

Arenavirus NPs contain a highly conserved DEDDh ExoN motif, through which LASV NP degrades virus-derived, immunostimulatory dsRNA in biochemical assays to eliminate the danger signal and inhibit the innate immune response. Nevertheless, the function of NP ExoN in arenavirus infection remains to be defined. In this study, we discovered that LASV potently restricts dsRNA accumulation during infection and minigenome replication. In contrast, although the NPs of JUNV and MACV also harbor the ExoN motif, dsRNA readily formed during JUNV and MACV infections, accompanied by IFN and PKR responses. Interestingly, LASV NP alone was not sufficient to limit dsRNA accumulation. Instead, both LASV NP and L protein were required to restrict immunostimulatory dsRNA accumulation. Our findings provide novel and important insights into the mechanism for the distinct innate immune response to these highly pathogenic arenaviruses and open new directions for future studies.
KEYWORDS: Junín virus, Lassa virus, Machupo virus, PKR, pathogen-associated molecular patterns, RIG-I, arenavirus, dsRNA, hemorrhagic fever-causing virus, innate immune
ABSTRACT
The arenaviruses Lassa virus (LASV), Junín virus (JUNV), and Machupo virus (MACV) can cause severe and fatal diseases in humans. Although these pathogens are closely related, the host immune responses to these virus infections differ remarkably, with direct implications for viral pathogenesis. LASV infection is immunosuppressive, with a very low-level interferon response. In contrast, JUNV and MACV infections stimulate a robust interferon (IFN) response in a retinoic acid-inducible gene I (RIG-I)-dependent manner and readily activate protein kinase R (PKR), a known host double-stranded RNA (dsRNA) sensor. In response to infection with RNA viruses, host nonself RNA sensors recognize virus-derived dsRNA as danger signals and initiate innate immune responses. Arenavirus nucleoproteins (NPs) contain a highly conserved exoribonuclease (ExoN) motif, through which LASV NP has been shown to degrade virus-derived immunostimulatory dsRNA in biochemical assays. In this study, we for the first time present evidence that LASV restricts dsRNA accumulation during infection. Although JUNV and MACV NPs also have the ExoN motif, dsRNA readily accumulated in infected cells and often colocalized with dsRNA sensors. Moreover, LASV coinfection diminished the accumulation of dsRNA and the IFN response in JUNV-infected cells. The disruption of LASV NP ExoN with a mutation led to dsRNA accumulation and impaired LASV replication in minigenome systems. Importantly, both LASV NP and RNA polymerase L protein were required to diminish the accumulation of dsRNA and the IFN response in JUNV infection. For the first time, we discovered a collaboration between LASV NP ExoN and L protein in limiting dsRNA accumulation. Our new findings provide mechanistic insights into the differential host innate immune responses to highly pathogenic arenavirus infections.
IMPORTANCE Arenavirus NPs contain a highly conserved DEDDh ExoN motif, through which LASV NP degrades virus-derived, immunostimulatory dsRNA in biochemical assays to eliminate the danger signal and inhibit the innate immune response. Nevertheless, the function of NP ExoN in arenavirus infection remains to be defined. In this study, we discovered that LASV potently restricts dsRNA accumulation during infection and minigenome replication. In contrast, although the NPs of JUNV and MACV also harbor the ExoN motif, dsRNA readily formed during JUNV and MACV infections, accompanied by IFN and PKR responses. Interestingly, LASV NP alone was not sufficient to limit dsRNA accumulation. Instead, both LASV NP and L protein were required to restrict immunostimulatory dsRNA accumulation. Our findings provide novel and important insights into the mechanism for the distinct innate immune response to these highly pathogenic arenaviruses and open new directions for future studies.
INTRODUCTION
Arenaviruses are enveloped, single-stranded, negative-sense RNA viruses (1). The family Arenaviridae currently consists of four genera, Mammarenavirus, Reptarenavirus, Hartmanivirus, and Antennavirus (2, 3). All human-pathogenic arenaviruses belong to the Mammarenavirus genus, the members of which are further classified into the Old World (OW) and New World (NW) arenaviruses (2). Except for the trisegmented Antennavirus genus, arenavirus genomes are bisegmented, with one large (L) segment of around 7.2 kb and one small (S) segment of around 3.4 kb. The S segment encodes the viral glycoprotein (GP) precursor and the nucleoprotein (NP), which is the major structural component of the nucleocapsid (1). The L segment encodes the RNA-dependent RNA polymerase L protein and a small, zinc finger protein (Z), which drives the assembly and budding of virus particles. The NP and L protein are minimal viral trans-acting factors responsible for viral RNA synthesis (4).
Mammarenaviruses often persistently infect their rodent reservoirs without causing overt disease and are shed via excreta from infected animals. Some of the arenaviruses may infect humans through aerosol exposure or direct contact and cause severe/fatal diseases for which vaccines and treatments are limited (5–8). Major human pathogens include the OW Lassa fever virus (LASV) and the NW Junín virus (JUNV) and Machupo virus (MACV). LASV is the causative agent of Lassa fever (LF), which is estimated to cause up to 300,000 infections and 5,000 deaths annually in West Africa (9). NW JUNV and MACV cause Argentine hemorrhagic fever (AHF) and Bolivian hemorrhagic fever (BHF), respectively, in South America (6, 10, 11). JUNV, MACV, and LASV are classified as category A priority pathogens in the United States. The World Health Organization has put LF on the Blueprint list of priority diseases for which there is an urgent need for accelerated research and development (https://www.who.int/news-room/events/detail/2018/02/06/default-calendar/2018-annual-review-of-diseases-prioritized-under-the-research-anddevelopment-blueprint).
Despite similar pathogenicities, the innate and adaptive immune responses to these highly pathogenic OW and NW arenaviruses differ remarkably (12, 13). Clinical and animal studies also indicate that distinct innate immune responses to these hemorrhagic fever-causing arenaviruses have profound impacts on pathogenesis and disease outcomes. Severe LASV cases are generally characterized by high-level viremia, a weak interferon (IFN)/cytokine response, a deficiency in the activation of dendritic cells, macrophages, and T cells, and a lack of neutralizing antibody production (14–19). LASV suppression of the immune response, particularly in the early stage of infection, is of great importance for pathogenicity, as early type I IFN and T-cell responses correlate with LASV clearance and host survival (20, 21). On the other hand, severe and fatal AHF cases are marked by extremely high levels of serum alpha interferon (IFN-α) (2,000 to 64,000 IU/ml), probably the highest IFN levels documented for human viral diseases, and elevated levels of proinflammatory cytokines, which correlates with the severity of the disease outcomes (22, 23). High levels of IFN/proinflammatory cytokines also correlate with the symptomatology (fever and myalgia) in AHF patients (22). Through the IFN pathway, JUNV infection impairs platelet formation and platelet release in vitro, which has been linked to the severe thrombocytopenia and hemorrhagic manifestations in AHF patients (24, 25). In lethal animal models of MACV infection, elevated IFN and proinflammatory cytokine responses have been noted (26–28). Interestingly, JUNV exhibits low sensitivity to IFN treatment in human cells (29–31), indicating that JUNV is relatively resistant to IFN-mediated antiviral effects in human cells.
The mechanism underlying the innate immune response to pathogenic arenavirus infection is largely unknown, partly due to the need for biosafety level 4 (BSL4) facilities for infection experiments. RNA virus infections often produce pathogen-associated molecular patterns (PAMPs), such as double-stranded RNA (dsRNA), that can be sensed by host pattern recognition receptors (PRRs), including retinoic acid-inducible gene I (RIG-I) and protein kinase R (PKR). The sensing of PAMPs by PRRs will trigger signaling cascades that ultimately lead to an innate immune response (including IFN/cytokine production) to virus infection. Studies with expression system and biochemical approaches demonstrated that arenaviruses utilize several strategies to counteract innate immune machinery. Comparative studies using recombinant arenavirus NPs revealed stronger type I IFN suppression and NF-κB inhibition by NPs of OW arenaviruses and the pathogenic JUNV and MACV compared to the NP of the nonpathogenic Tacaribe virus (TCRV) (32). Furthermore, arenavirus NPs contain a highly conserved DEDDh exoribonuclease (ExoN) motif (33, 34). Biochemical studies have established that the NPs of LASV, lymphocytic choriomeningitis virus (LCMV), TCRV, Pichinde virus (PICV), and Mopeia virus (MOPV) degrade virus-derived, immunostimulatory dsRNA (33–38). In the LASV NP, the amino acid residues D389, E391, D466, D533, and H528 constitute the DEDDh motif and are critical for ExoN activity. Some residues proximal to the DEDDh motif, including G392, are also essential for the ExoN activity (33). It is conceivable that arenavirus NPs can eliminate virus-derived dsRNA during infection, which otherwise could be sensed by host PRRs as a danger signal of virus infection. However, as suggested by the variation in IFN responses to LASV, JUNV, and MACV infections, it remains to be investigated whether and how effectively these highly pathogenic arenaviruses prevent dsRNA formation in the context of virus infection. We and other groups have found that NW JUNV and MACV infections, but not OW LASV, stimulated a robust IFN response in a RIG-I-dependent manner (29, 31, 39–41). Additionally, JUNV and MACV, but not LASV, readily activated PKR, a classical host dsRNA sensor (42). Moreover, LASV coinfection abrogated MACV-induced IFN and PKR responses (40, 42). We reasoned that infection with highly pathogenic NW arenaviruses (JUNV and MACV) may lead to an accumulation of dsRNA that is sensed by PRRs and eventually stimulate IFN and PKR responses; in contrast, LASV limits dsRNA accumulation to evade the host innate immune response.
