

The inflammatory response to influenza A virus (IAV) participates in infection control but contributes to disease severity. After viral detection, intracellular pathways are activated, initiating cytokine production, but these pathways are incompletely understood. We show that interferon regulatory factor 5 (IRF5) mediates IAV-induced inflammation and, in mice, drives pathology. This was independent of antiviral type 1 IFN and virus replication, implying that IRF5 could be specifically targeted to treat influenza virus-induced inflammation. We show for the first time that human iPSC technology can be exploited in genetic studies of virus-induced immune responses. Using this technology, we deleted IRF5 in human myeloid cells. These IRF5-deficient cells exhibited impaired influenza virus-induced cytokine production and revealed that IRF5 acts downstream of Toll-like receptor 7 and possibly retinoic acid-inducible gene I. Our data demonstrate the importance of IRF5 in influenza virus-induced inflammation, suggesting that genetic variation in the IRF5 gene may influence host susceptibility to viral diseases.
KEYWORDS: dendritic cells, IRF5, iPSCs, inflammation, influenza
ABSTRACT
Recognition of influenza A virus (IAV) by the innate immune system triggers pathways that restrict viral replication, activate innate immune cells, and regulate adaptive immunity. However, excessive innate immune activation can exaggerate disease. The pathways promoting excessive activation are incompletely understood, with limited experimental models to investigate the mechanisms driving influenza virus-induced inflammation in humans. Interferon regulatory factor 5 (IRF5) is a transcription factor that plays important roles in the induction of cytokines after viral sensing. In an in vivo model of IAV infection, IRF5 deficiency reduced IAV-driven immune pathology and associated inflammatory cytokine production, specifically reducing cytokine-producing myeloid cell populations in Irf5−/− mice but not impacting type 1 interferon (IFN) production or virus replication. Using cytometry by time of flight (CyTOF), we identified that human lung IRF5 expression was highest in cells of the myeloid lineage. To investigate the role of IRF5 in mediating human inflammatory responses by myeloid cells to IAV, we employed human-induced pluripotent stem cells (hIPSCs) with biallelic mutations in IRF5, demonstrating for the first time that induced pluripotent stem cell-derived dendritic cells (iPS-DCs) with biallelic mutations can be used to investigate the regulation of human virus-induced immune responses. Using this technology, we reveal that IRF5 deficiency in human DCs, or macrophages, corresponded with reduced virus-induced inflammatory cytokine production, with IRF5 acting downstream of Toll-like receptor 7 (TLR7) and, possibly, retinoic acid-inducible gene I (RIG-I) after viral sensing. Thus, IRF5 acts as a regulator of myeloid cell inflammatory cytokine production during IAV infection in mice and humans and drives immune-mediated viral pathogenesis independently of type 1 IFN and virus replication.
IMPORTANCE The inflammatory response to influenza A virus (IAV) participates in infection control but contributes to disease severity. After viral detection, intracellular pathways are activated, initiating cytokine production, but these pathways are incompletely understood. We show that interferon regulatory factor 5 (IRF5) mediates IAV-induced inflammation and, in mice, drives pathology. This was independent of antiviral type 1 IFN and virus replication, implying that IRF5 could be specifically targeted to treat influenza virus-induced inflammation. We show for the first time that human iPSC technology can be exploited in genetic studies of virus-induced immune responses. Using this technology, we deleted IRF5 in human myeloid cells. These IRF5-deficient cells exhibited impaired influenza virus-induced cytokine production and revealed that IRF5 acts downstream of Toll-like receptor 7 and possibly retinoic acid-inducible gene I. Our data demonstrate the importance of IRF5 in influenza virus-induced inflammation, suggesting that genetic variation in the IRF5 gene may influence host susceptibility to viral diseases.
INTRODUCTION
During infection with influenza A virus (IAV), the host immune system must calibrate immune responses to control viral infection while minimizing damage to host tissues. Disease manifestations are often associated with the host inflammatory response to the virus (1, 2), and clinical outcomes vary widely between individuals (3). The inflammatory response is initiated when pattern recognition receptors (PRRs) on innate immune cells recognize IAV pathogen-associated molecular patterns (PAMPs), which trigger signaling cascades resulting in the expression of specific inflammatory cytokines and chemokines (4, 5). Cytokines play various roles, such as directly inhibiting viral replication and activating the cytolytic functions of T cells, whereas chemokines recruit innate immune cells such as macrophages, neutrophils, NK cells, and inflammatory monocytes to the lungs and airways (6).
Interferon regulatory factor 5 (IRF5) is a member of the IRF family of transcription factors, whose members have a shared N-terminal DNA binding domain and bind consensus interferon-stimulated response element (ISRE) motifs. As ISREs are enriched in the regulatory regions of immune genes, IRFs play key roles as master regulators in the innate immune response (7) and may provide a mechanism for conferring signal specificity to target gene sets downstream of Toll-like receptor (TLR) signaling (8). While IRF3 and IRF7 are necessary for the induction of type I interferons (9, 10), IRF5 has been shown to be key in regulating inflammatory cytokine responses, generally acting downstream of TLR-MyD88 pathways (11, 12). Genetic polymorphisms in the IRF5 gene in humans have been linked to various autoimmune conditions, including systemic lupus erythematosus, rheumatoid arthritis, Sjogren’s syndrome, multiple sclerosis, and inflammatory bowel disease (13). IRF5 has also been shown to be important in regulating immune responses to various pathogens in murine and human cell models (14–17). Additionally, Irf5−/− mice are resistant to systemic shock induced by CpG ligands and lipopolysaccharide (LPS) (12). The extent to which IRF5 contributes to inflammation-induced pathologies, however, is unclear.
IRF5 is expressed predominantly by myeloid cells such as dendritic cells (DCs) and macrophages, in addition to B cells (13, 18). Myeloid cells can have protective and immunopathogenic roles during IAV infection, producing inflammatory cytokines and initiating adaptive immune responses (19). Furthermore, inflammatory monocytes and monocyte-derived DCs have been identified to drive inflammation and lung pathology during IAV infection (19, 20).
Studying inflammatory cytokine responses in human myeloid cells is challenging. Human DCs are difficult to culture in vitro and, although DCs can be induced from blood-derived monocytes, these cells display morphological and functional differences from human primary DCs, for example, differing in their capacities for T cell stimulation in comparison to CD11c+ blood-derived DCs (21). Primary myeloid cells are also difficult to genetically manipulate, meaning that studies addressing the effect of host genetics on myeloid cell responses can be challenging. Human induced pluripotent stem cells (hIPSCs) offer a useful system for studying host-pathogen variations because these cells are amenable to genetic manipulation, can be differentiated toward multiple cellular lineages, and are self-renewing, allowing for the production of sufficient quantities of cells of the same genetic background. hIPSC-derived macrophages (iPSDMs) have already been used to successfully model the interactions of pathogens with host cells (16, 22). However, to date, hIPSC technology has not been used to perform genetic investigations of virus-induced immune responses. To study the impact of IRF5 on human myeloid IAV-induced immune responses, we utilized hIPSCs generated from a healthy donor or with mutations in IRF5 generated by CRISPR-Cas9 engineering differentiated into dendritic cells and macrophages as a human model system to assess the role of IRF5 in the regulation of immune responses to IAV. Using these tools in combination with studies of human lung cells, in addition to Irf5−/− mice, we show that IRF5 drives IAV-induced inflammatory cytokine responses in mice and humans without impacting virus replication and type 1 interferon (IFN) secretion, and this process mediates viral pathogenesis in vivo.
RESULTS
IRF5 mediates inflammatory cytokine and myeloid cell responses to influenza A virus infection in mice.
The mouse is the primary experimental model for studying the immunological response to IAV, where it has been demonstrated that excessive inflammatory cytokine and cellular immune responses promote lung pathology (2, 23, 24). We first used this model to assess whether IRF5 impacts influenza virus-induced immune responses during IAV infection in vivo, using the low-pathogenicity murine-adapted H3N2 influenza A virus (A/X-31). Prior studies have indicated that viral infections of Irf5−/− mice lead to reduced cytokine production in comparison to wild-type (WT) controls (14, 17, 25). In accordance, we observed a significant reduction in early cytokine release in Irf5−/− mice, with interleukin 23 (IL-23), IFN-γ, tumor necrosis factor alpha (TNF-α), methyl-accepting chemotaxis protein 1 (MCP-1), IL-6, IL-17A, IL-1α, IL-12p70, granulocyte-macrophage colony-stimulating factor (GM-CSF), IL-1β, and IL-27 all significantly reduced in the bronchoalveolar lavage (BAL) fluid of Irf5−/− mice in comparison to WT controls 2 days postinfection (p.i.) (Fig. 1A), with some cytokines remaining significantly reduced in Irf5−/− mice 4 days p.i. (Fig. 1A). In contrast to other viral infections (17), IFN-α or IFN-β production in response to influenza infection was unaltered (Fig. 1B) at a time point (day 2 p.i.) previously demonstrated to represent the time of significant A/X-31 influenza virus-induced type 1 IFN secretion in this model (26). These data therefore imply that IRF5 selectively modulates the expression of certain influenza virus-induced inflammatory cytokines independently of type I IFNs in mice.
FIG 1.

IRF5 alters cytokine responses to influenza A virus in a murine infection model. WT and Irf5−/− mice were infected intranasally with 3 × 103 A/X-31 influenza virus. (A) Inflammatory cytokine expression in BAL fluid was measured using multiplex assays 2, 4, and 7 days p.i. Data shown are the mean ± SEM using 7 WT and 5 Irf5−/− mice (day 2) or five mice per genotype (day 4 and day 7) and represent the results from two independent experiments. (B) IFN-α and IFN-β levels in BAL fluid measured by ELISA in Irf5−/− and WT naive and IAV-infected mice at 2 days p.i. Data shown are the mean ± SEM of the results from 3 to 6 mice per group at 2 days p.i.
