

Influenza A viruses (IAV) are lytic viruses that have recently been found to activate necroptosis in many of the cell types they infect. Necroptotic cell death is potently immunogenic and limits IAV spread by directly eliminating infected cells and by mobilizing both innate and adaptive immune responses. The benefits of necroptosis to the host, however, may sometimes be outweighed by the potentially deleterious hyperinflammatory consequences of activating this death modality in pulmonary and other tissues.
KEYWORDS: caspase-8, MLKL, RIPK3, ZBP1, apoptosis, influenza, necroptosis
ABSTRACT
Influenza A viruses (IAV) are lytic viruses that have recently been found to activate necroptosis in many of the cell types they infect. Necroptotic cell death is potently immunogenic and limits IAV spread by directly eliminating infected cells and by mobilizing both innate and adaptive immune responses. The benefits of necroptosis to the host, however, may sometimes be outweighed by the potentially deleterious hyperinflammatory consequences of activating this death modality in pulmonary and other tissues.
INTRODUCTION
Viruses, regardless of their genomic organization, reproduction strategy, and cellular targets, are obligate intracellular parasites. Virus families, although remarkably diverse, each depend on cellular proteins, metabolites, and other building blocks to complete the replication process. As the profiles and abundance of these cellular cofactors may vary among susceptible cell types, virus replication is inextricably linked to the biology of the cell and often dictates if the infected cell will live or die. Some viruses are highly lytic and trigger cell destruction in hours, while others establish persistent infections that do not appreciably affect host cell viability. The same virus, however, can be lytic in one cell type but not in another. Virus-induced diseases are equally varied; they may have no overt impact on the infected host, or they can kill the host in days.
For many years, the ability of a virus to trigger cell death was directly associated with its pathogenic potential. For example, variola virus, measles virus, and poliovirus are highly lytic viruses that cause substantial illness in their human hosts. Indeed, poxes and rashes, common manifestations of viral infections, are due to lysis of infected cells. Distinct from this long-standing view that infected cell death was harmful for the host, we now appreciate that the death of an infected cell may be beneficial; viral factories are eliminated, and viral antigens that spill out of the dying cell trigger an early alert for the induction of host immunity. Therefore, the study of if and how cells undergo death upon viral infection is central to our understanding of virus-cell interactions and host immunity and affords unique insights into potential therapeutic interventions.
In this Gem, we describe emerging results from studies of necroptosis during IAV infection to illustrate how a cell death modality is beneficial for the host when well controlled, but also how such cell death can become detrimental if triggered in excess or in the wrong cell type or tissue.
MECHANISM OF IAV-ACTIVATED NECROPTOSIS AND ITS UPSIDE
Programmed necrosis, now called necroptosis, was first reported over 30 years ago, when the cytokine tumor necrosis factor-α (TNF-α), considered until that point an inducer of apoptosis, was found to activate necrotic cell death, not apoptosis, in certain murine fibroblast-derived cell lines (1). Following exposure to TNF-α, dying cells manifested such classic features of necrosis as cytoplasmic “boiling,” a swollen balloon-like appearance, and plasma membrane rupture (1). About 10 years later, several labs carrying out studies on apoptosis signaling made the somewhat paradoxical observation that, in some cell types (such as primary murine embryo fibroblasts), inhibiting apoptotic caspases did not prevent cell death (2–5). Instead, caspase blockade switched the fate of the cell from apoptosis to necrosis. Such necrotic death, now recognized as necroptosis, was seen primarily in response to innate immune stimuli, most notably the virus mimetic double-stranded RNA (dsRNA) and the cytokines gamma interferon (IFN-γ) and tumor necrosis factor alpha (TNF-α), foreshadowing a role for necroptosis in antiviral host defense (2, 4–11). In the last decade, receptor-interacting protein kinase 3 (RIPK3) and its substrate, mixed-lineage kinase-like (MLKL) protein, have emerged as being central to necroptosis, and we now have a fairly clear picture of the key steps by which this form of cell death is executed. Multiple innate immune pathways, including those initiated by retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs), Toll-like receptors (TLRs), IFNs, and death receptor ligands, trigger the activation of RIPK3, which then phosphorylates and activates MLKL. Once activated, MLKL traffics to the plasma membrane, where MLKL multimers insert into the lipid bilayer and form pores. These pores disrupt membrane integrity and cytosolic osmolarity, causing the cell to swell and undergo necrotic death (12–14). It is noteworthy that the phosphorylation of MLKL by RIPK3 is not an automatic death sentence; after MLKL becomes phosphorylated, the prenecroptotic cell becomes permissive to an influx of Ca2+ ions and undergoes additional biochemical changes that result in the exposure of phosphatidylserine to the outer leaflet of the plasma membrane, before membrane integrity is fully compromised (15, 16). If either RIPK3 or MLKL is inactivated at the phosphatidylserine exposure step (before membrane rupture), a substantial proportion of cells can be resuscitated from necroptotic demise. In these cells, endosomal sorting complexes required for transport-III (ESCRT-III)-driven membrane repair mechanisms quarantine and “pinch off” damaged areas of the plasma membrane, thereby preserving plasma membrane integrity (17, 18). Phosphorylated MLKL, moreover, requires association with higher-order inositol phosphates for oligomerization and subsequent execution of necroptosis; if the production of these inositol phosphates is prevented, the cell fails to undergo necroptosis, despite MLKL phosphorylation (19). These, and likely other, regulatory mechanisms ensure that the execution of necroptosis is tightly controlled (12, 20).