In cases of negative-sense RNA virus infection, it is known that dsRNA is not readily detectable with the commonly used J2 mouse monoclonal antibody, probably due to a lower level of dsRNA formation than that in positive-sense RNA virus infections (43). Recently, Son et al. found that the 9D5 monoclonal antibody (MAb), which was developed initially for diagnosis of panenterovirus, had a high affinity for dsRNA and could detect viral dsRNA in negative-sense RNA virus infection, including the prototypic arenavirus LCMV (44). To test our hypothesis, we have established an imaging methodology using the 9D5 MAb for visualization and measurement of dsRNA at the single-cell level during arenavirus infection in BSL2 labs (45, 46). A main discovery was that in vaccine strain (Candid#1) JUNV infection, dsRNA is readily detected and colocalizes with the PRRs PKR and RIG-I (46). We have validated the specificity of the 9D5 MAb to dsRNA by treating samples with RNase III, which degrades dsRNA, and RNase I, which preferentially digests single-stranded RNA (ssRNA) (46). RNase III treatment diminished the dsRNA signals in Candid#1-infected cells, while the viral NP level was at the same level as those in infected cells without RNase treatment. RNase I treatment did not affect the levels of dsRNA signals. Therefore, the specificity of 9D5 MAb to dsRNA has been confirmed. By using this assay, we further studied dsRNA formation during highly pathogenic arenavirus infections in BSL4 labs. Hereby, we present evidence that dsRNA was accumulated in highly pathogenic NW arenavirus infections but not in OW LASV infection. In JUNV- and MACV-infected cells, the dsRNA signals often colocalized with PRRs (RIG-I and PKR), consistent with previous findings that JUNV and MACV infections readily activate IFN and PKR responses (42). In contrast, dsRNA was not detectable in LASV infection. Further investigation with minigenome replication systems revealed that dsRNA was formed during JUNV minigenome replication but not LASV minigenome replication. Moreover, LASV coinfection diminished dsRNA accumulation in JUNV-infected cells. Despite the NP ExoN activity that effectively degrades virus-derived dsRNA in biochemical assays, the expression of LASV NP alone was not sufficient to reduce the dsRNA level in JUNV infection. Instead, both the LASV NP and L protein were required to limit dsRNA accumulation. For the first time, our data demonstrate a cooperation between LASV L and NP to restrict immunostimulatory dsRNA accumulation, and this novel finding opens new directions for future studies.
RESULTS
dsRNA accumulated during pathogenic NW arenavirus infections but not in LASV infection.
We have established a methodology to visualize dsRNA formed in arenavirus-infected cells in a BSL2 setting. Using this assay, we further investigated dsRNA accumulation in pathogenic arenavirus infections in a BSL4 facility. Human A549 lung epithelial cells were infected with JUNV (pathogenic Romero strain), MACV, or LASV at a multiplicity of infection (MOI) of 1.0. All viruses used in our study are recombinant viruses that were only passaged once in cultured cells after rescue using reverse-genetics systems. At 48 hours postinfection (hpi), cells were fixed and stained for dsRNA. dsRNA signals were readily detected in JUNV and MACV but not LASV infection (Fig. 1A). To quantify the dsRNA levels, 50 infected cells were randomly selected and measured for the mean fluorescence intensity (MFI) of the dsRNA signal. The data clearly showed a significant upregulation of dsRNA signals in JUNV- and MACV-infected cells, whereas the dsRNA level in LASV infection was similar to that in the mock infection (Fig. 1B). Assessment of viral RNA replication via reverse transcription-quantitative PCR (RT-qPCR) revealed that the LASV RNA level was higher than those of JUNV and MACV (Fig. 1C). Pearson’s correlation coefficient was used to quantitatively analyze the colocalization of dsRNA and NP signals detected in the immunofluorescence assay (IFA). Ranging from −1 to 1, the Pearson’s correlation coefficient indicates no significant correlation when it is 0 and a negative correlation when it is −1. A coefficient from 0.49 to 0.84 indicates a strong colocalization (47). Fifty infected cells were randomly selected from three separate experiments and measured for the Pearson’s colocalization coefficients (P coefficients) using the FIJI software (NIH). The result indicated a strong colocalization between NP and dsRNA for both JUNV and MACV (P coefficient = 0.71). In cells infected by pathogenic strains of JUNV and MACV, the NP often formed punctate structures. Similar punctate NP structures have been identified as the viral RNA replication sites in Candid#1 JUNV and LCMV infections (48, 49). Interestingly, in LASV infection, LASV NP seemed to locate in unique bubble-like structures (Fig. 1A).
FIG 1.

dsRNA formation during pathogenic NW arenavirus infection. (A) A549 cells were infected with rJUNV (Romero strain), rMACV, or rLASV at an MOI of 1.0. Cells were fixed and stained for dsRNA (green), JUNV, MACV, or anti-LASV NP (red) and nucleus (DAPI, blue) at 48 hpi. The images are representative of 3 separate experiments. (B) The mean fluorescence intensity (MFI) of dsRNA from of 50 infected cells that are randomly selected in different fields (FIJI, NIH). The mean and 95% confidence interval (CI) are presented. (C) Real-time RT-qPCR analysis to quantify virus RNA level (NP region). An equal amount of total RNA was used for each sample. The threshold cycle (CT) values were normalized to the CT values of β-actin. The mean and standard error of the mean (SEM) of the results from three experiments are presented (****, P < 0.0001; n.s., no significant difference, with one-way ANOVA).
Distribution of dsRNA and PRRs in pathogenic arenavirus-infected cells.
NW JUNV and MACV infections induce a type I IFN response in a RIG-I-dependent manner. It is very likely that the dsRNA formed in JUNV- and MACV-infected cells is recognized by RIG-I as PAMPs and triggers an IFN response. Next, we assessed the distribution of RIG-I and dsRNA at the single-cell level in JUNV, MACV, and LASV infections. A549 cells were infected with JUNV, MACV, and LASV for 48 h and stained for dsRNA, RIG-I, and NP (Fig. 2A). Analysis of the JUNV sample using a line plot profile indicated that the peaks of dsRNA signals were very often colocalized or in close proximity to RIG-I signals (Fig. 2A). The line plot profiles of the MACV sample also demonstrated that the dsRNA signal peaks were often colocalized or in close proximity to those of the RIG-I signals. Analysis of 50 infected cells by measuring Pearson’s colocalization coefficient indicated a strong colocalization of dsRNA and RIG-I in JUNV infection (0.83) and MACV infection (0.83) (Fig. 2B). In mock-infected cells, RIG-I was diffusedly distributed across the cytoplasm (Fig. 2A). In JUNV-infected cells, a change in RIG-I distribution could be noted, as the RIG-I sometimes appeared to concentrate in areas where dsRNA and NP were also detected (Fig. 2A, arrow and plot line profiles). In MACV infection, colocalization of dsRNA, RIG-I, and NP was also observed in some regions (Fig. 2A, arrow). The Pearson’s colocalization coefficients of NP and RIG-I were 0.69 for JUNV and 0.70 for MACV (n = 50). Consistent with our previous results (40), the expression of interferon-stimulated gene 15 (ISG15) was substantially upregulated at both the mRNA and protein levels in JUNV and MACV samples (Fig. 2C and D), demonstrating activation of the IFN response by these viruses. In LASV infection, the distribution of RIG-I changed remarkably compared with that in the mock-infected cells and appeared to colocalize with NP in some areas, as indicated in line plot profiles (Fig. 2A). The P coefficient for RIG-I and LASV NP was 0.68 (n = 50). No upregulation of ISG15 at either the mRNA level or protein level was identified in LASV infection (Fig. 2C and D).
FIG 2.

The distribution of dsRNA, RIG-I, and NP in pathogenic arenavirus-infected cells. A549 cells were infected with rJUNV (pathogenic Romero strain), rMACV, or rLASV (MOI, 1.0). (A) At 48 hpi, cells were fixed and stained for dsRNA (green), RIG-I (red), JUNV, MACV, or LASV NPs (magenta), and nucleus (blue). Images are representative of 3 separate experiments. Arrows indicate areas showing dsRNA, RIG-I, and NP triple colocalization. White lines are drawn in the JUNV, MACV, and LASV samples. The fluorescence plot profiles of signal intensities for NP (purple), dsRNA (green), and RIG-I (red) around the white lines are shown. (B) Pearson’s colocalization coefficients of dsRNA and RIG-I in JUNV and MACV infections. (C) RT-qPCR analysis of ISG15 mRNA levels. The mRNA levels were normalized to β-actin mRNA levels and are presented as fold changes to mock samples (mean + standard deviation [SD]) from three experiments. (D) Western blotting of LASV, JUNV, and MACV NPs, ISG15, and β-actin.