Early reduction in inflammatory cytokine production in Irf5−/− mice was accompanied by a moderate amelioration of IAV-induced weight loss (Fig. 2A). Interestingly, a recent study reported that reduced IAV-induced cytokine production in Irf5−/− mice was associated with reduced virus replication (25). However, at a time point where we observed substantially reduced cytokine production (day 2 p.i.), we observed no alteration in IAV load in Irf5−/− mice (Fig. 2B), nor did we observe altered virus load in Irf5−/− mice at a later time point of 4 days p.i. (Fig. 2B). Thus, our data demonstrate for the first time that IRF5 promotes IAV-induced weight loss independently of an impact on influenza virus replication.
FIG 2.

IRF5 enhances influenza A virus-induced inflammatory response in a murine infection model. (A) Weight loss of WT and Irf5−/− mice was assessed over time, and comparable results were observed in 4 independent experiments, with 4 to 5 WT or Irf5−/− mice in each group per experiment. Data shown are the mean ± SEM. (B) Replication of virus in the lungs was quantified using a plaque assay. Data shown are the mean ± SEM using 7 WT and 5 Irf5−/− mice for day 2 and 5 mice of each genotype for day 4. (C) Recruitment of specific myeloid cell populations (mDCs, monocyte-derived DCs; cDCs, conventional DCs; pDCs, plasmacytoid DCs; Inflam. mon, inflammatory monocytes) in WT and Irf5−/− mice was assessed by flow cytometry 2 days p.i. Populations were defined by the following markers: alveolar macrophages (Alveolar macs), SiglecF+ CD11b+ CD64+ Ly6C−; mDCs, SiglecF− CD11b+ MHC-II+ CD11c+ CD64+ Ly6C+; interstitial macrophages, SiglecF− CD11b+ MHC-II+ CD11c− CD64+ Ly6C+; inflammatory monocytes, SiglecF− CD11b+ MHC-II− Ly6C+ CD64+; cDCs, MHC-II+ CD11c+ Ly6C−; pDCs, B220+ SiglecH+ MHC-IIlow CD11clow; and eosinophils, SiglecF+ CD11c− CD11b+ Ly6C−. Data shown are the mean ± SEM using 11 WT and 10 Irf5−/− mice from multiple replicates. (D) The total number of each individual myeloid cell population (unstimulated, ex vivo) positive for IL-6 and TNF-α expression was detected by flow cytometry, with data presented representing the mean total cell number per 105 cells of each cell type ± SEM. Data represent two experiments.
Given the established role for myeloid cells in pulmonary inflammation during IAV infection (27, 28), we used polychromatic flow cytometry panels to assess whether Irf5 influenced myeloid cell accumulation during infection. Reductions in monocyte-derived DCs, interstitial macrophages, inflammatory monocytes, and conventional DCs in the lungs of Irf5−/− mice were observed at 2 days p.i. (Fig. 2C). Importantly, lower cytokine responses in Irf5−/− mice were accompanied by significant reductions in IL-6+ cDCs and TNF-α+ cDCs, interstitial macrophages, and cDCs and pDCs in the airways (Fig. 2D). Thus, these data suggested that Irf5 plays a key role in shaping the early innate inflammatory response during influenza virus infection and point to a central role for myeloid cells in promoting IRF5-driven viral disease.
Myeloid cells in the human lung express high levels of IRF5.
Although the mouse is a useful model for probing immune responses to IAV, numerous differences exist between the mouse and human immune systems. It was therefore important to investigate the role of human IRF5 in IAV-induced immune responses. We first measured IRF5 expression in multiple cell subsets in human lung samples using CyTOF (Fig. 3A). Using lung samples from four independent donors, we identified significantly different IRF5 expression levels dependent on cell subset (P = 0.0252, R2 = 0.9098) (Fig. 3B), with cells of the myeloid lineage, particularly eosinophils, basophils, and monocytes, displaying the highest expression of IRF5 in the human lung. Expression was higher in CD1c+ DCs and CD141+ DCs than in lung resident macrophages, where expression was relatively low. Furthermore, when IRF5 expression data were combined for all myeloid cell subsets and all lymphoid cell subsets (Fig. 3C), the expression of IRF5 was significantly higher in myeloid cells than in lymphoid cells (median expression of myeloid cells, 6.01; median expression of lymphoid cells, 1.79; P < 0.0001), suggesting that IRF5 expression is highest in the myeloid compartment.
FIG 3.

IRF5 expression in human lung cells. IRF5 expression by multiple cellular subsets derived from human lung tissue from independent donors was analyzed by CyTOF. (A) UMAP based on downsampled, concatenated files from lung samples from four donors using phenotypic markers. Post-UMAP analysis, populations (colored by cell type as identified by lung CyTOF) were defined via the following markers: CD4+ T cells, CD3+ CD4+ CD20−; CD8+ T cells, CD3+ CD20− CD8+; B cells, CD3− CD20+; NK cells, CD3− CD20− CD56+; CD14+ monocytes, CD16− CD11b+ CD14+ HLA-DR+; CD16+ monocytes, CD14− CD11b+ CD16+ HLA-DR+; macrophages, CD11b+ CD68+ HLA-DR+; pDCs, CD123+ CD11b+ HLA-DR+; CD141+ cDCs, CD11b+ HLA-DR+ CD1c− CD141+; CD1c+ cDCs, CD11b+ HLA-DR+ CD1c+ CD141−; eosinophils, Siglec8+ CD123−; and basophils, Siglec8+ CD123+. (B) Median IRF5 expression in populations identified in panel A from lung samples taken from four independent donors, corrected for nonspecific staining using unpermeabilized controls for each sample, and error bars represent the SEM. (C) Median IRF5 expression in myeloid versus lymphoid cell subsets, and error bars represent the SEM.
iPSCs with a biallelic mutation in IRF5 can be differentiated into conventional dendritic cells.
Given that, in the human lung, IRF5 expression was highest in cells of the myeloid lineage and that in mice Irf5 promoted proinflammatory cytokine production by myeloid cells in response to IAV infection, we next sought to establish a human myeloid cell model to scrutinize the role of IRF5 in the myeloid cell cytokine response to IAV. We differentiated an hIPSC line with a biallelic mutation in IRF5 generated using CRISPR-Cas9 genome editing (16) and the parental line Kolf2 into iPS-DCs using a published differentiation strategy (29). We also generated a complemented isogenic control line for IRF5−/− (here, IRF5Comp) to confirm the IRF5 dependency of any phenotypes observed (30). To confirm gene editing strategies, we examined the expression of IRF5 in Kolf2, IRF5−/−, and IRF5Comp iPS-DCs. IRF5 mRNA was detected in Kolf2 iPS-DCs but not IRF5−/− iPS-DCs, with expression restored in IRF5Comp iPS-DCs (Fig. 4A). Similar restoration of IRF5 protein expression in IRF5Comp iPS-DCs was observed (Fig. 4B). Furthermore, after infection of iPS-DCs with IAV, IRF5 was detected in Kolf2 iPS-DCs and not IRF5−/− iPS-DCs by immunostaining (Fig. 4C). We then compared IAV-induced cytokine production by iPS-DCs from our healthy control iPSC line Kolf2 with monocyte-derived DCs from the blood of three healthy donors which were left either immature or matured for 48 h with LPS. iPS-DCs demonstrated cytokine profiles after IAV infection similar to those of immature monocyte-derived DCs (Fig. 4D), validating iPS-DCs as an experimental system for examining virus-induced cytokine responses.
FIG 4.

IRF5−/− iPSCs, IRF5Comp iPSCs, and Kolf2 iPSCs can be differentiated into iPS-DCs which lack or express IRF5. CRISPR-Cas9 was used to generate biallelic mutations in IRF5 in the Kolf2 background. IRF5Comp iPSCs were generated using TALEN-mediated integration of IRF5 into the IRF5−/− background. (A) Relative expression of IRF5 in iPSCs and iPS-DCs relative to GAPDH. Data are shown as four technical replicates per assay, with assays repeated three times from independent iPS-DC batches. (B) Flow cytometry showing IRF5 expression in iPS-DCs generated from IRF5−/−, IRF5Comp, and Kolf2 iPSCs. (C) Immunostaining for IRF5 in A/X-31 influenza (IAV)-infected Kolf2 and IRF5−/− iPS-DCs (DAPI, blue; IRF5, green). (D) IL-6 and TNF-α production 24 h p.i. by IAV-challenged Kolf2 iPS-DCs and monocyte-derived DCs generated from human peripheral blood, either with or without 48 h LPS maturation, were assayed by ELISA. Data represented show the mean ± SEM from three independent Kolf2 differentiations for iPS-DCs, and from three independent healthy donors for monocyte-derived DCs.
It has previously been shown that IRF5 deficiency or transcription activator-like effector nuclease (TALEN)-based engineering targeting the adeno-associated virus integration site 1 (AAVS1) does not affect the differentiation capacity of iPSCs to iPS-derived macrophages (iPSDMs) (16, 31). To ensure that genome editing strategies had not altered the differentiation capacity of iPSCs to dendritic cells, we assessed the differentiation efficiencies of Kolf2, IRF5−/−, and IRF5Comp iPSCs. We observed similar numbers of cells harvested from embryoid bodies (EBs) from days 19 to 24 of differentiation, with no significant difference in the numbers of cells harvested from eight independent differentiations per line (Fig. 5A). After the completion of the 25-day DC differentiation, DC marker expression was examined by flow cytometry, with CD141, CLEC9A, CD11c, major histocompatibility complex class II (MHC-II), and CD86 similarly expressed in all three iPS-DC lines (Fig. 5B). There are three main subsets of human DCs, pDCs and two subsets of myeloid (conventional) DCs, CD1c+ and CD141+, with DC hematopoiesis distinct from the development of monocytes (32). iPS-DCs express markers of human conventional DCs, including CD11c and CD141 (Fig. 5B), as well as HLA-DR, CD86, and CLEC9A, which have been shown to be expressed by human CD141+ DCs (33). We did not detect CD303 expression, a marker for pDCs, or CD1c, the marker for the other subset of human conventional dendritic cells (34). As observed by Sachamitr et al. (29), we also detected CD14 and dendritic cell-specific intercellular adhesion molecule-3-grabbing nonintegrin (DC-SIGN) expression by iPS-DCs (J. L. Forbester and I. R. Humphreys, unpublished data). Gene expression analysis confirmed similar induction of the DC markers CD83 and CD86 in all three iPSC lines after differentiation to iPS-DCs and the loss of expression of the pluripotency markers NANOG and POU5F1 (Fig. 5C). The morphologies of Kolf2, IRF5−/−, and IRF5Comp iPS-DCs in culture were indistinguishable (Fig. 5D). Taken together, these data suggest that neither IRF5 deficiency nor TALEN-based engineering influences iPSC differentiation.
