

Pseudorabies virus (PRV) is an alphaherpesvirus related to human pathogens herpes simplex viruses 1 and 2 and varicella-zoster virus. Alphaherpesviruses are neuroinvasive pathogens that establish lifelong latent infections in the host peripheral nervous system (PNS). Following reactivation from latency, infection spreads from the PNS back via axons to the peripheral mucosal tissues, a process mediated by kinesin motors. Here, we unveil and characterize the underlying mechanisms for a PRV-induced, accelerated degradation of KIF1A, a kinesin-3 motor promoting the sorting and transport of PRV virions in axons. We show that PRV infection disrupts the synthesis of KIF1A and simultaneously promotes the degradation of intrinsically unstable KIF1A proteins by proteasomes in axons. Our work implies that the timing of motor reduction after reactivation would be critical because progeny particles would have a limited time window for sorting into and transport in axons for further host-to-host spread.
KEYWORDS: alphaherpesvirus, pseudorabies virus, kinesin, KIF1A, proteasomal degradation, axonal sorting, anterograde spread
ABSTRACT
Alphaherpesviruses, including pseudorabies virus (PRV), are neuroinvasive pathogens that establish lifelong latency in peripheral ganglia following the initial infection at mucosal surfaces. The establishment of latent infection and subsequent reactivations, during which newly assembled virions are sorted into and transported anterogradely inside axons to the initial mucosal site of infection, rely on axonal bidirectional transport mediated by microtubule-based motors. Previous studies using cultured peripheral nervous system (PNS) neurons have demonstrated that KIF1A, a kinesin-3 motor, mediates the efficient axonal sorting and transport of newly assembled PRV virions. Here we report that KIF1A, unlike other axonal kinesins, is an intrinsically unstable protein prone to proteasomal degradation. Interestingly, PRV infection of neuronal cells leads not only to a nonspecific depletion of KIF1A mRNA but also to an accelerated proteasomal degradation of KIF1A proteins, leading to a near depletion of KIF1A protein late in infection. Using a series of PRV mutants deficient in axonal sorting and anterograde spread, we identified the PRV US9/gE/gI protein complex as a viral factor facilitating the proteasomal degradation of KIF1A proteins. Moreover, by using compartmented neuronal cultures that fluidically and physically separate axons from cell bodies, we found that the proteasomal degradation of KIF1A occurs in axons during infection. We propose that the PRV anterograde sorting complex, gE/gI/US9, recruits KIF1A to viral transport vesicles for axonal sorting and transport and eventually accelerates the proteasomal degradation of KIF1A in axons.
IMPORTANCE Pseudorabies virus (PRV) is an alphaherpesvirus related to human pathogens herpes simplex viruses 1 and 2 and varicella-zoster virus. Alphaherpesviruses are neuroinvasive pathogens that establish lifelong latent infections in the host peripheral nervous system (PNS). Following reactivation from latency, infection spreads from the PNS back via axons to the peripheral mucosal tissues, a process mediated by kinesin motors. Here, we unveil and characterize the underlying mechanisms for a PRV-induced, accelerated degradation of KIF1A, a kinesin-3 motor promoting the sorting and transport of PRV virions in axons. We show that PRV infection disrupts the synthesis of KIF1A and simultaneously promotes the degradation of intrinsically unstable KIF1A proteins by proteasomes in axons. Our work implies that the timing of motor reduction after reactivation would be critical because progeny particles would have a limited time window for sorting into and transport in axons for further host-to-host spread.
INTRODUCTION
Alphaherpesviruses, including human pathogens herpes simplex viruses 1 and 2 (HSV-1 and -2) and varicella-zoster virus and the veterinary pathogen pseudorabies virus (PRV), are neuroinvasive pathogens that establish and maintain lifelong latency in the sensory and autonomic ganglia of the peripheral nervous system (PNS) of their mammalian hosts following initial infection at mucosal surfaces (1–3). Latently infected neurons in peripheral ganglia undergo occasional reactivation, during which viral genomes are actively replicated and new viral particles assembled. Spread of infection to new hosts requires that these progeny virions be transported to the initial site of epithelial infection, where replication and subsequent host-to-host transmission occur. On rare occasions, infection spreads from the PNS to the central nervous system, often with serious consequences, such as herpes simplex encephalitis (HSE).
Axonal bidirectional transport mechanisms are crucial for the virions and virus proteins to move efficiently through the PNS axons, which are often centimeters to meters long (4). In uninfected neurons, microtubule-based motors, including minus-end-directed cytoplasmic dynein and plus-end-directed kinesin superfamily motors, mediate fast axonal transport of RNAs, proteins, organelles, and vesicles between cell bodies and axonal termini for the maintenance and proper functioning of axons (5, 6). Neuroinvasive viruses such as the alphaherpesviruses and rabies virus have evolved to adopt existing axonal transport machinery for efficient neuroinvasion and subsequent spread to new hosts. A notable example is rabies virus (RABV), whose phosphoprotein (P) binds to LC8 dynein light chain and was shown to hijack the P75NTR-dependent transport to facilitate fast retrograde axonal transport in sensory dorsal root ganglion (DRG) neurons (7).
Initially after infection of peripheral tissues, alphaherpesvirus virions bind to PNS axon terminals, and viral capsids are released into axons, where they recruit cytoplasmic dynein via inner tegument proteins UL36 and UL37 (8, 9). This capsid-dynein complex then moves in the microtubule (MT) minus-end direction toward the cell body and deposits the viral DNA inside the nucleus. After reactivation of a latent PNS infection, kinesin superfamily motors are recruited both for viral egress at cell bodies and for sorting into axons and subsequent transport of viral structural proteins and newly assembled virions (10–14). Previous studies with sympathetic PNS neurons revealed that a subpopulation of fully enveloped PRV virions in transport vesicles recruit KIF1A, a kinesin-3 motor, through the interactions with a gE/gI/US9 tripartite membrane protein complex in lipid rafts within the late endosomes or the trans-Golgi network (TGN) (14, 15). After this interaction, fully enveloped virions in transport vesicles are sorted into axons, followed by unidirectional anterograde transport before egressing along the axonal shaft (16). In PRV-infected neurons, ectopic expression of dominant negative KIF1A proteins significantly reduced the number of PRV particles transported in the anterograde direction, demonstrating the critical role of KIF1A in sorting virus particles into axons and also in subsequent transport (14). In this report, we focus on a curious observation made by Kramer et al., who noted that the amount of KIF1A in infected cultured neuronal cells decreased significantly late after PRV infection (14).
In uninfected neurons, the kinesin motor KIF1A mediates fast axonal transport of membranous vesicles, including synaptic vesicle precursors and dense core vesicles, and is critical in dendrite morphogenesis and synaptogenesis (17–19). Neurons lacking KIF1A showed impaired transport in synaptic vesicle precursors, marked neuronal degeneration, and death both in cultured neurons and in vivo (20). Despite the functional importance of KIF1A, several lines of evidence suggest that KIF1A motors are degraded upon the completion of their axonal transportation runs and are not recycled for further use. Studies with a ligature applied to the mouse sciatic nerve suggested that more KIF1A was present in the proximal (close to the cell body), but not distal (close to the axonal terminal), region of the ligature, suggesting that once KIF1A motors entered axons, they did not return to the cell bodies (e.g., with dyneins) (21). UNC-104, the KIF1A homolog in Caenorhabditis elegans, was shown to be degraded by ubiquitin-related pathways at synapses of mechanosensory neurons (22). More importantly, the stability of UNC-104 protein strongly correlates with the motor’s specific binding to cargo, implying a potential mechanistic link between motor degradation and cargo release (22).
