 4 × 104 cells/well in 96-well flat-bottom tissue culture plates. Following incubation, the supernatant was removed and the infected cell monolayers were washed once with RMPI 1640 containing 10% FBS, HEPES, penicillin, streptomycin, l-glutamine, and gentamicin (R10 medium) before addition of NK cells. Primary human NK cells were isolated from peripheral blood mononuclear cells (PBMC) after an overnight rest in R10 medium with 10-ng/ml interleukin-15 (Miltenyi Biotech) by depletion of magnetically labeled cells (human NK cell isolation kit; Miltenyi Biotech). Live NK cells (5 × 104) were added to each well containing HCMV-infected MRC-5 cell monolayers. Plasma samples were diluted in R10 medium and added to the cells at a final dilution of 1:50 in duplicate. Brefeldin A (1 μl/ml; GolgiPlug; BD Biosciences), monensin (4 μl/6 ml; GolgiStop; BD Biosciences), and CD107a-fluorescein isothiocyanate (clone H4A3; BD Biosciences) were added to each well, and the plates were incubated for 6 h at 37°C in a humidified 5% CO2 incubator. The NK cells were then gently resuspended, taking care not to disturb the MRC-5 cell monolayer, and the NK cell-containing supernatant was collected and transferred to 96-well V-bottom plates. The recovered NK cells were washed with PBS and stained with LIVE/DEAD Aqua dead cell stain at a 1:1,000 dilution for 20 min at room temperature. The cells were then washed with 1% FBS–PBS and stained for 20 min at room temperature with the following panel of fluorescently conjugated antibodies diluted in 1% FBS–PBS: CD56-PE-Cy7 (clone NCAM16.2; BD Biosciences), CD16-Pacific Blue (clone 3G8; BD Biosciences), and CD69-Brilliant Violet 785 (clone FN50; BioLegend). The cells were then washed twice and resuspended in 1% paraformaldehyde fixative for flow cytometric analysis. Data analysis was performed using FlowJo software (v9.9.6). Data are reported as the percentage of CD107a-positive live NK cells (singlets, lymphocytes, Aqua Blue negative, CD56+, and/or CD16+ CD107a+). CD69 was not used in the final analysis due to the low frequency of CD107a+ responses. Parallel assays were performed with uninfected MRC-5 cells as a control for the identification of non-CMV-specific responses, and final data were presented after subtraction of the background activity observed against uninfected cells.
Soluble protein bead coupling and antibody depletion.
Cyanogen bromide (CNBr)-activated Sepharose beads (GE Healthcare) were rehydrated with 1 mM HCl and then suspended in coupling buffer (0.1 M NaHCO3, 0.5 M NaCl, pH 8.3). Every 100 μl of bead slurry was combined with 200 μg of soluble gB ectodomain (postfusion conformation; courtesy of Jason McLellan, University of Texas, Austin, TX). Coupling proceeded for 12 h at 4°C on an inversion rotator. Excess soluble protein was washed off with 5 column volumes of coupling buffer. Unbound CNBr-activated groups were blocked with quenching buffer (0.1 M Tris HCl, pH 8.0) for 2 h at room temperature. Protein-conjugated beads were washed with 3 cycles of alternating-pH washes with 0.1 M acetic acid, pH 4.0, and 0.1 M Tris HCl, pH 8. For depletion of gB postfusion-specific antibodies, 100 μg of protein-coupled bead slurry was loaded into a spin microelution column (Thermo Fisher Scientific; Pierce). Four hundred microliters of filtered (SpinX) 1:50-diluted plasma from vaccinated rabbits was then added to each column. The plasma was centrifuged through the column 10 times with bound IgG elution (0.2 M glycine elution buffer, pH 2.5) and bead recalibration (3 cycles of alternating-pH washes) after spins 5 and 10. Adequate specific depletion was confirmed by ELISA against the depleted protein. Mock-depleted samples underwent an identical depletion procedure in the presence of HIV-1 gp120-conjugated CNBr-activated beads.