FIG 5.

IRF5−/− iPSCs, IRF5Comp iPSCs, and Kolf2 iPSCs can be differentiated into iPS-DCs that display similar morphologies. IRF5−/− iPSCs, IRF5Comp iPSCs, and Kolf2 iPSCs were differentiated into dendritic cells using defined concentrations of growth factors to generate embryoid bodies (EBs) and GM-CSF and IL-4 to generate immature DCs from these EBs. (A) Total cell numbers of DC precursors harvested from DC differentiation plates. Data shown are from 8 independent differentiations per iPSC line. (B) Surface expression of DC markers was examined via flow cytometry in Kolf2, IRF5−/−, and IRF5Comp iPS-DCs. Representative plots are presented from one experiment, with experiments performed at least three times. (C) Gene expression of DC markers CD83 and CD86 and iPSC markers NANOG and POU5F1 by iPS-DCs relative to GAPDH was quantified using TaqMan gene expression assays. The data shown represent four technical replicates per assay, with assays repeated at least twice from independent iPS-DC batches. (D) Morphologies of iPS-DCs generated from Kolf2, IRF5Comp, and IRF5−/− iPSCs.
IRF5 enhances IAV-induced inflammatory cytokine production in iPS-DCs.
After confirming that IRF5 deficiency did not alter iPS-DC surface phenotype or morphology (Fig. 5) and that iPS-DCs exhibit cytokine profiles similar to those of human monocyte-derived DCs after IAV stimulation (Fig. 4D), we next used iPS-DCs to determine whether IRF5 has a cell-intrinsic role in human DC cytokine production, in particular, the proinflammatory cytokines IL-6 and TNF-α. Despite a protective role for IL-6 being reported in murine models of IAV infection (35, 36), high production of IL-6 is linked to the severity of symptoms in human patient cohorts (37, 38), whereas TNF-α has been shown to enhance cellular inflammation and pathology during IAV infection (23).
Twenty-four hours after stimulation of iPS-DCs with IAV, IL-6 and TNF-α production by IRF5−/− iPS-DCs was significantly reduced in comparison to that in Kolf2 iPS-DCs, whereas cytokine production was restored in IRF5Comp iPS-DCs (Fig. 6A, iPS-DCs). IRF5 deficiency had no impact on virus entry, as indicated by comparable influenza nucleoprotein (NP) staining after 24 h (Fig. 6B and C). In addition, there was no significant difference in cell viability after IAV stimulation, as measured by live viability dye and flow cytometry 24 h p.i. (mean ± standard error of the mean [SEM] percent of live cells: 65.767% ± 0.353% for IRF5Comp, 63.95% ± 1.655% for IRF5−/−, and 70% ± 1.654% for Kolf2). Surface analysis of DC markers by flow cytometry showed that the number of iPS-DCs expressing DC maturation markers after IAV infection was similarly significantly increased in Kolf2, IRF5−/−, and IRF5Comp iPS-DCs (Fig. 6D). Moreover, gene complementation has previously been used to inhibit immune responsiveness in the context of restoration of expression of the inhibitory IL-10 receptor into IL10RB−/− iPS-derived myeloid cells (31). Thus, we do not believe that the restored cytokine responsivess of complemented IRF5−/− cells is a consequence of nonspecific induction of cytokine secretion by the complementation process but instead is due to IRF5 itself. Collectively, these data suggest that IRF5 deficiency selectively alters iPS-DC cytokine production after exposure to IAV. In addition, to probe IRF5 deficiency in a different myeloid cell lineage, we differentiated IRF5−/−, Kolf2, and IRF5Comp iPSCs to macrophages (iPSDMs) using a slightly modified version of a previously published protocol (22), demonstrating a significant reduction in IL-6 and TNF-α production similar to that observed in iPS-DCs (Fig. 6A, iPSDMs).
FIG 6.

IRF5 enhances IAV-induced inflammatory cytokine production in iPS-DCs and iPSDMs. (A) IL-6 and TNF-α were measured by ELISA in supernatants harvested from iPS-DCs and iPSDMs generated from an iPSC line with a biallelic mutation in IRF5, compared to the parent line Kolf2, and a line with a functional IRF5 gene was reintroduced into the AAVS1 integration site by TALEN engineering after infection with IAV at an MOI of 1. Supernatants were harvested at 24 h for assays; data shown represent the mean ± SEM for triplicate wells from at least 3 independent experiments. (B) IRF5−/−, IRF5Comp, and Kolf2 iPS-DCs were infected with IAV at an MOI of 1 and then stained for IAV NP 24 h postinfection and analyzed via flow cytometry. SSC-H, side scatter height. (C) Percentage of positive NP iPS-DCs 24 h postinfection with IAV, with data presented showing the mean ± SEM from three independent experiments. (D) Expression of DC maturation surface markers for iPS-DCs generated from IRF5−/−, Kolf2, or IRF5Comp hIPSCs 24 h postinfection with A/X-31 influenza (IAV) at an MOI of 1, as measured by flow cytometry, with data presented showing the mean ± SEM from three independent experiments.
IRF5 acts downstream of TLR7 and, possibly, RIG-I signaling pathways to drive human myeloid cell cytokine responses to IAV.
In some experimental systems, IRF5 mediates virus-induced production of type I IFN (17, 39). Given that type I IFN is implicated in driving influenza virus-induced inflammatory cytokine responses (26), we assessed whether IRF5 deficiency impacted influenza virus-induced IFN production. Blocking type I IFN reduced IAV-induced IL-6 and TNF-α production, albeit, in the case of IL-6 not to levels produced by IRF5−/− iPS-DCs (Fig. 7A). Furthermore, IRF5 had no impact on type I IFN secretion by iPS-DCs (Fig. 7B). Thus, although type I IFN enhanced IRF5-induced proinflammatory cytokine secretion, the production of type I IFN by iPS-DCs was not an IRF5-regulated process.
FIG 7.
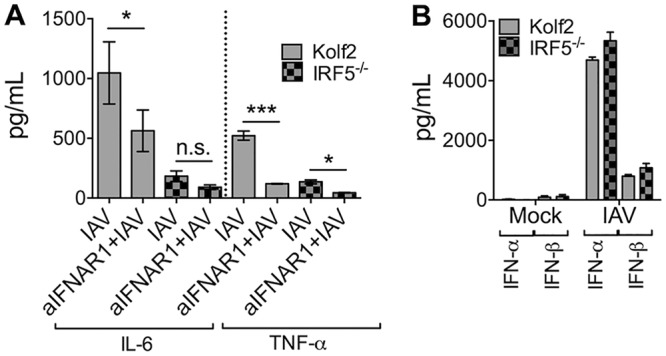
Type I IFN signaling enhances IL-6 and TNF-α production by iPS-DCs. A total of 2 × 104 iPS-DCs were challenged as stated below for each assay, and supernatants were harvested after 24 h, unless otherwise stated. A/X-31 influenza (IAV) was used at an MOI of 1. (A) Cells were preincubated for 1 h with anti-IFNAR1 antibody or left untreated prior to viral infection. Data shown represent the mean ±SEM for triplicate wells from at least 3 experiments. Supernatants were harvested and assayed for IL-6 and TNF-α by ELISA. (B) Supernatants from mock or IAV-infected Kolf2 or IRF5−/− iPS-DCs were harvested at 24 h and assayed for IFN-α and IFN-β by ELISA. Data shown represent 2 separate experiments.
We next investigated which PRRs require IRF5 to elicit cytokine responses following IAV stimulation of iPS-DCs. IAV is detected by endosomal TLR7 and members of the DExDC helicase family and RIG-I in dendritic cells (40–42). Consistent with data from human and murine macrophages (12, 43), IRF5−/− iPS-DCs produced significantly less IL-6 in response to agonists of TLR7 (and TLRs 4, 3, and 9, Fig. 8A). Moreover, stimulation of IRF5−/− iPS-DCs with the RIG-I-specific agonist 3p-hpRNA led to a significant reduction in IL-6 production (Fig. 8B), demonstrating that IRF5 mediates RIG-I- and TLR7-induced responses in iPS-DCs. Substantial IFN-dependent induction of the RIG-I-encoding gene DDX58 and, to a lesser extent, TLR7, was observed upon IAV stimulation of iPS-DCs (Fig. 8C and D), and IRF5 deficiency did not impair baseline PRR expression (Fig. 8E), suggesting that the reduced expression of proinflammatory cytokines in response to TLR7, RIG-I, and IAV stimulation in IRF5−/− iPS-DCs was not a consequence of restricted PRR expression by these cells.
FIG 8.

IRF5 acts downstream of TLR7 and RIG-I to drive inflammatory cytokine responses in iPS-DCs. A total of 2 × 104 iPS-DCs were challenged as stated below for each condition in each assay, and supernatants were harvested after 24 h. A/X-31 influenza virus (IAV) was used at an MOI of 1. For blocking assays, cells were either preincubated for 1 h with inhibitor (IMD 0354, IKKβ inhibitor), or inhibitor was added directly with viral inoculum (ODN 20958, TLR7 inhibitor). Data shown represent the mean ± SEM of the results for triplicate wells from at least 3 experiments, unless otherwise stated. (A) IL-6 production by Kolf2 and IRF5−/− iPS-DCs in response to stimulation with various TLR ligands (TLR2, Pam3CSK4, 300 ng/ml; TLR3, poly(I·C), 50 μg/ml; TLR4, LPS, 50 μg/ml; TLR7, imiquimod, 50 μg/ml; TLR9, ODN 2216, 3 μg/ml) was measured by ELISA. Data shown represent four wells per condition for one iPS-DC batch per line, with assays replicated in two independent experiments. (B) IL-6 response as measured by ELISA in Kolf2 and IRF5−/− iPS-DCs to RIG-I ligand 3p-hpRNA with or without IKKβ inhibitor IMD 0354 and to IAV with or without IMD 0354. (C) Fold change in mRNA levels for TLR7 and DDX58, measured by RT-qPCR using GAPDH as an endogenous control. (D) DDX58 and TLR7 mRNA levels in iPS-DCs after IAV infection with or without blocking of type I IFN signaling using anti-IFNAR1. Data shown represent four technical replicates per assay, with assays repeated at least twice from independent iPS-DC batches. (E) Relative mRNA levels of TLR7 in iPS-DCs generated from IRF5−/− iPSCs or parent Kolf2 iPSCs, measured using RT-qPCR. (F) IL-6 response as measured by ELISA in Kolf2 and IRF5−/− iPS-DCs to A/X-31 influenza virus with or without TLR7 inhibitor ODN 20958.