In this study, we investigated KIF1A protein dynamics during PRV infection both in neuronal cell lines and in primary neurons. Here, we report that KIF1A (kinesin-3), unlike kinesin-1 and kinesin-2 motors, is intrinsically prone to proteasomal degradation in uninfected neuronal cells. We further showed that PRV infection blocks new synthesis of this motor and that viral anterograde-sorting complex US9/gE/gI specifically targets KIF1A for proteasomal degradation, resulting in a near depletion of the protein 24 h following infection. Using compartmented neuronal cultures that fluidically and physically separate axons from cell bodies, we found that the proteasomal degradation of KIF1A proteins occurs in axons. Moreover, using a series of PRV mutants deficient in anterograde spread, and using adenovirus vectors to express the viral proteins in neuronal cells, we discovered that the PRV US9/gE/gI protein complex targets specifically KIF1A motors for proteasomal degradation. Together, these results suggest that progeny PRV particles recruit KIF1A motors for efficient axonal sorting and anterograde transport that lead to the degradation of the motor protein in axons. Since new synthesis of KIF1A is also blocked by PRV infection, these results suggest that progeny particles have a limited time window for sorting into and transport in axons for further host-to-host spread.
RESULTS
In uninfected, differentiated PC12 cells, the steady-state concentration of KIF1A protein is achieved through rapid protein synthesis and proteasomal degradation.
We tested the hypothesis that in differentiated PC12 cells axonal kinesins undergo rapid degradation while protein synthesis continues. To determine the role of the proteasome in this process, we treated uninfected differentiated PC12 cells with the proteasomal inhibitor MG132 for 2, 4, 6, and 8 h and monitored the protein concentration of three different kinesin motors: KIF5 (kinesin-1, the conventional kinesin), KIF3A (kinesin-2), and KIF1A (kinesin-3). MG132 treatment led to a >2-fold increase in KIF1A concentration within 6 h posttreatment, indicating that KIF1A protein rapidly accumulated upon proteasome inhibition (Fig. 1A and B). However, this increase in KIF1A concentration was ablated when PC12 cells were simultaneously treated with MG132 and cycloheximide (CHX), a protein synthesis inhibitor, and KIF1A concentration remained at levels similar to those in untreated cells. These experiments demonstrated that KIF1A proteins are degraded by the proteasomes with new proteins efficiently made to maintain protein steady-state levels. Consistent with this idea, in cells treated with CHX alone, KIF1A concentration dropped by over 50% within 4 h of treatment (Fig. 1A and B).
FIG 1.

KIF1A protein undergoes rapid accumulation and degradation in neuronal cells upon MG132 and CHX treatments. (A) Differentiated PC12 cells were treated with the translation inhibitor cycloheximide, proteasome inhibitor MG132, or both cycloheximide and MG132 for 2, 4, 6, or 8 h. Cells were harvested at the indicated time points and lysates were analyzed by Western blotting to monitor protein levels of KIF1A, KIF5, KIF3A, and actin. (B to D) KIF1A, KIF5, and KIF3A protein levels were measured by band intensities and normalized to actin protein levels for each time point. Normalized values were again normalized to those of mock-infected 0-h samples. Values are means plus SEMs (error bars) from three independent experiments. n.s., not statistically significant. **, P < 0.01; ***, P < 0.001; ****, P < 0.0001 for the indicated comparison at 8 h posttreatment.
In contrast to KIF1A, neither KIF5 nor KIF3A protein concentration increased significantly during MG132 treatment (Fig. 1A, C, and D). With both MG132 and CHX treatments, KIF5 and KIF3A concentrations appeared unchanged in the 8-h treatment period (Fig. 1A, C, and D), suggesting that while both proteins are degraded by proteasomes, their rate of degradation is much lower than that of KIF1A. As a result, the half-lives (t1/2s) of both KIF5 and KIF3A proteins were much longer (>8 h) than the half-life of KIF1A in CHX-treated PC12 cells.
PRV infection induces specific reduction of KIF1A protein late in infection.
The inherent instability of KIF1A in uninfected cells prompted us to explore an observation previously made in our laboratory: the concentration of KIF1A protein, the major kinesin facilitating axonal sorting and transport of PRV virions, decreased late in PRV infection of sympathetic neuronal cells. This observation might explain the significant numbers of immobile PRV particles in axons late in infection (23). Accordingly, we infected differentiated PC12 cells with PRV Becker at a multiplicity of infection (MOI) of 20 for 3, 8, 12, 16, 20, or 24 h and monitored KIF1A protein levels by quantitative Western blot methods. Similar to the case with the previous study, we found that the KIF1A protein concentration dropped by ∼90% at 24 h postinfection (hpi) (Fig. 2). To determine if PRV infection affects the steady-state concentrations of other kinesin motors, we monitored the protein levels of KIF5 and KIF3A, two kinesins well characterized in mediating axonal transport. Neither KIF5 nor KIF3A protein levels were affected by PRV infection (Fig. 2).
FIG 2.

KIF1A protein concentration is reduced during PRV infection in neuronal cells. Differentiated PC12 cells were mock infected or infected with PRV Becker for 3, 8, 12, 16, 20, or 24 h at an MOI of 20. Cells were harvested at the time points shown, and lysates were analyzed by Western blotting to monitor protein levels of KIF1A, KIF5, KIF3A, and actin. Kinesin levels were measured by band intensities and normalized with respect to actin levels at each time point. Normalized values were again normalized to those of mock-infected 0-h samples. Viral protein expressions (PRV early gene EP0 and late genes MCP and US9) were also monitored by Western blotting at different times. Values are means plus SEMs (error bars) from four independent experiments. ***, P < 0.001; ****, P < 0.0001.
PRV infection reduces both KIF1A and KIF5B transcripts.
Alphaherpesviruses promote the instability of many host cell transcripts early in infection, leading to reduced protein synthesis (24–27). We hypothesized that the loss of KIF1A proteins in PRV-infected PC12 cells resulted from such mRNA degradation in combination with the selective short half-life of KIF1A protein. We therefore measured the amounts of KIF1A transcripts in PRV-infected cells by reverse transcription-quantitative PCR (qRT-PCR). Differentiated PC12 cells were infected with PRV Becker and the attenuated vaccine strain PRV Bartha at an MOI of 20 for 3, 8, 12, 16, 20, or 24 h. Infected cells were collected for RNA extraction and qRT-PCR analysis of KIF1A, KIF5B, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA levels (Fig. 3). In PRV Becker-infected cells, we detected a significant and uniform reduction of KIF1A, KIF5B, and GAPDH transcripts as early as 8 hpi, and their respective concentrations dropped by more than 80% at 16 hpi (Fig. 3A).
FIG 3.

The abundances of KIF1A, KIF5B, and GAPDH transcripts are significantly reduced in PRV-infected neuronal cells. Differentiated PC12 cells were infected with PRV Becker (A) or PRV Bartha (B) for 3, 8, 12, 16, 20, or 24 h at an MOI of 20. Cells were harvested at the time points shown. Total RNAs were extracted and qRT-PCR quantification was performed to measure KIF1A, KIF5B, and GAPDH mRNA levels using 18S rRNA as an internal control at each time point. The normalized values were then normalized to those of mock-infected 0-h samples. (C) The data from panels A and B are replotted to show the difference in the rate at which KIF1A and KIF5B transcript levels were reduced in PC12 cells infected with PRV Becker and PRV Bartha. Values are means plus SEMs (error bars) from three independent experiments. *, P < 0.05; **, P < 0.01.
PRV Bartha infection decreased KIF1A transcripts at a lower rate than PRV Becker infection, starting at 12 hpi, with more than 80% loss at 20 hpi (Fig. 3B and C). Reduction of GAPDH and KIF5B transcripts occurred with rates and magnitude comparable to those of the KIF1A transcripts. These experiments showed that KIF1A transcripts were reduced in PRV-infected PC12 cells but with different kinetics after infection by different PRV strains. Such transcriptional regulation may be one reason why KIF1A protein levels drop significantly during PRV infection. However, this effect was not specific for KIF1A, because KIF5B transcripts were also reduced after infection. Therefore, the specific loss of KIF1A and not KIF5B proteins in infected neurons was not due to reduction of transcript levels.
KIF1A is degraded by the proteasome during PRV infection in differentiated PC12 cells and in primary SCG neurons.
