

Abstract
Background
Primary sclerosing cholangitis is a progressive chronic cholestatic liver disease that usually leads to the development of cirrhosis. Studies evaluating bile acids in the treatment of primary sclerosing cholangitis have shown a potential benefit of their use. However, no influence on patients survival and disease outcome has yet been proven.
Objectives
To assess the beneficial and harmful effects of bile acids for patients with primary sclerosing cholangitis.
Search methods
We searched The Cochrane Hepato‐Biliary Group Controlled Trials Register, The Cochrane Library, MEDLINE, EMBASE and Science Citation Index Expanded generally from inception through to October 2010.
Selection criteria
Randomised clinical trials comparing any dose of bile acids or duration of treatment versus placebo, no intervention, or another intervention were included irrespective of blinding, language, or publication status.
Data collection and analysis
Two authors extracted data independently. We evaluated the risk of bias of the trials using prespecified domains. We performed the meta‐analysis according to the intention‐to‐treat principle. We presented outcomes as relative risks (RR) or mean differences (MD), both with 95% confidence intervals (CI).
Main results
Eight trials evaluated ursodeoxycholic acid versus placebo or no intervention (592 patients). The eight randomised clinical trials have a high risk of bias. Patients were treated for three months to six years (median three years). The dosage of ursodeoxycholic acid used in the trials ranged from low (10 mg/kg body weight/day) to high (28 to 30 mg/kg body weight/day). Ursodeoxycholic acid did not significantly reduce the risk of death (RR 1.00; 95% CI 0.46 to 2.20); treatment failure including liver transplantation, varices, ascites, and encephalopathy (RR 1.22; 95% CI 0.91 to 1.64); liver histological deterioration (RR 0.89; 95% CI 0.45 to 1.74); or liver cholangiographic deterioration (RR 0.60; 95% CI 0.23 to 1.57). Ursodeoxycholic acid significantly improved serum bilirubin (MD ‐14.6 µmol/litre; 95% CI ‐18.7 to ‐10.6), alkaline phosphatases (MD ‐506 IU/litre; 95% CI ‐583 to ‐430), aspartate aminotransferase (MD ‐46 IU/litre; 95% CI ‐77 to ‐16), and gamma‐glutamyltranspeptidase (MD ‐260 IU/litre; 95% CI ‐315 to ‐205), but not albumin (MD ‐0.20 g/litre; 95% CI ‐1.91 to 1.50). Ursodeoxycholic acid was safe and well tolerated by patients with primary sclerosing cholangitis.
Authors' conclusions
We did not find enough evidence to support or refute the use of bile acids in the treatment of primary sclerosing cholangitis. However, bile acids seem to lead to a significant improvement in liver biochemistry. Therefore, more randomised trials are needed before any of the bile acids can be recommended for this indication.
Keywords: Humans; Cholagogues and Choleretics; Cholagogues and Choleretics/therapeutic use; Cholangitis, Sclerosing; Cholangitis, Sclerosing/drug therapy; Liver Diseases; Liver Diseases/drug therapy; Randomized Controlled Trials as Topic; Ursodeoxycholic Acid; Ursodeoxycholic Acid/therapeutic use
Plain language summary
Bile acids for primary sclerosing cholangitis
Primary sclerosing cholangitis (PSC) is a chronic cholestatic liver disease characterised by progressive inflammation and scarring of liver bile ducts. Destruction of bile ducts leads to incidence of bile flow to the gut, resulting in the development of biliary cirrhosis and end‐stage liver disease. PSC is most common in young males and its aetiology is still not fully understood. The disease is usually classified as an autoimmune disorder, but other aetiological factors cannot be excluded. There is a strong association of PSC with inflammatory bowel diseases, particularly ulcerative colitis, which coexists in approximately 70% of patients. Besides its progressive and irreversible nature, PSC is also associated with an increased risk for cholangiocarcinoma, which contributes to an even higher morbidity and mortality of this disease.
The diagnosis of primary sclerosing cholangitis is based on a combined approach that includes clinical, laboratory, and imaging findings. Since the disease is usually asymptomatic in its initial stage, early recognition and diagnosis is actually rather rare. Because of poor understanding of aetiology and pathogenesis, treatment of PSC is still not satisfactory. Numerous medications have been evaluated for therapy of PSC, but most of them showed none or minimal effect, and certain drugs were associated with serious adverse events. Bile acids have also been considered for the treatment of PSC demonstrating a possible beneficial effect. However, further investigation of their role in PSC therapy is needed. Therefore, the treatment of choice for patients with end‐stage liver disease due to PSC remains liver transplantation. Despite the relatively low incidence of PSC in the general population, PSC remains among the most common indications for liver transplantation in Europe and the United States.
Based on eight randomised clinical trials of high risk of bias, the administration of ursodeoxycholic acid to patients with primary sclerosing cholangitis did not significantly reduce mortality, hepatic decompensation, need for liver transplantation, liver histological deterioration, or radiological deterioration compared with placebo or no intervention. The use of ursodeoxycholic acid showed a statistically significant improvement of liver biochemistry. However, evidence of these beneficial effects is weak as it is produced from trials with high risk of bias and a rather small number of patients. Furthermore, these observations are at risk of outcome measure reporting bias as half or less than half of the trials reported on these outcomes. One trial assessed the self‐estimated quality of life of patients with primary sclerosing cholangitis treated with ursodeoxycholic acid. No significant difference was found in any of the studied components, physical as well as mental. Based on an analysis of six of the eight included trials, the use of ursodeoxycholic acid seemed safe and well tolerated, without reports of serious adverse events. We were unable to identify trials evaluating other bile acids for patients with primary sclerosing cholangitis. Accordingly, the evidence does neither support nor refute bile acids for primary sclerosing cholangitis.
Background
Primary sclerosing cholangitis (PSC) is a chronic cholestatic disease of progressive course that usually leads to the development of biliary cirrhosis (Silveira 2008). The aetiology of PSC is still unknown and genetic factors as well as environmental factors may be involved. There is increasing evidence that autoimmune processes may have an important role, but other components such as portal bacteraemia, viral infections, toxic and metabolic liver injury, and innate predispositions cannot be excluded (Wee 1985a; Donaldson 1991; Olerup 1995; Talwalkar 2005; Gordon 2008). The reported incidence of PSC in the United States and Northern Europe ranges from 0.9 to 1.3 per 100,000 population and prevalence from 8 to 14 per 100,000 population. However, a recent population‐based cohort study by Card 2008 showed appreciably lower rates of incidence and prevalence in the United Kingdom (0.41 and 3.85, respectively). PSC is more common in young middle‐aged men and a large proportion of patients have associated inflammatory bowel disease, especially ulcerative colitis. Seventy to eighty per cent of patients with PSC in the United States and Northern Europe will have or develop inflammatory bowel disease (Lee 1995; Gordon 2008; Silveira 2008).