Splenic T cell intracellular cytokine staining.
Primary spleen cells were thawed in RPMI plus 10% FBS with Benzonase (50 U/ml) and then measured for count and viability on a Muse cell counter (Luminex). The cells were coincubated in duplicate with ConA (5 μg/ml), HCMV UL155 (250 ng/ml; JPT), or medium for 20 to 24 h at 37°C. Samples were stained for the rabbit pan-T cell marker KEN-5 (Santa Cruz Biotechnology) and a LIVE/DEAD viability/cytotoxicity kit (Invitrogen) and then fixed and permeabilized using BD Cytofix/Cytoperm according to the manufacturer’s instructions. Cells were stained for rabbit IFN-γ (MAb Tech). Flow cytometry was performed on a BD LSR II flow cytometer, and data were analyzed with FlowJo software (v10). The gating strategy is shown in Fig. 8A to C.
Modeling of projected gB binding.
The gB-binding ED50 was calculated for each serum sample as described above. ED50 values were log10 transformed, and then the antibody decay for each group was modeled linearly. Using this linear model, values were projected out to 50 weeks following initial vaccination, and a 95% prediction interval was calculated. All modeling was carried out using the R statistical interface (version 3.3.1; www.r-project.org).
Statistical analysis.
Nonparametric tests were utilized because of the small group sizes (n = 6 per group). Furthermore, the 20-week time point was employed for all statistical comparisons between vaccination groups. The magnitudes of the immune responses between the 3 vaccine groups were compared first by the Kruskal-Wallis test. If P was <0.05, the post hoc Mann-Whitney U test was conducted for gB ecto and gB mRNA compared with gB FL. All statistical tests were carried out using the R statistical interface (version 3.3.1, www.r-project.org) and were two-tailed.
Ethics statement.
Animals were maintained in accordance with the American Association for Accreditation of Laboratory Animal Care standards and the Guide for the Care and Use of Laboratory Animals (63). Efforts were made to minimize stress and provide enrichment opportunities when possible (social housing when possible, objects to manipulate in the cage, varied food supplements, interaction with caregivers and research staff). All protocols were reviewed and approved by the Duke University Animal Care and Use Committee (IACUC) prior to the initiation of the study (protocol number A314-15-12).
Data availability.
Source data are available for download and may be found in Data Set S1 in the supplemental material.
Supplementary Material
ACKNOWLEDGMENTS
We thank Diego Zapata and the Duke Division of Laboratory and Animal Resources for assistance with rabbit handling. Furthermore, we acknowledge Sanofi Pasteur, Merck, and Trellis Biosciences for the generous gift of research materials.
This work was supported by an NIH/NIAID R21 grant to S.R.P. (grant R21AI136556) and an NIH/NICHD F30 grant to C.S.N. (grant F30HD089577).
The funders had no role in study design, data collection and interpretation, the decision to publish, or preparation of the manuscript. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
S.R.P. provides consulting services to Pfizer Inc., Merck, Moderna, and Sanofi Pasteur for their preclinical HCMV vaccine programs. The other authors have no competing interests to declare.