It is now becoming apparent that RIPK3-activated necroptosis is essential to host defense against a number of DNA and RNA viruses, including IAV (21, 22). For many years, IAV was thought to activate mainly apoptosis in cultured cells (23, 24). In 2016, however, we identified RIPK3 as a host factor essential for IAV-induced cell death in primary murine cell types, including airway epithelial cells and embryo fibroblasts, and showed that IAV was also a potent activator of necroptosis (25). Most cell lines commonly used in biomedical research, as well as in IAV studies (e.g., HeLa and A549), do not express RIPK3, providing a straightforward explanation for why IAV-activated necroptosis, and indeed the core necroptosis signaling machinery itself, went undiscovered for as long as it did (26–29). That same year, we and others identified Z-form nucleic acid binding protein/DNA activator of interferons (ZBP1/DAI) as the host protein that senses replicating IAV, activates RIPK3, and initiates cell death signaling (30, 31). ZBP1 is a nucleic acid sensor that detects IAV genomic and subgenomic RNAs, following which it interacts with and activates RIPK3. Once activated, RIPK3 triggers two pathways of cell death, only one of which is necroptosis. The second pathway is mediated by caspase-8 and results in apoptosis. Curiously, while IAV-activated necroptosis requires the kinase activity of RIPK3, apoptosis does not; instead, RIPK3 functions as a nonkinase scaffold protein to stimulate caspase-8 activity (Fig. 1). Thus, ZBP1-RIPK3 signaling can activate two very different cell fates depending on whether RIPK3 functions as a kinase (for necroptosis) or as an adaptor (for apoptosis). Preventing necroptosis signaling (for example, by ablating MLKL or blocking RIPK3 activity) does not significantly affect the magnitude of IAV-activated cell death. Instead, the mode of cell death simply switches to exclusively apoptosis. Similarly, blocking apoptosis converts all IAV-induced cell death to necroptosis. Only the combined elimination of apoptosis and necroptosis signaling, such as via ablation of ZBP1 or RIPK3 or by codeletion of MLKL and caspase-8, prevents cell death. The infected cell is then unable to undergo altruistic cell death and becomes a “factory” for virus reproduction (25).
FIG 1.
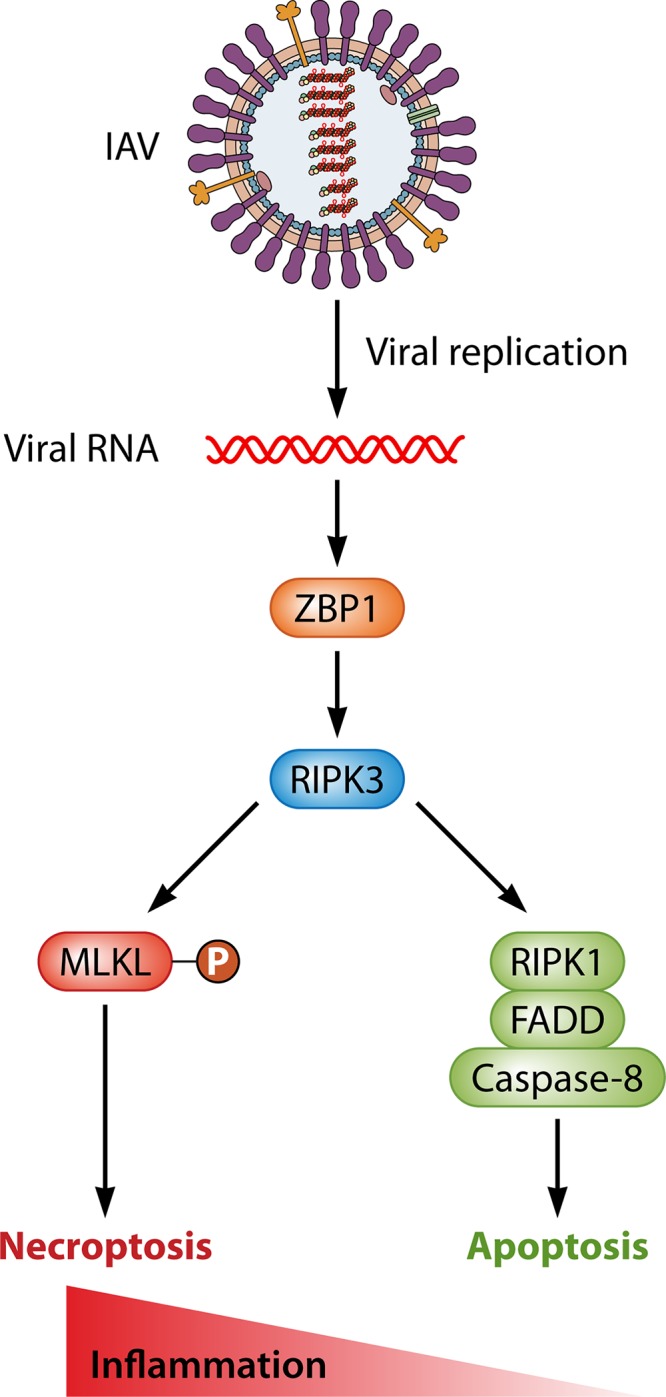
IAV-triggered cell death pathways. The host protein ZBP1 senses genomic and subgenomic RNAs produced by replicating IAV, following which it activates RIPK3. Once activated, RIPK3 can induce two modes of cell death, necroptosis (left) and apoptosis (right). Necroptosis is mediated by MLKL and requires the kinase function of RIPK3, whereas apoptosis is driven by a RIPK1-FADD-caspase-8 axis, and does not require RIPK3 activity. These pathways are active in renewable lung cell types, such as type I airway epithelial cells and fibroblasts, but whether they operate analogously in irreplaceable cell types (such as neurons) is currently unknown. As necroptosis is more inflammatory than is apoptosis, the degree to which RIPK3 activates necroptosis may dictate if ZBP1-activated cell death produces beneficial or pathological outcomes during IAV infections.
The importance of ZBP1-RIPK3 cell death signaling to host defense is underscored by the finding that ZBP1- and RIPK3-deficient mice are highly susceptible to IAV-induced lethality by the intranasal route (25, 31, 32). Whereas wild-type mice limit IAV from spreading throughout the infected lung and can ultimately clear the virus from pulmonary tissue within 10 days of infection, neither Zbp1−/− nor Ripk3−/− mice can curb virus spread. Rather, uncontrolled progeny virus production occurs in the lung tissues of these mice, long after virus has been eradicated from the respiratory tract in wild-type mice. In other words, infected pulmonary cells in Zbp1−/− and Ripk3−/− mice have become virus factories. Ultimately, because cells with compromised death signaling cannot stop virus replication and consequent transmission to surrounding uninfected cells, gas exchange is likely compromised, and respiratory failure ensues. Interestingly, as with cells in culture, mice singly deficient in either necroptosis (Mlkl−/−) or apoptosis (Casp8DA/DA; these mice carry an inactivating point mutation in caspase-8 [33, 34]) show no detectable defects in their capacity to control IAV reproduction, indicating that apoptosis and necroptosis are redundant processes in anti-IAV host defense. Only when both pathways are simultaneously ablated, as in Zbp1−/−, Ripk3−/−, or Mlkl−/−/Casp8DA/DA mice, is antiviral host defense compromised.