We found that KIF1A (but not other kinesins) is rapidly degraded by the proteasome in uninfected PC12 cells (Fig. 1). We also knew from previous studies that complexes of PRV US9 and KIF1A contain the E3-ubiquitin ligases NEDD4 and EP0, suggesting that KIF1A may be degraded via the ubiquitin-proteasome pathway after PRV infection (14). To examine this possibility, we infected differentiated PC12 cells with PRV Becker at an MOI of 20, followed by treatment with the proteasomal inhibitor MG132 at 12 hpi. In mock-infected cells, KIF1A proteins accumulated rapidly, doubling in concentration after MG132 treatment for 8 h (Fig. 4A and C). However, in PRV Becker-infected cells, MG132 treatment led to an effective stabilization of KIF1A protein levels where the protein concentration remained mostly unchanged during the treatment period (Fig. 4B and C). This finding suggests that in the late phase of infection, new KIF1A synthesis is blocked but proteasomal degradation continues, leading to the accelerated loss of KIF1A during PRV infection.
FIG 4.

KIF1A is degraded by the proteasome in the late phase of PRV infection. Differentiated PC12 cells were mock infected (A) or infected with PRV Becker (B) for 8, 12, 16, 20, or 24 h at an MOI of 20. In a parallel experiment, the proteasomal inhibitor MG132 was added at 12 h postinfection at a concentration of 2.5 μM. (C) KIF1A levels were analyzed by Western blotting and normalized to actin levels at each time point. Normalized values were again normalized to those of mock-infected 0-h samples. Values are means plus SEMs (error bars) from three independent experiments. **, P < 0.01 for comparison between PRV-infected samples and PRV-infected samples with MG132 added.
These studies were done with differentiated PC12 cells that have attributes similar to those of sympathetic neurons. We performed similar experiments with primary rat superior cervical ganglionic (SCG) neurons cultured in compartmented chambers where we could examine kinesin motor concentration in isolated axons. We cultured primary sympathetic rat SCG neurons in trichambers, in which axons (in the neurite compartment [N]) were fluidically and physically separated from cell bodies (in the soma compartment [S]) (Fig. 5A). SCG cell bodies in the S compartment were infected with PRV Becker for 24 h and extracts from the S and N compartments were collected for Western blot analysis. The concentration of KIF1A protein decreased dramatically in both cell bodies and axons after PRV infection (Fig. 5B).
FIG 5.
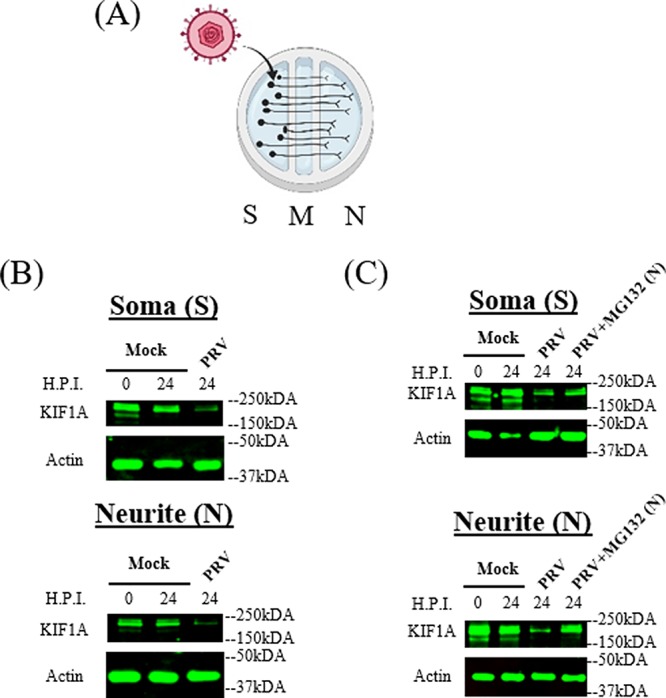
PRV infection of compartmented primary neuronal cultures leads to reduction of KIF1A protein and proteasomal degradation in axons. (A) Illustration of in vitro culture of primary superior cervical ganglion neurons using modified Campenot chambers that include the S (soma), M (methocel), and N (neurite, axon termini) compartments. A total of 106 PFU of PRV Becker were added in the S compartment. (B and C) SCG neurons were infected at the S compartment with PRV Becker for 24 h. (B) Cell bodies and axons from the S and N compartments were collected at the time points shown, and lysates were analyzed by Western blotting to monitor the levels of KIF1A and actin. (C) DMSO or MG132 (2.5 μM) was added to the N compartment at 8 hpi. Cell bodies and axons from the S and N compartments were collected at the time points shown, and lysates were analyzed by Western blotting to monitor the levels of KIF1A and actin.
We further determined if the loss of KIF1A protein is mediated by proteasomes in isolated SCG axons during infection. SCG neurons cultured in trichambers were infected with PRV Becker in the S compartment, followed by MG132 treatment in the N compartment at 8 hpi. KIF1A protein levels were reduced significantly in both S and N compartments after PRV Becker infection (Fig. 5C). Moreover, axonal KIF1A levels were partially restored by MG132 treatment in the N compartment (Fig. 5C). We concluded that PRV infection accelerates the degradation of KIF1A proteins by proteasomes in axons.
Expression of both PRV early and late genes is involved in accelerated KIF1A degradation during infection.
We next searched for viral proteins that might accelerate KIF1A protein degradation. First, we determined if early events such as virion entry and tegument protein delivery would be enough to promote KIF1A degradation. We infected PC12 cells either with UV-treated PRV Becker virions (UVPRV) or with a PRV mutant that lacks IE180 (PRV HKO146), the single master immediate early transcriptional activator of viral genes required for RNA transcription and DNA replication (28, 29). Both UVPRV and PRV HKO146 (grown in IE180-complementing cells) enter the cells, release outer tegument proteins (including vhs, the virion host shutoff protein), and deposit viral DNA in the nucleus, but neither can initiate de novo viral protein synthesis. We found that after either infection, KIF1A was not degraded (Fig. 6A). We concluded that PRV entry into PC12 cells is not sufficient to accelerate KIF1A protein degradation.
FIG 6.

Accelerated proteasomal degradation of KIF1A during infection requires the expression of both PRV early and late proteins. (A) De novo PRV viral protein synthesis is required for KIF1A degradation. Differentiated PC12 cells were mock infected or infected with PRV Becker, UV’d PRV Becker, or IE180-null PRV (HKO146) for 24 h at an MOI of 20. Cells were harvested and lysates were analyzed by Western blotting to monitor the levels of KIF1A, actin, and VP5 at the indicated time points. (B) Differentiated PC12 cells were mock treated or treated with AraT (100 μg/ml) starting 15 min before being infected with PRV Becker for 0, 8, 12, 16, 20, or 24 h. Samples were collected at the indicated time points. Lysates were subjected to Western blot analysis to measure protein levels of KIF1A, actin, VP5, and EP0. (C) For every infection, KIF1A protein abundance was measured by band intensities and normalized with respect to actin levels at each time point. Normalized values were then normalized to those for mock-infected 0-h samples. Values are means plus SEMs (error bars) from three independent experiments. *, P < 0.05 for PRV Becker versus PRV Becker with AraT comparison at the specified time point.
We next determined if PRV immediate early and early or late genes were involved in KIF1A protein degradation. We pretreated differentiated PC12 cells with the herpesvirus DNA replication inhibitor AraT for 15 min and infected them with PRV Becker for 8, 12, 16, 20, or 24 h. AraT is a nucleoside analog that inhibits herpesvirus DNA replication following its phosphorylation by the herpesvirus thymidine kinase. AraT inhibition should have minimal effects on early viral gene expression but should severely diminish late gene expression. As expected, we detected comparable amounts of the early protein EP0 in both AraT-treated and untreated cells during infection, whereas the accumulation of the late protein VP5 (also major capsid protein [MCP]) was severely affected by the AraT treatment (Fig. 6B). AraT treatment led to a minor, though statistically significant, stabilization of KIF1A protein in the later stage of infection (Fig. 6B and C). This experiment demonstrated that while PRV IE and/or E gene products could promote a reduction in KIF1A protein levels (e.g., potentially through the reduction of KIF1A transcripts), viral proteins accumulating in the late phase of infection are also required for efficient degradation of KIF1A.
PRV anterograde-sorting complex US9/gE/gI promotes the accelerated degradation of KIF1A proteins.
