

Abstract
Introduction:
Chiari malformations are a group of congenital anomalies which involve the hindbrain and the cervical spinal canal.
Case presentation:
Here, we describe a patient who presented with acute diplopia and gait unsteadiness which was first deigned with Chiari malformation type-1. However due to progression of the ataxia the full neurologic evaluation was considered which established the diagnosis of spinocerebellar ataxia type 3 (Machado-Joseph-Disease).
Conclusion:
We aim to highlight the importance of careful examination in order to avoid misdiagnosis of even rare diseases.
Keywords: Ataxia, Case Reports, Chiari Malformation, Spinocerebellar Atrophy
Introduction
Chiari malformations are a group of congenital anomalies which involve the hindbrain and the cervical spinal canal. Based on the anatomy of the brain tissue which is displaced to the spinal canal and accompanied developmental central nervous system anomalies, it is divided into three main groups (1–3). Chiari malformation type-1 is defined as tonsilar herniation of at least 3–5 mm below the foramen magnum. The majority of patients are asymptomatic. However the clinical manifestation usually occur in late childhood or adolescence with a wide variety of symptoms as headache which is the characteristic feature to drop attack, spasticity, balance impairment, blurred vision or diplopia, lower cranial nerve involvement, sleep apnea and syrinx related symptoms (4, 5).
Spinocerebellar ataxia type-3 (SCA-3) is an autosomal dominant inherited neurodegenerative disorder which is characterized by progressive cerebellar ataxia associated with various symptoms as peripheral neuropathy, pyramidal and extrapyramidal signs(6, 7).
To the best of our knowledg, there are very rare reports of the simultaneous accompaniment of Chiari malformation type-1 and neurodegenerative cerebellar ataxia in the literature. Here in, we present a case with Chiari malformation type-1 accompained with SCA.Informed consent was obtained by the patient for publication of the case report.
Case Presentation
The patient was a 20-year-old right handed, healthy man presented to our emergency department due to acute diplopia since 2 days before admission following a powerful sneeze. He had always been well except for a history of slight unsteadiness since a month before admission. Additionally, he reported a daily dull, non-incapacitating generalized pain aggravated by coughing or sneezing. On examination, the patient was alert and conscious with normal verbal communication. Examination of the head and neck revealed inward deviation of both eyes and the inability to move outwards. Noting taken, no papilledema was observed on fundoscopic examination. No other abnormal neurologic signs were observed.
The patient had been under brain and cervical magnetic resonance imaging (MRI) which revealed a descent of the cerebellar tonsils and part of the brainstem in the foramen magnum (10 mm), which was not accompanied by cervical syringomyelia (figure 1). Subsequently, the patient had been under posterior fossa decompression with suboccipital craniotomy and duraplasty which resulted to complete resolution of the chirai. The surgery was followed by partial remission of the diplopia. However, the unsteadiness had gradually worsened in a way that the patient got severely ataxic over 6 months. The neurological examination was characterized by slurred speech, gait ataxia without a tilt, bilateral dysmetria at the index-nose and heel to shin test. The cranial examination revealed right 6th nerve palsy which was the remainder of the recent diplopia (figure 2), nystagmus in all gaze direction, more evident on the right side. Other neurologic examination was remarkable.
Figure 1:
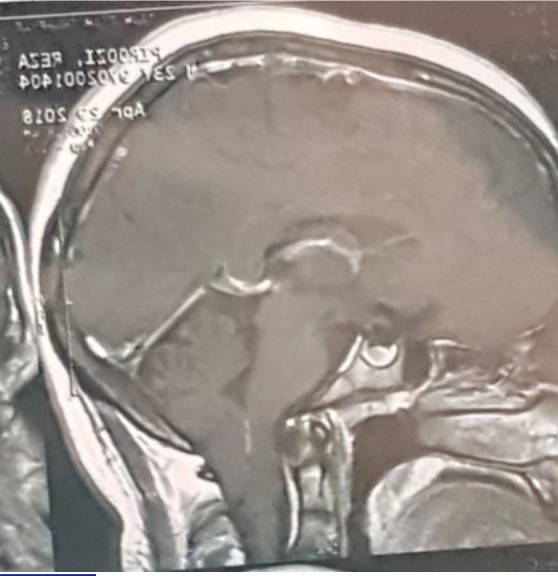
Patient’s brain MRI (T1 sagittal view) shows evidence of descent of the cerebellar tonsils and part of the brainstem in the foramen magnum (red arrow)
Figure 2:
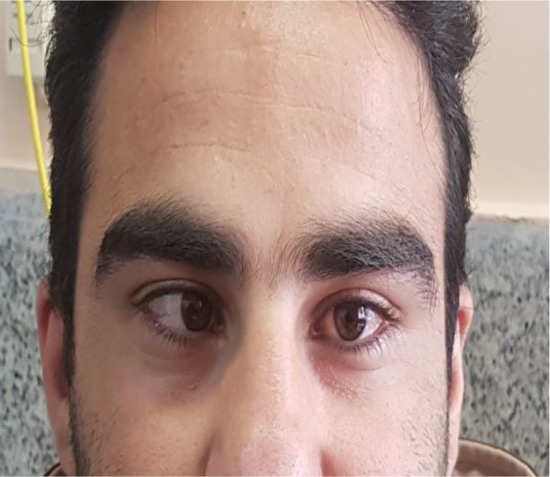
Patient’s face revealed that the right eye is inwardly deviated
In a more précised history, the family history was negative for any neurological disorder and the patient did not make biographies of any problem except for ataxia. Consequently, the patient underwent a complete screening for acquired ataxia including vitamin B1 deficiency due to alcohol abuse, autoimmune and paraneoplastic ataxia, ataxia associated with celiac disease, demyelinating disorder and autoimmune thyroiditis, nutritional deficiency and infections which were all unrevealing. Again the patient had been under brain MRI which showed a significant bilateral cerebellar hemispheres atrophy.