Finally, we wanted to determine which PRRs mediated IAV-induced cytokine responses in iPS-DCs. Because (i) there is no selective antagonist of RIG-I and (ii) human CD141+ DCs express TLR7 (44), we focused on the role of TLR7 in mediating IAV-induced iPS-DC cytokine responses. The addition of the specific TLR7 antagonist ODN 20958 to IAV-stimulated Kolf2 iPS-DCs significantly abated IL-6 production, whereas TLR7 inhibition in IRF5−/− iPS-DCs did not further reduce IAV-induced IL-6 responses (Fig. 8F). These data suggest that IRF5 promotes TLR7-mediated cytokine responses following IAV detection by human DCs. However, IAV-induced cytokine secretion was incompletely inhibited by TLR7 blockade in Kolf2 iPS-DCs, suggesting that other PRRs contribute to IRF5-mediated responses. IκB kinase beta (IKKβ) has been shown to play a crucial role in IRF5 and NF-κB activation (11). In support of this, pretreatment of IAV-stimulated iPC-DCs with the IKKβ inhibitor IMD 0354 dramatically reduced IAV-induced cytokine production by iPS-DCs (Fig. 8B), implying that other PRRs, including (possibly) RIG-I, contribute to IAV-induced cytokine responses by iPS-DCs in addition to TLR7.
DISCUSSION
Here, using iPS-DCs as a model system, we have shown that IRF5 expression by myeloid cells is important in driving the inflammatory response to IAV without impacting viral uptake by iPS-DCs or DC maturation. Using various blocking assays and stimulation with TLR/RIG-I-like receptor (TLR/RLR) ligands, we show that IRF5 is most likely acting downstream of TLR7 and, possibly, RIG-I signaling to drive the production of proinflammatory cytokines.
Given that IFN-stimulated genes contribute to anti-influenza immunity (5), to identify whether IFR5 and/or related pathways could be safely exploited to dampen inflammatory cytokine responses to influenza virus, it is important to understand the relationship between IRF5 and virus-induced type 1 IFN. We found that IRF5−/− iPS-DCs and Irf5−/− mice were not deficient in type I IFN production but that type I IFN enhances the IRF5-mediated inflammatory cytokine response, a process associated with IFN-mediated induction of TLR7 and RIG-I. Although certain studies have reported a role for IRF5 in type I IFN induction directly in certain contexts (25, 43, 45), functional redundancies between IRF proteins may exist (46). Also, although a role for IRF5 in promoting type 1 IFN secretion following influenza virus infection in vivo has been reported, the same studies observed reduced virus replication in Irf5−/− mice, precluding the possibility of uncoupling decreased virus replication and subsequent pattern recognition receptor stimulation from a direct influence of IRF5 on type 1 IFN expression. Furthermore, early in vivo studies of Irf5 responses to viruses may have been confounded by a Dock2 mutation prevalent in Irf5−/− mouse colonies (46). Also, it has been demonstrated in vitro that unlike IRF3 and IRF7, IRF5 does not bind to the virus-response elements in IFN promoters (47). IRF5-mediated induction of type I IFN may also be virus specific, at least in vitro, with Newcastle disease virus (NDV), vesicular stomatitis virus (VSV), and herpes simplex virus 1 (HSV-1) infection shown to activate IRF5 but lead to the induction of distinct IFNA gene subtypes in human cell lines (48). IRF3 and IRF7 are activated by IAV, and these transcription factors have been shown to be necessary for inducing type I IFN after IAV infection (49, 50). Therefore, we suggest that after sensing of IAV by dendritic cells, IRF3, IRF5, and IRF7 are induced, resulting in the production of type I IFN and inflammatory cytokines, with the type I IFN induced by IRF3 and IRF7 exacerbating IRF5 activation, likely in part through the induction of TLR7 and, possibly, RIG-I expression. In the context of influenza virus pathogenesis, our data imply that IRF5 could be safely targeted to limit virus-induced proinflammatory cytokine production without affecting IFN production and the associated induction of antiviral effector genes.
The phenotype of reduced inflammatory cytokines observed in our iPS-DC model was also evident in vivo using Irf5−/− mice. Although we observed no mortality in either WT or Irf5−/− mice (J. L. Forbester and I. R. Humphreys, unpublished data), we observed reduced cytokine production by myeloid cells that correlated with reduced cellular pulmonary infiltration and a moderate impact on virus-induced weight loss. Although Irf5−/− mice have previously been shown to be less susceptible to IAV-induced pathology (25), in this study, we were able to decouple viral load and inflammatory cytokines in the early stages of infection, demonstrating that the enhanced pathology in WT compared to Irf5−/− mice was immune-mediated rather than a consequence of heightened virus replication. Why differences exist between our data and those obtained by Chen et al. (25) is unclear, although this may reflect the different influenza virus strains (H3N2 versus H1N1) used in these experiments. Irrespective, the data presented herein demonstrate that IRF5 modulates viral pathogenesis via the regulation of inflammation and not virus replication, and that targeting this pathway as an adjunct therapy to antiviral drug treatment may represent an effective therapeutic approach to the treatment of influenza virus pathogenesis.
Although the mouse provides a useful model to study viral pathogenesis, there are inherent immunological differences between mice and humans (51). Therefore, we wanted to establish a human cell system amenable to genetic manipulation, so that gene function can be understood in the context of a human cellular environment. Given that primary human myeloid cells are difficult to genetically manipulate and access in large numbers, hIPSCs offer a solution; once hIPSCs are generated, they can be differentiated down multiple cellular lineages, providing the opportunity to study gene function in multiple different cell types, with a defined genetic background. In addition, as hIPSCs are self-renewing, the starting material is an unlimited resource. Furthermore, many research groups have shown that hIPSCs can be relatively easily genetically manipulated using tools such as zinc finger nucleases, TALENs, and CRISPR-Cas9 systems (52). Here, we show that iPSCs can be differentiated into DCs expressing markers of human CD141+ myeloid DCs. However, the levels of the specific lineage markers for CD141+ DCs, CLEC9A and XCR1, were quite low in our DC populations, which has been previously described (29). The complex environment and array of signals to which DC progenitors are exposed during development present a challenge to replication in vitro. However, a fundamental understanding of human DC development is expanding, and in the future, knowledge of detailed changes in the transcriptional profiles of these cells during development can be applied to help refine differentiation protocols. However, the iPS-DCs described herein expressed multiple DC lineage markers, suggesting that our differentiation protocol is sufficient to derive DC-like cells. In addition, we demonstrated that after differentiation into iPSDMs, IRF5−/− cells are also deficient in IL-6 and TNF-α production, demonstrating for the first time that virus-induced immune responses, including cytokine secretion, can be investigated in iPS-derived cells of multiple myeloid lineages that contain biallelic mutations, thus demonstrating the flexibility of iPSCs as tools to study immune responses to pathogens in multiple cell types.
In two independent studies, response expression quantitative trait loci (reQTLs) were found in IRF5 after stimulation of cells with virus or TLR ligands (53, 54), suggesting that variation within the IRF5 locus may be important in driving differences in expression. It would be interesting in future studies to see if single-nucleotide polymorphisms (SNPs) which drive higher IRF5 expression in human macrophages and DCs also correlate with a heightened inflammatory response to viruses such as IAV, as our data suggest that such individuals may be preferentially susceptible to influenza virus pathogenesis and imply that targeting high IRF5 levels in these individuals could reduce inflammation without impacting virus control. As well as being a useful tool to knock out genes to assess cell-specific function as we have shown here, iPS-DCs could also be used as a tool to explore how common human genetic variants are associated with immune cell responses to various pathogens.
MATERIALS AND METHODS
Mice and viral infections.
Irf5−/− mice were bred in-house on a C57BL/6 background, and their generation has been described previously (12). C57BL/6 WT mice were purchased from Charles River or Envigo. Age- and sex-matched mice between 7 and 12 weeks of age were used in the experiments. Mice were infected intranasally with 3 × 103 PFU A/X-31 influenza virus in 50 μl of sterile phosphate-buffered saline (PBS). Mouse weights were recorded daily, and mice were further monitored for signs of illness.
Plaque assays.
Influenza virus from the lungs of WT and Irf5−/− mice was quantified on Madin-Darby canine kidney (MDCK) cell monolayers after a 5-h incubation at 37°C. Cell layers were then overlaid with methylcellulose (Sigma-Aldrich) and incubated at 37°C for a further 48 h. The medium was then removed, and cell layers were washed, fixed, blocked, and further incubated with anti-influenza A nucleoprotein (clone AA5H; Serotec) and then with anti-mouse IgG-horseradish peroxidase (IgG-HRP; Bio-Rad). Plaques were developed using 3-amino-9-ethylcarbazole (AEC) peroxidase substrate solution and subsequently counted via light microscopy.
Leukocyte isolation, intracellular cytokine staining, and flow cytometry.