PRV particles in transport vesicles use KIF1A for sorting into axons and subsequent transport. The recruitment of KIF1A to transport vesicles containing enveloped PRV particles at the TGN requires viral membrane protein US9 (14). This interaction is stabilized by the heterodimer of gE/gI (15, 30). We had noted previously that two ubiquitin ligases, NEDD4 and viral EP0, copurified with US9 and KIF1A (14). Since KIF1A instability results from proteasomal degradation, we hypothesized that the tripartite complex US9/gE/gI might be involved in accelerating KIF1A degradation. We infected differentiated PC12 cells with PRV Becker and two PRV mutants (PRV Bartha and PRV Babe) that have deletions of the US9, gE, and gI genes and are deficient in axonal sorting and anterograde spread: PRV Bartha and PRV BaBe (PRV Becker with the Us region of Bartha). Infection with either mutant lacking US9, gE, and gI partially restored KIF1A proteins during PRV infection (Fig. 7A and B). As both of these mutant viruses degrade host transcripts presumably through early proteins, KIF1A levels show a decrease between 8 and 12 hpi comparable to those with Becker infection. However, we did not detect the rapid decrease in KIF1A protein levels after 12 h in Bartha and BaBe infections, suggesting that the tripartite complex helps accelerate the degradation of KIF1A by proteasomes (Fig. 1A and B). We further infected PC12 cells with two PRV US9 mutants deficient in anterograde spread, PRV 161 (US9-null) and PRV 172 (Y49-50A US9, functionally null). In PRV 172 infections, all US9, gE, and gI proteins were present, but the dityrosine mutation within the acidic cluster of US9 gene disrupted the formation of the tripartite complex and made the virus particles incapable of recruiting KIF1A and anterograde spread (14, 31, 32). Both PRV 161 and PRV 172 infections restored KIF1A proteins in magnitudes comparable to those with infections with PRV Bartha and BaBe (Fig. 7A and B). These results suggested that the formation of the tripartite complex is necessary for the accelerated loss of KIF1A proteins.
FIG 7.

PRV anterograde-spread complex US9/gE/gI promotes the accerlated degradation of KIF1A proteins. (A) Differentiated PC12 cells were infected with PRV Becker or different PRV mutants defective in anterograde spread (results from PRV BaBe and PRV161 infections are shown) for 0, 8, 12, 16, 20, or 24 h. Cells were harvested and lysates were analyzed by Western blotting to monitor KIF1A, MCP, US9, and actin protein levels. (B) For every infection, KIF1A levels were measured by band intensities and normalized with respect to actin levels at each time point. Normalized values were then normalized to those for mock-infected 0-h samples. Values are means plus SEMs (error bars) from seven independent experiments. *, P < 0.05 for all mutants versus PRV Becker at the specified time point. (C) Differentiated PC12 cells were transduced with adenoviral vectors expressing GFP, GFP-US9, or GFP-US9/gE-mCherry/gI-mTurquoise 2. Cells were collected at the indicated days posttransduction (d.p.t) and subjected to Western blot analysis to monitor KIF1A, gE, gI, US9, and actin levels.
We further determined if the US9/gE/gI complex could target KIF1A for degradation independent of PRV infection. We transduced differentiated PC12 cells with adenovirus vector expressing the US9 protein or simultaneously with adenovirus (Ad) vectors expressing US9, gE, and gI protein for 1, 2, 3, 4, or 5 days. Neither the control Ad-green fluorescent protein (GFP) nor Ad-GFP-US9 transduction resulted in KIF1A protein instability, but when PC12 cells were transduced with equal units of the three adenovirus vectors expressing US9, gE, and gI, the KIF1A protein was completely degraded by 5 days posttransduction (Fig. 7C). We concluded that a functional anterograde sorting complex could augment the proteasomal degradation of KIF1A protein independent of infection by PRV.
Together, these results suggest that PRV infection leads to the depletion of KIF1A proteins in infected neurons both by blocking new protein synthesis and also by specifically targeting the motor for anterograde spread of progeny late in infection, which eventually accelerates proteasomal degradation of KIF1A.
DISCUSSION
A major distinction of alphaherpesviruses from most of other neuroinvasive viruses is their ability to travel in both anterograde and retrograde directions in axons and in neuronal circuits (4). The anterograde transport of newly assembled herpesvirus virions is crucial for the viral life cycle because it ensures that virus particles are sorted into axons to facilitate interhost transmission. KIF1A mediates the sorting and transport of PRV egressing virions in axons, with the dynamics of transport involving long segment runs with high velocity (Vmax ∼2.4 μm/s) (16). The anterograde transport of PRV virions was greatly hindered in the neurons expressing the dominant negative form of KIF1A protein (14). In this study, we demonstrated that KIF1A is intrinsically prone to degradation by the proteasomes and that PRV infection accelerates such degradation and results in near depletion late in infection. The steady-state concentration of KIF1A protein in uninfected neuronal cells is achieved through rapid protein synthesis and proteasomal degradation. Upon PRV infection, KIF1A mRNA was degraded in a nonspecific manner, reducing KIF1A protein synthesis, while the existing proteins were gradually degraded by the proteasome. In addition, adenovirus vectors expressing proteins constituting the PRV anterograde sorting complex US9/gE/gI induced a decline in KIF1A protein level independent of PRV infection, suggesting that the US9/gE/gI tripartite complex accelerates the proteasomal degradation of KIF1A and contributes, alongside PRV host shutoff, to the decline in KIF1A protein during PRV infection. Importantly, using compartmented primary neuron cultures, we showed that proteasomal degradation of KIF1A is accelerated in axons after PRV infection.
The intrinsic instability of the KIF1A protein.
The kinesin motor KIF1A mediates fast axonal transport of membranous vesicles, including synaptic vesicle precursors and dense core vesicles, and is critical in synaptogenesis (17, 18). In differentiated PC12 cells, KIF1A protein levels decreased rapidly upon treatment with the protein synthesis inhibitor cycloheximide (CHX) and showed an apparent half-life (t1/2) of approximately 4 h. The half-lives of kinesin-1 and -2 motors were longer under the same condition (>8 h). Our results are consistent with a previous proteomics study using rat cortical neurons, where KIF1A had the 8th shortest half-life in among ∼2,800 proteins measured (t1/2 = 0.42 day) (33), suggesting that this is a consistent characteristic of KIF1A in both central nervous system (CNS) and PNS neurons. The decline in KIF1A concentration was blocked when PC12 cells were treated simultaneously with CHX and MG132, demonstrating that the loss of KIF1A protein was predominately mediated by proteasomes. Our results confirmed another previous proteomics study in which the half-life of the KIF1A protein increased and the degradation rate dropped upon inhibition of proteasomes (34).
UNC-104, the KIF1A homolog in C. elegans, was shown to be degraded at synapse regions in vivo in an ubiquitin-dependent manner. Moreover, the stability of the motor was linked to its capacity for cargo binding (22). It was also proposed that the degradation of Liprinα1, an cargo adaptor for KIF1A, by either proteasome or CaMKII phosphorylation was required for the “unloading” of LAR (leukocyte common antigen-related) family of receptor protein tyrosine phosphatases at synapses (35, 36). These studies imply a potential mechanistic link between motor degradation and cargo release. Since we also observed proteasomal degradation of KIF1A in fluidically and physically separated axons during PRV infection, KIF1A may be actively degraded in axons by proteasomes upon release of viral cargo.
The relative stabilities of KIF5 and KIF3A proteins were unexpected, since several lines of evidence suggested that both motors are actively degraded in axons (37–40). However, unlike KIF1A, both KIF5 and KIF3A also mediate short distance transport of cellular cargoes within cell bodies and may be recycled for future use. The particular short half-life of KIF1A is consistent with the idea that KIF1A is a specialized motor for axonal transport.
The reduction of KIF1A mRNA upon PRV infection.