Notably, despite higher lung virus burden, Ripk3−/− mice manifest markedly reduced infiltration of T cells into the infected lung. IAV-specific CD8+ cytotoxic T lymphocyte (CTL) numbers were also significantly diminished in the lungs of these mice, and these CTLs were not as capable of mounting an efficient effector response as those from wild-type mice (25). RIPK3-dependent cell death thus serves at least two important functions in anti-IAV host defense, as follows: (i) death of infected cells mitigates unabated virus progeny production and spread, and (ii) the dying cell alerts and mobilizes adaptive immune responses by supplying damage-associated molecular patterns (DAMPs), viral antigens, and other immunological cues. With regard to sounding the immunological alarm, necroptosis is considered more immunogenic than is apoptosis (35, 36). Indeed, necroptosis on its own may actually be a more effective antiviral mechanism than apoptosis, as mice in which necroptosis is the primary IAV-triggered cell death pathway (e.g., Casp8DA/DA) clear virus more efficiently than do even wild-type animals in which both processes are operative. Thus, cell death, and necroptosis in particular, has clear benefits as an anti-IAV host defense mechanism.
THE DOWNSIDE TO NECROPTOSIS DURING IAV INFECTIONS
Programmed cell death pathways can be considered beneficial in anti-IAV immunity only insofar as they facilitate virus clearance without compromising lung function and subsequent recovery. As we have outlined above, altruistic cell death, when well controlled, limits IAV spread within pulmonary tissue. However, when cell death is unchecked, it can drive pathogenesis, and even mortality, despite virus clearance. In fact, the death of more than ∼10% of type I alveolar epithelial cells (which mediate gas exchange during respiration) is highly correlated with mortality in mice infected with IAV (37, 38). Similarly, in humans, bronchoalveolar lesions with areas of cellular necrosis are hallmarks of severe IAV disease, including IAV-induced acute respiratory distress syndrome (ARDS). In these scenarios, unfettered cell death not only directly compromises lung function but also overwhelms mechanisms that mediate the clearance of cellular debris and promote the repair of damaged tissue. Ultimately, such hyperinflammation provokes infiltration of immune cell types, such as monocytes and neutrophils, which can themselves drive pathogenesis. Neutrophils, in particular, have been implicated in orchestrating a feed-forward pathogenic program in severe IAV disease (39–43). Finally, infiltrating CTLs are also potent inducers of cell death, and CTL-mediated destruction of infected cells can come at the price of irreversible lung damage (44). There are thus clear dangers to unleashing unrestricted cell death during acute IAV infections. This is particularly true of necroptosis; by its very nature, necrotic lysis of the cell releases numerous inflammatory mediators, including DNA itself, into the extracellular milieu, serving as beacons for immune cells. Apoptosis, in contrast, results in the orderly disassembly of the dying cell and its dismantlement in discrete membrane-bound “apoptotic bodies” for immunologically silent disposal. It is therefore not surprising that necroptosis also drives disease severity during IAV infection.
The first indication that necroptosis might be pathogenic following IAV infections preceded its discovery as a beneficial virus clearance mechanism. In 2014, Saleh and colleagues reported that mice deficient in the E3 ubiquitin ligase cellular inhibitor of apoptosis protein 2 (cIAP2) manifested aberrantly high RIPK3 activity upon IAV infection, resulting in severe bronchiolar degradation and eventual lethality (45). When RIPK3 was coablated in cIAP2-deficient mice, both tissue pathology and lethality were prevented. Interestingly, neither CTL responses nor virus loads were significantly affected by cIAP2 loss, suggesting that the damage to pulmonary tissue likely arose from an immune cell-mediated “bystander” effect on uninfected lung epithelium. Using histological and immunofluorescence approaches, the authors showed that this effect was very likely necroptosis (45). Although these findings were observed when the threshold for RIPK3 activation was artificially lowered (by cIAP2 ablation), they presaged the possibility that necroptosis can have pathological, even fatal, consequences when hyperactivated during IAV infections. Indeed, a recent study has shown that lower cIAP2 levels (which presumably result in increased RIPK3 activity) are correlated with the severity of ARDS outcomes in humans following H7N9 infections (46). Conversely, RIPK3 deficiency was found to be protective following infection of mice with an H7N9 strain of virulent IAV (47). Thus, RIPK3-activated necroptosis can be protective in mild-to-moderate disease but becomes pathogenic in severe cases of influenza. Analogously, ZBP1 is protective against sublethal (or modestly lethal) doses of intranasally administered virus but exacerbates morbidity when the virus is delivered intratracheally, allowing heightened replication in the distal lung (31, 32). Our own unpublished results echo these findings, where germ line deletion of MLKL limits neutrophil recruitment to the lung and can protect mice from IAV-mediated lethality. Thus, although necroptosis is beneficial for host antiviral immunity, there is a dark side to this mode of death that is revealed in virulent IAV disease.
CELL TYPE-SPECIFIC DIFFERENCES IN IAV-ACTIVATED NECROPTOSIS?
The results described above were obtained from studies of fibroblasts, epithelial cells, and other structural cell types, most of which are readily renewable. Activation of necroptosis in these cell populations is an effective antiviral strategy, with tolerable, and largely reversible, collateral damage to the host under most circumstances. But what about IAV infections of nonrenewable cell types, such as neurons? Although IAV is not typically considered a neurotropic pathogen, the presence of replicating virus in the central nervous system (CNS) is well documented in case studies of cerebrospinal fluid from severely affected patients and in brain tissues upon autopsy (48–54). Moreover, CNS complications can ensue following pulmonary infection, and such sequelae could result from either direct infection of CNS-resident cells, or indirectly due to systemic inflammation and cytokine production (48, 55–57). The prevalence of severe CNS complications is often associated with pandemic strains of IAV, including the H1N1 1918 Spanish flu, the 2009 H1N1 swine flu, and highly pathogenic H5N1 avian influenza viruses with pandemic potential (54, 58–65).