PSC is characterised by inflammation and fibrosis of intrahepatic and/or extrahepatic bile ducts and can be classified into large‐duct or small‐duct variants according to the involvement of the biliary tree (Ludwig 1981; Kaplan 2007). Large‐duct PSC principally involves the extrahepatic ducts and those parts of the intrahepatic biliary ductal system that can be visualised cholangiographically. Characteristic histological findings may or may not be found on liver biopsy. Small‐duct PSC involves the intrahepatic ducts at the microscopic level and is characterised by typical findings on liver biopsy, while the bile ducts visible by imaging methods are normal (Ludwig 1986; Ludwig 1991).
PSC progresses slowly, perhaps over decades, and leads to liver fibrosis. This may cause portal hypertension and, eventually, death from liver failure (Wee 1985b). Diagnosis usually relies on a combination of clinical, laboratory and imaging findings. Approximately 15% to 55% of the patients are asymptomatic at presentation. The most common symptoms are fatigue, pruritus, jaundice, abdominal discomfort and fever (Kaplan 2007; Silveira 2008). There are no specific biochemical changes, although a cholestatic picture with serum alkaline phosphatase elevation and fluctuations of serum bilirubin levels are frequently present. Due to lack of clinical findings in the early stage of disease the diagnosis of PSC is made by a mean delay of four years from the first presentation of biochemical abnormalities (Tischendorf 2008). Cholangiography and endoscopic retrograde cholangiopancreatography (ERCP) have been the golden diagnostic standards in PSC patients. Recently, magnetic resonance cholangiopancreatography (MRCP) has become the leading diagnostic technique in PSC patients, mainly because of its non‐invasiveness and cost‐effectiveness (Talwalkar 2004; Meagher 2007). The major radiological criterion is diffuse, multifocal stricturing characterised by irregularity of both the intrahepatic and extrahepatic bile ducts (Chen 1984). Histological findings are frequently non‐specific, therefore the role of liver biopsy in PSC evaluation is questionable. Pathognomonic concentric periductal fibrosis ('onion‐skinning'), narrowing, obliteration and disappearing of small bile ducts are found in less than 15% of patients (Burak 2003).
Treatment of PSC, as well as diagnostic evaluation, is also based mainly on a combined approach that includes medical, endoscopic, and surgical interventions (Lindor 1987). Endoscopic treatment includes balloon dilation, stenting and nasobiliary catheter perfusion. It is mostly reserved for patients with dominant strictures that involve larger extrahepatic biliary ducts. Stenting of such lesions has been associated with a greater number of intervention‐related complications, therefore balloon dilation of strictures alone is preferred (Stiehl 1997;Linder 2001; Michaels 2008;Silveira 2008). The actual benefits and harms of endoscopic procedures in PSC are still unknown, since there are no published randomised clinical trials evaluating this subject. Surgical procedure of choice in PSC patients is liver transplantation. Liver transplantation remains the only long‐term treatment in PSC (Cullen 2005). Resection of extrahepatic biliary tree and long‐term transhepatic stenting are reserved for carefully selected non‐cirrhotic patients with pronounced cholestasis or recurrent cholangitis caused by extrahepatic strictures that cannot be managed endoscopically (Angulo 1999; Michaels 2008).
Medical approaches for the treatment of PSC have included the use of cupriuretic, immunosuppressive, anti‐fibrotic, and choleretic agents (LaRusso 1999). A variety of drugs have been evaluated (eg, budesonide, colchicines, cyclosporine, methotrexate, mycofenolate mofetil, prednisone, tacrolimus) and despite certain encouraging results none of them showed convincing evidence of benefit and some of them were accompanied by major adverse effects (Beuers 2009). Considerable interest has been directed towards the potential benefit of ursodeoxycholic acid (UDCA) in the treatment of chronic cholestatic liver diseases. Other bile acids, besides UDCA, include chenodeoxycholic acid, deoxycholic acid, lithocholic and tauro‐ursodeoxycholic acid. Endogenous bile acids, such as deoxycholic and chenodeoxycholic acid, accumulate in the liver in the course of cholestasis and induce liver injury by damaging cellular membranes with their hydrophobic detergent‐like properties (Palmer 1972; Sholmerich 1984; Perez 2009). The hepatocyte toxicity of hydrophobic bile acids has been observed both in animals and humans (Jaeschke 2002). The addition of UDCA is associated with several potentially protective mechanisms of action. UDCA increases the fraction of hydrophilic bile acids which directly stabilises cell membranes (Perez 2009), competitively inhibits the absorption of toxic endogenous bile acids in the terminal ileum (Gordon 2008), and has potential immunomodulatory effects by inhibiting the expression of abnormal major histocompatibility complex class I molecules, thus resulting in suppression of cytokine and immunoglobulin production and T‐cell mediated cytotoxicity (Calmus 1990; Yoshikawa 1998).
Several randomised clinical trials have shown that UDCA improves serum biochemical indices of cholestasis and cytolysis in patients with primary biliary cirrhosis (Gong 2008), another cholestatic liver disease, and those with viral hepatitis (Chen 2007). Similar effects have been shown in PSC patients on UDCA (Stiehl 1996). Additional trials have been performed to evaluate the effect of other bile acids like tauro‐UDCA for primary biliary cirrhosis and chronic hepatitis (Crosignani 1996; Podda 1996), but we are not aware of trials examining tauro‐UDCA for patients with PSC. Our previous Cochrane systematic review on the topic (New Reference), including six trials that had enrolled 223 patients, was unable to support or refute the bile acids treatment for PSC patients. We have been able to identify a meta‐analysis by Shi 2009 addressing this issue. The meta‐analysis included eight randomised clinical trials with a total of 465 patients showing a statistically significant improvement in liver biochemistry and histology in patients treated with UDCA. However, no significant effect was confirmed on the incidence of death and/or liver transplantation, as well as on pruritus and fatigue.
Objectives
To evaluate the beneficial and harmful effects of bile acids in the treatment of patients with PSC by comparing bile acids versus placebo, no intervention or another intervention in randomised clinical trials.
Methods