The dynamic steady-state level of KIF1A protein in uninfected cells suggests that in PRV-infected cells, KIF1A protein concentration may be sensitive to viral host shutoff or mRNA instability induced by infection. Alphaherpesviruses reduce/block the synthesis of some cellular proteins after infection to facilitate viral gene expression and to evade host antiviral responses. The virion host shutoff protein (vhs) is an endoribonuclease encoded by the viral UL41 gene (41–43). HSV-1 vhs enters the infected cells as a part of incoming virion tegument and subsequently interacts with translation initiation factors eIF4H, eIF4B, and eIF4A to degrade host mRNAs and halt host protein translation (44, 45). Infection of PC12 cells with inactivated virions (UV’d, IE180-null PRV) did not lead to the decline in KIF1A protein. We conclude that tegument proteins like vhs delivered by entering particles are not sufficient for the reduction in KIF1A protein during infection.
We further analyzed the accumulation of cellular mRNAs during infection and compared the levels of kinesin motor transcripts. qRT-PCR analysis of PRV-infected PC12 cells revealed that the levels of KIF1A, KIF5B, and GAPDH transcripts all decreased significantly at similar rates, starting as early as 3 hpi and reaching almost full depletion at 12 hpi. The reduction of KIF1A mRNA reduced the rate of new protein synthesis. This reduction of new KIF1A proteins, coupled with the active degradation of existing KIF1A motors by proteasomes, leads to the significant decrease in KIF1A protein concentration in infected neuronal cells.
The PRV anterograde sorting complex US9/gE/gI directs more KIF1A motors to be degraded by proteasomes.
US9 forms a complex with two other membrane proteins, gE and gI, to sort progeny PRV virions in transport vesicles into axons (14, 15, 30). We have previously identified KIF1A as an interaction partner of PRV US9 protein (14). In addition, we had noted that two ubiquitin ligases, NEDD4 and viral EP0, copurified with US9 and KIF1A (14). Since we also showed accelerated proteasomal degradation of KIF1A during infection, we hypothesized that the PRV anterograde sorting complex, gE/gI/US9, could independently recruit KIF1A to viral transport vesicles for axonal sorting and transport and eventually promote the proteasomal degradation of KIF1A in axons. We investigated this hypothesis using adenovirus vectors to express the components of the anterograde sorting complex (US9 alone or the whole complex) in neuronal cells without PRV infection. In cells transduced with adenovirus vectors expressing US9, gE, and gI, US9 interacts with the gE/gI heterodimer in lipid rafts, and the US9, gE, and gI complex was observed to undergo axonal anterograde transport together with long-range motility characteristic of anterogradely moving PRV virions in axons (30). PRV envelope proteins US9/gE/gI, when expressed together in neuronal cells, induced a significant decline in KIF1A protein concentration. Therefore, we concluded that the viral anterograde sorting complex US9/gE/gI promotes the accelerated loss of KIF1A proteins.
This result is consistent with experiments in which PC12 cells were infected with PRV strains that fail to express a functional US9/gE/gI complex to recruit KIF1A to undergo anterograde spread. These PRV mutants still promote host shutoff, leading to the decline of KIF1A proteins late in infection. However, the rate of decline is lower than that observed after wild-type PRV infection, suggesting that the US9/gE/gI complex accelerates the degradation of KIF1A by the proteasomes. The inhibition of late gene expression (e.g., US9) by AraT treatment also led to a minor yet statistically significant stabilization of KIF1A protein concentration. We concluded that the PRV anterograde sorting complex US9/gE/gI directs more KIF1A motors to be degraded by proteasomes. This might shed light on the mechanisms underlying the intrinsic instability of KIF1A protein, which potentially stem from the interaction of the motor with its cellular cargoes.
PRV infection induces the accelerated loss of KIF1A protein through two separate mechanisms.
Our model to account for our results is shown in Fig. 8. KIF1A motors, mediating the anterograde transport of the vesicles in axons critical for synapse formation and function, are inherently prone to proteasomal degradation. Such intrinsic instability results in the dynamic steady-state level of KIF1A protein maintained through rapid protein synthesis and proteasomal degradation in neuronal cells (Fig. 8A). The balance between protein synthesis and degradation is disrupted by PRV infection through two separate mechanisms (Fig. 8B). First, PRV infection promotes the nonspecific loss of KIF1A mRNAs and reduces the synthesis of KIF1A protein. The concentration of existing KIF1A protein continues to decline through default proteasomal degradation. Second, PRV infection accelerates the degradation of KIF1A protein through the PRV anterograde sorting complex US9/gE/gI. We propose that US9 recruits KIF1A to transport vesicles containing enveloped virions or light particles, as well as vesicles with viral glycoproteins for axonal sorting and transport, and eventually this complex promotes the proteasomal degradation of KIF1A in axons.
FIG 8.

PRV infection induces the accelerated loss of KIF1A protein through two separate mechanisms. (A) In uninfected cells, KIF1A protein is inherently prone to be degraded by the proteasome. The steady-state concentration of KIF1A protein is achieved through rapid protein synthesis and proteasomal degradation. (B) PRV infection induces KIF1A mRNA degradation and reduced protein synthesis, while the existing KIF1A proteins undergo default proteasomal degradation. Furthermore, the proteasomal degradation of the existing KIF1A proteins is accelerated by the PRV gE/gI/US9 complex in the late phase of infection. Images were created with BioRender.
Our work implies that after reactivation from latently infected PNS neurons, the targeted loss of KIF1A motor proteins leads to a limited time window for sorting into and transport of viral particles in axons for further host-to-host spread. Although the egress of newly made PRV particles can be observed as early as 5 hpi from neuronal cell bodies, the axonal sorting and transport of PRV particles occurs during the late stage of the infection (8 to 14 hpi), coinciding with the decline in KIF1A protein levels (I. Hogue and L. W. Enquist, unpublished data) (23). In the meantime, stalled particles in axons begin to be seen after 10 hpi (23). Indeed, the loss of available KIF1A motors over the course of PRV infection results in reduced recruitment of the motor by US9 (14). These findings suggest that the level of KIF1A proteins correlates with the number of particles that enter axons late in infection. While our work has focused on PRV, it would be important to know if infection by other alphaherpesviruses promotes differential degradation of axonal motor proteins. The anterograde transport of HSV-1 has been shown to be mediated by kinesin-1 (KIF5A, KIF5B, and KIF5C) and kinesin-3 (KIF1A) motors (11, 46) (E. Engel and L. W. Enquist, unpublished data). It is therefore of clinical importance to determine if HSV-1 infection induces similar loss in human neurons in vitro. Such a loss of kinesin-1 and kinesin-3 motors after reactivation from PNS neurons could have significant consequence for long-term neuronal function. Further work is required to understand whether PNS neurons can tolerate the loss of specific kinesin motors during reactivation episodes and whether the motor concentrations are restored after reactivation if those neurons survive.
MATERIALS AND METHODS
Cell lines and viruses.
Porcine kidney epithelial cells (PK15; ATCC) were maintained in Dulbecco modified Eagle medium (DMEM; HyClone) supplemented with 10% fetal bovine serum (FBS; HyClone) and 1% penicillin-streptomycin (HyClone). PK15 cells were used to propagate and determine titers of all PRV strains used in this study. 293A cells (Invitrogen) were used to propagate and determine the titers of adenovirus vectors used in this study. The neuronal PC12 cell line has been used extensively to model primary neurons infection of PRV and to reproduce the US9-associated transport phenotype (14, 47). PC12 cells were cultured on dishes coated with type 1 rat tail collagen (BD Bioscience, Bedford, MA) in 85% RPMI 1640 (Thermo Fisher) supplemented with 10% horse serum (Life Technologies) and 5% FBS (48). PC12 cells were differentiated in RPMI 1640 supplemented with 1% horse serum and nerve growth factor (NGF; 2.5S; Invitrogen) at 100 ng/ml (48). Differentiation medium was replaced every third day for 8 days before infection.
The PRV strains used in this study include both wild-type laboratory stain and recombinants. PRV Becker is a wild-type strain (25, 26). PRV Bartha is an attenuated vaccine strain (49, 50). PRV BaBe is a viral recombinant in the PRV Becker background with deletions from the unique short (US) region of PRV Bartha (51). HKO146 lacks the only PRV immediate early gene, IE180, and expresses hSyn Cre-t2a-venus in US9 loci in the PRV Becker background (H. Oyibo, P. Znamenskiy, H. V. Oviedo, L. W. Enquist, and A. Zador, unpublished data). PRV 161 harbors a complete deletion of the US9 gene in the PRV Becker background (52). PRV 172 harbors two tyrosine-to-alanine substitutions (Y49A and Y50A) in the PRV Becker background (31). Individual replication-deficient adenovirus vectors expressing GFP, GFP-US9, gE-mCherry, ad gI-mTurquoise 2 were previously reported (14, 53).