PKR is a classical dsRNA sensor which undergoes autophosphorylation upon binding to dsRNA and becomes enzymatically activated (p-PKR) (50). Thus, PKR phosphorylation has been widely used to assess PKR activation. A549 cells infected by JUNV, MACV, and LASV were stained for PKR (Fig. 3A) or p-PKR (Fig. 4A), dsRNA, and NP. In the case of PKR, line plot profile analysis indicated that the peaks of dsRNA signals frequently colocalized with the peaks of PKR signals in JUNV infection, while colocalization of dsRNA and PKR could also be seen in some regions in MACV infection (Fig. 3B). Different from JUNV and MACV infection, line plot profile analysis of the LASV sample suggested a tendency that the peaks of PKR signals were located in areas where the NP level was low or between two peaks of NP signals. Pearson’s colocalization analysis of 50 cells randomly selected from three experiments indicated that LASV NP was moderately colocalized with PKR (0.35). In comparison, the P coefficients of NP and PKR for JUNV and MACV were 0.77 and 0.75, respectively. The p-PKR level was determined by measuring the MFI of p-PKR signals from 50 randomly selected, virus-infected (NP-positive) cells. The data revealed a significant increase in p-PKR levels in JUNV- and MACV-infected cells (Fig. 4B), demonstrating PKR activation in these samples. In JUNV- or MACV-infected cells, p-PKR was distributed diffusedly across the cytoplasm (Fig. 4A) and sometimes colocalized with dsRNA in regions where the NP level was below the detection limit (Fig. 4C). The Pearson’s colocalization coefficients for p-PKR and dsRNA signals were 0.53 for JUNV and 0.52 for MACV. Interestingly, JUNV and MACV NPs only moderately colocalized with p-PKR (0.32 and 0.23, respectively). In LASV infection, a slight upregulation of p-PKR was identified (Fig. 4B). Notably, LASV NP seemed to locate close to p-PKR (Fig. 4A), which was also evident in line plot profile analysis (Fig. 4C). Pearson’s colocalization coefficients indicated that LASV NP colocalized with p-PKR (0.52) more strongly than with PKR (0.35).
FIG 3.
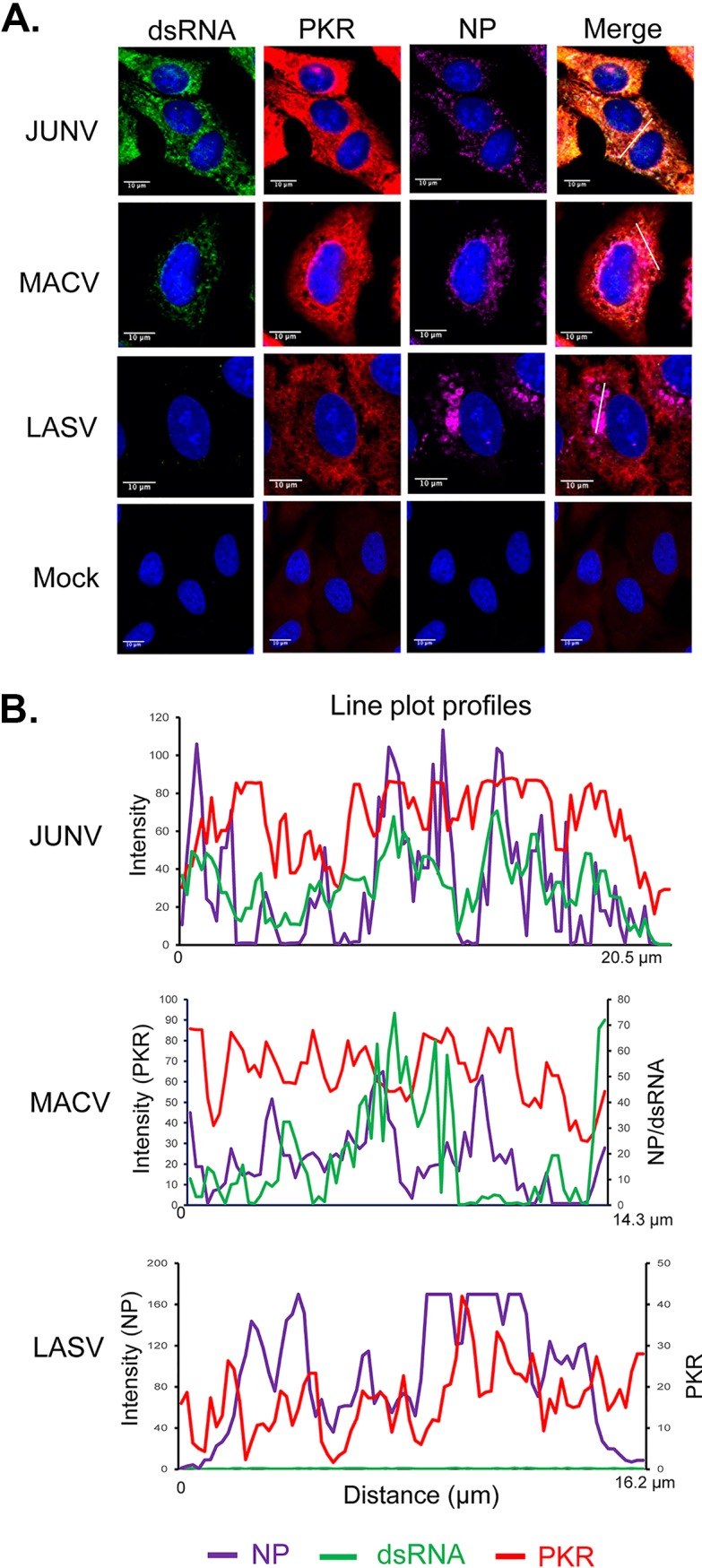
Distribution of dsRNA and PKR during pathogenic arenavirus-infected cells. A549 cells were infected with rJUNV (Romero strain), rMACV, or rLASV at an MOI of 1.0. (A) At 48 hpi, cells were stained with antibodies for (A) PKR (red), dsRNA (green), JUNV NP, MACV NP or LASV NP (magenta), and for nucleus with DAPI (blue). Images are representative from 3 separate experiments. (B) The fluorescence plot profiles of signal intensities for NP (purple), dsRNA (green) and PKR (red) around the white lines drawn in panel A for JUNV, MACV, and LASV samples are shown.
FIG 4.

Distribution of dsRNA and p-PKR during pathogenic arenavirus-infected cells. A549 cells were infected with rJUNV (Romero strain), rMACV, or rLASV at an MOI of 1.0. (A) At 48 hpi, cells were stained with antibodies for p-PKR (red), dsRNA (green), JUNV NP, MACV NP, or LASV NP (magenta), and nucleus with DAPI (blue). The images are representative of 3 separate experiments. White lines are drawn in JUNV, MACV, and LASV samples. (C) The fluorescence plot profiles of signal intensities for NP (purple), dsRNA (green), and p-PKR (red) around the white lines are shown. (B) For each virus, the MFI of p-PKR was calculated from 50 infected cells. The mean and 95% CI are presented (****, P < 0.0001; n.s., no significant difference, with one-way ANOVA).
LASV coinfection diminished dsRNA accumulation and the IFN response in JUNV-infected cells.
LASV coinfection diminishes the MACV-stimulated IFN response and PKR response (42). Based on our new data of dsRNA accumulation in pathogenic NW arenavirus (MACV and JUNV) infections but not in OW LASV infection, we postulated that LASV coinfection might reduce dsRNA accumulation in JUNV- and MACV-infected cells. To test this, A549 cells were infected with JUNV, LASV, or together with JUNV and LASV at an MOI of 3.0 and stained for dsRNA and NP. To assess the virus infectivity in coinfection, the percentage of virus-infected cells was assessed by counting the NP-positive cells among 500 cells from 2 separate experiments. At 24 hpi, approximately 80% of the cells were NP positive in JUNV or LASV single-infection samples. In JUNV and LASV coinfection, 66% of the cells were infected by both viruses, 8% were infected by JUNV only, and 10% were infected by LASV only. At 48 hpi, approximately 91% of the cells were positive for JUNV NP or LASV NP in JUNV or LASV singly infected cells. In coinfection, 95% of the cells were infected by both JUNV and LASV, 3% by JUNV only, and 2% by LASV only. These data confirmed that coinfection did not affect individual virus infection. Next, we examined dsRNA accumulation in coinfection. At 24 hpi, dsRNA accumulated during JUNV infection but not during LASV and JUNV coinfection or during LASV infection alone (Fig. 5A). At 48 hpi, while dsRNA continued to accumulate in JUNV infection, it was still not detectable in cells coinfected with LASV and JUNV or infected with LASV alone (Fig. 5B). The dsRNA level was quantified by measuring the dsRNA MFI from 50 infected or coinfected cells at 24 hpi (Fig. 5C) and 48 hpi (Fig. 5D). LASV coinfection drastically and significantly reduced dsRNA levels in JUNV-infected cells at both time points. At 48 hpi, the dsRNA level in LASV- and JUNV-coinfected cells was not differentiable from those in mock- or LASV-infected cells (Fig. 5D). Along with the diminished dsRNA signals in coinfected cells, the expression levels of the ISG15 protein (Fig. 5E) and ISG15 mRNA (Fig. 5F) also declined to levels comparable with those in LASV-infected cells, indicating that the IFN response was also abrogated by LASV coinfection. The expression levels of JUNV and LASV viral RNA and NP protein in LASV and JUNV coinfection were similar to those in JUNV or LASV infection alone at 24 and 48 hpi (Fig. 5E and G). This result further indicated that JUNV and LASV coinfection did not affect the infection of each virus. Taken together, the data clearly showed that LASV coinfection potently suppresses dsRNA accumulation and the IFN response in JUNV-infected cells without affecting JUNV replication.