Criteria for considering studies for this review
Types of studies
We included randomised clinical trials irrespective of blinding, publication status, year of publication and language. Quasi‐randomised studies or observational studies, retrieved with the search results, were considered for inclusion of data reporting harm, but not for data on benefit.
Types of participants
Patients with PSC diagnosed by any method according to, or compatible with, at least two of the three following definitions were included: 1. Biochemical criteria including one or more of the following: elevated serum activities of alkaline phosphatases, gamma‐glutamyltranspeptidase, alanine aminotransferase, aspartate aminotransferase, and/or raised serum bilirubin concentration. 2. Radiological criteria including cholangiographic demonstration of diffusely distributed, multifocal, annular strictures with intervening segments of normal or ectatic ducts; short and band‐like strictures; and diverticulum‐like out‐pouchings of the biliary tree. 3. Hepatic histological criteria including: (a) stage 1 changes: cholangitis or portal hepatitis; (b) stage 2 changes: periportal hepatitis with periportal fibrosis; (c) stage 3 changes: septal fibrosis or necrosis, or both; (d) stage 4 change: biliary cirrhosis.
Types of interventions
Any dose or duration of a bile acid treatment versus placebo, no intervention, or another intervention. Co‐interventions were allowed if received by both intervention groups within a trial.
Types of outcome measures
The primary outcome measures were: 1. All‐cause mortality (at the end of treatment). 2. Number of treatment failures including liver transplantation, biopsy‐proven cirrhosis, adenocarcinoma of the bile duct, and signs of decompensated liver cirrhosis such as varices, encephalopathy, or ascites. 3. Adverse events. Adverse events defined as any untoward medical occurrence not necessarily having a causal relationship with the treatment but resulting in a dose reduction or discontinuation of treatment. Severe adverse events were defined according to the ICH guidelines (ICH‐GCP 1997) as any event that would increase mortality; is life‐threatening; requires inpatient hospitalisation; results in a persistent or significant disability; or any important medical event which may jeopardise the patient or requires intervention to prevent it. 4. Quality of life.
The secondary outcome measures were: 1. Clinical symptoms, ie, number of patients with pruritus and fatigue or fatigue severity scale changes, or both. 2. Biochemical responses: serum activities of alkaline phosphatases, alanine aminotransferase, aspartate aminotransferase, and gamma‐glutamyl transpeptidase, and serum bilirubin and albumin concentrations and/or number of patients with abnormal values of these biochemical variables. 3. Radiological response: number of patients with radiological deterioration. 4. Histological response: number of patients with histological deterioration.
Search methods for identification of studies
Electronic searches
We searched The Cochrane Hepato‐Biliary Group Controlled Trials Register (Gluud 2010), The Cochrane Central Register of Controlled Trials (CENTRAL) in The Cochrane Library, MEDLINE, EMBASE, and Science Citation Index Expanded (Royle 2003). The search strategies with the time span of the searches are given in Appendix 1.
Searching other resources
We contacted the Chinese Cochrane Centre regarding the search of The Chinese Biomedical Database and received a reply that they were unable to help us with this search. Therefore, the latter database could not be included in the search strategy of this update. More trials were identified by reading the reference list of the identified studies. We contacted the principal authors and co‐authors of the identified randomised clinical trials for information about additional trials, which they might know of. The first group of authors had also written to pharmaceutical companies involved in the production of bile acids to obtain information on published or unpublished randomised clinical trials in 2003, but no information had been received at that time (New Reference).
Data collection and analysis
The update of this review was conducted according to the protocol, previously published in The Cochrane Library (New Reference) and following the recommendations given by the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2009) and the Cochrane Hepato‐Biliary Group Module (Gluud 2010).
Selection of studies
Identified trials were listed and two authors (GP and VG) evaluated whether the trials met the inclusion criteria. Excluded trials were listed with the reasons for exclusion. DS and CG provided consultation, supervision and final evaluation. Disagreements were resolved by discussion.
Data extraction and management
GP and VG extracted and validated the data independently. Disagreements were resolved by discussion or by CG. We extracted the following characteristics from each trial: primary author, number of patients randomised, patient inclusion and exclusion criteria, risk of bias, sample size calculation, intention‐to‐treat analysis, intervention regimens, mean age, proportion of males and females, proportion of patients with cirrhosis, proportion of patients with large‐duct and small‐duct PSC, proportion of patients with inflammatory bowel disease, time to follow‐up, number of outcomes, and number and type of adverse events in the intervention and the control group. Additional information was sought by correspondence with the principal investigators or co‐investigators of trials in cases where the relevant data were not published.
Assessment of risk of bias in included studies
Risk of bias was defined as the confidence that the design and reporting of the trial restricted bias in the intervention comparison (Schulz 1995; Moher 1998; Kjaergard 2001; Wood 2008). Due to the risk of overestimation of intervention effects in randomised trials with unclear or inadequate components, we assessed the risk of bias using the definitions in the following domains of risk of bias.
Allocation sequence generation ‐ Low risk of bias: sequence generation was achieved using computer random number generation or a random number table. Drawing lots, tossing a coin, shuffling cards and throwing dice are adequate if performed by an independent adjudicator. ‐ Uncertain risk of bias: the trial was described as randomised, but the method of sequence generation was not specified. ‐ High risk of bias: the sequence generation method is not, or may not be, random. Quasi‐randomised studies, those using dates, names, or admittance numbers in order to allocate patients are inadequate and were excluded for the assessment of benefits but not for harms. Allocation concealment ‐ Low risk of bias: allocation was controlled by a central and independent randomisation unit, sequentially numbered, opaque and sealed envelopes or similar, so that intervention allocations could not have been foreseen in advance of, or during, enrolment. ‐ Uncertain risk of bias: the trial was described as randomised but the method used to conceal the allocation was not described, so that intervention