Primary neuronal culture.
Embryonic superior cervical ganglion (SCG) neurons were isolated and cultured as previously described (54). Briefly, superior cervical ganglia (SCG) were isolated from day 17 Sprague-Dawley rat (Hilltop Labs) embryos and plated and maintained on poly-dl-ornithine (Sigma-Aldrich)- and laminin (Invitrogen)-coated dishes (Falcon or MatTek) in neuronal medium made with neurobasal medium (Gibco) supplemented with 1% penicillin-streptomycin with 2 mM glutamine (Invitrogen), 2% B-27 (Gibco), and 100 ng/ml of NGF (2.5S; Invitrogen). At 2 days postplating, 1 mM cytosine-d-arabinofuranoside (AraC; Sigma-Aldrich) was added to selectively eliminate dividing nonneuronal cells in culture for 2 days. SCG neurons were allowed to differentiate for at least 14 days before infection.
Compartmented neuronal cultures were prepared on poly-ornithine- and laminin-coated plastic dishes. Parallel grooves were etched into the plastic dish, and 1% methylcellulose in neuronal medium was added to the region that would underlie the middle compartment of the isolator rings. CAMP320 isolator rings (Tyler Research) were coated with autoclaved silicone vacuum grease on one side and gently applied to the dish so that the etched grooves extended across three compartments. Dissociated SCG neurons were plated in the soma compartment (S) and were allowed to extend axons along the grooves through the M compartment and into the neurite (N) compartment for 14 to 28 days.
Viral infection in neuronal culture.
Triplicates of differentiated PC12 cells were infected with the indicated PRV strain at a multiplicity of infection (MOI) of 20 at the desired hours postinfection. Infected cells from each triplicate were harvested and combined into a single sample for further biochemical analysis. Compartmented SCG neuronal cultures were infected in the soma (S) compartment for 24 h. Neuronal cell bodies from the S compartments and axons from the N compartments were harvested for biochemical analysis. The proteasome inhibitor MG132 (Millipore Sigma) was prepared in dimethyl sulfoxide (DMSO) and added at a concentration of 2.5 μM. The herpesvirus DNA replication inhibitor thymine 1-β-d-arabinofuranoside (AraT; Millipore Sigma) was added at a concentration of 100 μg/ml 15 min prior to infection.
Adenovirus vector transduction.
The adenovirus vectors expressing GFP, GFP-US9, gE-mCherry, and gI-mTurquoise-2 were reported previously (14, 53). Adenovirus vectors were propagated in 293A cells, and cell-associated virus was harvested in serum-free DMEM. Adenoviral transduction of differentiated PC12 cells was performed for up to 5 days to ensure the expression of fluorescent transgene(s) in the majority of cells (>90%).
Quantitative Western blotting.
Neuronal cell lysates were prepared in radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris-HCl, 150 mM NaCl, 5 mM EDTA, 1% NP-40, 0.1% SDS, 0.1% Triton X-100, and 1% sodium deoxycholate, pH 8.0) supplemented with 1 mM dithiothreitol (DTT; Sigma-Aldrich) and protease inhibitor cocktail (Sigma-Aldrich). Cell lysates were kept on ice for 1 h and centrifuged at 13,200 rpm at 4°C for 10 min. Cleared cell lysates were transferred to a new tube and mixed with 4× LDS sample loading buffer (Invitrogen) supplemented with 240 mM DTT. The samples were heated at 95°C for 10 min and centrifuged at 4°C for 10 min to remove potential protein aggregates before proteins were separated in gradient polyacrylamide gels (4 to 12%) (Invitrogen). Proteins were transferred onto nitrocellulose membranes (Whatman) by semidry transfer (Bio-Rad). Membranes were blocked in 5% nonfat dry milk in Tris-buffered saline with 0.1% Tween 20 (TBS-T) for 1 h at room temperature. Primary and secondary antibodies for immunoblot analysis were prepared in 1% milk in TBS-T. Protein bands were imaged with a LI-COR Odyssey CLx imaging system. The signal intensity was quantified using LI-COR Image Studio Lite software.
Antibodies used in this study included a mouse monoclonal KIF1A antibody (clone 16 at 1:2,000; BD), a mouse monoclonal kinesin heavy chain antibody (MAB-1614 at 1:1,000; Millipore Sigma), a mouse monoclonal KIF3A antibody (clone K2.4 at 1:1,000; Covance Inc.), a mouse monoclonal β-actin antibody (1:10,000; Sigma), a monoclonal US9 antibody (IA8, DSHB, at 1:100), a rabbit polyclonal GST-EP0 antibody at 1:2,000 (55), a mouse monoclonal PRV VP5 antibody at 1:1,000 (56), a mouse monoclonal PRV US3 antibody at 1:10,000 (57), a rabbit polyclonal gE cytoplasmic tail antibody at 1:1,000 (58), and a rabbit polyclonal gI antibody (a generous gift from K. Bienkowska-Szewczyk) at 1:1,000 (59). IRDye secondary antibodies included 800CW donkey anti-mouse IgG, 800CW donkey anti-rabbit IgG, and 680RD donkey anti-rabbit at 1:15,000 (LI-COR).
qRT-PCR analysis.
Differentiated PC12 cells infected with PRV were harvested and subjected to total RNA extraction using an RNeasy Plus minikit (Qiagen). First-strand cDNA synthesis of isolated RNAs was performed with a SuperScript III first-strand synthesis kit with oligo(dT) primer (Invitrogen). Quantitative reverse transcription-PCR was performed using Eppendorf Realplex2 Mastercycler with reaction mixtures prepared with a KAPA SYBR fast universal qPCR kit (KAPA Biosystems). The PCR primers for KIF1A, KIF5B, GAPDH, and 18S rRNA were reported elsewhere (60). The qRT-PCR samples were prepared in triplicates. The relative abundance of RNA in each sample was calculated using the comparative threshold cycle (–ΔΔCT) method normalized to 18S rRNA and subsequently to uninfected samples.
Statistical analysis.
One-way analysis of variance (ANOVA) with Tukey’s posttest and two-way ANOVA with Sidak’s posttest were performed using GraphPad Prism 6. Values in the text, graphs, and figure legends throughout are means ± standard errors of the means (SEMs).
Image production.
Cartoon illustrations were made with the web application BioRender under the Princeton University license for education use.
Ethics statement.
All animal work was performed in accordance with the Princeton Institutional Animal Care and Use Committee (protocols 1947-16). Princeton personnel are required to adhere to applicable federal, state, local and institutional laws and policies governing animal research, including the Animal Welfare Act and Regulations (AWA), the Public Health Service Policy on Humane Care and Use of Laboratory Animals, the Principles for the Utilization and Care of Vertebrate Animals Used in Testing, Research and Training, and the Health Research Extension Act of 1985.
ACKNOWLEDGMENTS
We thank Halina Staniszewska-Goraczniak for excellent technical and logistical support and the members of the Enquist Lab for their critical comments on the projects.
This project was funded by NIH, National Institute of Neurological Disorders and Stroke (NINDS), grants RO1 NS033506 and RO1 NS060699 (L.W.E.).