According to the new findings, the inherited atatxia was strongly suggested aside the underlying chari malformation. At this time the patient had been under genetic test. Ataxia repeat expansion panel test was checked which revealed a pathogenic (full penetrance) CAG repeats (64 repeats) in the ATX3 gene. Eventually the patient was diagnosed with SCA-3 (Machado-Joseph-Disease).
Discussion
In a case described, a cerebellar tonsils and brain stem descent was compatible with chari malformation type-1 which resulted to decompression. Although diplopia and ataxia are both manifestations of chiari, the progression of ataxia despite successful decompression made a possibility of other neurologic disorder. The chiari malformation is a rare disease with a prevalence of approximately 1 per 1000 (4). On the other hand, SCA_3 is the most common autosomal dominant ataxia worldwide with variable prevalence in a range of 0.26 −0.5 per 100000. It is a polyglutamine neurodegenerative disorder caused by expansion of CAG repeats in the ATX3 gene (8). There is a variety of both manifestation and onset in which the cerebellar ataxia is the hallmark presentation. Other clinical features could be pyramidal signs, extrapyramidal features as parkinsonism or dystonia, oculomotor paresis, neuropathy and sleep disorder (9, 10).
In early stages, the ataxia might be misdiagnosed and contributed to other disorders as in our patient who was initially diagnosed with chiari malformation. However, taking into account the probability of the occurrence of both disorders simultaneously is approximately considered to be less than 5 per 100000000. In 2013 Maria Petracca reported a 35 years old healthy woman who was diagnosed with chiari malformation type1 which was resolved by surgical decompression. Three years later, he developed progressive ataxia with evidence of cerebelalr atrophy in brain MRI which was not attributed to previous chiari malformation. She eventually was diagnosed with ILOCA according to genetic evaluation (11).
Conclusions
According to the variability of the onset and clinical manifestation of the spinocerebellar ataxia, here in we aim to highlight the importance of scrupulous neurologic evaluation to prevent misdiagnosis due to attributing all the clinical features of the patient to the underlying known disease.
Acknowledgements
None.
Footnotes
Authors’ contribution
All the authors fulfil the criteria of authorship based on the recommendations of the International Committee of Medical Journal Editors (ICMJE).
Conflict of interest
None declared
Funding
None declared
References
- 1.Tubbs RS, Shoja MM, Ardalan MR, Shokouhi G, Loukas M. Hindbrain herniation: A review of embryological theories. Ital J Anat Embryol. 2008;113(1):37–46. [PubMed] [Google Scholar]
- 2.Masson C, Colombani JM. Chiari type 1 malformation and magnetic resonance imaging. Presse Med. 2005;34(21):1662–7. [DOI] [PubMed] [Google Scholar]
- 3.Riveira C, Pascual J. Is Chiari type I malformation a reason for chronic daily headache. Curr Pain Headache Rep. 2007;11(1):53–5. [DOI] [PubMed] [Google Scholar]
- 4.Wani AM, Zayyani NR, Al Miamini W, Khoujah AM, Alharbi Z, Diari MS. Arnold-Chiari malformation type 1 complicated by sudden onset anterior spinal artery thrombosis, tetraparesis and respiratory arrest. BMJ Case Rep. 2011;2011:bcr0720103170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Speer MC, Enterline DS, Mehltretter L, Hammock P, Joseph J, Dickerson M, et al. Review Article: Chiari Type I Malformation with or Without Syringomyelia: Prevalence and Genetics. J Genet Couns. 2003;12(4):297–311. [DOI] [PubMed] [Google Scholar]
- 6.Durr A. Autosomal dominant cerebellar ataxias: polyglutamine expansions and beyond. Lancet Neurol. 2010;9(9):885–94. [DOI] [PubMed] [Google Scholar]
- 7.Schols L, Bauer P, Schmidt T, Schulte T, Riess O. Autosomal dominant cerebellar ataxias: clinical features, genetics, and pathogenesis. Lancet Neurol. 2004;3(5):291–304. [DOI] [PubMed] [Google Scholar]
- 8.Bettencourt C, Lima M. Machado-Joseph. Disease: from first descriptions to new perspectives. Orphanet J Rare Dis. 2011;6:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Margolis RL. The spinocerebellar ataxias: order emerges from chaos. Curr Neurol Neurosci Rep. 2002;2(5):447–56. [DOI] [PubMed] [Google Scholar]
- 10.Wang YG, Du J, Wang JL, Chen J, Chen C, Luo YY, et al. Six cases of SCA3/MJD patients that mimic hereditary spastic paraplegia in clinic. J Neurol Sci. 2009;285(1–2):121–4. [DOI] [PubMed] [Google Scholar]
- 11.Petracca M, Cerillo I, Montella S, Cerullo G, Carrieri PB. Idiopathic late-onset cerebellar ataxia with cerebellar atrophy in a patient diagnosed with Chiari I malformation: a case report. Neurol Sci. 2013;34(12):2235–7. [DOI] [PubMed] [Google Scholar]