allocations may have been foreseen in advance of, or during, enrolment. ‐ High risk of bias: if the allocation sequence was known to the investigators who assigned participants or if the study was quasi‐randomised. Quasi‐randomised studies were excluded for the assessment of benefits but not for harms. Blinding ‐ Low risk of bias: the trial was described as blinded, the parties that were blinded, and the method of blinding was described, so that knowledge of allocation was adequately prevented during the trial. ‐ Uncertain risk of bias: the trial was described as blind, but the method of blinding was not described, so that knowledge of allocation was possible during the trial. ‐ High risk of bias, the trial was not blinded, so that the allocation was known during the trial. Incomplete outcome data ‐ Low risk of bias: the numbers and reasons for dropouts and withdrawals in all intervention groups were described or if it was specified that there were no dropouts or withdrawals. ‐ Uncertain risk of bias: the report gave the impression that there had been no dropouts or withdrawals, but this was not specifically stated. ‐ High risk of bias: the number or reasons for dropouts and withdrawals were not described.
Selective outcome reporting ‐ Low risk of bias: pre‐defined, or clinically relevant and reasonably expected outcomes were reported on. ‐ Uncertain risk of bias: not all pre‐defined, or clinically relevant and reasonably expected outcomes were reported on or were not reported fully, or it is unclear whether data on these outcomes were recorded or not. ‐ High risk of bias: one or more clinically relevant and reasonably expected outcomes were not reported on; data on these outcomes were likely to have been recorded.
Trials assessed as having 'low risk of bias' in all individual domains above were considered 'trials with low risk of bias'. Trials assessed as having 'uncertain risk of bias' or 'high risk of bias' in one or more of the specified above individual domains were considered trials with 'high risk of bias'.
Furthermore, we registered whether the randomised clinical trials had used an intention‐to‐treat analysis (Gluud 2001) and had calculated a sample size estimate.
Measures of treatment effect
We performed the analysis in RevMan 5 (RevMan 2008). We presented dichotomous data as relative risk (RR) with 95% confidence interval (CI) and continuous outcome measures by mean differences (MD) with 95% CI. Results of analyses including only one trial obtained with RevMan 5 were compared to the recommended Fisher's exact test, Chi2 test, or t‐test.
Unit of analysis issues
Randomised clinical trials.
Dealing with missing data
For any missing data we tried to contact the original investigators to request missing data (eg, the missing sex ratio in the Stiehl 1994 trial). Analyses for binary outcomes included all patients irrespective of compliance or follow‐up, according to the intention‐to‐treat principle, using the last reported observed response ('carry forward'). The analyses for continuous outcomes included only the patients with available data. In the assessment of histological responses and quality of life, per protocol analyses were performed.
Assessment of heterogeneity
We explored the presence of statistical heterogeneity by chi‐squared test with significance set at P < 0.10 and measured the quantities of heterogeneity by I2 (Higgins 2002).
Assessment of reporting biases
We planned to explore bias by funnel plot analyses (Egger 1997), but as we had less than 10 trials, we have not performed it.
Data synthesis
The data were analysed with both a random‐effects model (DerSimonian 1986) and a fixed‐effect model (Demets 1987). If there was no difference between the results of the two models, we reported only the results of the fixed‐effect model analysis.
Subgroup analysis and investigation of heterogeneity
We planned to perform subgroup analyses regarding: 1. Risk of bias of the randomised clinical trials ‐ comparing the intervention effect for trials with low risk of bias components to the intervention effect in trials with unclear or high risk of bias components. 2. Dose and duration of treatment with bile acids ‐ comparing the intervention effect in trials administrating bile acid at or above the dose multiplied by duration to the intervention effect of trials administrating bile acid at less than the median dose multiplied by duration. 3. Co‐interventions: comparing the intervention effect of trials with co‐interventions to the intervention effect of trials without co‐interventions. 4. Trials including mainly or exclusively large‐duct PSC with trials including mainly or exclusively small‐duct PSC. 5. Publication status ‐ comparing full manuscript trials with all other identified trials.
Sensitivity analysis
We were unable to perform several of the planned sensitivity analyses for a number of reasons: the bias risk of all eight trials was high; co‐interventions were not used in any of the trials; we were unable to identify whether the included patients had large‐duct or small‐duct PSC, with the exception of one trial; and we did not find any unpublished studies. The sensitivity analyses that we could perform are regarding duration of treatment and dose of UDCA administered.
According to the review protocol, we defined short treatment duration as being less than 24 months and long treatment duration as being 24 months or longer. Two trials (Beuers 1992; Stiehl 1994) had short treatment duration while the other six trials (Lo 1992; de Maria 1996; Lindor 1997; Mitchell 2001; Olsson 2005; Lindor 2009) had long treatment duration. We also defined a low dose of UDCA (less than 13 mg/kg body weight/day) and a high dose (13 mg/kg body weight/day or more) by the median dose of UDCA used in the trials included in this review. The low dose of UDCA was applied in three trials (Lo 1992; Stiehl 1994; de Maria 1996), and the high dose of UDCA in the other five trials (Beuers 1992; Lindor 1997; Mitchell 2001; Olsson 2005; Lindor 2009).
Regarding the number of deaths at the end of treatment, we did not find any significant difference between the short treatment duration (RR 0.43; 95% CI 0.02 to 9.00) and long treatment duration (RR 1.08; 95% CI 0.48 to 2.44) (Analysis 2.1) or between a low dose of UDCA (no deaths in this comparison) and a high dose of UDCA (RR 1.00; 95% CI 0.46 to 2.20) (Analysis 2.2). In order to explore the relationship between treatment duration and dose of UDCA, we conducted a post hoc sensitivity analysis by dividing trials into four groups: long treatment duration with high dose of UDCA (Lindor 1997; Mitchell 2001; Olsson 2005; Lindor 2009); short treatment duration with high dose of UDCA (Beuers 1992); long treatment duration with low dose of UDCA (Lo 1992; de Maria 1996); and short treatment duration with low dose of UDCA (Stiehl 1994). No statistically significant differences in mortality were found between the treatment groups in this analysis (Analysis 2.3).
2.1. Analysis.
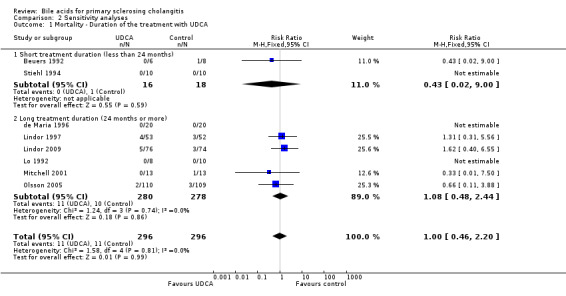
Comparison 2 Sensitivity analyses, Outcome 1 Mortality ‐ Duration of the treatment with UDCA.
2.2. Analysis.