Most adult neurons are irreplaceable; the majority of the neurons of the mammalian brain are generated from neuroepithelial cells during development and do not divide thereafter (66), although some regions, including the neural circuits of the olfactory bulb and dentate gyrus of the hippocampus, may undergo neurogenesis throughout life (67). Therefore, the consequences of activating cell death, particularly a death modality as potently inflammatory as necroptosis, in the CNS following viral neuroinvasion could be lethal to the host. Consequently, it is possible that the necroptosis machinery either is muted in nonrenewable cells or functions in a unique manner. As cases in point, studies by Oberst and colleagues have demonstrated that RIPK3 restricts West Nile virus and Zika virus, both neurotropic flaviviruses, without activating MLKL-dependent necroptosis (68, 69). Instead, RIPK3 protects mice against West Nile virus by inducing antiviral chemokine expression in neurons (68), and ZBP1-RIPK3 signaling limits Zika virus by altering the metabolic state in neurons and making them less permissive for virus reproduction (69).
These findings parallel observations made when otherwise-cytolytic RNA virus infections occur in neurons. For example, we have shown that measles virus (MV) efficiently infects both fibroblasts and neurons but with vastly different outcomes. In fibroblasts, infection is characterized by extensive production of extracellular progeny, syncytium formation, and cell death, whereas MV replication and spread occur in neurons in the absence of infectious progeny release, cell fusion, or appreciable neuronal loss (70–72). Similarly, Sindbis virus is lytic in most cell types, including fibroblasts, but establishes a persistent infection in primary rodent neurons. Remarkably, the expression of a single host protein, Bcl-2, can change the Sindbis virus reproduction cycle in fibroblasts to one resembling that observed in neurons (73). That is, Sindbis virus infection of Bcl-2-expressing fibroblasts resulted in a noncytolytic, persistent infection rather than in cell death. Thus, simply blocking virus-triggered programmed cell death can “convert” a lytic virus to a persistent one, and, as Bcl-2 is expressed in neurons but not in fibroblasts, differences in the expression or function of host cell survival and cell death proteins among different cell types can dramatically affect both virus reproduction and cell fate. Consequently, facts about the replication and pathogenesis of IAV in the brain cannot be reliably inferred from what we know about its replication in the respiratory tract. Given the risks associated with neurotropic infections, IAV’s unique biology in the brain and its interaction with the necroptosis machinery in neurons strongly warrant further study.
In sum, while necroptotic death can eliminate infected cells, activate antiviral immune responses, and hasten virus clearance, these benefits come not only at the cost of potentially pathogenic hyperinflammatory responses but also of potentially irreversible cell loss. If cell death is activated in the wrong cell type, such as by human immunodeficiency virus type 1 (HIV-1) infection of T cells, or by many RNA virus infections of nonrenewable mature neurons, the loss of these cells could have irreversible consequences for the host. It is therefore likely that neurons and other nonrenewable cell types may have evolved nonnecroptotic modes of IAV control, not only to prevent this loss, but also to dampen potentially catastrophic CNS inflammation.
AN EVOLUTIONARY VIEW OF IAV-ACTIVATED NECROPTOSIS
The RIPK3-driven cell death machinery is only found in vertebrates and then too is remarkably poorly conserved between classes of this subphylum (74). For example, ZBP1, RIPK3, and MLKL are all absent in marsupials, and MLKL is not seen in carnivorous placentals (74). Intriguingly, neither ZBP1 nor RIPK3 is found in birds. Species of aquatic birds and shorebirds are the major natural hosts of IAV and represent the primary reservoir of IAV diversity in the wild. In birds, IAV infection is mostly restricted to the gastrointestinal tract and is often asymptomatic or produces only mild symptoms (75). Could the absence of ZBP1-initiated necroptosis signaling in birds allow IAV to replicate in gut epithelial tissue without triggering hyperinflammation and severe disease? In other words, might the risks associated with a robust necroptotic program have outweighed the potential benefits in waterfowl as they coevolved with the great diversity of IAV they currently host? And, by extension, might the necroptosis machinery have presented a selective disadvantage during host-virus coevolution in the other vertebrate classes in which it is absent? A corollary of this line of thinking is that ZBP1-RIPK3 cell death signaling may also represent an effective barrier to prevent cross-species transfer of IAV and other viruses. For example, herpes simplex virus 1 and 2 (HSV-1 and HSV-2, respectively) are both restricted to humans, and recent evidence indicates that RIPK3-driven necroptosis signaling may be a key mediator of their restricted species distribution (76–78). Whereas both HSVs encode proteins that block necroptosis in human cells (in which the viruses can establish persistent infections), the same viral proteins trigger premature necroptosis and prevent productive infection in murine cells. With IAV, when strains jump from birds to humans (which possess intact ZBP1-RIPK3 signaling), they are often unfit for replication or, in rare instances, cause very severe disease. In these scenarios, the activation of ZBP1-initiated necroptosis in humans and other mammals may preclude the establishment of productive infection by prematurely eliminating infected cells. In those instances where severe disease is observed, IAV strains that are suboptimal activators of ZBP1 may replicate to higher levels in pulmonary tissues and drive lung pathology when they leap from one species to another. Future studies comparing ZBP1-dependent necroptotic responses between avian strains of IAV that have yet to successfully transmit to or among humans and those IAV strains (e.g., seasonal H1N1 and H3N2 viruses) that are well adapted to their human hosts will begin to illuminate the role of necroptosis in IAV species restriction. It will also be interesting to determine if similar ZBP1-driven mechanisms limit the transmissibility (and/or affect the pathogenic potential) of other emerging viruses, such as the coronaviruses responsible for multiple outbreaks this millennium. There is much yet to learn, but at least one important principle has emerged: that trade-offs between the virus clearance benefits of necroptosis and the potential risks of activating (or hyperactivating) this form of cell death have likely driven its regulation not only between cell types within species but also across vertebrate species.
ACKNOWLEDGMENTS
We thank Ting Zhang and Patrick Lane (Sceyence Studios) for figure preparation.
Work in the S.B. laboratory is supported by NIH grants AI135025 and AI144400. The G.F.R. laboratory is supported by NIH grants NS060701 and NS099788 and by a gift from the F. M. Kirby Foundation. Both S.B. and G.F.R. are supported by Cancer Center Support grant P30CA006927.