BAL fluid and lungs were collected from Irf5−/− and WT mice at days 2, 4, and 7 p.i. Lung digestions were performed by incubation with collagenase solution (RPMI supplemented with 5% fetal bovine serum [FBS], 1 mg/ml collagenase D [Roche], 5 mM CaCl2, 50 mg/ml DNase I [Sigma-Aldrich]), and single-cell suspensions were generated by passing through 100-μm filters. Cells were stained with Zombie Aqua fixable dye, incubated with anti-mouse CD16/CD32 (both BioLegend), and stained for surface markers with a combination of the following antibodies. For murine myeloid panels, cells were stained for surface markers using anti-mouse CD11b-fluorescein isothiocyanate (CD11b-FITC) (M1/70; BioLegend), Ly6C-PerCP/Cy5.5 (HK1.4; BioLegend), SiglecF-PeVio770 (ES22-10D8; Miltenyi Biotec), CD64-Pe/Dazzle (X54-5/7.1; BioLegend), CD45R/B220-APC/Cy7 (RA3-6B2; BioLegend), MHC-II-BV711 (M5/114.15.2; BioLegend), CD11c-BV605 (N418; BioLegend), and SiglecH-Pacific Blue (551; BioLegend). Following surface staining, some cells were fixed and permeabilized with fixation/permeabilization solution (BD Biosciences) and stained with anti-IL-6-phycoerythrin (anti-IL-6-PE) (MP5-20F3; BioLegend) and TNF-α-allophycocyanin (TNF-α-APC) (MP6-XT22; BioLegend). All data were acquired using an Attune NxT flow cytometer (Thermo Fisher Scientific). Electronic compensation was performed with antibody (Ab) capture beads stained separately with individual monoclonal antibodies (MAbs) used in the experimental panel. Data were analyzed using the FlowJo software (TreeStar, Inc.). The total numbers of different cell populations were calculated by multiplying the total number of viable leukocytes (assessed by trypan blue exclusion) by the percentage of positive cells, as detected by flow cytometry.
Mass cytometry staining for IRF5 expression.
Para-tumor lung tissue samples from metastatic cancer or fibrosis patients were extracted and deemed to show no visible signs of inflammation as assessed by a pathologist. Peripheral blood mononuclear cells (PBMCs) from one donor were run with each lung sample to control for interrun variability. One hundred milliliters of heparinized blood was drawn from a healthy control donor, PBMCs were isolated, and aliquots were frozen until use. Directly conjugated antibodies (CD45-89Y, clone HI30; EpCAM-141Pr, clone 9C4; CD31-151Eu, clone EPR3094; CD68-159Tb, clone KP1; Siglec8-164Dy, clone 7C9) were all purchased from Fluidigm. Conjugations for other antibodies (CD11b-142Nd, clone ICRF44; CD4-145Nd, clone RPA-T4; CD20-147Sm, clone 2H7; CD115-152Sm, clone 12-3A3-1810; CD123-155Gd, clone 6H6; CD14-160Gd, clone M5P2; CD56-166Er, clone NCM/HCD56; CD8-168Er, clone SK1; HLA-DR-169Tm, clone L243; CD3-170Er, clone UCHT1; CD1c-171Yb, clone L161; and CD141-173Yb, clone M80) were performed with the Maxpar X8 multimetal labeling kit (Fluidigm), according to the manufacturer’s instructions.
Cells resuspended at 1 × 107 cells/ml were stained with 5 mmol/liter cisplatin (live/dead; Fluidigm) and surface antibody cocktail before permeabilization with Maxpar nuclear antigen staining buffer and staining with anti-IRF5 (conjugate, 175Lu; clone, EPR17067). An unpermeabilized control was treated with cell staining buffer and stained with intracellular antibodies. Cells were stained with 125 nM Ir-Intercalator according to the manufacturer’s protocols (Fluidigm) and fixed with 1.6% formaldehyde. Cells were counted on a BD Accuri C6 cytometer and resuspended at 2 × 106 cells/ml in 0.1× EQ four-element calibration beads (Fluidigm). Cells were acquired using a CyTOF Helios cytometer (Fluidigm). Data were processed and normalized using the CyTOF software v6.7 (Fluidigm). Data were analyzed using FlowJo (TreeStar, Inc.).
Mass cytometry analysis.
CyTOF data were analyzed using FlowJo 10.5.2 (TreeStar, Inc.). After gating on live, intact, singlet cells, CD45 versus EpCAM expression was used to identify epithelial cells (CD45− EpCAM+) or immune cells (CD45+ EpCAM−). CD45+ EpCAM− cells were downsampled to a maximum of 200,000 per sample, with stained and control samples concatenated into one file. The concatenated file was run through a uniform manifold approximation and projection (UMAP) analysis of the surface markers CD11b, CD4, CD20, CD123, CD68, CD14, Siglec8, CD56, CD8, HLA-DR, CD3, CD1c, CD141, and CD16. Post-UMAP analysis, distinct cell subsets were identified by mapping the expression of specific subset markers back onto the UMAP to define populations of immune cells. Subsets were defined as follows: CD4+ T cells, CD3+ CD4+ CD20−; CD8+ T cells, CD3+ CD20− CD8+; B cells, CD3− CD20+; NK cells, CD3− CD20− CD56+; CD14+ monocytes, CD16− CD11b+ CD14+ HLA-DR+; CD16+ monocytes, CD14− CD11b+ CD16+ HLA-DR+; macrophages, CD11b+ CD68+ HLA-DR+; pDCs, CD123+ CD11b+ HLA-DR+; CD141+ cDCs, CD11b+ HLA-DR+ CD1c− CD141+; CD1c+ cDCs, CD11b+ HLA-DR+ CD1c+ CD141−; eosinophils, Siglec8+ CD123−; basophils, Siglec8+ CD123+; and epithelial cells, CD45− EpCAM+. Individual samples were identified by gating on event length versus sample identifier (ID), and the median value was determined for IRF5 for each individual sample.
Generation of blood-derived human dendritic cells.
PBMCs from three independent donors were isolated from leukapheresis products using Lymphoprep density gradient centrifugation and SepMate PBMC isolation tubes (StemCell Technologies), under the Weatherall Institute of Molecular Medicine, University of Oxford Human Tissue Authority license 12433. Human CD14 microbeads were used in combination with LS columns (both Miltenyi Biotec) to positively select CD14+ blood monocytes. CD14+ cells were seeded at a density of 3 × 106 to 5 × 106 isolated monocytes in 3 ml of RPMI medium supplemented with 10% heat-inactivated fetal bovine serum (FBS; Sigma-Aldrich), 250 IU/ml IL-4, and 800 IU/ml GM-CSF into a 6-well plate and incubated at 37°C for 2 days. After 2 days, 1.5 ml of medium was removed from each well, and 1.5 ml of fresh medium supplemented with 500 IU/ml IL-4 and 1,600 IU/ml GM-CSF was added. After a further 3-day incubation, cells were harvested at the immature phenotype and assayed or further matured with LPS at 10 μg/ml for 24 h.
hIPSCs.
The healthy control hIPSC line Kolf2 was acquired through the Human Induced Pluripotent Stem Cells Initiative Consortium (HipSci; http://www.hipsci.org/), through which it was also characterized (55). The generation of IRF5−/− iPSCs has been previously described (16). Briefly, Kolf2 iPSCs were dissociated to single cells and nucleofected (Amaxa2b nucleofector; Lonza) with Cas9 coding plasmid (hCas9; Addgene 41815), single guide RNA (sgRNA) plasmid (CRISPR guides, left CRISPR_IRF5, CCAAGTGGAAGGCCAACCTGCGC, and right CRISPR_IRF5, GACTTCCGCCTCATCTACGACGG), and donor plasmid containing 5′ and 3′ homology arms for IRF5 targeting and the pL1-EF1αPuro-L2 cassette. Postnucleofection, cells were selected for up to 11 days with 0.25 μg/ml puromycin, after which individual colonies were picked onto 96-well plates, grown to confluence, and then replica plated. To genotype individual clones from 96-well replica plates, cells were lysed and used for PCR amplification with LongAmp Taq DNA polymerase (NEB). Insertion of the cassette into the correct locus was confirmed by visualizing on 1% E-gel (Life Technologies). PCR products were generated by gene-specific and cassette-specific primers, with single integration of cassette confirmed by a quantitative PCR (qPCR) copy number assay. Positive clones were then screened for damage to the nontargeted allele via PCR and Sanger sequencing. To generate our complemented IRF5 iPSC line (IRF5Comp) and restore the expression of functional IRF5 in the IRF5−/− iPSCs, we generated the AAVS1 EF1a-IRF5-PGK-puro targeting vector by Gibson Assembly. The Gibson Assembly product was transformed into One Shot TOP10 chemically competent Escherichia coli cells (Thermo Fisher Scientific), and positive colonies were picked. Isolated plasmids from the positive colonies were taken to confirm the presence and sequence of elongation factor 1 alpha (EF1α)-IRF5 in the targeting vector by restriction digestions, PCR, and sequencing. Subsequently, the targeting vector was transformed into competent E. coli cells to isolate endotoxin-free plasmids to transform into the IRF5−/− iPSCs. We transfected the mutant human hIPSCs with TALEN-L (CCCCTCCACCCCACAGT), TALEN-R (AGGATTGGTGACAGAAA), and targeting vector via nucleofection (Amaxa Biosystems). The resultant targeted cells were selected on puromycin for 7 days, and the surviving colonies were picked and expanded. The positive clones were confirmed by reverse transcription-quantitative PCR (RT-qPCR) for IRF5 expression and flow cytometry for protein expression. Prior to differentiation, iPSCs were grown feeder free using the Essential 8 Flex medium kit (Thermo Fisher Scientific) on vitronectin (VTN-N; Thermo Fisher Scientific)-coated plates, as per the manufacturer’s instructions, to 70 to 80% confluence. iPSCs were harvested for differentiation using Versene solution (Thermo Fisher Scientific).
Differentiation of iPSCs to dendritic cells and macrophages.