Comparison 2 Sensitivity analyses, Outcome 2 Mortality ‐ Dose of UDCA in the treatment.
2.3. Analysis.

Comparison 2 Sensitivity analyses, Outcome 3 Mortality ‐ Different duration of the treatment with different dose of UDCA.
Results
Description of studies
See: Characteristics of included studies; Characteristics of excluded studies.
Results of the search
We performed electronic searches resulting in a total of 484 references. After reading the titles and abstracts, we excluded 462 of these articles because they were duplicates or irrelevant to our study. A total of 22 articles were retrieved for further assessment. Of these, 14 publications were excluded because they were observational studies, case‐series, or did not meet our inclusion criteria. They are listed under Characteristics of excluded studies with reasons for exclusion. Besides the six trials already included in the previous version of this review, we included two new trials (Olsson 2005; Lindor 2009) and four other references referring to three previously included trials (Beuers 1992; Stiehl 1994; Mitchell 2001). We were not able to identify more trials by reading the reference lists of the identified studies, contacting the principle authors and co‐authors of the identified trials, or approaching pharmaceutical companies for information on additional published or unpublished randomised clinical trials.
Included studies
The eight included publications in our review (seven full publications and one abstract) were randomised clinical trials that reported the random allocation of patients with PSC into groups receiving bile acids versus placebo or no intervention. These trials are listed in the table Characteristics of included studies. All eight trials were published in English. Three randomised clinical trials were from Germany (Beuers 1992; Stiehl 1994; Mitchell 2001), three from the United States of America (de Maria 1996; Lindor 1997; Lindor 2009), one from the United Kingdom (Lo 1992), and one was a Swedish‐Norwegian‐Danish trial (Olsson 2005).
Patients Patients with PSC diagnosed by standard biochemical, histological, and radiological features in the absence of secondary cholangitis, hepato‐biliary malignancy, and viral, metabolic, or autoimmune liver disease were included in the review. In total, 592 patients were randomised with a median size for the eight trials of 33 patients (range from 14 to 219). The mean age of the patients in all included trials ranged from 31 to 52 years. The male to female ratio was 370:200, while for the 22 patients that were lost to follow‐up in the Olsson 2005 trial no specification of sex was reported.
Concomitant inflammatory bowel disease was common. The proportion of patients with inflammatory bowel disease was 42.5% in the de Maria 1996 trial, 61% in the Lo 1992 trial, and over 71% in the other four trials (Beuers 1992; Stiehl 1994; Lindor 1997; Mitchell 2001). Only one trial (Stiehl 1994) reported the ratio of patients with intrahepatic (10 patients) and extrahepatic (9 patients) PSC according to endoscopic retrograde cholangiopancreatography findings.
Bile acids and collateral interventions UDCA was the only bile acid that was evaluated in the eight trials. Seven trials compared UDCA versus placebo (Beuers 1992; Lo 1992; Stiehl 1994; Lindor 1997; Mitchell 2001; Olsson 2005; Lindor 2009) and one trial compared UDCA versus no treatment (de Maria 1996). Patients were treated for two years in three of the trials (Lo 1992; de Maria 1996; Mitchell 2001) and for one year in one trial (Beuers 1992). Patients in the Olsson 2005 and Lindor 2009 trials were treated for up to five years. The duration of treatment in the Lindor 1997 trial was up to six years, with a median duration of 2.2 years. The Stiehl 1994 trial was discontinued after three months because of a more than two‐fold increase in alanine and aspartate aminotransferase activities in eight out of ten patients in the placebo group. Accordingly, the median duration of UDCA administration was three years (range from three months to six years).
Patients received 10 mg/kg body weight/day of UDCA in the Lo 1992 trial, and a dose of 13 to 15 mg/kg body weight/day was used in four trials (Beuers 1992; Stiehl 1994; de Maria 1996; Lindor 1997). Patients in the Mitchell 2001 and Olsson 2005 trials received UDCA 20 mg/kg body weight/day, and 17 to 23 mg/kg body weight/day, respectively. The highest dose of 28 to 30 mg/kg body weight/day UDCA was administered in the Lindor 2009 trial.
Outcome measures None of the included trials reported follow‐up after the end of treatment. Accordingly, the outcomes were reported at the end of treatment.
The outcome measures reported by most trials were mortality, histological and radiological changes, clinical symptoms, biochemical variables, and adverse events. Four trials (Lindor 1997; Mitchell 2001; Olsson 2005; Lindor 2009) reported the number of treatment failures, including liver transplantation, varices, ascites, and encephalopathy.
Excluded studies
The excluded trials are listed in the Table of excluded studies and the reason for the exclusion is given.
Risk of bias in included studies
Four trials (Beuers 1992; Stiehl 1994; Lindor 1997; Lindor 2009) reported adequate generation of the allocation sequence. None of the trials reported adequate allocation concealment. Five trials (Beuers 1992; Lindor 1997; Mitchell 2001; Olsson 2005; Lindor 2009) used a placebo, which was considered adequate by their authors to achieve the successful blinding of both patients and investigators. Five trials (Beuers 1992; Lo 1992; de Maria 1996; Olsson 2005; Lindor 2009) reported adequate description of incomplete outcome data by the number of withdrawals, the reasons for withdrawal, or no patients dropped out. The trials by Beuers 1992; Mitchell 2001; Lindor 2009 are the only considered free of selective reporting. Three trials did not perform sample size calculation (Lo 1992; de Maria 1996; Mitchell 2001). The trial by Olsson 2005 performed sample size calculation, but did not attained the pre‐specified sample size. Reasons for early termination of the trial were not reported. Another trial Beuers 1992 was terminated early, because an interim analysis performed six months after study initiation showed a significant difference in activity of alkaline‐phosphatases between the two groups of patients. Two trials fairly used intention‐to‐treat analysis (de Maria 1996; Lindor 2009). Three trials (Beuers 1992; Lo 1992; de Maria 1996) did not provide enough information to clearly assess the possibility of baseline imbalance in the trials. Accordingly, none of the trials were of low risk of bias, that is, being judged with having low risk of bias in generation of the allocation sequence, allocation concealment, double blinding, incomplete data and outcome reporting, and no other potential sources of bias (Figure 1; Figure 2).
1.
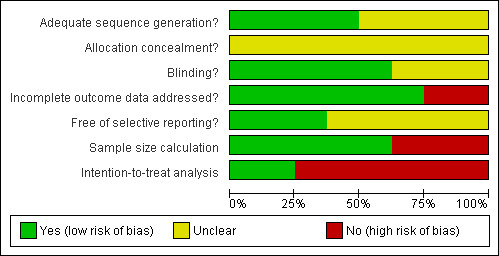
Methodological quality graph: review authors' judgements about each methodological quality item presented as percentages across all included studies.
2.
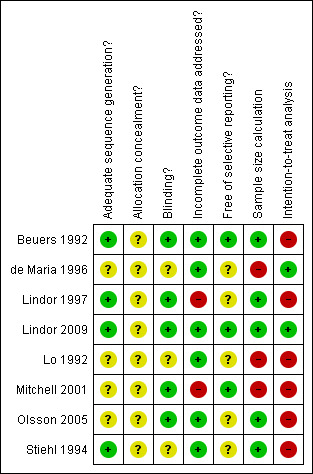
Methodological quality summary: review authors' judgements about each methodological quality item for each included study.
Effects of interventions
We included eight trials in this review. Seven trials with a total of 552 patients compared UDCA versus placebo, and one trial compared UDCA versus no intervention in a total of 40 patients. In all cases of analyses containing a single trial, the results obtained by RevMan 5 were identical or very similar to the results obtained by recommended Fisher's exact test, Chi2 test, or t‐test.
All‐cause mortality All included trials reported on all‐cause mortality at the end of treatment. UDCA did not significantly reduce overall mortality (RR 1.00, 95% CI 0.46 to 2.20) when compared with the control group. There were 11 deaths out of 296 patients (3.7%) in the UDCA group as well as in the control group, with no statistically significant heterogeneity (I2 = 0%) (Analysis 1.1). In the Lindor 1997 trial, causes of death included cholangiocarcinoma or gallbladder cancer in three patients, and liver failure or complications of portal hypertension in four patients. One patient (in the placebo group) from Beuers 1992 reported that one patient in the placebo group died of decompensated cirrhosis with gastrointestinal bleeding, while Mitchell 2001 reported that the one death in the placebo group was unrelated to liver disease. Five deaths occurred in the Olsson 2005 trial, one caused by hepatic failure and four due to cholangiocarcinoma. We were not able to find the causes of the eight deaths occurred in the Lindor 2009 trial, while no deaths were reported in the remaining three trials.
1.1. Analysis.