FIG 5.

LASV coinfection diminished dsRNA accumulation in JUNV infection. A549 cells were infected with rJUNV (Romero strain), rLASV, or rJUNV and rLASV at an MOI of 3.0. (A and B) At 24 hpi (A) or 48 hpi (B), cells were fixed and stained with anti-dsRNA antibody (green), anti-JUNV NP antibody (red), anti-LASV NP antibody (magenta), and DAPI (blue). The images are representative of 2 separate experiments. (C and D) MFI of dsRNA from 50-infected cells at 24 hpi (C) and 48 hpi (D). The mean and 95% CI are presented. (E) Western blotting of JUNV NP, LASV NP, ISG-15, and β-actin. (F) RT-qPCR analysis of ISG15 mRNA level. The data were normalized to β-actin and are expressed as fold changes of the ISG15 levels in mock samples at 48 h. The mean and SD are presented (n = 3). (G). Real-time RT-qPCR analysis of the JUNV RNA and LASV RNA copy numbers (NP region). An equal amount of total RNA was used. The data were normalized to β-actin and are shown as the mean and SD (****, P < 0.0001; n.s., no significant difference, with one-way ANOVA).
Both LASV NP and L protein were required for restricting dsRNA accumulation in JUNV infection.
To test the role of LASV NP in diminishing dsRNA accumulation in LASV and JUNV coinfection, HEK293T cells were transfected with plasmid expressing LASV NP for 24 h and then infected with the Candid#1 strain of JUNV (MOI of 1.0). As controls, HEK293T cells were also transfected with plasmids expressing LASV L protein, LASV NP and L protein, or both JUNV NP and L protein prior to JUNV infection. As expected, dsRNA was readily detected in mock-transfected cells after JUNV infection (Fig. 6). Surprisingly, LASV NP alone did not affect dsRNA accumulation in JUNV-infected cells, nor did LASV L protein alone (Fig. 6A and B). Only when both LASV NP and LASV L protein were expressed was the dsRNA level reduced to a level similar to that in mock-infected cells (Fig. 6A and B). Plasmid expression of JUNV NP and L protein did not reduce dsRNA accumulation in JUNV infection. Consistent with the dsRNA data, the IFN-β mRNA expression was abrogated only when both LASV NP and LP were expressed (Fig. 6C). RT-qPCR analysis confirmed that LASV NP and L protein coexpression did not affect JUNV RNA synthesis (Fig. 6D).
FIG 6.

Both the LASV NP and L protein are required for restricting dsRNA formation. (A) HEK293T cells were transfected with plasmids to express both LASV NP and LASV L protein (LP), LASV NP, LASV LP, or both JUNV NP and LP as indicated. After 24 h, cells were infected with recombinant Candid (rCandid) JUNV at an MOI of 1.0. At 48 hpi, cells were fixed and stained with anti-dsRNA (green), anti-JUNV NP (red), and anti-LASV NP (magenta) antibodies and with DAPI (blue) for the nucleus. The images are representative of 2 separate experiments. (B) The MFI of dsRNA from 50 infected and transfected cells is shown (mean + 95% CI). (C and D) Real-time RT-qPCR analysis of IFN-β mRNA (C) and JUNV RNA copy numbers (NP locus) (D). Data were normalized to β-actin and are presented as the mean + SD.
dsRNA formed during JUNV but not LASV minigenome replication.
The accumulation of dsRNA in JUNV- and MACV-infected cells, but not in LASV-infected cells, was in line with the stimulation of IFN and PKR responses by JUNV and MACV infection but not LASV infection. To further understand the mechanism, we investigated dsRNA formation using JUNV and LASV minigenome replication systems. The coding regions in the JUNV or LASV S segment genome were replaced by firefly luciferase (FFL) and green fluorescent protein (GFP) reporter genes. The PCR products were inserted between human polymerase I promoter (hPol-I) and terminator sequences. The resulting hPol-I-driven minigenome plasmids (phPol-I-JUNV/FFL/GFP for the JUNV minigenome and phPol-I-LASV/FFL/GFP for the LASV minigenome) were transfected into HEK293T cells along with homologous NP and L protein-expressing plasmids. The pRL-SV40 plasmid expressing renilla luciferase was also included as control for normalization. At 40 hpi, the minigenome reporter FFL activity was measured. When NP and L proteins were expressed, LASV or JUNV minigenome (MG)-driven FFL reporter expression was upregulated by 2-log or 1.4-log, respectively, relative to the control sample that was transfected with corresponding minigenome plasmids only, demonstrating an efficient replication of LASV or JUNV minigenome in these systems (Fig. 7A). dsRNA accumulation was identified in JUNV minigenome replicating cells (Fig. 7B) but not in LASV minigenome replicating cells. The data of dsRNA MFI from 50 transfected cells indicated that the dsRNA level In JUNV minigenome replicating cells was significantly higher than those in control cells (JUNV MG only, without NP and L protein) and LASV minigenome replicating cells (Fig. 7C). Consistent with LASV infection, the dsRNA level in LASV minigenome replicating cells was the same as that in mock-transfected cells (Fig. 7C). No upregulation in dsRNA level could be found in cells transfected with JUNV or LASV minigenome plasmid alone compared with the dsRNA level in mock-transfected samples. Taken together, JUNV and LASV minigenome replication systems recapitulated the phenotypes of dsRNA formation seen in JUNV and LASV infections.
FIG 7.

dsRNA formed during JUNV minigenome replication but not in LASV minigenome replication. (A) HEK-293T cells were transfected with the JUNV or LASV minigenome plasmid along with plasmids expressing homologous NP and L protein (LP). The expression of the minigenome reporter FFL was measured and normalized to the rluc control and is presented as the fold change relative to the expression in cells transfected with minigenome plasmid only. Data shown are the average FFL from three independent experiments and SD. (B) HEK293T cells were infected with rCandid JUNV at an MOI of 1.0 (JUNV), transfected with JUNV minigenome plasmid (JUNV MG) with JUNV LP and NP (JUNV LP + NP) or JUNV MG plasmid alone (−), or transfected with LASV minigenome plasmid (LASV MG) with LASV LP and NP (LASV LP + NP) or MG plasmid alone (−). Forty-eight hours after transfection or infection, cells were fixed and stained with antibodies for dsRNA (green), JUNV NP or LASV NP (red), and DAPI (blue). Images are representative from 3 separate experiments. (C) The dsRNA MFI for each sample was calculated from 50 transfected cells. The mean and 95% CI are presented (****, P < 0.0001; n.s., no significant difference, one-way ANOVA).
Mutations that disrupt LASV NP ExoN also led to dsRNA accumulation in LASV minigenome replication.
D389A and D389A-G392A mutations abolish the exoribonuclease activity of LASV NP (33). By utilizing the LASV minigenome replication system, we further investigated if disruption of NP ExoN has an impact on dsRNA accumulation. HEK293T cells were transfected with plasmids expressing LASV NP ExoN mutants or wild-type (wt) NP, along with plasmids expressing LASV LP and the LASV minigenome. At 40 hpi, cells were lysed and measured for the reporter FFL activity. The LASV NP D389A mutant was substantially less efficient in supporting minigenome replication, while the additional G392A mutation partially restored minigenome replication (Fig. 8A). This result is consistent with a previous report that NP D389A and D389T/G392A ExoN mutants less efficiently support LASV minigenome replication (51). The imaging assay showed that both NP D389A and D389A-G392A mutations resulted in dsRNA accumulation during LASV minigenome replication (Fig. 8B). Quantitative analysis of dsRNA levels by measuring the dsRNA MFI of 50 transfected cells from 3 separate experiments clearly showed that D389A and D389A-G392A mutations caused a significant increase in dsRNA levels in LASV minigenome replication. In comparison, the dsRNA levels in wt LASV NP-expressing cells were comparable to the dsRNA levels in mock-transfected cells. These data demonstrated the critical role of LASV NP ExoN activity in limiting dsRNA accumulation.
FIG 8.