To differentiate iPSCs to dendritic cells, slight modifications were made to a previously published protocol (29). Briefly, upon reaching confluence, iPSCs were harvested and plated into Essential 8 Flex medium supplemented with 50 ng/ml bone morphogenetic protein 4 (BMP-4; Bio-Techne), 20 ng/ml stem cell factor (SCF; Bio-Techne), 50 ng/ml vascular endothelial growth factor (VEGF; Peprotech EC Ltd.), and 50 ng/ml GM-CSF (Peprotech EC Ltd.) in ultralow attachment (ULA) plates (Corning). The medium was changed to X-VIVO-15 (Lonza), with sequential removal of BMP-4 by day 5, VEGF by approximately day 14, and SCF by approximately day 19. In addition, IL-4 (Peprotech EC Ltd.) was added sequentially in increasing concentrations, starting from approximately day 12 at 25 ng/ml and increasing to 100 ng/ml by approximately day 20. By day 20, floating immature DCs were harvested from ULA plates, filtered through 70-μm filters (Corning), counted, and seeded at 1 × 106 per well of 6-well CellBind plates (Corning) in X-VIVO-15 medium supplemented with 100 ng/ml IL-4 and 50 ng/ml GM-CSF. iPS-DCs were used for assays at the immature phase between 4 and 5 days postseeding in CellBind plates. In addition, iPS-DCs could be matured for a further 48 h at 5 days postplating by the addition of 50 ng/ml GM-CSF, 100 ng/ml IL-4, 20 ng/ml IFN-γ (Bio-Techne), 50 ng/ml TNF-α (Bio-Techne), 10 ng/ml IL-1β (Bio-Techne), and 1 μg/ml prostaglandin E2 (PGE2; Sigma-Aldrich) to induce further expression of CD141+ DC lineage markers. For the assays, floating iPS-DCs were harvested from differentiation plates, washed with PBS, counted, and seeded in X-VIVO-15 medium without cytokines at an assay-dependent concentration. To differentiate iPSCs to macrophages, the approaches of Hale et al. (22) and van Wilgenburg et al. (56) were modified to allow for feeder-free differentiation. Briefly, upon reaching confluence, human iPSCs were collected and transferred into Essential 8 Flex medium supplemented with 50 ng/ml BMP-4 (Bio-Techne), 20 ng/ml SCF (Bio-Techne), and 50 ng/ml VEGF (Peprotech EC Ltd.) in ultralow attachment plates (Corning) for 4 days to generate embryoid bodies (EBs). On day 5, EBs were used for the generation of myeloid precursor cells by plating into 6-well tissue culture-treated plates (Corning) coated for 2 h at room temperature with 0.1% gelatin in X-VIVO-15 medium supplemented with 25 ng/ml IL-3 (Bio-Techne) and 50 ng/ml M-CSF (Bio-Techne). After several weeks, floating myeloid precursors were harvested and terminally differentiated into matured macrophages (iPSDMs) in the presence of higher concentrations of macrophage colony-stimulating factor (M-CSF; 100 ng/ml) for 7 days. For the experiments, macrophages were detached using Lidocaine solution (4 mg/ml lidocaine-HCl with 10 mM EDTA in PBS) and seeded at 2 × 105 cells per well (24-well plate) or 1 × 106 cells per well (six-well plate).
Preparation of RNA and RT-qPCR.
iPS-DCs were harvested from the plates, and RNA was prepared using the RNeasy minikit (Qiagen). RNA was reverse transcribed with the QuantiTect reverse transcription (RT) kit (Qiagen), according to the manufacturer’s protocol. All RT-qPCR experiments were performed with TaqMan gene expression assays and TaqMan gene expression master mix (Applied Biosystems) on the Applied Biosystems StepOne real-time PCR system. RT-qPCR data were analyzed via the comparative threshold cycle (CT) method with glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as an endogenous control.
Flow cytometric analysis of iPS-DCs.
For analysis of surface markers on iPS-DCs, cells were stained with Zombie Aqua fixable dye (BioLegend), Fc receptors were blocked using human TruStain FcX (BioLegend), and cells were then subsequently stained for surface markers with a combination of the following antibodies: anti-human HLA-DR-Alexa Fluor 488 (AF488) (L243; BioLegend) or CD14-FITC (M5E2; BioLegend), CD83-PerCP/Cy5.5 (HB15e; BioLegend) or CD1c-PerCP/Cy5.5 (L161; BioLegend), CD141-PE/Cy7 (M80; BioLegend) or DC-SIGN-PE/Cy7 (9E9A8; BioLegend), or XCR1-PE (FAB8571; Bio-Techne), CD11c-APC/Cy7 (Bu15; BioLegend), CLEC9A-APC (8F9; BioLegend), CD86-BV711 (IT2.2; BioLegend), CD303-BV785 (201A; BioLegend) or HLA-DR-BV785 (L243; BioLegend), and HLA-A,B,C-Pacific Blue (W6/32; BioLegend). For the detection of IRF5 or IAV nucleoprotein, cells were stained with Zombie Aqua fixable dye, fixed with 4% paraformaldehyde, and permeabilized with 0.5% Triton X-100, followed by incubation with human TruStain FcX and staining with IRF5-AF647 (EPR6094; Abcam) or anti-influenza A virus nucleoprotein antibody (431; Abcam) in 0.1% Triton X-100 solution. All data were acquired using an LSRFortessa flow cytometer (BD Biosciences). Electronic compensation was performed with Ab capture beads stained separately with individual MAbs used in the experimental panel. Data were analyzed using the FlowJo software (TreeStar, Inc.).
Infection of iPS-DCs and iPSDMs with IAV.
iPS-DCs, iPSDMs, or human monocyte-derived DCs (mDCs) were infected with A/X-31 influenza virus at a multiplicity of infection (MOI) of 1 by the addition of virus to culture supernatant and centrifugation at 630 × g for 20 min at room temperature, after which the medium was replaced with fresh culture medium.
Immunostaining for confocal microscopy.
iPS-DCs were harvested from plates and spun onto slides coated with 0.01% poly-l-lysine using a Cytospin cytocentrifuge. Samples were blocked and permeabilized in 2% Triton X-100 (Sigma-Aldrich) in 5% FBS diluted in PBS. Primary antibodies were applied at room temperature in 0.25% Triton X-100 in 5% FBS diluted in PBS for 1 h and then rinsed 3 times with PBS. Secondary antibodies were applied in the same manner. Nuclei were counterstained with 10 nM DAPI (4′,6-diamidino-2-phenylindole) dilactate diluted in PBS for 30 min, and samples were rinsed 6 times with PBS, mounted in ProLong Gold reagent with added DAPI (Invitrogen), and analyzed using a Leica SP8 digital light sheet (DLS) microscope.
TLR/RIG-I stimulations.
iPS-DCs were plated at 1 × 104 cells per well in 200 μl of X-VIVO-15 medium without cytokines. TLR ligands were added directly to the medium, and supernatants were harvested after a 24-h incubation at 37°C. For the assays, TLR ligands were used at the following concentrations: Pam3CSK4, 300 ng/ml (InvivoGen); poly(I·C), 50 μg/ml (InvivoGen); lipopolysaccharide, 500 ng/ml (Sigma-Aldrich); imiquimod, 50 μg/ml (InvivoGen); and ODN 2216, 3 μg/ml (Miltenyi Biotech). For RIG-I stimulation, 1 μg of 3p-hpRNA was complexed with LyoVec (InvivoGen) for 15 min at room temperature and then added to iPS-DCs at 10 ng/ml.
Cytokine and chemokine analysis.
Human IL-6 and TNF-α protein were measured by an enzyme-linked immunosorbent assay (ELISA) (BioLegend). ELISAs for human IFN-α and IFN-β were performed on supernatants harvested from mock-infected and IAV-infected iPS-DCs using the VeriKine human interferon alpha/interferon beta ELISA kit (PBL Assay Science). Murine BAL fluid cytokines were detected using the LEGENDPlex mouse inflammation panel (13-plex; BioLegend), as per the manufacturer’s instructions, at 2, 4, and 6 days p.i. and analyzed using the LEGENDPlex analysis software. ELISAs for mouse IFN-α and IFN-β were performed on BAL fluid from naive mice and mice at 2 days p.i. using VeriKine mouse interferon alpha/interferon beta ELISA kits (PBL Assay Science).
Blocking assays.
For blocking assays, cells were either preincubated for 1 h with 5 μg/ml inhibitor (IMD 0354, IKKβ inhibitor; Santa Cruz Biotechnology), or inhibitor was added directly with 5 μM viral inoculum (TLR7 inhibitor, ODN 20958; Miltenyi Biotech). For type I IFN blocking, cells were preincubated with 5 μg/ml anti-IFNAR1 antibody (Sigma-Aldrich) for 1 h prior to infection with A/X-31 influenza virus without removal of antibody.
Statistical analysis.
Statistical significance was performed using the GraphPad Prism software. The Mann-Whitney U or Student’s t test was used for two-group comparisons. For comparison of IRF5 expression levels between lung cell subsets identified via CyTOF, a repeated-measures one-way analysis of variance (ANOVA) was used. A P value of ≤0.05 was considered to be significant. For all tests performed, P values are reported as follows: n.s., >0.05; *, ≤0.05; **, ≤0.01; ***, ≤0.001; and ****, ≤0.0001.
Ethics statement.
All animal studies were performed at Cardiff University (Heath Park research support facility) under UK Home Office Project License number P7867DADD, as approved by the UK Home Office, London, United Kingdom. Written consent was obtained for the use of cell lines for the HipSci project from healthy volunteers. A favorable ethical opinion was granted by the National Research Ethics Service (NRES) Research Ethics Committee Yorkshire and The Humber-Leeds West, under reference number 15/YH/0391. Lung tissue samples were obtained from lung cancer and fibrosis patients from Oxford Radcliffe Biobank with written consent; a favorable ethical opinion was granted by the South Central-Oxford C Research Ethics Committee for the collection and frozen storage of both tumor and para-tumor lung samples (reference number 09/H0606/5 + 5).
ACKNOWLEDGMENTS
This work was funded by a Wellcome Trust Senior Research Fellowship to I.R.H. (grant 207503/Z/17/Z); the Medical Research Council, United Kingdom (grant MR/L018942/1 and MRC Human Immunology Unit Core); and the Chinese Academy of Medical Sciences (CAMS) Innovation Fund for Medical Sciences (CIFMS), China (grant 2018-I2M-2-002). The Wellcome Trust Sanger Institute was the source of the Kolf2 human induced pluripotent cell line, which was generated under the Human Induced Pluripotent Stem Cell Initiative funded by a grant from the Wellcome Trust and the Medical Research Council, supported by the Wellcome Trust (grant WT098051) and the NIHR/Wellcome Trust Clinical Research Facility.