C.S.N. and S.R.P. designed the research; C.S.N., J.A.J., N.P., H.R., M.G., and W.E. performed the research; C.S.N. analyzed the data; D.W., J.S.M., and J.P. contributed reagents and expertise; C.S.N. and S.R.P. wrote the paper.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Swanson EC, Gillis P, Hernandez-Alvarado N, Fernandez-Alarcon C, Schmit M, Zabeli JC, Wussow F, Diamond DJ, Schleiss MR. 2015. Comparison of monovalent glycoprotein B with bivalent gB/pp65 (GP83) vaccine for congenital cytomegalovirus infection in a guinea pig model: Inclusion of GP83 reduces gB antibody response but both vaccine approaches provide equivalent protection against pup mortality. Vaccine 33:4013–4018. doi: 10.1016/j.vaccine.2015.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Manicklal S, Emery VC, Lazzarotto T, Boppana SB, Gupta RK. 2013. The “silent” global burden of congenital cytomegalovirus. Clin Microbiol Rev 26:86–102. doi: 10.1128/CMR.00062-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kenneson A, Cannon MJ. 2007. Review and meta-analysis of the epidemiology of congenital cytomegalovirus (CMV) infection. Rev Med Virol 17:253–276. doi: 10.1002/rmv.535. [DOI] [PubMed] [Google Scholar]
- 4.Ross SA, Boppana SB. 2005. Congenital cytomegalovirus infection: outcome and diagnosis. Semin Pediatr Infect Dis 16:44–49. doi: 10.1053/j.spid.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 5.Legendre C, Pascual M. 2008. Improving outcomes for solid-organ transplant recipients at risk from cytomegalovirus infection: late-onset disease and indirect consequences. Clin Infect Dis 46:732–740. doi: 10.1086/527397. [DOI] [PubMed] [Google Scholar]
- 6.Ljungman P, Griffiths P, Paya C. 2002. Definitions of cytomegalovirus infection and disease in transplant recipients. Clin Infect Dis 34:1094–1097. doi: 10.1086/339329. [DOI] [PubMed] [Google Scholar]
- 7.Bialas KM, Permar SR. 2016. The march towards a vaccine for congenital CMV: rationale and models. PLoS Pathog 12:e1005355. doi: 10.1371/journal.ppat.1005355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Griffiths PD. 2012. Burden of disease associated with human cytomegalovirus and prospects for elimination by universal immunisation. Lancet Infect Dis 12:790–798. doi: 10.1016/S1473-3099(12)70197-4. [DOI] [PubMed] [Google Scholar]
- 9.Schleiss MR. 2018. Recombinant cytomegalovirus glycoprotein B vaccine: rethinking the immunological basis of protection. Proc Natl Acad Sci U S A 115:6110–6112. doi: 10.1073/pnas.1806420115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vanarsdall AL, Johnson DC. 2012. Human cytomegalovirus entry into cells. Curr Opin Virol 2:37–42. doi: 10.1016/j.coviro.2012.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zydek M, Petitt M, Fang-Hoover J, Adler B, Kauvar LM, Pereira L, Tabata T. 2014. HCMV infection of human trophoblast progenitor cells of the placenta is neutralized by a human monoclonal antibody to glycoprotein B and not by antibodies to the pentamer complex. Viruses 6:1346–1364. doi: 10.3390/v6031346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pass RF, Zhang C, Evans A, Simpson T, Andrews W, Huang ML, Corey L, Hill J, Davis E, Flanigan C, Cloud G. 2009. Vaccine prevention of maternal cytomegalovirus infection. N Engl J Med 360:1191–1199. doi: 10.1056/NEJMoa0804749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bernstein DI, Munoz FM, Callahan ST, Rupp R, Wootton SH, Edwards KM, Turley CB, Stanberry LR, Patel SM, McNeal MM, Pichon S, Amegashie C, Bellamy AR. 2016. Safety and efficacy of a cytomegalovirus glycoprotein B (gB) vaccine in adolescent girls: a randomized clinical trial. Vaccine 34:313–319. doi: 10.1016/j.vaccine.2015.11.