REFERENCES
- 1.Steiner I, Kennedy PGE, Pachner AR. 2007. The neurotropic herpes viruses: herpes simplex and varicella-zoster. Lancet Neurol 6:1015–1028. doi: 10.1016/S1474-4422(07)70267-3. [DOI] [PubMed] [Google Scholar]
- 2.Koelle DM, Corey L. 2008. Herpes simplex: insights on pathogenesis and possible vaccines. Annu Rev Med 59:381–395. doi: 10.1146/annurev.med.59.061606.095540. [DOI] [PubMed] [Google Scholar]
- 3.Pomeranz LE, Reynolds AE, Hengartner CJ. 2005. Molecular biology of pseudorabies virus: impact on neurovirology and veterinary medicine. Microbiol Mol Biol Rev 69:462–500. doi: 10.1128/MMBR.69.3.462-500.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Smith G. 2012. Herpesvirus transport to the nervous system and back again. Annu Rev Microbiol 66:153–176. doi: 10.1146/annurev-micro-092611-150051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hirokawa N, Noda Y, Tanaka Y, Niwa S. 2009. Kinesin superfamily motor proteins and intracellular transport. Nat Rev Mol Cell Biol 10:682–696. doi: 10.1038/nrm2774. [DOI] [PubMed] [Google Scholar]
- 6.Hirokawa N, Niwa S, Tanaka Y. 2010. Molecular motors in neurons: transport mechanisms and roles in brain function, development, and disease. Neuron 68:610–638. doi: 10.1016/j.neuron.2010.09.039. [DOI] [PubMed] [Google Scholar]
- 7.Gluska S, Zahavi EE, Chein M, Gradus T, Bauer A, Finke S, Perlson E. 2014. Rabies virus hijacks and accelerates the p75NTR retrograde axonal transport machinery. PLoS Pathog 10:e1004348. doi: 10.1371/journal.ppat.1004348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Smith GA, Pomeranz L, Gross SP, Enquist LW. 2004. Local modulation of plus-end transport targets herpesvirus entry and egress in sensory axons. Proc Natl Acad Sci U S A 101:16034–16039. doi: 10.1073/pnas.0404686101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zaichick SV, Bohannon KP, Hughes A, Sollars PJ, Pickard GE, Smith GA. 2013. The herpesvirus VP1/2 protein is an effector of dynein-mediated capsid transport and neuroinvasion. Cell Host Microbe 13:193–203. doi: 10.1016/j.chom.2013.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Buch A, Muller O, Ivanova L, Dohner K, Bialy D, Bosse JB, Pohlmann A, Binz A, Hegemann M, Nagel CH, Koltzenburg M, Viejo-Borbolla A, Rosenhahn B, Bauerfeind R, Sodeik B. 2017. Inner tegument proteins of herpes simplex virus are sufficient for intracellular capsid motility in neurons but not for axonal targeting. PLoS Pathog 13:e1006813. doi: 10.1371/journal.ppat.1006813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.DuRaine G, Wisner TW, Paul Howard P, Johnson DC. 2018. Kinesin-1 proteins KIF5A, -5B, and -5C promote anterograde transport of herpes simplex virus enveloped virions in axons. J Virol 92: e01269-18. doi: 10.1128/JVI.01269-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Radtke K, Kieneke D, Wolfstein A, Michael K, Steffen W, Scholz T, Karger A, Sodeik B. 2010. Plus- and minus-end directed microtubule motors bind simultaneously to herpes simplex virus capsids using different inner tegument structures. PLoS Pathog 6:e1000991. doi: 10.1371/journal.ppat.1000991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wolfstein A, Nagel CH, Radtke K, Dohner K, Allan VJ, Sodeik B. 2006. The inner tegument promotes herpes simplex virus capsid motility along microtubules in vitro. Traffic 7:227–237. doi: 10.1111/j.1600-0854.2005.00379.x. [DOI] [PubMed] [Google Scholar]
- 14.Kramer T, Greco TM, Taylor MP, Ambrosini AE, Cristea IM, Enquist LW. 2012. Kinesin-3 mediates axonal sorting and directional transport of alphaherpesvirus particles in neurons. Cell Host Microbe 12:806–814. doi: 10.1016/j.chom.2012.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kratchmarov R, Kramer T, Greco TM, Taylor MP, Ch'ng TH, Cristea IM, Enquist LW. 2013. Glycoproteins gE and gI are required for efficient KIF1A-dependent anterograde axonal transport of alphaherpesvirus particles in neurons. J Virol 87:9431–9440. doi: 10.1128/JVI.01317-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Scherer J, Yaffe ZA, Vershinin M, Enquist LW. 2016. Dual-color herpesvirus capsids discriminate inoculum from progeny and reveal axonal transport dynamics. J Virol 90:9997–10006. doi: 10.1128/JVI.01122-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Siddiqui N, Straube A. 2017. Intracellular cargo transport by kinesin-3 motors. Biochemistry (Mosc) 82:803–815. doi: 10.1134/S0006297917070057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kern JV, Zhang YV, Kramer S, Brenman JE, Rasse TM. 2013. The kinesin-3, unc-104 regulates dendrite morphogenesis and synaptic development in Drosophila. Genetics 195:59–72. doi: 10.1534/genetics.113.151639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Klopfenstein DR, Tomishige M, Stuurman N, Vale RD. 2002. Role of phosphatidylinositol(4,5)bisphosphate organization in membrane transport by the Unc104 kinesin motor. Cell 109:347–358. doi: 10.1016/s0092-8674(02)00708-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yonekawa Y, Harada A, Okada Y, Funakoshi T, Kanai Y, Takei Y, Terada S, Noda T, Hirokawa N. 1998. Defect in synaptic vesicle precursor transport and neuronal cell death in KIF1A motor protein-deficient mice. J Cell Biol 141:431–441. doi: 10.1083/jcb.141.2.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Okada Y, Yamazaki H, Sekine-Aizawa Y, Hirokawa N. 1995. The neuron-specific kinesin superfamily protein KIFIA is a unique monomeric motor for anterograde axonal transport of synaptic vesicle precursors. Cell 81:769–780. doi: 10.1016/0092-8674(95)90538-3. [DOI] [PubMed] [Google Scholar]
- 22.Kumar J, Choudhary BC, Metpally R, Zheng Q, Nonet ML, Ramanathan S, Klopfenstein DR, Koushika SP. 2010. The Caenorhabditis elegans kinesin-3 motor UNC-104/KIF1A is degraded upon loss of specific binding to cargo. PLoS Genet 6:e1001200. doi: 10.1371/journal.pgen.1001200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smith GA, Gross SP, Enquist LW. 2001. Herpesviruses use bidirectional fast-axonal transport to spread in sensory neurons. Proc Natl Acad Sci U S A 98:3466–3470. doi: 10.1073/pnas.061029798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Becker Y, Tavor E, Asher Y, Berkowitz C, Moyal M. 1993. Effect of herpes simplex virus type-1 UL41 gene on the stability of mRNA from the cellular genes: beta-actin, fibronectin, glucose transporter-l, and docking protein, and on virus intraperitoneal pathogenicity to newborn mice. Virus Genes 7:133–143. doi: 10.1007/bf01702393. [DOI] [PubMed] [Google Scholar]
- 25.Smibert CA, Johnson DC, Smiley JR. 1992. Identification and characterization of the virion-induced host shutoff product of herpes simplex virus gene UL41. J Gen Virol 73:467–470. doi: 10.1099/0022-1317-73-2-467. [DOI] [PubMed] [Google Scholar]
- 26.Hardy WR, Sandri-Goldin RM. 1994. Herpes simplex virus inhibits host cell splicing, and regulatory protein ICP27 is required for this effect. J Virol 68:7790–7799. doi: 10.1128/JVI.68.12.7790-7799.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hardwicke MA, Sandri-Goldin RM. 1994. The herpes simplex virus regulatory protein ICP27 contributes to the decrease in cellular mRNA levels during infection. J Virol 68:4797–4810. doi: 10.1128/JVI.68.8.4797-4810.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu BW, Engel EA, Enquist LW. 2014. Characterization of a replication-incompetent pseudorabies virus mutant lacking the sole immediate early gene IE180. mBio 5:e01850-14. doi: 10.1128/mBio.01850-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oyibo HK, Znamenskiy P, Oviedo HV, Enquist LW, Zador AM. 2014. Long-term Cre-mediated retrograde tagging of neurons using a novel recombinant pseudorabies virus. Front Neuroanat 8:86. doi: 10.3389/fnana.2014.00086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Scherer J, Hogue IB, Yaffe ZA, Tanneti NS, Winer BY, Vershinin M, Enquist L. 2020. A kinesin-3 recruitment complex facilitates axonal sorting of enveloped alpha herpesvirus capsids. PLoS Pathog 16:e1007985. doi: 10.1371/journal.ppat.1007985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brideau AD, Eldridge M, Enquist LW. 2000. Directional transneuronal infection by pseudorabies virus is dependent on an acidic internalization motif in the Us9 cytoplasmic tail. J Virol 74:4549–4561. doi: 10.1128/jvi.74.10.4549-4561.