Comparison 1 UDCA versus control (placebo or no treatment), Outcome 1 Mortality at the end of treatment.
Treatment failures There was no significant difference between UDCA and placebo with respect to treatment failures such as liver transplantation, varices, ascites, and encephalopathy (RR 1.22, 95% CI 0.91 to 1.64) (Analysis 1.2). Four trials (Lindor 1997; Mitchell 2001; Olsson 2005; Lindor 2009) reported the number of treatment failures. A total of 66 treatment failures among 252 patients in the UDCA groups (26.2%) (liver transplantation = 23, cirrhosis = 6, varices = 23, ascites = 3, encephalopathy = 3, liver failure = 3, cholangiocarcinoma = 5), and 53 among 248 patients in the control groups (21.4%) (liver transplantation = 21, cirrhosis = 4, varices = 12, ascites = 8, encephalopathy = 0, liver failure = 2, cholangiocarcinoma = 6) were reported.
1.2. Analysis.

Comparison 1 UDCA versus control (placebo or no treatment), Outcome 2 Number of treatment failures at the end of treatment.
Adverse events Six trials among all the included and excluded trials reported on adverse events (Beuers 1992; Lo 1992; Lindor 1997; Mitchell 2001; Olsson 2005; Charatcharoenwitthaya '08) on a total of 444 patients. However, we analysed only the data from included trials on a total of 402 patients. In the Beuers 1992 trial, one patient in the UDCA group developed diarrhoea. In the Lindor 1997 trial, in the placebo group one patient experienced a flare up of chronic ulcerative colitis while another developed diarrhoea. The trials by Lo 1992 and Mitchell 2001 stated that no adverse events occurred during the study period. There was no statistically significant difference between treatment with UDCA versus placebo or no intervention with respect to the number of adverse events (RR 1.10, 95% CI 0.76 to 1.60) (Analysis 1.3). The excluded trial by Charatcharoenwitthaya '08 reported a flare of ulcerative colitis in the studied cohort of patients. No other excluded trials reported on adverse events (see Table 1). Quality of life and cost‐effectiveness Only the trial (Olsson 2005) reported the analysis of self‐estimated quality of life, assessing separately physical and mental components. No significant differences were found between the groups of patients for both, physical (MD ‐0.80; 95% CI ‐3.19 to 1.59) and mental (MD ‐0.20; 95% CI ‐2.69 to 2.29) components (Analysis 1.4; Analysis 1.5).
1.3. Analysis.