Defects in LASV NP exonuclease lead to dsRNA formation in LASV MG replication. HEK-293T cells were transfected with plasmids for LASV MG (LASV phPol-I/FFL/GFP), LASV L protein (LP), and LASV NP, LASV NP D389A, or LASV NP D389A-G392A as indicated, along with pRL-SV40 expressing the rluc reporter for normalization. (A) At 40 hours posttransfection (hpt), the MG reporter firefly luciferase activity was measured and normalized to the rluc activity. Fold change was calculated based on the FFL value over that in cells transfected with LASV MG plasmid alone. Data shown as the mean and SD of the results from three experiments. (B) At 48 hpi, cells were fixed and stained with anti-dsRNA antibody (green), anti-LASV NP antibody (red), and DAPI (blue). The images are representative of 3 separate experiments. (C) The MFI of dsRNA of 50 transfected cells from 3 separate experiments was calculated. The mean and 95% CI are presented (****, P < 0.0001, one-way ANOVA).
DISCUSSION
Our study, for the first time, presents evidence for dsRNA accumulation in highly pathogenic NW arenavirus (JUNV and MACV) infections but not in LASV infection (Fig. 1A). To ensure the reproducibility of results, we used rescued recombinant viruses with minimal passage history (P1) throughout the studies. This finding is further- confirmed using JUNV and LASV minigenome replication systems (Fig. 7), which indicates that the phenotypic difference in dsRNA formation is unlikely to be caused by differences in virus stocks and more likely is associated with viral replication. The LASV RNA level was always higher than those of JUNV and MACV during infection (Fig. 1). Nevertheless, the dsRNA level is below the detection limit in LASV samples. These data ruled out the possibility that the lack of dsRNA formation in LASV infection was due to a lower level of viral RNA synthesis. Furthermore, the disruption of LASV NP ExoN function affected LASV minigenome replication and also resulted in dsRNA formation (Fig. 8), suggesting a connection between virus replication and dsRNA accumulation. Interestingly, LASV coinfection diminished dsRNA accumulation in JUNV-infected cells (Fig. 5). Future studies are warranted to elucidate the mechanism by which LASV limits dsRNA accumulation. Another interesting finding is that although the ExoN motif is highly conserved among all arenavirus NPs, dsRNA readily accumulates in JUNV and MACV infections, in clear contrast to LASV infection. Future studies need to be performed to better understand the role of NP ExoN in virus infection (discussed below). Overall, our study reveals a fundamental difference in PAMP dsRNA formation during the infections of these highly pathogenic arenaviruses, which provides a mechanistic insight into the phenotypical differences in IFN and PKR responses to JUNV, MACV, and LASV infections.
In RNA virus infection, host PRRs (e.g., RIG-I and PKR) recognize virus-derived dsRNA as PAMPs and initiate a cascade of signaling transduction that eventually leads to innate immune responses. JUNV infection induces the IFN response in a RIG-I-dependent manner (29), clearly indicating successful RIG-I sensing of JUNV infection. Our imaging data support the proposed RIG-I recognition of dsRNA in JUNV- and MACV-infected cells, as concentrated RIG-I signals can be found in proximity to dsRNA signals (Fig. 2). In comparison, RIG-I is distributed diffusedly in the cytoplasm of mock-infected cells. Line plot profile analysis also indicated that the dsRNA signals and RIG-I signals often colocalized in JUNV- and MACV-infected cells. Consistently, and as expected and previously reported, ISG expression was also upregulated during JUNV and MACV infections (Fig. 2), demonstrating that JUNV and MACV stimulated the IFN pathway.
Our data also provide evidence that PKR senses dsRNA molecules and becomes activated in JUNV and MACV infection. Elevated levels of enzymatically activated PKR (p-PKR) were identified in JUNV- and MACV-infected cells (Fig. 4). In contrast, the p-PKR level in LASV-infected cells was not distinguishable from that in mock-infected cells. Line plot profile analysis clearly showed colocalization of p-PKR and dsRNA in JUNV and MACV infections, particularly in regions where viral NPs were below the detection limit (Fig. 4C). Additionally, analysis with Pearson’s colocalization coefficient indicated a colocalization of p-PKR and dsRNA signals during JUNV infection (0.53) and in MACV infection (0.52). Both results supported PKR sensing of dsRNA and becoming activated in JUNV and MACV infections. We have previously identified the phosphorylation of translation eukaryotic initiation factor 2 alpha (eIF2α) and inhibition of global translation during JUNV (pathogenic strain) and MACV infections, indicating that p-PKR is fully functional (42). In vaccine Candid#1 JUNV infection, PKR is also colocalized with dsRNA and phosphorylated (31, 46, 52). However, unlike in pathogenic JUNV infection, there is no upregulation of p-eIF2α nor translation inhibition (31, 52), indicating that p-PKR is not functional in cells infected by the JUNV vaccine Candid#1 strain. The present study provides an explanation for the apparent difference in PKR functionality between JUNV Candid#1 infection and pathogenic JUNV infection. During Candid#1 JUNV infection, it is clear that PKR senses dsRNA and becomes phosphorylated. The p-PKR also strongly colocalized with NP (P coefficient = 0.79) (46). It is conceivable that NP efficiently interacts with p-PKR, suppresses its activity, and consequently prevents eIF2 phosphorylation and host translation inhibition in the case of Candid#1 infection. On the other hand, p-PKR only moderately colocalized with NP in cells infected by the pathogenic strain JUNV (P coefficient = 0.23) and by MACV (P coefficient = 0.32) (Fig. 4), indicating a weaker colocalization of p-PKR and NP during infection with the pathogenic strains of JUNV and MACV. Consistently, line plot profiles showed that dsRNA sometimes colocalized with p-PKR where the NP level was below the detection limit in pathogenic JUNV and MACV infections (Fig. 4C). Thus, a large proportion of p-PKR is not bound to NPs and remains functional in phosphorylating eIF2α and inhibiting global protein translation in cells infected by the pathogenic strains of JUNV and MACV. Of note, PKR activation does not exhibit an antiviral effect on pathogenic strain JUNV and MACV infections (42). Replication of the pathogenic strains of JUNV and MACV is slightly augmented in wild-type cells compared with that in PKR-deficient cells, concomitant with attenuated ISG expression.
We noticed a striking change in the distribution of RIG-I (Fig. 2) and p-PKR (Fig. 4) in LASV infection. LASV NP was found in unique, bubble-like structures. The RIG-I signals in LASV-infected cells were located in proximity to NP, unlike the diffused distribution pattern of RIG-I in mock-infected cells. A strong colocalization of LASV NP with RIG-I was further supported by Pearson’s colocalization coefficient (0.68, n = 50) and by the line plot profile data (Fig. 2A). These results imply that LASV NP might interact with components of the RIG-I signalosome and suppress the RIG-I signaling cascade, which remains to be investigated in future studies. We observed a moderate colocalization of LASV NP with PKR (P coefficient = 0.35, n = 50), which is consistent with a previous report that LASV NP is not coimmunoprecipitated with PKR in expressing cells (52). In line plot profiles of PKR and NP distribution in LASV-infected cells, it is evident that the peaks of PKR signals often located in areas where the NP level was low or between two peaks of NP signals (Fig. 3C). A modest increase in p-PKR was found in LASV infection (Fig. 4B). Interestingly, LASV NP appeared to be more preferentially colocalized with the p-PKR than PKR, as evidenced by the NP and p-PKR localization plot profile data (Fig. 4C) and a higher P coefficient with p-PKR (0.52) than that with PKR (0.35). Taken together, the change in distribution of RIG-I and p-PKR during LASV infection implies that a low level of PAMP dsRNA was formed and sensed by PRRs (RIG-I and PKR). As RIG-I also recognizes other PAMP RNA species, in particular, ssRNA containing 5′-di- and -triphosphates, RIG-I might also detect 5′-triphosphate-containing ssRNA in arenavirus infection as well. This possibility awaits to be addressed in future studies. Nevertheless, LASV effectively shut down the IFN and PKR pathways at downstream steps, as IFN and PKR responses are not measurable in LASV infection. Thus, LASV targets the IFN and PKR pathways by (i) restricting dsRNA accumulation to minimize PRR recognition of PAMP, and (ii) inhibiting downstream IFN and PKR signaling to shut down the IFN and PKR responses.
The mechanism underlying dsRNA accumulation in JUNV and MACV infections remains to be defined in future studies. The DEDDh ExoN motif is highly conserved among all arenavirus NPs. Additionally, the ExoN activity to digest dsRNA has also been established in biochemical assays for NPs from many arenaviruses, including LASV. Intriguingly, dsRNA is readily accumulated during JUNV and MACV infections as well as in JUNV minigenome replication. The ExoN activity of JUNV and MACV NPs has not been determined experimentally. A crystal structure study suggests that JUNV NP lacks ExoN activity (53), which is controversial in the field (38). Further studies are required to determine the ExoN activity for JUNV and MACV NPs. It is possible that there is variation in ExoN activity or its regulation among arenavirus NPs.