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Griffiths PD, Stanton A, McCarrell E, Smith C, Osman M, Harber M, Davenport A, Jones G, Wheeler DC, O'Beirne J, Thorburn D, Patch D, Atkinson CE, Pichon S, Sweny P, Lanzman M, Woodford E, Rothwell E, Old N, Kinyanjui R, Haque T, Atabani S, Luck S, Prideaux S, Milne RSB, Emery VC, Burroughs AK. 2011. Cytomegalovirus glycoprotein-B vaccine with MF59 adjuvant in transplant recipients: a phase 2 randomised placebo-controlled trial. Lancet 377:1256–1263. doi: 10.1016/S0140-6736(11)60136-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nelson CS, Huffman T, Jenks JA, Cisneros de la Rosa E, Xie G, Vandergrift N, Pass RF, Pollara J, Permar SR. 2018. HCMV glycoprotein B subunit vaccine efficacy mediated by nonneutralizing antibody effector functions. Proc Natl Acad Sci U S A 115:6267–6272. doi: 10.1073/pnas.1800177115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baraniak I, Kropff B, Ambrose L, McIntosh M, McLean GR, Pichon S, Atkinson C, Milne RSB, Mach M, Griffiths PD, Reeves MB. 2018. Protection from cytomegalovirus viremia following glycoprotein B vaccination is not dependent on neutralizing antibodies. Proc Natl Acad Sci U S A 115:6273–6278. doi: 10.1073/pnas.1800224115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pollara J, Jones DI, Huffman T, Edwards RW, Dennis M, Li SH, Jha S, Goodman D, Kumar A, LaBranche CC, Montefiori DC, Fouda GG, Hope TJ, Tomaras GD, Staats HF, Ferrari G, Permar SR. 2019. Bridging vaccine-induced HIV-1 neutralizing and effector antibody responses in rabbit and rhesus macaque animal models. J Virol 93:e02119-18. doi: 10.1128/JVI.02119-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nelson CH, Cisneros de la Rosa E, Xi G, Vandergrift N, Pass RF, Pollara J, Permar SR. 2018. HCMV glycoprotein B subunit vaccine efficacy was mediated by non-neutralizing antibody effector functions. bioRxiv doi: 10.1101/246884. [DOI] [PMC free article] [PubMed]
- 19.Baraniak I, Kropff B, McLean GR, Pichon S, Piras-Douce F, Milne RSB, Smith C, Mach M, Griffiths PD, Reeves MB. 2018. Epitope-specific humoral responses to human cytomegalovirus glycoprotein-B vaccine with MF59: anti-AD2 levels correlate with protection from viremia. J Infect Dis 217:1907–1917. doi: 10.1093/infdis/jiy102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bialas KM, Westreich D, Cisneros de la Rosa E, Nelson CS, Kauvar LM, Fu TM, Permar SR. 2016. Maternal antibody responses and nonprimary congenital cytomegalovirus infection of HIV-1-exposed infants. J Infect Dis 214:1916–1923. doi: 10.1093/infdis/jiw487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pardi N, Hogan MJ, Naradikian MS, Parkhouse K, Cain DW, Jones L, Moody MA, Verkerke HP, Myles A, Willis E, LaBranche CC, Montefiori DC, Lobby JL, Saunders KO, Liao HX, Korber BT, Sutherland LL, Scearce RM, Hraber PT, Tombacz I, Muramatsu H, Ni H, Balikov DA, Li C, Mui BL, Tam YK, Krammer F, Kariko K, Polacino P, Eisenlohr LC, Madden TD, Hope MJ, Lewis MG, Lee KK, Hu SL, Hensley SE, Cancro MP, Haynes BF, Weissman D. 2018. Nucleoside-modified mRNA vaccines induce potent T follicular helper and germinal center B cell responses. J Exp Med 215:1571–1588. doi: 10.1084/jem.20171450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Macagno