REFERENCES
- 1.Laster SM, Wood JG, Gooding LR. 1988. Tumor necrosis factor can induce both apoptic and necrotic forms of cell lysis. J Immunol 141:2629–2634. [PubMed] [Google Scholar]
- 2.Jäättelä M, Tschopp J. 2003. Caspase-independent cell death in T lymphocytes. Nat Immunol 4:416–423. doi: 10.1038/ni0503-416. [DOI] [PubMed] [Google Scholar]
- 3.Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. 2010. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol 11:700–714. doi: 10.1038/nrm2970. [DOI] [PubMed] [Google Scholar]
- 4.Li M, Beg AA. 2000. Induction of necrotic-like cell death by tumor necrosis factor alpha and caspase inhibitors: novel mechanism for killing virus-infected cells. J Virol 74:7470–7477. doi: 10.1128/jvi.74.16.7470-7477.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vercammen D, Beyaert R, Denecker G, Goossens V, Van Loo G, Declercq W, Grooten J, Fiers W, Vandenabeele P. 1998. Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor. J Exp Med 187:1477–1485. doi: 10.1084/jem.187.9.1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vercammen D, Brouckaert G, Denecker G, Van de Craen M, Declercq W, Fiers W, Vandenabeele P. 1998. Dual signaling of the Fas receptor: initiation of both apoptotic and necrotic cell death pathways. J Exp Med 188:919–930. doi: 10.1084/jem.188.5.919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Khwaja A, Tatton L. 1999. Resistance to the cytotoxic effects of tumor necrosis factor alpha can be overcome by inhibition of a FADD/caspase-dependent signaling pathway. J Biol Chem 274:36817–36823. doi: 10.1074/jbc.274.51.36817. [DOI] [PubMed] [Google Scholar]
- 8.Kalai M, Van Loo G, Vanden Berghe T, Meeus A, Burm W, Saelens X, Vandenabeele P. 2002. Tipping the balance between necrosis and apoptosis in human and murine cells treated with interferon and dsRNA. Cell Death Differ 9:981–994. doi: 10.1038/sj.cdd.4401051. [DOI] [PubMed] [Google Scholar]
- 9.Hirsch T, Marchetti P, Susin SA, Dallaporta B, Zamzami N, Marzo I, Geuskens M, Kroemer G. 1997. The apoptosis-necrosis paradox. Apoptogenic proteases activated after mitochondrial permeability transition determine the mode of cell death. Oncogene 15:1573–1581. doi: 10.1038/sj.onc.1201324. [DOI] [PubMed] [Google Scholar]
- 10.Li M, Shillinglaw W, Henzel WJ, Beg AA. 2001. The Rela(p65) subunit of NF-kappaB is essential for inhibiting double-stranded RNA-induced cytotoxicity. J Biol Chem 276:1185–1194. doi: 10.1074/jbc.M006647200. [DOI] [PubMed] [Google Scholar]
- 11.Hitomi J, Christofferson DE, Ng A, Yao J, Degterev A, Xavier RJ, Yuan J. 2008. Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell 135:1311–1323. doi: 10.1016/j.cell.2008.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moriwaki K, Chan FK. 2013. RIP3: a molecular switch for necrosis and inflammation. Genes Dev 27:1640–1649. doi: 10.1101/gad.223321.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pasparakis M, Vandenabeele P. 2015. Necroptosis and its role in inflammation. Nature 517:311–320. doi: 10.1038/nature14191. [DOI] [PubMed] [Google Scholar]
- 14.Zhang J, Yang Y, He W, Sun L. 2016. Necrosome core machinery: MLKL. Cell Mol Life Sci 73:2153–2163. doi: 10.1007/s00018-016-2190-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gong YN, Crawford JC, Heckmann BL, Green DR. 2019. To the edge of cell death and back. FEBS J 286:430–440. doi: 10.1111/febs.14714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gong YN, Guy C, Crawford JC, Green DR. 2017. Biological events and molecular signaling following MLKL activation during necroptosis. Cell Cycle 16:1748–1760. doi: 10.1080/15384101.2017.1371889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gong YN, Guy C, Olauson H, Becker JU, Yang M, Fitzgerald P, Linkermann A, Green DR. 2017. ESCRT-III acts downstream of MLKL to regulate necroptotic cell death and its consequences. Cell 169:286–300.e216. doi: 10.1016/j.cell.2017.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zargarian S, Shlomovitz I, Erlich Z, Hourizadeh A, Ofir-Birin Y, Croker BA, Regev-Rudzki N, Edry-Botzer L, Gerlic M. 2017. Phosphatidylserine externalization, “necroptotic bodies” release, and phagocytosis during necroptosis. PLoS Biol 15:e2002711. doi: 10.1371/journal.pbio.2002711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dovey CM, Diep J, Clarke BP, Hale AT, McNamara DE, Guo H, Brown NW Jr, Cao JY, Grace CR, Gough PJ, Bertin J, Dixon SJ, Fiedler D, Mocarski ES, Kaiser WJ, Moldoveanu T, York JD, Carette JE. 2018. MLKL requires the inositol phosphate code to execute necroptosis. Mol Cell 70:936–948.e937. doi: 10.1016/j.molcel.2018.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aachoui Y, Kajiwara Y, Leaf IA, Mao D, Ting JP, Coers J, Aderem A, Buxbaum JD, Miao EA. 2015. Canonical inflammasomes drive IFN-gamma to prime caspase-11 in defense against a cytosol-invasive bacterium. Cell Host Microbe 18:320–332. doi: 10.1016/j.chom.2015.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mocarski ES, Kaiser WJ, Livingston-Rosanoff D, Upton JW, Daley-Bauer LP. 2014. True grit: programmed necrosis in antiviral host defense, inflammation, and immunogenicity. J Immunol 192:2019–2026. doi: 10.4049/jimmunol.1302426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Upton JW, Shubina M, Balachandran S. 2017. RIPK3-driven cell death during virus infections. Immunol Rev 277:90–101. doi: 10.1111/imr.12539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hinshaw VS, Olsen CW, Dybdahl-Sissoko N, Evans D. 1994. Apoptosis: a mechanism of cell killing by influenza A and B viruses. J Virol 68:3667–3673. doi: 10.1128/JVI.68.6.3667-3673.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Herold S, Ludwig S, Pleschka S, Wolff T. 2012. Apoptosis signaling in influenza virus propagation, innate host defense, and lung injury. J Leukoc Biol 92:75–82. doi: 10.1189/jlb.1011530. [DOI] [PubMed] [Google Scholar]