Comparison 1 UDCA versus control (placebo or no treatment), Outcome 3 Adverse events.
1. Adverse events.
| Study | Pts. in experimental group | Pts. in control group | AE in experimental group | AE in control group | Author's conclusion |
| Beuers 1992 | 6 | 8 | Diarrhoea (1 pt.) | No Ae | The symptoms ceased after UDCA treatment termination. |
| Lo 1992 | 8 | 10 | No AE | No AE | UDCA seemed to be well tolerated. |
| Stiehl 1994 | 10 | 10 | Diarrhoea (2 pts.) | No AE | After a reduction of the dose to 500 mg and 250 mg/day, UDCA was well tolerated. Re‐exposure to the higher dose (750 mg) re‐induced diarrhoea. |
| Lindor 1997 | 53 | 52 | No AE | Diarrhoea (1 pt.), flare of ulcerative colitis (1 pt.) | Ursodiol was well tolerated. |
| Mitchell 2001 | 13 | 13 | No AE | No AE | No patients required alteration in the prescribed trial medication for diarrhoea or any other cause. |
| Olsson 2005 | 110 | 109 | 37 | 34 | There seemed to be no difference between the two groups in the overall frequency of reported side effects attributed to the capsules (UDCA, 38.1%; placebo 33.7%) |
| Charatcharoenwitthaya 2008 | 42 | No‐controls | Flare of ulcerative colitis (1 pt.) | No controls | UDCA was well tolerated, but one patient stopped medication after four years of therapy because of a flare of ulcerative colitis. |
pt(s) = patient(s). AE = adverse events.
1.4. Analysis.

Comparison 1 UDCA versus control (placebo or no treatment), Outcome 4 Quality of life ‐ physical component.
1.5. Analysis.

Comparison 1 UDCA versus control (placebo or no treatment), Outcome 5 Quality of life ‐ mental component.
Liver histology deterioration Four trials (Beuers 1992; de Maria 1996; Lindor 1997; Mitchell 2001) reported the number of patients with deterioration of liver histology. Twelve out of 92 patients in the UDCA groups and 14 out of 93 patients in the control groups had deterioration of liver histology. UDCA did not significantly decrease the risk of liver histology deterioration (RR 0.89, 95% CI 0.45 to 1.74) (Analysis 1.6).
1.6. Analysis.