It will be of great interest to determine the mechanism of LASV restriction of dsRNA accumulation, which may provide new and important insights into the mechanism for LASV evasion of the host immune response. Several observations are noted in the present study. (i) The LASV NP ExoN activity is necessary for controlling dsRNA formation, as the disruption of NP ExoN activity by introducing D389A and D389A-G392A mutations led to dsRNA accumulation in LASV minigenome replication. Our minigenome data are also in line with a previous report that infection with the LASV NP D389A-G392A mutant stimulates a strong IFN response (51). (ii) The LASV NP alone is not sufficient to limit dsRNA accumulation, nor is the LASV L protein alone; both LASV NP and L are required to abrogate dsRNA formation in JUNV infection. (iii) The LASV minigenome data also shows an inverse correlation between dsRNA formation and viral replication efficiency. Replication of the LASV minigenome was impaired by the D389A and D389A-G392A mutations (Fig. 8), which is the same as what has been found by others in similar LASV minigenome experiments (51). Conversely, dsRNA levels were substantially upregulated at the same time (Fig. 8). (iv) In general, LASV RNA replication is consistently more efficient than are JUNV and MACV during infections (Fig. 1) (42); nevertheless, the dsRNA signal is always below the detection limit in LASV infection. All these data suggest that LASV restricts dsRNA accumulation through a mechanism that involves viral replication.
The fact that the NP DEDDh ExoN motif is highly conserved among arenavirus NPs also suggests that it is critical for the arenavirus life cycle. The LASV NP D389A mutant readily reverts to the wild-type sequence in cultured cells (51). Additionally, some LASV and PICV NP ExoN mutants are either not rescuable or exhibit impaired replication capacity (36, 51). All of these findings support an important role of NP ExoN in virus replication, which warrants future investigation.
In summary, our present study revealed PAMP dsRNA accumulation during highly pathogenic NW arenavirus (JUNV and MACV) infection but not in LASV infection. LASV efficaciously restricted dsRNA accumulation to minimize host PRR sensing of virus infection through a mechanism that required both NP and L protein. These new and important findings provide mechanistic insights into the distinct innate immune response to these viruses and help better elucidate arenavirus and host interaction. Further studies are warranted to determine how LASV NP and L protein collaborate to control dsRNA accumulation to evade the host immune response, the role of NP ExoN in arenavirus infection, as well as the mechanism of dsRNA formation in JUNV and MACV infection.
MATERIALS AND METHODS
Cells and viruses.
A549 (CCL-185; ATCC), HEK-293T cells (CRL-3216; ATCC), and Vero cells (CCL-81; ATCC) were maintained in Dulbecco’s modified eagle medium (HyClone) supplemented with 10% fetal bovine serum (FBS; Atlanta Bio) and 1% penicillin-streptomycin solution (HyClone). The JUNV (vaccine Candid#1 strain and pathogenic Romero strain), LASV (Josiah strain), and MACV (Carvallo strain) used in studies were recombinant viruses that were rescued using reverse-genetic systems and passaged only once in Vero cells, as previously described (54). All plasmids used for virus rescue were sequencing confirmed. All infection work with pathogenic arenaviruses was performed at the BSL4 facilities at the Galveston National Laboratory in the University of Texas Medical Branch in accordance with institutional health and safety guidelines and federal regulations.
Antibodies.
The antibody for dsRNA was the murine MAb panenterovirus clone 9D5 purchased from Millipore Sigma (catalog no. 3361, ready for use, further diluted 1:2 in our lab). The primary antibodies used in the Western blotting assay were rabbit anti-PKR (12297S, 1:1,000; Cell Signaling), rabbit anti-p-PKR (T451, 1:1,000; Abcam), rabbit anti-ISG15 (2734S, 1:1,000; Cell Signaling), mouse ascetic fluid against LASV (1:1,000; Robert Tesh, UTMB), mouse anti-JUNV NP (AG12, 1:1,000, cross-reactive to MACV NP; BEI), and mouse anti-β-actin (sc-47778, 1:1,000; Santa Cruz). The secondary antibodies were horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG (1:3,000; Cell Signaling) and HRP-conjugated goat anti-rabbit IgG (1:5,000; SouthernBiotech).
In IFA, the rabbit MAbs against PKR (ab32506, 1:200; Abcam) and p-PKR (ab81303, 1:100; Abcam) were used. The secondary antibodies used were goat-anti-mouse Alexa Fluor 488 (1:2,000; Invitrogen) and donkey-anti-rabbit Alexa Fluor 594 (1:2,000; Invitrogen). The Alexa Fluor 594-conjugated RIG-I antibody was purchased from Santa Cruz (sc-376845, 1:1,000). The mouse anti-JUNV NP (AG12, 1:1,000; BEI) was conjugated to Alexa Fluor 555 or Alexa Fluor 647, and the mouse anti-LASV NP (01-04-0105, 1:1,000; Cambridge Bio) was conjugated to Alexa Fluor 647, using the corresponding antibody labeling kits (Invitrogen). The cross-reactivities of mouse anti-LASV NP antibody to JUNV NP and of mouse anti-JUNV NP antibody to LASV NP tested negative in our validation assay.
Immunofluorescence assay.
A549 or HEK-293T cells were grown on poly-d-lysine (PDL)-coated coverslips (Neuvitro) and infected with JUNV, MACV, and/or LASV at an MOI of 1.0 or 3.0 or transfected with LASV or JUNV minigenome plasmid phPol-I-S-FFL/GFP, LASV NP, JUNV NP, LASV LP, and/or JUNV NP as indicated in each experiment. Transfection was performed using the calcium phosphate-mediated protocol, according to the manufacturer’s instructions (ProFection mammalian transfection system; Promega). At a specified time point, cells were fixed and stained, as previously described (45, 46). Briefly, cells were fixed with 100% ice-cold methanol for 15 min at −20°C and then washed for 5 min with phosphate-buffered saline (PBS; Corning) at 4°C three times. Cells were then washed with 0.2% Triton X-100 (Sigma) for 5 min and with PBS 4 times. Unconjugated primary antibodies (to dsRNA, PKR, or p-PKR) were diluted in 3% bovine serum albumin (BSA; Sigma) and incubated at 4°C overnight. Cells were then washed 5 times in PBS. Secondary antibodies were diluted in 3% BSA and incubated at room temperature for 1 h. Cells were washed 5 times in PBS. For double or triple staining, conjugated antibodies (to LASV NP or RIG-I) were diluted in 3% BSA and incubated at room temperature (RT) for 2 h or 4°C overnight. Cells were washed five times in PBS and counterstained with 1 μg/ml 4′,6-diamidino-2-phenylindole (DAPI) in PBS. After three washes with PBS containing 0.05% Triton X-100, two washes with PBS for 5 min each time, and one wash with double-distilled water (ddH2O) for 1 min, coverslips were inverted onto ProLong Gold Antifade reagent (Invitrogen) on glass microscope slides (Fisher Scientific) and then left to cure overnight. Imaging was performed using the Olympus FV1000D BX61 upright confocal scanning microscope under an oil immersion lens (60×/1.42 numerical aperture). Laser emission was set at 385 to 405 nm (DAPI), 470 to 530 nm (Alexa 488), 530 to 575 (Alexa 560), 560 to 615 nm (Alexa 594), and 615 to 690 nm (Alexa 647). The same emission was used for each sample, and only linear changes were made to all samples when necessary using the FIJI software (NIH). All statistical analysis was performed with one-way analysis of variance (ANOVA) with Brown’s Forsythe test and Games-Howell correction for multiple comparisons (Prism GraphPad).
Minigenome replication reporter assay.
LASV and JUNV minigenome plasmids were constructed by replacing the viral glycoprotein complex (GPC) and NP open reading frames (ORFs) on the S segment with firefly luciferase (FFL) gene and a green fluorescent protein (GFP) gene, respectively, with recombinant PCR. The amplicons were cloned into a plasmid between the human polymerase (Pol) I promoter and terminator to generate the phPol-I-S-FFL/GPF minigenome plasmids. HEK-293T cells were seeded at 2.5 × 105 in 12-well plates. For each virus, pC-NP, pC-LP, and phPol-I-S-FFL/GFP were transfected into cells at equal copy numbers (total, 2 μg/DNA per well). A pRL-SV40 plasmid (Promega) expressing renilla luciferase (rluc) was cotransfected as a control. Transfection was performed using Lipofectamine 2000 (Invitrogen), as per the manufacturer’s instruction. Forty-eight hours posttransfection, luminescence was measured using the Dual-Glo luciferase kit (Promega) with a GloMax-Multi luminometer (Promega). All experiments were performed three times. The reporter FFL activities were normalized to the rluc activity.
RNA extraction and real-time RT-qPCR.
A549 or HEK-293T cells were seeded into a 12-well plate at a density of 2.0 × 105 cells per well and infected with recombinant JUNV (rJUNV), recombinant MACV (rMACV), and/or recombinant LASV (rLASV), as indicated for each experiment, at an MOI of 1.0 or 3.0. RNA lysates were prepared using the TRIzol reagent (Life Technologies). RNA was purified using the RNeasy minikit (Qiagen) and treated with DNase I (Qiagen). An equal amount of total RNA (1 μg) was reverse transcribed to cDNA using the iScript Advanced cDNA synthesis kit (Bio-Rad). Real-time reverse transcription-quantitative PCR (RT-qPCR) was performed using SsoAdvanced Universal SYBR green supermix (Bio-Rad) on a CFX96 real-time PCR detection system (Bio-Rad), as described before (42). For analysis, the expression level of each target gene was normalized to the β-actin housekeeping gene. Validated primers were purchased from Bio-Rad for β-actin, ISG15, and IFN-β mRNAs. The primers used for the NPs were JUNV NP forward primer (5′-GGCAGTAAGCCGATCACGTA), JUNV NP reverse primer (5′-TCGACATTGAAGGACCAGCC), MACV NP forward primer (5′-GCCCTTCAATGTCAAGCCAC), MACV NP reverse primer (5′-GACCGAGACAACCCGAGAAA), LASV NP forward primer (5′-GAAGGGCCTGGGAAAACACT), and LASV NP reverse primer (5′-AGGTAAGCCCAGCGGTAAAC). To evaluate the copy numbers of viral RNAs in each sample, plasmid DNA templates that contained 102 to 109 copies of corresponding viral NP genes were used as standards.