- 25.Nogusa S, Thapa RJ, Dillon CP, Liedmann S, Oguin TH III, Ingram JP, Rodriguez DA, Kosoff R, Sharma S, Sturm O, Verbist K, Gough PJ, Bertin J, Hartmann BM, Sealfon SC, Kaiser WJ, Mocarski ES, Lopez CB, Thomas PG, Oberst A, Green DR, Balachandran S. 2016. RIPK3 activates parallel pathways of MLKL-driven necroptosis and FADD-mediated apoptosis to protect against influenza A virus. Cell Host Microbe 20:13–24. doi: 10.1016/j.chom.2016.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.He S, Wang L, Miao L, Wang T, Du F, Zhao L, Wang X. 2009. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 137:1100–1111. doi: 10.1016/j.cell.2009.05.021. [DOI] [PubMed] [Google Scholar]
- 27.Morgan MJ, Kim YS. 2015. The serine threonine kinase RIP3: lost and found. BMB Rep 48:303–312. doi: 10.5483/bmbrep.2015.48.6.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koo GB, Morgan MJ, Lee DG, Kim WJ, Yoon JH, Koo JS, Kim SI, Kim SJ, Son MK, Hong SS, Levy JM, Pollyea DA, Jordan CT, Yan P, Frankhouser D, Nicolet D, Maharry K, Marcucci G, Choi KS, Cho H, Thorburn A, Kim YS. 2015. Methylation-dependent loss of RIP3 expression in cancer represses programmed necrosis in response to chemotherapeutics. Cell Res 25:707–725. doi: 10.1038/cr.2015.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Najafov A, Zervantonakis IK, Mookhtiar AK, Greninger P, March RJ, Egan RK, Luu HS, Stover DG, Matulonis UA, Benes CH, Yuan J. 2018. BRAF and AXL oncogenes drive RIPK3 expression loss in cancer. PLoS Biol 16:e2005756. doi: 10.1371/journal.pbio.2005756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kuriakose T, Man SM, Subbarao Malireddi RK, Karki R, Kesavardhana S, Place DE, Neale G, Vogel P, Kanneganti T-D. 2016. ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Sci Immunol 1:aag2045. doi: 10.1126/sciimmunol.aag2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thapa RJ, Ingram JP, Ragan KB, Nogusa S, Boyd DF, Benitez AA, Sridharan H, Kosoff R, Shubina M, Landsteiner VJ, Andrake M, Vogel P, Sigal LJ, tenOever BR, Thomas PG, Upton JW, Balachandran S. 2016. DAI senses influenza A virus genomic RNA and activates RIPK3-dependent cell death. Cell Host Microbe 20:674–681. doi: 10.1016/j.chom.2016.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Momota M, Lelliott P, Kubo A, Kusakabe T, Kobiyama K, Kuroda E, Imai Y, Akira S, Coban C, Ishii KJ. 20 October 2019. ZBP1 governs the inflammasome-independent IL-1alpha and neutrophil inflammation that play a dual role in anti-influenza virus immunity. Int Immunol. doi: 10.1093/intimm/dxz070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kang TB, Oh GS, Scandella E, Bolinger B, Ludewig B, Kovalenko A, Wallach D. 2008. Mutation of a self-processing site in caspase-8 compromises its apoptotic but not its nonapoptotic functions in bacterial artificial chromosome-transgenic mice. J Immunol 181:2522–2532. doi: 10.4049/jimmunol.181.4.2522. [DOI] [PubMed] [Google Scholar]
- 34.Philip NH, DeLaney A, Peterson LW, Santos-Marrero M, Grier JT, Sun Y, Wynosky-Dolfi MA, Zwack EE, Hu B, Olsen TM, Rongvaux A, Pope SD, Lopez CB, Oberst A, Beiting DP, Henao-Mejia J, Brodsky IE. 2016. Activity of uncleaved caspase-8 controls anti-bacterial immune defense and TLR-induced cytokine production independent of cell death. PLoS Pathog 12:e1005910. doi: 10.1371/journal.ppat.1005910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yatim N, Jusforgues-Saklani H, Orozco S, Schulz O, Barreira da Silva R, Reis e Sousa C, Green DR, Oberst A, Albert ML. 2015. RIPK1 and NF-kappaB signaling in dying cells determines cross-priming of CD8+ T cells. Science 350:328–334. doi: 10.1126/science.aad0395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ren J, Jia X, Zhao Y, Shi W, Lu J, Zhang Y, Wu J, Liang B, Wu R, Fu G, Han J. 2017. The RIP3-RIP1-NF-kappaB signaling axis is dispensable for necroptotic cells to elicit cross-priming of CD8+ T cells. Cell Mol Immunol 14:639–642. doi: 10.1038/cmi.2017.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Davidson S, Crotta S, McCabe TM, Wack A. 2014. Pathogenic potential of interferon alphabeta in acute influenza infection. Nat Commun 5:3864. doi: 10.1038/ncomms4864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sanders CJ, Vogel P, McClaren JL, Bajracharya R, Doherty PC, Thomas PG. 2013. Compromised respiratory function in lethal influenza infection is characterized by the depletion of type I alveolar epithelial cells beyond threshold levels. Am J Physiol Lung Cell Mol Physiol 304:L481–L488. doi: 10.1152/ajplung.00343.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brandes M, Klauschen F, Kuchen S, Germain RN. 2013. A systems analysis identifies a feedforward inflammatory circuit leading to lethal influenza infection. Cell 154:197–212. doi: 10.1016/j.cell.2013.