A, Bernasconi NL, Vanzetta F, Dander E, Sarasini A, Revello MG, Gerna G, Sallusto F, Lanzavecchia A. 2010. Isolation of human monoclonal antibodies that potently neutralize human cytomegalovirus infection by targeting different epitopes on the gH/gL/UL128-131A complex. J Virol 84:1005–1013. doi: 10.1128/JVI.01809-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fouts AE, Chan P, Stephan JP, Vandlen R, Feierbach B. 2012. Antibodies against the gH/gL/UL128/UL130/UL131 complex comprise the majority of the anti-cytomegalovirus (anti-CMV) neutralizing antibody response in CMV hyperimmune globulin. J Virol 86:7444–7447. doi: 10.1128/JVI.00467-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nelson CS, Herold BC, Permar SR. 2018. A new era in cytomegalovirus vaccinology: considerations for rational design of next-generation vaccines to prevent congenital cytomegalovirus infection. NPJ Vaccines 3:38. doi: 10.1038/s41541-018-0074-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bialas KM, Tanaka T, Tran D, Varner V, Cisneros De La Rosa E, Chiuppesi F, Wussow F, Kattenhorn L, Macri S, Kunz EL, Estroff JA, Kirchherr J, Yue Y, Fan Q, Lauck M, O'Connor DH, Hall AHS, Xavier A, Diamond DJ, Barry PA, Kaur A, Permar SR. 2015. Maternal CD4+ T cells protect against severe congenital cytomegalovirus disease in a novel nonhuman primate model of placental cytomegalovirus transmission. Proc Natl Acad Sci U S A 112:13645–13650. doi: 10.1073/pnas.1511526112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Itell HL, Kaur A, Deere JD, Barry PA, Permar SR. 2017. Rhesus monkeys for a nonhuman primate model of cytomegalovirus infections. Curr Opin Virol 25:126–133. doi: 10.1016/j.coviro.2017.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bourne N, Schleiss MR, Bravo FJ, Bernstein DI. 2001. Preconception immunization with a cytomegalovirus (CMV) glycoprotein vaccine improves pregnancy outcome in a guinea pig model of congenital CMV infection. J Infect Dis 183:59–64. doi: 10.1086/317654. [DOI] [PubMed] [Google Scholar]
- 28.Chatterjee A, Harrison CJ, Britt WJ, Bewtra C. 2001. Modification of maternal and congenital cytomegalovirus infection by anti-glycoprotein B antibody transfer in guinea pigs. J Infect Dis 183:1547–1553. doi: 10.1086/320714. [DOI] [PubMed] [Google Scholar]
- 29.Auerbach MR, Yan D, Vij R, Hongo JA, Nakamura G, Vernes JM, Meng YG, Lein S, Chan P, Ross J, Carano R, Deng R, Lewin-Koh N, Xu M, Feierbach B. 2014. A neutralizing anti-gH/gL monoclonal antibody is protective in the guinea pig model of congenital CMV infection. PLoS Pathog 10:e1004060. doi: 10.1371/journal.ppat.1004060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nelson CS, Cruz DV, Tran D, Bialas KM, Stamper L, Wu H, Gilbert M, Blair R, Alvarez X, Itell H, Chen M, Deshpande A, Chiuppesi F, Wussow F, Diamond DJ, Vandergrift N, Walter MR, Barry PA, Cohen-Wolkowiez M, Koelle K, Kaur A, Permar SR. 2017. Preexisting antibodies can protect against congenital cytomegalovirus infection in monkeys. JCI Insight 2:94002. doi: 10.1172/jci.insight.94002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nelson CS, Baraniak I, Lilleri D, Reeves MB, Griffiths PD, Permar SR. 2020. Immune correlates of protection against human cytomegalovirus acquisition, replication, and disease. J Infect Dis 221(Suppl 1):S45–S59. doi: 10.1093/infdis/jiz428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boppana SB, Britt WJ. 1995. Antiviral antibody responses and intrauterine transmission after primary maternal cytomegalovirus infection. J Infect Dis 171:1115–1121. doi: 10.1093/infdis/171.5.1115. [DOI] [PubMed] [Google Scholar]