Western blotting.
A549 cells were seeded into a 12-well plate at a density of 2.0 × 105 cells per well. Twenty-four hours later, cells were infected with rJUNV, rMACV, and/or rLASV at an MOI of 1.0 or 3.0. At the desired time point, protein lysates were prepared with 200 μl of 2× Laemmli buffer (Bio-Rad) containing a protease inhibitor cocktail for mammalian cells (Sigma) and phosphatase inhibitor cocktails (Sigma) and incubated at 95°C for 10 min. The equal loading of protein samples was confirmed by running each sample onto 4% to 20% SDS-PAGE gels, followed with Coomassie G250 staining (Bio-Safe Coomassie; Bio-Rad). Western blot (WB) assays were performed as described before (42).
ACKNOWLEDGMENTS
S.P. was supported by Public Health Service grants RO1AI093445 and RO1AI129198 and the John S. Dunn Distinguished Chair in Biodefense endowment. E.J.M. was supported by NIH/NIAID T32 training grant AI007526. C.H. was supported by UTMB Commitment Fund P84373 and acknowledges the Galveston National Laboratory (supported by the Public Health Service award 5UC7AI094660) for research activity support. J.M. was supported by an Overseas Research Fellowship (20170296) from the Japan Society for the Promotion of Science.
We thank Maxim Ivannikov and the UTMB Imaging Core for microscope assistance and use of facilities.
REFERENCES
- 1.Buchmeier MJ, de la Torre JC, Peters CJ. 2007. Arenaviridae: the viruses and their replication, 5th ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Maes P, Adkins S, Alkhovsky SV, Avšič-Županc T, Ballinger MJ, Bente DA, Beer M, Bergeron É, Blair CD, Briese T, Buchmeier MJ, Burt FJ, Calisher CH, Charrel RN, Choi IR, Clegg JCS, de la Torre JC, de Lamballerie X, DeRisi JL, Digiaro M, Drebot M, Ebihara H, Elbeaino T, Ergünay K, Fulhorst CF, Garrison AR, Gāo GF, Gonzalez J-PJ, Groschup MH, Günther S, Haenni A-L, Hall RA, Hewson R, Hughes HR, Jain RK, Jonson MG, Junglen S, Klempa B, Klingström J, Kormelink R, Lambert AJ, Langevin SA, Lukashevich IS, Marklewitz M, Martelli GP, Mielke-Ehret N, Mirazimi A, Mühlbach H-P, Naidu R, Nunes MRT, et al. . 2019. Taxonomy of the order Bunyavirales: second update 2018. Arch Virol 164:927–941. doi: 10.1007/s00705-018-04127-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shi M, Lin XD, Chen X, Tian JH, Chen LJ, Li K, Wang W, Eden JS, Shen JJ, Liu L, Holmes EC, Zhang YZ. 2018. The evolutionary history of vertebrate RNA viruses. Nature 556:197–202. doi: 10.1038/s41586-018-0012-7. [DOI] [PubMed] [Google Scholar]
- 4.Pinschewer DD, Perez M, de la Torre JC. 2003. Role of the virus nucleoprotein in the regulation of lymphocytic choriomeningitis virus transcription and RNA replication. J Virol 77:3882–3887. doi: 10.1128/jvi.77.6.3882-3887.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Geisbert TW, Jahrling PB. 2004. Exotic emerging viral diseases: progress and challenges. Nat Med 10:S110–S121. doi: 10.1038/nm1142. [DOI] [PubMed] [Google Scholar]
- 6.Grant A, Seregin A, Huang C, Kolokoltsova O, Brasier A, Peters C, Paessler S. 2012. Junin virus pathogenesis and virus replication. Viruses 4:2317–2339. doi: 10.3390/v4102317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Günther S, Lenz O. 2004. Lassa Virus. Crit Rev Clin Lab Sci 41:339–390. doi: 10.1080/10408360490497456. [DOI] [PubMed] [Google Scholar]
- 8.Lukashevich IS. 2012. Advanced vaccine candidates for Lassa fever. Viruses 4:2514–2557. doi: 10.3390/v4112514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McCormick JB, Webb PA, Krebs JW, Johnson KM, Smith ES. 1987. A prospective study of the epidemiology and ecology of Lassa fever. J Infect Dis 155:437–444. doi: 10.1093/infdis/155.3.437. [DOI] [PubMed] [Google Scholar]
- 10.Gómez RM, Jaquenod de Giusti C, Sanchez Vallduvi MM, Frik J, Ferrer MF, Schattner M. 2011. Junín virus. A XXI century update. Microbes Infect 13:303–311. doi: 10.1016/j.micinf.2010.12.006. [DOI] [PubMed] [Google Scholar]
- 11.Patterson M, Grant A, Paessler S. 2014. Epidemiology and pathogenesis of Bolivian hemorrhagic fever. Curr Opin Virol 5:82–90. doi: 10.1016/j.coviro.2014.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mantlo E, Paessler S, Huang C. 2019. Differential immune responses to hemorrhagic fever-causing arenaviruses. Vaccines (Basel) 7:138. doi: 10.3390/vaccines7040138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ly H. 2017. Differential immune responses to New World and Old World mammalian arenaviruses. Int J Mol Sci 18. doi: 10.3390/ijms18051040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mahanty S, Bausch DG, Thomas RL, Goba A, Bah A, Peters CJ, Rollin PE. 2001. Low levels of interleukin-8 and interferon-inducible protein-10 in serum are associated with fatal infections in acute Lassa fever. J Infect Dis 183:1713–1721. doi: 10.1086/320722. [DOI] [PubMed] [Google Scholar]
- 15.Yun NE, Walker DH. 2012. Pathogenesis of Lassa fever. Viruses 4:2031–2048. doi: 10.3390/v4102031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pannetier D, Faure C, Georges-Courbot MC, Deubel V, Baize S. 2004. Human macrophages, but not dendritic cells, are activated and produce alpha/beta interferons in response to Mopeia virus infection. J Virol 78:10516–10524. doi: 10.1128/JVI.78.19.10516-10524.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pannetier D, Reynard S, Russier M, Journeaux A, Tordo N, Deubel V, Baize S. 2011. Human dendritic cells infected with the nonpathogenic Mopeia virus induce stronger T-cell responses than those infected with Lassa virus. J Virol 85:8293–8306. doi: 10.1128/JVI.02120-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Russier M, Pannetier D, Baize S. 2012. Immune responses and Lassa virus infection. Viruses 4:2766–2785. doi: 10.3390/v4112766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baize S, Kaplon J, Faure C, Pannetier D, Georges-Courbot MC, Deubel V. 2004. Lassa virus infection of human dendritic cells and macrophages is productive but fails to activate cells. J Immunol 172:2861–2869. doi: 10.4049/jimmunol.172.5.2861. [DOI] [PubMed] [Google Scholar]
- 20.Baize S, Marianneau P, Loth P, Reynard S, Journeaux A, Chevallier M, Tordo N, Deubel V, Contamin H. 2009. Early and strong immune responses are associated with control of viral replication and recovery in Lassa virus-infected cynomolgus monkeys. J Virol 83:5890–5903. doi: 10.1128/JVI.01948-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McElroy AK, Akondy RS, Harmon JR, Ellebedy AH, Cannon D, Klena JD, Sidney J, Sette A, Mehta AK, Kraft CS, Lyon MG, Varkey JB, Ribner BS, Nichol ST, Spiropoulou CF. 2017. A case of human Lassa virus infection with robust acute T-cell activation and long-term virus-specific T-cell responses. J Infect Dis 215:1862–1872. doi: 10.1093/infdis/jix201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Levis SC, Saavedra MC, Ceccoli C, Falcoff E, Feuillade MR, Enria DA, Maiztegui JI, Falcoff R. 1984. Endogenous interferon in Argentine hemorrhagic fever. J Infect Dis 149:428–433. doi: 10.1093/infdis/149.3.428. [DOI] [PubMed] [Google Scholar]
- 23.Levis SC, Saavedra MC, Ceccoli C, Feuillade MR, Enria DA, Maiztegui JI, Falcoff R. 1985. Correlation between endogenous interferon and the clinical evolution of patients with Argentine hemorrhagic fever. J Interferon Res 5:383–389. doi: 10.1089/jir.1985.5.383. [DOI] [PubMed] [Google Scholar]
- 24.Pozner RG, Ure AE, Jaquenod de Giusti C, D'Atri LP, Italiano JE, Torres O, Romanowski V, Schattner M, Gómez RM. 2010. Junín virus infection of human hematopoietic progenitors impairs in vitro proplatelet formation and platelet release via a bystander effect involving type I IFN signaling. PLoS Pathog 6:e1000847. doi: 10.1371/journal.ppat.1000847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schattner M, Rivadeneyra L, Pozner RG, Gomez RM. 2013. Pathogenic mechanisms involved in the hematological alterations of arenavirus-induced hemorrhagic fevers. Viruses 5:340–351. doi: 10.3390/v5010340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bell TM, Bunton TE, Shaia CI, Raymond JW, Honnold SP, Donnelly GC, Shamblin JD, Wilkinson ER, Cashman KA. 2016. Pathogenesis of Bolivian hemorrhagic fever in guinea pigs. Vet Pathol 53:190–199. doi: 10.1177/0300985815588609. [DOI] [PubMed] [Google Scholar]