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Camp JV, Jonsson CB. 2017. A role for neutrophils in viral respiratory disease. Front Immunol 8:550. doi: 10.3389/fimmu.2017.00550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Narasaraju T, Yang E, Samy RP, Ng HH, Poh WP, Liew AA, Phoon MC, van Rooijen N, Chow VT. 2011. Excessive neutrophils and neutrophil extracellular traps contribute to acute lung injury of influenza pneumonitis. Am J Pathol 179:199–210. doi: 10.1016/j.ajpath.2011.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pillai PS, Molony RD, Martinod K, Dong H, Pang IK, Tal MC, Solis AG, Bielecki P, Mohanty S, Trentalange M, Homer RJ, Flavell RA, Wagner DD, Montgomery RR, Shaw AC, Staeheli P, Iwasaki A. 2016. Mx1 reveals innate pathways to antiviral resistance and lethal influenza disease. Science 352:463–466. doi: 10.1126/science.aaf3926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bradley LM, Douglass MF, Chatterjee D, Akira S, Baaten BJ. 2012. Matrix metalloprotease 9 mediates neutrophil migration into the airways in response to influenza virus-induced toll-like receptor signaling. PLoS Pathog 8:e1002641. doi: 10.1371/journal.ppat.1002641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hatta Y, Hershberger K, Shinya K, Proll SC, Dubielzig RR, Hatta M, Katze MG, Kawaoka Y, Suresh M. 2010. Viral replication rate regulates clinical outcome and CD8 T cell responses during highly pathogenic H5N1 influenza virus infection in mice. PLoS Pathog 6:e1001139. doi: 10.1371/journal.ppat.1001139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rodrigue-Gervais IG, Labbe K, Dagenais M, Dupaul-Chicoine J, Champagne C, Morizot A, Skeldon A, Brincks EL, Vidal SM, Griffith TS, Saleh M. 2014. Cellular inhibitor of apoptosis protein cIAP2 protects against pulmonary tissue necrosis during influenza virus infection to promote host survival. Cell Host Microbe 15:23–35. doi: 10.1016/j.chom.2013.12.003. [DOI] [PubMed] [Google Scholar]
- 46.Qin C, Sai XY, Qian XF, Wu Y, Zou LF, Wang HM, Bian T, Yan Z. 2019. Close relationship between cIAP2 and human ARDS induced by severe H7N9 infection. Biomed Res Int 2019:2121357. doi: 10.1155/2019/2121357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xu YL, Tang HL, Peng HR, Zhao P, Qi ZT, Wang W. 2017. RIP3 deficiency ameliorates inflammatory response in mice infected with influenza H7N9 virus infection. Oncotarget 8:27715–27724. doi: 10.18632/oncotarget.16016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ito Y, Ichiyama T, Kimura H, Shibata M, Ishiwada N, Kuroki H, Furukawa S, Morishima T. 1999. Detection of influenza virus RNA by reverse transcription-PCR and proinflammatory cytokines in influenza virus-associated encephalopathy. J Med Virol 58:420–425. doi:. [DOI] [PubMed] [Google Scholar]
- 49.Fujimoto S, Kobayashi M, Uemura O, Iwasa M, Ando T, Katoh T, Nakamura C, Maki N, Togari H, Wada Y. 1998. PCR on cerebrospinal fluid to show influenza-associated acute encephalopathy or encephalitis. Lancet 352:873–875. doi: 10.1016/S0140-6736(98)12449-2. [DOI] [PubMed] [Google Scholar]
- 50.Santini M, Kutlesa M, Zarkovic K, Drazenovic V, Barsic B. 2012. Influenza A 2009 H1N1 encephalitis in adults with viral RNA in cerebrospinal fluid. Scand J Infect Dis 44:992–996. doi: 10.3109/00365548.2012.689849. [DOI] [PubMed] [Google Scholar]
- 51.de Jong MD, Bach VC, Phan TQ, Vo MH, Tran TT, Nguyen BH, Beld M, Le TP, Truong HK, Nguyen VVC, Tran TH, Do QH, Farrar J. 2005. Fatal avian influenza A (H5N1) in a child presenting with diarrhea followed by coma. N Engl J Med 352:686–691. doi: 10.1056/NEJMoa044307. [DOI] [PubMed] [Google Scholar]
- 52.Morishima T, Togashi T, Yokota S, Okuno Y, Miyazaki C, Tashiro M, Okabe N, Collaborative Study Group on Influenza-Associated Encephalopathy in Japan. 2002. Encephalitis and encephalopathy associated with an influenza epidemic in Japan. Clin Infect Dis 35:512–517. doi: 10.1086/341407. [DOI] [PubMed] [Google Scholar]
- 53.Steininger C, Popow-Kraupp T, Laferl H, Seiser A, Gödl I, Djamshidian S, Puchhammer-Stöckl E. 2003. Acute encephalopathy associated with influenza A virus infection. Clin Infect Dis 36:567–574. doi: 10.1086/367623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Takahashi M, Yamada T, Nakashita Y, Saikusa H, Deguchi M, Kida H, Tashiro M, Toyoda T. 2000. Influenza virus-induced encephalopathy: clinicopathologic study of an autopsied case. Pediatr Int 42:204–214. doi: 10.1046/j.1442-200x.2000.01203.x. [DOI] [PubMed] [Google Scholar]
- 55.Ichiyama T, NIshikawa M, Yoshitomi T, Hayashi T, Furukawa S. 1998. TNF-alpha, IL-1 beta, and IL-6 in cerebrospinal fluid from children with prolonged febrile seizures. Comparison with acute encephalitis/encephalopathy. Neurology 50:407–411. doi: 10.1212/wnl.50.2.407. [DOI] [PubMed] [Google Scholar]
- 56.Short KR, Veeris R, Leijten LM, van den Brand JM, Jong VL, Stittelaar K, Osterhaus ADME, Andeweg A, van Riel D. 2017. Proinflammatory cytokine responses in extra-respiratory tissues during severe influenza. J Infect Dis 216:829–833. doi: 10.1093/infdis/jix281. [DOI] [PubMed] [Google Scholar]
- 57.Aiba H, Mochizuki M, Kimura M, Hojo H. 2001. Predictive value of serum IL-6 level in influenza virus-associated encephalopathy. Neurology 57:295–299. doi: 10.1212/wnl.57.2.295. [DOI] [PubMed] [Google Scholar]