- 33.Bootz A, Karbach A, Spindler J, Kropff B, Reuter N, Sticht H, Winkler TH, Britt WJ, Mach M. 2017. Protective capacity of neutralizing and non-neutralizing antibodies against glycoprotein B of cytomegalovirus. PLoS Pathog 13:e1006601. doi: 10.1371/journal.ppat.1006601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Forthal DN, Phan T, Landucci G. 2001. Antibody inhibition of cytomegalovirus: the role of natural killer and macrophage effector cells. Transpl Infect Dis 3(Suppl 2):31–34. doi: 10.1034/j.1399-3062.2001.00006.x. [DOI] [PubMed] [Google Scholar]
- 35.Chen SF, Holmes TH, Slifer T, Ramachandran V, Mackey S, Hebson C, Arvin AM, Lewis DB, Dekker CL. 2016. Longitudinal kinetics of cytomegalovirus-specific T-cell immunity and viral replication in infants with congenital cytomegalovirus infection. J Pediatr Infect Dis Soc 5:14–20. doi: 10.1093/jpids/piu089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hansen SG, Powers CJ, Richards R, Ventura AB, Ford JC, Siess D, Axthelm MK, Nelson JA, Jarvis MA, Picker LJ, Fruh K. 2010. Evasion of CD8+ T cells is critical for superinfection by cytomegalovirus. Science 328:102–106. doi: 10.1126/science.1185350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gamadia LE, Rentenaar RJ, van Lier RA, ten Berge IJ. 2004. Properties of CD4(+) T cells in human cytomegalovirus infection. Hum Immunol 65:486–492. doi: 10.1016/j.humimm.2004.02.020. [DOI] [PubMed] [Google Scholar]
- 38.Gerna G, Lilleri D, Fornara C, Bruno F, Gabanti E, Cane I, Furione M, Revello MG. 2015. Differential kinetics of human cytomegalovirus load and antibody responses in primary infection of the immunocompetent and immunocompromised host. J Gen Virol 96:360–369. doi: 10.1099/vir.0.070441-0. [DOI] [PubMed] [Google Scholar]
- 39.Widmann T, Sester U, Gartner BC, Schubert J, Pfreundschuh M, Kohler H, Sester M. 2008. Levels of CMV specific CD4 T cells are dynamic and correlate with CMV viremia after allogeneic stem cell transplantation. PLoS One 3:e3634. doi: 10.1371/journal.pone.0003634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lilleri D, Fornara C, Furione M, Zavattoni M, Revello MG, Gerna G. 2007. Development of human cytomegalovirus-specific T cell immunity during primary infection of pregnant women and its correlation with virus transmission to the fetus. J Infect Dis 195:1062–1070. doi: 10.1086/512245. [DOI] [PubMed] [Google Scholar]
- 41.Revello MG, Lilleri D, Zavattoni M, Furione M, Genini E, Comolli G, Gerna G. 2006. Lymphoproliferative response in primary human cytomegalovirus (HCMV) infection is delayed in HCMV transmitter mothers. J Infect Dis 193:269–276. doi: 10.1086/498872. [DOI] [PubMed] [Google Scholar]
- 42.Saldan A, Forner G, Mengoli C, Gussetti N, Palu G, Abate D. 2015. Strong cell-mediated immune response to human cytomegalovirus is associated with increased risk of fetal infection in primarily infected pregnant women. Clin Infect Dis 61:1228–1234. doi: 10.1093/cid/civ561. [DOI] [PubMed] [Google Scholar]
- 43.Lilleri D, Zelini P, Fornara C, Zavaglio F, Rampino T, Perez L, Gabanti E, Gerna G. 2018. Human cytomegalovirus (HCMV)-specific T cell but not neutralizing or IgG binding antibody responses to glycoprotein complexes gB, gHgLgO, and pUL128L correlate with protection against high HCMV viral load reactivation in solid-organ transplant recipients. J Med Virol 90:1620–1628. doi: 10.1002/jmv.25225. [DOI] [PubMed] [Google Scholar]