- 27.Bell TM, Shaia CI, Bunton TE, Robinson CG, Wilkinson ER, Hensley LE, Cashman KA. 2015. Pathology of experimental Machupo virus infection, Chicava strain, in cynomolgus macaques (Macaca fascicularis) by intramuscular and aerosol exposure. Vet Pathol 52:26–37. doi: 10.1177/0300985814540544. [DOI] [PubMed] [Google Scholar]
- 28.Stephen EL, Scott SK, Eddy GA, Levy HB. 1977. Effect of interferon on togavirus and arenavirus infections of animals. Tex Rep Biol Med 35:449–454. [PubMed] [Google Scholar]
- 29.Huang C, Kolokoltsova OA, Yun NE, Seregin AV, Poussard AL, Walker AG, Brasier AR, Zhao Y, Tian B, de la Torre JC, Paessler S. 2012. Junín virus infection activates the type I interferon pathway in a RIG-I-dependent manner. PLoS Negl Trop Dis 6:e1659. doi: 10.1371/journal.pntd.0001659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang C, Walker AG, Grant AM, Kolokoltsova OA, Yun NE, Seregin AV, Paessler S. 2014. Potent inhibition of Junin virus infection by interferon in murine cells. PLoS Negl Trop Dis 8:e2933. doi: 10.1371/journal.pntd.0002933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moreno H, Moller R, Fedeli C, Gerold G, Kunz S. 2019. Comparison of the innate immune responses to pathogenic and nonpathogenic clade B New World arenaviruses. J Virol 93:e00148-19. doi: 10.1128/JVI.00148-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martínez-Sobrido L, Giannakas P, Cubitt B, García-Sastre A, de la Torre JC. 2007. Differential inhibition of type I interferon induction by arenavirus nucleoproteins. J Virol 81:12696–12703. doi: 10.1128/JVI.00882-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hastie KM, Kimberlin CR, Zandonatti MA, MacRae IJ, Saphire EO. 2011. Structure of the Lassa virus nucleoprotein reveals a dsRNA-specific 3′ to 5′ exonuclease activity essential for immune suppression. Proc Natl Acad Sci U S A 108:2396–2401. doi: 10.1073/pnas.1016404108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Qi X, Lan S, Wang W, Schelde LM, Dong H, Wallat GD, Ly H, Liang Y, Dong C. 2010. Cap binding and immune evasion revealed by Lassa nucleoprotein structure. Nature 468:779–783. doi: 10.1038/nature09605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Carnec X, Mateo M, Page A, Reynard S, Hortion J, Picard C, Yekwa E, Barrot L, Barron S, Vallve A, Raoul H, Carbonnelle C, Ferron F, Baize S. 2018. A vaccine platform against arenaviruses based on a recombinant hyperattenuated Mopeia virus expressing heterologous glycoproteins. J Virol 92:e02230-17. doi: 10.1128/JVI.02230-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang Q, Shao J, Lan S, Zhou Y, Xing J, Dong C, Liang Y, Ly H. 2015. In vitro and in vivo characterizations of Pichinde viral nucleoprotein exoribonuclease functions. J Virol 89:6595–6607. doi: 10.1128/JVI.00009-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jiang X, Huang Q, Wang W, Dong H, Ly H, Liang Y, Dong C. 2013. Structures of arenaviral nucleoproteins with triphosphate dsRNA reveal a unique mechanism of immune suppression. J Biol Chem 288:16949–16959. doi: 10.1074/jbc.M112.420521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.West BR, Hastie KM, Saphire EO. 2014. Structure of the LCMV nucleoprotein provides a template for understanding arenavirus replication and immunosuppression. Acta Crystallogr D Biol Crystallogr 70:1764–1769. doi: 10.1107/S1399004714007883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cuevas CD, Lavanya M, Wang E, Ross SR. 2011. Junin virus infects mouse cells and induces innate immune responses. J Virol 85:11058–11068. doi: 10.1128/JVI.05304-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang C, Kolokoltsova OA, Yun NE, Seregin AV, Ronca S, Koma T, Paessler S. 2015. Highly pathogenic New World and Old World human arenaviruses induce distinct interferon responses in human cells. J Virol 89:7079–7088. doi: 10.1128/JVI.00526-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Negrotto S, Mena HA, Ure AE, Jaquenod De Giusti C, Bollati-Fogolín M, Vermeulen EM, Schattner M, Gómez RM. 2015. Human plasmacytoid dendritic cells elicited different responses after infection with pathogenic and nonpathogenic Junin virus strains. J Virol 89:7409–7413. doi: 10.1128/JVI.01014-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huang C, Kolokoltsova OA, Mateer EJ, Koma T, Paessler S. 2017. Highly pathogenic New World arenavirus infection activates the pattern recognition receptor protein kinase R without attenuating virus replication in human cells. J Virol 91:e01090-17. doi: 10.1128/JVI.01090-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Weber F, Wagner V, Rasmussen SB, Hartmann R, Paludan SR. 2006. Double-stranded RNA is produced by positive-strand RNA viruses and DNA viruses but not in detectable amounts by negative-strand RNA viruses. J Virol 80:5059–5064. doi: 10.1128/JVI.80.10.5059-5064.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Son KN, Liang Z, Lipton HL. 2015. Double-stranded RNA is detected by immunofluorescence analysis in RNA and DNA virus infections, including those by negative-stranded RNA viruses. J Virol 89:9383–9392. doi: 10.1128/JVI.01299-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mateer E, Paessler S, Huang C. 2019. Confocal imaging of double-stranded RNA and pattern recognition receptors in negative-sense RNA virus infection. J Vis Exp doi: 10.3791/59095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mateer EJ, Paessler S, Huang C. 2018. Visualization of double-stranded RNA colocalizing with pattern recognition receptors in arenavirus infected cells. Front Cell Infect Microbiol 8:251. doi: 10.3389/fcimb.2018.00251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zinchuk V, Grossenbacher-Zinchuk O. 2014. Quantitative colocalization analysis of fluorescence microscopy images. Curr Protoc Cell Biol Chapter 4:Unit 4.19. doi: 10.1002/0471143030.cb0419s39. [DOI] [PubMed] [Google Scholar]
- 48.Baird NL, York J, Nunberg JH. 2012. Arenavirus infection induces discrete cytosolic structures for RNA replication. J Virol 86:11301–11310. doi: 10.1128/JVI.01635-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Knopp KA, Ngo T, Gershon PD, Buchmeier MJ. 2015. Single nucleoprotein residue modulates arenavirus replication complex formation. mBio 6:e00524-15. doi: 10.1128/mBio.00524-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.García MA, Meurs EF, Esteban M. 2007. The dsRNA protein kinase PKR: virus and cell control. Biochimie 89:799–811. doi: 10.1016/j.biochi.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 51.Carnec X, Baize S, Reynard S, Diancourt L, Caro V, Tordo N, Bouloy M. 2011. Lassa virus nucleoprotein mutants generated by reverse genetics induce a robust type I interferon response in human dendritic cells and macrophages. J Virol 85:12093–12097. doi: 10.1128/JVI.00429-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.King BR, Hershkowitz D, Eisenhauer PL, Weir ME, Ziegler CM, Russo J, Bruce EA, Ballif BA, Botten J. 2017. A map of the arenavirus nucleoprotein-host protein interactome reveals that Junin virus selectively impairs the antiviral activity of double-stranded RNA-activated protein kinase (PKR). J Virol 91:e00763-17. doi: 10.1128/JVI.00763-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang Y, Li L, Liu X, Dong S, Wang W, Huo T, Guo Y, Rao Z, Yang C. 2013. Crystal structure of Junin virus nucleoprotein. J Gen Virol 94:2175–2183. doi: 10.1099/vir.0.055053-0. [DOI] [PubMed] [Google Scholar]
- 54.Emonet SF, Seregin AV, Yun NE, Poussard AL, Walker AG, de la Torre JC, Paessler S. 2011. Rescue from cloned cDNAs and in vivo characterization of recombinant pathogenic Romero and live-attenuated Candid #1 strains of Junin virus, the causative agent of Argentine hemorrhagic fever disease. J Virol 85:1473–1483. doi: 10.1128/JVI.02102-10. [DOI] [PMC free article] [PubMed] [Google Scholar]