The Life Science Technologies Corporation provided Cytotune for reprogramming. We thank the Wellcome Trust Sanger Institute Gene editing pipeline for generation of IRF5−/− iPSCs and the Mass Spectrometry Facility at the Weatherall Institute of Molecular Medicine for help with CyTOF experiments.
J.L.F., I.R.H., M.C., S.D., M.M., and L.C. performed experiments in mice. J.L.F., E.L., E.L.C., C.T., S.C., and C.H. performed experiments using human iPSCs. A.Y. generated TALEN-engineered IRF5−/− iPSCs. J.L.F. and D.W. performed experiments using human blood-derived DCs. D.W. conducted CyTOF experiments and analyzed the CyTOF data. I.U. provided Irf5−/− mice. J.L.F., I.R.H., I.U., M.C., and D.W. designed the experiments. I.R.H., T.D., and G.D. supervised the study. J.L.F. and I.R.H. wrote the manuscript. All authors approved the final manuscript.
REFERENCES
- 1.de Jong MD, Simmons CP, Thanh TT, Hien VM, Smith GJD, Chau TNB, Hoang DM, Van Vinh Chau N, Khanh TH, Dong VC, Qui PT, Van Cam B, Ha DQ, Guan Y, Peiris JSM, Chinh NT, Hien TT, Farrar J. 2006. Fatal outcome of human influenza A (H5N1) is associated with high viral load and hypercytokinemia. Nat Med 12:1203–1207. doi: 10.1038/nm1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Humphreys IR, Walzl G, Edwards L, Rae A, Hill S, Hussell T. 2003. A critical role for OX40 in T cell-mediated immunopathology during lung viral infection. J Exp Med 198:1237–1242. doi: 10.1084/jem.20030351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.GeurtsvanKessel CH, Willart MAM, van Rijt LS, Muskens F, Kool M, Baas C, Thielemans K, Bennett C, Clausen BE, Hoogsteden HC, Osterhaus A, Rimmelzwaan GF, Lambrecht BN. 2008. Clearance of influenza virus from the lung depends on migratory langerin+ CD11b− but not plasmacytoid dendritic cells. J Exp Med 205:1621–1634. doi: 10.1084/jem.20071365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu J, Cao X. 2016. Cellular and molecular regulation of innate inflammatory responses. Cell Mol Immunol 13:711–721. doi: 10.1038/cmi.2016.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Iwasaki A, Pillai PS. 2014. Innate immunity to influenza virus infection. Nat Rev Immunol 14:315–328. doi: 10.1038/nri3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tavares LP, Teixeira MM, Garcia CC. 2017. The inflammatory response triggered by influenza virus: a two edged sword. Inflamm Res 66:283–302. doi: 10.1007/s00011-016-0996-0. [DOI] [PubMed] [Google Scholar]
- 7.Khoyratty TE, Udalova IA. 2018. Diverse mechanisms of IRF5 action in inflammatory responses. Int J Biochem Cell Biol 99:38–42. doi: 10.1016/j.biocel.2018.03.012. [DOI] [PubMed] [Google Scholar]
- 8.Krausgruber T, Saliba D, Ryzhakov G, Lanfrancotti A, Blazek K, Udalova IA. 2010. IRF5 is required for late-phase TNF secretion by human dendritic cells. Blood 115:4421–4430. doi: 10.1182/blood-2010-01-263020. [DOI] [PubMed] [Google Scholar]
- 9.Douagi I, McInerney GM, Hidmark AS, Miriallis V, Johansen K, Svensson L, Karlsson Hedestam GB. 2007. Role of interferon regulatory factor 3 in type I interferon responses in rotavirus-infected dendritic cells and fibroblasts. J Virol 81:2758–2768. doi: 10.1128/JVI.01555-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, Shimada N, Ohba Y, Takaoka A, Yoshida N, Taniguchi T. 2005. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature 434:772–777. doi: 10.1038/nature03464. [DOI] [PubMed] [Google Scholar]
- 11.Ren J, Chen X, Chen ZJ. 2014. IKKβ is an IRF5 kinase that instigates inflammation. Proc Natl Acad Sci U S A 111:17438–17443. doi: 10.1073/pnas.1418516111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Takaoka A, Yanai H, Kondo S, Duncan G, Negishi H, Mizutani T, Kano S, Honda K, Ohba Y, Mak TW, Taniguchi T. 2005. Integral role of IRF-5 in the gene induction programme activated by Toll-like receptors. Nature 434:243–249. doi: 10.1038/nature03308. [DOI] [PubMed] [Google Scholar]
- 13.Eames HL, Corbin AL, Udalova IA. 2016. Interferon regulatory factor 5 in human autoimmunity and murine models of autoimmune disease. Transl Res 167:167–182. doi: 10.1016/j.trsl.2015.06.018. [DOI] [PubMed] [Google Scholar]
- 14.Thackray LB, Shrestha B, Richner JM, Miner JJ, Pinto AK, Lazear HM, Gale M, Diamond MS. 2014. Interferon regulatory factor 5-dependent immune responses in the draining lymph node protect against West Nile virus infection. J Virol 88:11007–11021. doi: 10.1128/JVI.01545-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Paun A, Bankoti R, Joshi T, Pitha PM, Stäger S. 2011. Critical role of IRF-5 in the development of T helper 1 responses to Leishmania donovani infection. PLoS Pathog 7:e1001246. doi: 10.1371/journal.ppat.1001246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yeung ATY, Hale C, Lee AH, Gill EE, Bushell W, Parry-Smith D, Goulding D, Pickard D, Roumeliotis T, Choudhary J, Thomson N, Skarnes WC, Dougan G, Hancock R. 2017. Exploiting induced pluripotent stem cell-derived macrophages to unravel host factors influencing Chlamydia trachomatis pathogenesis. Nat Commun 8:15013. doi: 10.1038/ncomms15013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yanai H, Chen H.-m, Inuzuka T, Kondo S, Mak TW, Takaoka A, Honda K, Taniguchi T. 2007. Role of IFN regulatory factor 5 transcription factor in antiviral immunity and tumor suppression. Proc Natl Acad Sci U S A 104:3402–3407. doi: 10.1073/pnas.0611559104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mancl ME, Hu G, Sangster-Guity N, Olshalsky SL, Hoops K, Fitzgerald-Bocarsly P, Pitha PM, Pinder K, Barnes BJ. 2005. Two discrete promoters regulate the alternatively spliced human interferon regulatory factor-5 isoforms. J Biol Chem 280:21078–21090. doi: 10.1074/jbc.M500543200. [DOI] [PubMed] [Google Scholar]
- 19.Lin KL, Suzuki Y, Nakano H, Ramsburg E, Gunn MD. 2008. CCR2+ monocyte-derived dendritic cells and exudate macrophages produce influenza-induced pulmonary immune pathology and mortality. J Immunol 180:2562–2572. doi: 10.4049/jimmunol.180.4.2562. [DOI] [PubMed] [Google Scholar]
- 20.Ellis GT, Davidson S, Crotta S, Branzk N, Papayannopoulos V, Wack A. 2015. TRAIL+ monocytes and monocyte‐related cells cause lung damage and thereby increase susceptibility to influenza-Streptococcus pneumoniae coinfection. EMBO Rep 16:1203–1218. doi: 10.15252/embr.201540473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Osugi Y, Vuckovic S, Hart D. 2002. Myeloid blood CD11c+ dendritic cells and monocyte-derived dendritic cells differ in their ability to stimulate T lymphocytes. Blood 100:2858–2866. doi: 10.1182/blood.V100.8.2858. [DOI] [PubMed] [Google Scholar]
- 22.Hale C, Yeung A, Goulding D, Pickard D, Alasoo K, Powrie F, Dougan G, Mukhopadhyay S. 2015. Induced pluripotent stem cell derived macrophages as a cellular system to study Salmonella and other pathogens. PLoS One 10:e0124307. doi: 10.1371/journal.pone.0124307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hussell T, Pennycook A, Openshaw P. 2001. Inhibition of tumor necrosis factor reduces the severity of virus-specific lung immunopathology. Eur J Immunol 31:2566–2573. doi:. [DOI] [PubMed] [Google Scholar]
- 24.Vlahos R, Stambas J, Bozinovski S, Broughton BRS, Drummond GR, Selemidis S. 2011. Inhibition of Nox2 oxidase activity ameliorates influenza A virus-induced lung inflammation. PLoS Pathog 7:e1001271. doi: 10.1371/journal.ppat.1001271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen X, Zhou L, Peng N, Yu H, Li M, Cao Z, Lin Y, Wang X, Li Q, Wang J, She Y, Zhu C, Lu M, Zhu Y, Liu S. 2017. MicroRNA-302a suppresses influenza A virus-stimulated interferon regulatory factor-5 expression and cytokine storm induction. J Biol Chem 292:21291–21303. doi: 10.1074/jbc.M117.805937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Davidson S, Crotta S, McCabe TM, Wack A. 2014. Pathogenic potential of interferon αβ in acute influenza infection. Nat Commun 5:3864. doi: 10.1038/ncomms4864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu L, Yoon H, Zhao MQ, Liu J, Ramana CV, Enelow RI. 2004. Cutting edge: pulmonary immunopathology mediated by antigen-specific expression of TNF-α by antiviral CD8+ T cells. J Immunol 173:721–725. doi: 10.4049/jimmunol.173.2.721. [DOI] [PubMed] [Google Scholar]
- 28.Song M-S, Cho Y-H, Park S-J, Pascua PNQ, Baek YH, Kwon H-I, Lee O-J, Kong B-W, Kim H, Shin E-C, Kim C-J, Choi YK. 2013. Early regulation of viral infection reduces inflammation and rescues Mx-positive mice from lethal avian influenza infection. Am J Pathol 182:1308–1321. doi: 10.1016/j.ajpath.2012.12.022. [DOI] [PubMed] [Google Scholar]