- 44.Aubert G, Hassan-Walker AF, Madrigal JA, Emery VC, Morte C, Grace S, Koh MB, Potter M, Prentice HG, Dodi IA, Travers PJ. 2001. Cytomegalovirus-specific cellular immune responses and viremia in recipients of allogeneic stem cell transplants. J Infect Dis 184:955–963. doi: 10.1086/323354. [DOI] [PubMed] [Google Scholar]
- 45.Sacre K, Carcelain G, Cassoux N, Fillet AM, Costagliola D, Vittecoq D, Salmon D, Amoura Z, Katlama C, Autran B. 2005. Repertoire, diversity, and differentiation of specific CD8 T cells are associated with immune protection against human cytomegalovirus disease. J Exp Med 201:1999–2010. doi: 10.1084/jem.20042408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gabanti E, Bruno F, Lilleri D, Fornara C, Zelini P, Cane I, Migotto C, Sarchi E, Furione M, Gerna G. 2014. Human cytomegalovirus (HCMV)-specific CD4+ and CD8+ T cells are both required for prevention of HCMV disease in seropositive solid-organ transplant recipients. PLoS One 9:e106044. doi: 10.1371/journal.pone.0106044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Snyder LD, Chan C, Kwon D, Yi JS, Martissa JA, Copeland CA, Osborne RJ, Sparks SD, Palmer SM, Weinhold KJ. 2016. Polyfunctional T-cell signatures to predict protection from cytomegalovirus after lung transplantation. Am J Respir Crit Care Med 193:78–85. doi: 10.1164/rccm.201504-0733OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pardi N, Hogan MJ, Pelc RS, Muramatsu H, Andersen H, DeMaso CR, Dowd KA, Sutherland LL, Scearce RM, Parks R, Wagner W, Granados A, Greenhouse J, Walker M, Willis E, Yu JS, McGee CE, Sempowski GD, Mui BL, Tam YK, Huang YJ, Vanlandingham D, Holmes VM, Balachandran H, Sahu S, Lifton M, Higgs S, Hensley SE, Madden TD, Hope MJ, Kariko K, Santra S, Graham BS, Lewis MG, Pierson TC, Haynes BF, Weissman D. 2017. Zika virus protection by a single low-dose nucleoside-modified mRNA vaccination. Nature 543:248–251. doi: 10.1038/nature21428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pardi N, Hogan MJ, Porter FW, Weissman D. 2018. mRNA vaccines—a new era in vaccinology. Nat Rev Drug Discov 17:261–279. doi: 10.1038/nrd.2017.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pardi N, Parkhouse K, Kirkpatrick E, McMahon M, Zost SJ, Mui BL, Tam YK, Kariko K, Barbosa CJ, Madden TD, Hope MJ, Krammer F, Hensley SE, Weissman D. 2018. Nucleoside-modified mRNA immunization elicits influenza virus hemagglutinin stalk-specific antibodies. Nat Commun 9:3361. doi: 10.1038/s41467-018-05482-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Frey SE, Harrison C, Pass RF, Yang E, Boken D, Sekulovich RE, Percell S, Izu AE, Hirabayashi S, Burke RL, Duliege AM. 1999. Effects of antigen dose and immunization regimens on antibody responses to a cytomegalovirus glycoprotein B subunit vaccine. J Infect Dis 180:1700–1703. doi: 10.1086/315060. [DOI] [PubMed] [Google Scholar]
- 52.John S, Yuzhakov O, Woods A, Deterling J, Hassett K, Shaw CA, Ciaramella G. 2018. Multi-antigenic human cytomegalovirus mRNA vaccines that elicit potent humoral and cell-mediated immunity. Vaccine 36:1689–1699. doi: 10.1016/j.vaccine.2018.01.029. [DOI] [PubMed] [Google Scholar]