- 58.van Riel D, Leijten LM, Verdijk RM, GeurtsvanKessel C, van der Vries E, van Rossum AMC, Osterhaus ADME, Kuiken T. 2014. Evidence for influenza virus CNS invasion along the olfactory route in an immunocompromised infant. J Infect Dis 210:419–423. doi: 10.1093/infdis/jiu097. [DOI] [PubMed] [Google Scholar]
- 59.Fernández-Blázquez A, Castanon-Apilanez M, Alvarez-Arguelles ME, Sabater-Cabrera C, Rojo-Alba S, Boga JA, de la Tossa GM, Fernandez BQ, Melon S. 2019. Neuroinvasion of influenza A/H3N2: a fatal case in an immunocompetent adult. J Neurovirol 25:275–279. doi: 10.1007/s13365-018-0690-9. [DOI] [PubMed] [Google Scholar]
- 60.Zhang Z, Zhang J, Huang K, Li K, Yuen K, Guan Y, Chen H, Ng W. 2009. Systemic infection of avian influenza A virus H5N1 subtype in humans. Hum Pathol 40:735–739. doi: 10.1016/j.humpath.2008.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Simon M, Hernu R, Cour M, Casalegno J, Lina B, Argaud L. 2013. Fatal influenza A(H1N1)pdm09 encephalopathy in immunocompetent man. Emerg Infect Dis 19:1005–1007. doi: 10.3201/eid1906.130062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Franková V, Jirasek A, Tumova B. 1977. Type A influenza: postmortem virus isolation from different organs in human lethal cases. Arch Virol 53:265–268. doi: 10.1007/BF01314671. [DOI] [PubMed] [Google Scholar]
- 63.Gu J, Xie Z, Gao Z, Liu J, Korteweg C, Ye J, Lau L, Lu J, Gao Z, Zhang B, McNutt M, Lu M, Anderson V, Gong E, Yu A, Lipkin W. 2007. H5N1 infection of the respiratory tract and beyond: a molecular pathology study. Lancet 370:1137–1145. doi: 10.1016/S0140-6736(07)61515-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Price D, Postlethwaite R, Longson M. 1976. Influenzavirus A2 infection presenting with febrile convulstions and gastrointestinal symptoms in young children. Clin Pediatr (Phila) 15:361–367. doi: 10.1177/000992287601500408. [DOI] [PubMed] [Google Scholar]
- 65.Flewett T, Hoult J. 1958. Influenzal encephalopathy and postinfluenzal encephalitis. Lancet ii:11–15. doi: 10.1016/s0140-6736(58)90003-5. [DOI] [PubMed] [Google Scholar]
- 66.Rakic P. 1985. Limits of neurogenesis in primates. Science 227:1054–1056. doi: 10.1126/science.3975601. [DOI] [PubMed] [Google Scholar]
- 67.Gage FH. 2002. Neurogenesis in the adult brain. J Neurosci 22:612–613. doi: 10.1523/JNEUROSCI.22-03-00612.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Daniels BP, Snyder AG, Olsen TM, Orozco S, Oguin TH III, Tait SWG, Martinez J, Gale M Jr, Loo YM, Oberst A. 2017. RIPK3 restricts viral pathogenesis via cell death-independent neuroinflammation. Cell 169:301–313.e311. doi: 10.1016/j.cell.2017.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Daniels BP, Kofman SB, Smith JR, Norris GT, Snyder AG, Kolb JP, Gao X, Locasale JW, Martinez J, Gale M Jr, Loo YM, Oberst A. 2019. The nucleotide sensor ZBP1 and kinase RIPK3 induce the enzyme IRG1 to promote an antiviral metabolic state in neurons. Immunity 50:64–76.e64. doi: 10.1016/j.immuni.2018.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lawrence DM, Patterson CE, Gales TL, D'Orazio JL, Vaughn MM, Rall GF. 2000. Measles virus spread between neurons requires cell contact but not CD46 expression, syncytium formation, or extracellular virus production. J Virol 74:1908–1918. doi: 10.1128/jvi.74.4.1908-1918.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Makhortova NR, Askovich P, Patterson CE, Gechman LA, Gerard NP, Rall GF. 2007. Neurokinin-1 enables measles virus trans-synaptic spread in neurons. Virology 362:235–244. doi: 10.1016/j.virol.2007.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rall GF, Manchester M, Daniels LR, Callahan EM, Belman AR, Oldstone MB. 1997. A transgenic mouse model for measles virus infection of the brain. Proc Natl Acad Sci U S A 94:4659–4663. doi: 10.1073/pnas.94.9.4659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Levine B, Huang Q, Isaacs JT, Reed JC, Griffin DE, Hardwick JM. 1993. Conversion of lytic to persistent alphavirus infection by the bcl-2 cellular oncogene. Nature 361:739–742. doi: 10.1038/361739a0. [DOI] [PubMed] [Google Scholar]
- 74.Dondelinger Y, Hulpiau P, Saeys Y, Bertrand MJ, Vandenabeele P. 2016. An evolutionary perspective on the necroptotic pathway. Trends Cell Biol 26:721–732. doi: 10.1016/j.tcb.2016.06.004. [DOI] [PubMed] [Google Scholar]
- 75.Yoon SW, Webby RJ, Webster RG. 2014. Evolution and ecology of influenza A viruses. Curr Top Microbiol Immunol 385:359–375. doi: 10.1007/82_2014_396. [DOI] [PubMed] [Google Scholar]
- 76.Guo H, Omoto S, Harris PA, Finger JN, Bertin J, Gough PJ, Kaiser WJ, Mocarski ES. 2015. Herpes simplex virus suppresses necroptosis in human cells. Cell Host Microbe 17:243–251. doi: 10.1016/j.chom.2015.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Huang Z, Wu SQ, Liang Y, Zhou X, Chen W, Li L, Wu J, Zhuang Q, Chen C, Li J, Zhong CQ, Xia W, Zhou R, Zheng C, Han J. 2015. RIP1/RIP3 binding to HSV-1 ICP6 initiates necroptosis to restrict virus propagation in mice. Cell Host Microbe 17:229–242. doi: 10.1016/j.chom.2015.01.002. [DOI] [PubMed] [Google Scholar]
- 78.Wang X, Li Y, Liu S, Yu X, Li L, Shi C, He W, Li J, Xu L, Hu Z, Yu L, Yang Z, Chen Q, Ge L, Zhang Z, Zhou B, Jiang X, Chen S, He S. 2014. Direct activation of RIP3/MLKL-dependent necrosis by herpes simplex virus 1 (HSV-1) protein ICP6 triggers host antiviral defense. Proc Natl Acad Sci U S A 111:15438–15443. doi: 10.1073/pnas.1412767111. [DOI] [PMC free article] [PubMed] [Google Scholar]