- 29.Sachamitr P, Leishman AJ, Davies TJ, Fairchild PJ. 2018. Directed differentiation of human induced pluripotent stem cells into dendritic cells displaying tolerogenic properties and resembling the CD141+ subset. Front Immunol 8:1935. doi: 10.3389/fimmu.2017.01935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Forbester JL, Lees EA, Goulding D, Forrest S, Yeung A, Speak A, Clare S, Coomber EL, Mukhopadhyay S, Kraiczy J, Schreiber F, Lawley TD, Hancock REW, Uhlig HH, Zilbauer M, Powrie F, Dougan G. 2018. Interleukin-22 promotes phagolysosomal fusion to induce protection against Salmonella enterica Typhimurium in human epithelial cells. Proc Natl Acad Sci U S A 115:10118–10123. doi: 10.1073/pnas.1811866115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mukhopadhyay S, Heinz E, Porreca I, Alasoo K, Yeung A, Yang H-T, Schwerd T, Forbester JL, Hale C, Agu CA, Choi YH, Rodrigues J, Capitani M, Jostins-Dean L, Thomas DC, Travis S, Gaffney D, Skarnes WC, Thomson N, Uhlig HH, Dougan G, Powrie F. 2020. Loss of IL-10 signaling in macrophages limits bacterial killing driven by prostaglandin E2. J Exp Med 217:e20180649. doi: 10.1084/jem.20180649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Collin M, Bigley V. 2018. Human dendritic cell subsets: an update. Immunology 154:3–20. doi: 10.1111/imm.12888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Colletti NJ, Liu H, Gower AC, Alekseyev YO, Arendt CW, Shaw MH. 2016. TLR3 signaling promotes the induction of unique human BDCA-3 dendritic cell populations. Front Immunol 7:88. doi: 10.3389/fimmu.2016.00088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Breton G, Zheng S, Valieris R, Tojal da Silva I, Satija R, Nussenzweig MC. 2016. Human dendritic cells (DCs) are derived from distinct circulating precursors that are precommitted to become CD1c+ or CD141+ DCs. J Exp Med 213:2861–2870. doi: 10.1084/jem.20161135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lauder SN, Jones E, Smart K, Bloom A, Williams AS, Hindley JP, Ondondo B, Taylor PR, Clement M, Fielding C, Godkin AJ, Jones SA, Gallimore AM. 2013. Interleukin-6 limits influenza-induced inflammation and protects against fatal lung pathology. Eur J Immunol 43:2613–2625. doi: 10.1002/eji.201243018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dienz O, Rud JG, Eaton SM, Lanthier PA, Burg E, Drew A, Bunn J, Suratt BT, Haynes L, Rincon M. 2012. Essential role of IL-6 in protection against H1N1 influenza virus by promoting neutrophil survival in the lung. Mucosal Immunol 5:258–266. doi: 10.1038/mi.2012.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kaiser L, Fritz RS, Straus SE, Gubareva L, Hayden FG. 2001. Symptom pathogenesis during acute influenza: interleukin-6 and other cytokine responses. J Med Virol 64:262–268. doi: 10.1002/jmv.1045. [DOI] [PubMed] [Google Scholar]
- 38.Hayden FG, Fritz R, Lobo MC, Alvord W, Strober W, Straus SE. 1998. Local and systemic cytokine responses during experimental human influenza A virus infection. Relation to symptom formation and host defense. J Clin Invest 101:643–649. doi: 10.1172/JCI1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lopez-Pelaez M, Lamont DJ, Peggie M, Shpiro N, Gray NS, Cohen P. 2014. Protein kinase IKKβ-catalyzed phosphorylation of IRF5 at Ser462 induces its dimerization and nuclear translocation in myeloid cells. Proc Natl Acad Sci U S A 111:17432–17437. doi: 10.1073/pnas.1418399111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pang IK, Pillai PS, Iwasaki A. 2013. Efficient influenza A virus replication in the respiratory tract requires signals from TLR7 and RIG-I. Proc Natl Acad Sci U S A 110:13910–13915. doi: 10.1073/pnas.1303275110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang Z, Yuan B, Lu N, Facchinetti V, Liu Y-J. 2011. DHX9 pairs with IPS-1 to sense double-stranded RNA in myeloid dendritic cells. J Immunol 187:4501–4508. doi: 10.4049/jimmunol.1101307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kandasamy M, Suryawanshi A, Tundup S, Perez JT, Schmolke M, Manicassamy S, Manicassamy B. 2016. RIG-I signaling is critical for efficient polyfunctional T cell responses during influenza virus infection. PLoS Pathog 12:e1005754. doi: 10.1371/journal.ppat.1005754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schoenemeyer A, Barnes BJ, Mancl ME, Latz E, Goutagny N, Pitha PM, Fitzgerald KA, Golenbock DT. 2005. The interferon regulatory factor, IRF5, is a central mediator of Toll-like receptor 7 signaling. J Biol Chem 280:17005–17012. doi: 10.1074/jbc.M412584200. [DOI] [PubMed] [Google Scholar]
- 44.Silvin A, Yu CI, Lahaye X, Imperatore F, Brault J-B, Cardinaud S, Becker C, Kwan W-H, Conrad C, Maurin M, Goudot C, Marques-Ladeira S, Wang Y, Pascual V, Anguiano E, Albrecht RA, Iannacone M, García-Sastre A, Goud B, Dalod M, Moris A, Merad M, Palucka AK, Manel N. 2017. Constitutive resistance to viral infection in human CD141+ dendritic cells. Sci Immunol 2:eaai8071. doi: 10.1126/sciimmunol.aai8071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lazear HM, Lancaster A, Wilkins C, Suthar MS, Huang A, Vick SC, Clepper L, Thackray L, Brassil MM, Virgin HW, Nikolich-Zugich J, Moses AV, Gale M, Früh K, Diamond MS. 2013. IRF-3, IRF-5, and IRF-7 coordinately regulate the type I IFN response in myeloid dendritic cells downstream of MAVS signaling. PLoS Pathog 9:e1003118. doi: 10.1371/journal.ppat.1003118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Purtha WE, Swiecki M, Colonna M, Diamond MS, Bhattacharya D. 2012. Spontaneous mutation of the Dock2 gene in Irf5−/− mice complicates interpretation of type I interferon production and antibody responses. Proc Natl Acad Sci U S A 109:E898–E904. doi: 10.1073/pnas.1118155109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Andrilenas KK, Ramlall V, Kurland J, Leung B, Harbaugh AG, Siggers T. 2018. DNA-binding landscape of IRF3, IRF5 and IRF7 dimers: implications for dimer-specific gene regulation. Nucleic Acids Res 46:2509–2520. doi: 10.1093/nar/gky002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Barnes BJ, Kellum MJ, Field AE, Pitha PM. 2002. Multiple regulatory domains of IRF-5 control activation, cellular localization, and induction of chemokines that mediate recruitment of T lymphocytes. Mol Cell Biol 22:5721–5740. doi: 10.1128/mcb.22.16.5721-5740.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hatesuer B, Hoang HTT, Riese P, Trittel S, Gerhauser I, Elbahesh H, Geffers R, Wilk E, Schughart K. 2017. Deletion of Irf3 and Irf7 genes in mice results in altered interferon pathway activation and granulocyte-dominated inflammatory responses to influenza A infection. J Innate Immun 9:145–161. doi: 10.1159/000450705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ciancanelli MJ, Huang SXL, Luthra P, Garner H, Itan Y, Volpi S, Lafaille FG, Trouillet C, Schmolke M, Albrecht RA, Israelsson E, Lim HK, Casadio M, Hermesh T, Lorenzo L, Leung LW, Pedergnana V, Boisson B, Okada S, Picard C, Ringuier B, Troussier F, Chaussabel D, Abel L, Pellier I, Notarangelo LD, Garcia-Sastre A, Basler CF, Geissmann F, Zhang S-Y, Snoeck H-W, Casanova J-L. 2015. Life-threatening influenza and impaired interferon amplification in human IRF7 deficiency. Science 348:448–453. doi: 10.1126/science.aaa1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mestas J, Hughes C. 2004. Of mice and not men: differences between mouse and human immunology. J Immunol 172:2731–2738. doi: 10.4049/jimmunol.172.5.2731. [DOI] [PubMed] [Google Scholar]
- 52.Hockemeyer D, Jaenisch R. 2016. Induced pluripotent stem cells meet genome editing. Cell Stem Cell 18:573–586. doi: 10.1016/j.stem.2016.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lee MN, Ye C, Villani A-C, Raj T, Li W, Eisenhaure TM, Imboywa SH, Chipendo PI, Ran FA, Slowikowski K, Ward LD, Raddassi K, McCabe C, Lee MH, Frohlich IY, Hafler DA, Kellis M, Raychaudhuri S, Zhang F, Stranger BE, Benoist CO, De Jager PL, Regev A, Hacohen N. 2014. Common genetic variants modulate pathogen-sensing responses in human dendritic cells. Science 343:1246980. doi: 10.1126/science.1246980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Çalışkan M, Baker SW, Gilad Y, Ober C. 2015. Host genetic variation influences gene expression response to rhinovirus infection. PLoS Genet 11:e1005111. doi: 10.1371/journal.pgen.1005111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Leha A, HipSci Consortium, Moens N, Meleckyte R, Culley OJ, Gervasio MK, Kerz M, Reimer A, Cain SA, Streeter I, Folarin A, Stegle O, Kielty CM, Durbin R, Watt FM, Danovi D. 2016. A high-content platform to characterise human induced pluripotent stem cell lines. Methods 96:85–96. doi: 10.1016/j.ymeth.2015.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.van Wilgenburg B, Browne C, Vowles J, Cowley SA. 2013. Efficient, long term production of monocyte-derived macrophages from human pluripotent stem cells under partly-defined and fully-defined conditions. PLoS One 8:e71098. doi: 10.1371/journal.pone.0071098. [DOI] [PMC free article] [PubMed] [Google Scholar]