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Taylor MP, Kramer T, Lyman MG, Kratchmarov R, Enquist LW. 2012. Visualization of an alphaherpesvirus membrane protein that is essential for anterograde axonal spread of infection in neurons. mBio 3:e00063-12. doi: 10.1128/mBio.00063-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cohen LD, Zuchman R, Sorokina O, Muller A, Dieterich DC, Armstrong JD, Ziv T, Ziv NE. 2013. Metabolic turnover of synaptic proteins: kinetics, interdependencies and implications for synaptic maintenance. PLoS One 8:e63191. doi: 10.1371/journal.pone.0063191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hakim V, Cohen LD, Zuchman R, Ziv T, Ziv NE. 2016. The effects of proteasomal inhibition on synaptic proteostasis. EMBO J 35:2238–2262. doi: 10.15252/embj.201593594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hoogenraad CC, Feliu-Mojer MI, Spangler SA, Milstein AD, Dunah AW, Hung AY, Sheng M. 2007. Liprinalpha1 degradation by calcium/calmodulin-dependent protein kinase II regulates LAR receptor tyrosine phosphatase distribution and dendrite development. Dev Cell 12:587–602. doi: 10.1016/j.devcel.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 36.Schlager MA, Hoogenraad CC. 2009. Basic mechanisms for recognition and transport of synaptic cargos. Mol Brain 2:25. doi: 10.1186/1756-6606-2-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kondo S, Sato-Yoshitake R, Noda Y, Aizawa H, Nakata T, Matsuura Y, Hirokawa N. 1994. KIF3A is a new microtubule-based anterograde motor in the nerve axon. J Cell Biol 125:1095–1107. doi: 10.1083/jcb.125.5.1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yamazaki H, Nakata T, Okada Y, Hirokawa N. 1995. KIF3A/B: a heterodimeric kinesin superfamily protein that works as a microtubule plus end-directed motor for membrane organelle transport. J Cell Biol 130:1387–1399. doi: 10.1083/jcb.130.6.1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li J-Y, Pfister KK, Brady S, Dahlstrom A. 1999. Axonal transport and distribution of immunologically distinct kinesin heavy chains in rat neurons. J Neuroscience Res 58:226–241. doi:. [DOI] [PubMed] [Google Scholar]
- 40.Hirokawa N, Sato-Yoshitake R, Kobayashi N, Pfister KK, Bloom GS, Brady ST. 1991. Kinesin associates with anterogradely transported membranous organelles in vivo. J Cell Biol 114:295–302. doi: 10.1083/jcb.114.2.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Everly DN Jr, Feng P, Mian IS, Read GS. 2002. mRNA degradation by the virion host shutoff (Vhs) protein of herpes simplex virus: genetic and biochemical evidence that Vhs is a nuclease. J Virol 76:8560–8571. doi: 10.1128/jvi.76.17.8560-8571.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Perez-Parada J, Saffran HA, Smiley JR. 2004. RNA degradation induced by the herpes simplex virus vhs protein proceeds 5′ to 3′ in vitro. J Virol 78:13391–13394. doi: 10.1128/JVI.78.23.13391-13394.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Taddeo B, Roizman B. 2006. The virion host shutoff protein (UL41) of herpes simplex virus 1 is an endoribonuclease with a substrate specificity similar to that of RNase A. J Virol 80:9341–9345. doi: 10.1128/JVI.01008-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Doepker RC, Hsu WL, Saffran HA, Smiley JR. 2004. Herpes simplex virus virion host shutoff protein is stimulated by translation initiation factors eIF4B and eIF4H. J Virol 78:4684–4699. doi: 10.1128/jvi.78.9.4684-4699.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Feng P, Everly DN Jr, Read GS. 2005. mRNA decay during herpes simplex virus (HSV) infections: protein-protein interactions involving the HSV virion host shutoff protein and translation factors eIF4H and eIF4A. J Virol 79:9651–9664. doi: 10.1128/JVI.79.15.9651-9664.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Diefenbach RJ, Davis A, Miranda-Saksena M, Fernandez MA, Kelly BJ, Jones CA, LaVail JH, Xue J, Lai J, Cunningham AL. 2016. The basic domain of herpes simplex virus 1 pUS9 recruits kinesin-1 to facilitate egress from neurons. J Virol 90:2102–2111. doi: 10.1128/JVI.03041-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lyman MG, Curanovic D, Enquist LW. 2008. Targeting of pseudorabies virus structural proteins to axons requires association of the viral Us9 protein with lipid rafts. PLoS Pathog 4:e1000065. doi: 10.1371/journal.ppat.1000065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ch'ng TH, Flood EA, Enquist LW. 2005. Culturing primary and transformed neuronal cells for studying pseudorabies virus infection. Methods Mol Biol 292:299–316. doi: 10.1385/1-59259-848-x:299. [DOI] [PubMed] [Google Scholar]
- 49.Platt KB, Mare CJ, Hinz PN. 1979. Differentiation of vaccine strains and field isolates of pseudorabies (Aujeszky’s disease) virus: thermal sensitivity and rabbit virulence markers. Arch Virol 60:13–23. doi: 10.1007/bf01318093. [DOI] [PubMed] [Google Scholar]
- 50.Szpara ML, Tafuri YR, Parsons L, Shamim SR, Verstrepen KJ, Legendre M, Enquist LW. 2011. A wide extent of inter-strain diversity in virulent and vaccine strains of alphaherpesviruses. PLoS Pathog 7:e1002282. doi: 10.1371/journal.ppat.1002282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Card JP, Whealy ME, Robbins AK, Enquist LW. 1992. Pseudorabies virus envelope glycoprotein gI influences both neurotropism and virulence during infection of the rat visual system. J Virol 66:3032–3041. doi: 10.1128/JVI.66.5.3032-3041.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brideau AD, Card JP, Enquist LW. 2000. Role of pseudorabies virus Us9, a type II membrane protein, in infection of tissue culture cells and the rat nervous system. J Virol 74:834–845. doi: 10.1128/jvi.74.2.834-845.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hogue IB, Scherer J, Enquist LW. 2016. Exocytosis of alphaherpesvirus virions, light particles, and glycoproteins uses constitutive secretory mechanisms. mBio 7:e00820-16. doi: 10.1128/mBio.00820-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Curanovic D, Ch’ng TH, Szpara M, Enquist L. 2009. Compartmented neuron cultures for directional infection by alpha herpesviruses. Curr Protoc Cell Biol Chapter 26:Unit 26.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brukman A, Enquist LW. 2006. Pseudorabies virus EP0 protein counteracts an interferon-induced antiviral state in a species-specific manner. J Virol 80:10871–10873. doi: 10.1128/JVI.01308-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lyman MG, Feierbach B, Curanovic D, Bisher M, Enquist LW. 2007. Pseudorabies virus Us9 directs axonal sorting of viral capsids. J Virol 81:11363–11371. doi: 10.1128/JVI.01281-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Olsen LM, Ch'ng TH, Card JP, Enquist LW. 2006. Role of pseudorabies virus Us3 protein kinase during neuronal infection. J Virol 80:6387–6398. doi: 10.1128/JVI.00352-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tirabassi RS, Enquist LW. 2000. Role of the pseudorabies virus gI cytoplasmic domain in neuroinvasion, virulence, and posttranslational N-linked glycosylation. J Virol 74:3505–3516. doi: 10.1128/jvi.74.8.3505-3516.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tirabassi RS, Enquist LW. 1998. Role of envelope protein gE endocytosis in the pseudorabies virus life cycle. J Virol 72:4571–4579. doi: 10.1128/JVI.72.6.4571-4579.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Baptista FI, Pinto MJ, Elvas F, Almeida RD, Ambrosio AF. 2013. Diabetes alters KIF1A and KIF5B motor proteins in the hippocampus. PLoS One 8:e65515. doi: 10.1371/journal.pone.0065515. [DOI] [PMC free article] [PubMed] [Google Scholar]