- 53.Pardi N, Tuyishime S, Muramatsu H, Kariko K, Mui BL, Tam YK, Madden TD, Hope MJ, Weissman D. 2015. Expression kinetics of nucleoside-modified mRNA delivered in lipid nanoparticles to mice by various routes. J Control Release 217:345–351. doi: 10.1016/j.jconrel.2015.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lambert PH, Laurent PE. 2008. Intradermal vaccine delivery: will new delivery systems transform vaccine administration? Vaccine 26:3197–3208. doi: 10.1016/j.vaccine.2008.03.095. [DOI] [PubMed] [Google Scholar]
- 55.Pardi N, Muramatsu H, Weissman D, Kariko K. 2013. In vitro transcription of long RNA containing modified nucleosides. Methods Mol Biol 969:29–42. doi: 10.1007/978-1-62703-260-5_2. [DOI] [PubMed] [Google Scholar]
- 56.Thess A, Grund S, Mui BL, Hope MJ, Baumhof P, Fotin-Mleczek M, Schlake T. 2015. Sequence-engineered mRNA without chemical nucleoside modifications enables an effective protein therapy in large animals. Mol Ther 23:1456–1464. doi: 10.1038/mt.2015.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Weissman D, Pardi N, Muramatsu H, Kariko K. 2013. HPLC purification of in vitro transcribed long RNA. Methods Mol Biol 969:43–54. doi: 10.1007/978-1-62703-260-5_3. [DOI] [PubMed] [Google Scholar]
- 58.Maier MA, Jayaraman M, Matsuda S, Liu J, Barros S, Querbes W, Tam YK, Ansell SM, Kumar V, Qin J, Zhang X, Wang Q, Panesar S, Hutabarat R, Carioto M, Hettinger J, Kandasamy P, Butler D, Rajeev KG, Pang B, Charisse K, Fitzgerald K, Mui BL, Du X, Cullis P, Madden TD, Hope MJ, Manoharan M, Akinc A. 2013. Biodegradable lipids enabling rapidly eliminated lipid nanoparticles for systemic delivery of RNAi therapeutics. Mol Ther 21:1570–1578. doi: 10.1038/mt.2013.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jayaraman M, Ansell SM, Mui BL, Tam YK, Chen J, Du X, Butler D, Eltepu L, Matsuda S, Narayanannair JK, Rajeev KG, Hafez IM, Akinc A, Maier MA, Tracy MA, Cullis PR, Madden TD, Manoharan M, Hope MJ. 2012. Maximizing the potency of siRNA lipid nanoparticles for hepatic gene silencing in vivo. Angew Chem Int Ed Engl 51:8529–8533. doi: 10.1002/anie.201203263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang D, Li F, Freed DC, Finnefrock AC, Tang A, Grimes SN, Casimiro DR, Fu TM. 2011. Quantitative analysis of neutralizing antibody response to human cytomegalovirus in natural infection. Vaccine 29:9075–9080. doi: 10.1016/j.vaccine.2011.09.056. [DOI] [PubMed] [Google Scholar]
- 61.Wang D, Yu QC, Schroer J, Murphy E, Shenk T. 2007. Human cytomegalovirus uses two distinct pathways to enter retinal pigmented epithelial cells. Proc Natl Acad Sci U S A 104:20037–20042. doi: 10.1073/pnas.0709704104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Reed LJ, Muench H. 1938. A simple method of estimating fifty per cent endpoints. Am J Epidemiol 27:493–497. doi: 10.1093/oxfordjournals.aje.a118408. [DOI] [Google Scholar]
- 63.National Research Council. 2011. Guide for the care and use of laboratory animals, 8th ed National Academies Press, Washington, DC. [Google Scholar]
- 64.Burke HG, Heldwein EE. 2015. Crystal structure of the human cytomegalovirus glycoprotein B. PLoS Pathog 11:e1005227. doi: 10.1371/journal.ppat.1005227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chandramouli S, Ciferri C, Nikitin PA, Caló S, Gerrein R, Balabanis K, Monroe J, Hebner C, Lilja AE, Settembre EC, Carfi A. 2015. Structure of HCMV glycoprotein B in the postfusion conformation bound to a neutralizing human antibody. Nat Commun 6:8176. doi: 10.1038/ncomms9176. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Source data are available for download and may be found in Data Set S1 in the supplemental material.