Comparison 1 UDCA versus control (placebo or no treatment), Outcome 6 Number of patients with liver histological deterioration at the end of treatment.
One trial including 21 patients (Mitchell 2001) reported the histological inflammatory score at the end of treatment (mean 3.50, standard deviation = 2.10 in the UDCA group and mean 4.50, standard deviation = 3.00 in the placebo group). No significant difference was found between the two treatment groups for this outcome (Analysis 1.7).
1.7. Analysis.

Comparison 1 UDCA versus control (placebo or no treatment), Outcome 7 Histological inflammatory score at the end of treatment.
Cholangiographic deterioration Three trials (Stiehl 1994; de Maria 1996; Mitchell 2001) reported the number of patients with cholangiographic deterioration; four out of 43 patients in the UDCA group and ten out of 43 patients in the control group had cholangiographic deterioration. UDCA did not significantly affect the risk of cholangiographic deterioration (RR 0.43, 95% CI 0.18 to 1.02) (Analysis 1.8).
1.8. Analysis.

Comparison 1 UDCA versus control (placebo or no treatment), Outcome 8 Number of patients with cholangiographic deterioration at the end of treatment.
Clinical symptoms Three trials (Stiehl 1994; Lindor 1997; Mitchell 2001) reported four out of 76 patients in the UDCA group and six out of 75 patients in the control group having worsening clinical symptoms at the end of treatment. UDCA did not significantly decrease the number of patients with worsening fatigue and/or pruritus (RR 0.66, 95% CI 0.20 to 2.19) (Analysis 1.9). In the trial by Olsson 2005 incidence of PSC‐related symptoms during the study period were reported graphically, but no actual data were given. The authors report a decrease in the incidence of pruritus and abdominal pain, without any significant difference between groups of patients.
1.9. Analysis.

Comparison 1 UDCA versus control (placebo or no treatment), Outcome 9 Number of patients with worsening clinical symptoms (fatigue and/or pruritus) at the end of treatment.
Liver biochemistry Three trials (Stiehl 1994; Lindor 1997; Mitchell 2001) including 108 patients reported the serum bilirubin level at the end of treatment. UDCA significantly decreased the serum bilirubin concentration (MD ‐14.6 µmol/litre; 95% CI ‐18.7 to ‐10.6; reduction ranged from 33% to 60%) (Analysis 1.10).
1.10. Analysis.

Comparison 1 UDCA versus control (placebo or no treatment), Outcome 10 Serum bilirubin level (µmol/l) at the end of treatment.
Four trials (Beuers 1992; Stiehl 1994; Lindor 1997; Mitchell 2001) including 120 patients reported serum alkaline phosphatases activity. UDCA significantly decreased serum alkaline phosphatases activity (MD ‐506 IU/litre, 95% CI ‐583 to ‐430; reduction ranged from 45% to 67%) (Analysis 1.11).
1.11. Analysis.

Comparison 1 UDCA versus control (placebo or no treatment), Outcome 11 Serum alkaline phosphatases activity (IU/L) at the end of treatment.
Two trials (Lindor 1997; Mitchell 2001) including 88 patients reported serum aspartate aminotransferase activity. UDCA significantly decreased aspartate aminotransferase activity (MD ‐46 IU/litre, 95% CI ‐77 to ‐16; reduction ranged from 41% to 48%) (Analysis 1.12).
1.12. Analysis.

Comparison 1 UDCA versus control (placebo or no treatment), Outcome 12 Serum aspartate aminotransferase activity (IU/L) at the end of treatment.
Two trials (Stiehl 1994; Mitchell 2001) including 42 patients reported serum gamma‐glutamyl transpeptidase activity. UDCA significantly decreased gamma‐glutamyl transpeptidase activity (MD ‐260 IU/litre, 95% CI ‐315 to ‐205; reduction ranged from 70% to 79%) (Analysis 1.13).
1.13. Analysis.

Comparison 1 UDCA versus control (placebo or no treatment), Outcome 13 Serum gamma‐glutamyltranspeptidase activity (IU/L) at the end of treatment.
Three trials (Stiehl 1994; Lindor 1997; Mitchell 2001) including 108 patients reported serum albumin concentration. UDCA had no significant effect on the serum albumin concentration (MD ‐0.20 g/litre, 95% CI ‐1.91 to 1.50) (Analysis 1.14).
1.14. Analysis.

Comparison 1 UDCA versus control (placebo or no treatment), Outcome 14 Serum albumin concentration (g/l) at the end of treatment.
We found no cost‐effectiveness analysis in any of the included trials.
Discussion
This systematic review could not demonstrate any significant effects of UDCA on mortality, treatment failures (including liver transplantation, varices, ascites, and encephalopathy), clinical symptoms, liver histology, or cholangiography in patients with PSC compared with placebo or no intervention. However, we find a significant reduction in liver biochemical variables, including serum bilirubin and liver enzyme activities, following UDCA administration. This observation is, however, at risk of outcome reporting bias as half or less of the trials reported these outcomes. Moreover, other error risks may be operative (please see below). UDCA appeared to be safe and well tolerated since we did not observe any significant increase in the occurrence of serious or non‐serious adverse events in patients with PSC.
Four out of the eight trials reported the method used to generate the allocation sequence, but none of the trials reported the method for allocation concealment. Five trials, as considered by the study authors, used adequate blinding methods achieving successful blinding of both patients and study investigators. However, as we have stressed earlier, trials with UDCA may be difficult to blind (Gong 2008). 'Intention‐to‐treat analysis' was fairly performed only in two trials. Two other trials stated the use of 'intention‐to‐treat' analysis, while in fact all of the results were based on the patients who remained at the end of treatment. Based on these observations, we suggest that more attention ought to be paid to these important methodological issues in future trials. The dimensions of methodological quality of trials have significant influence on the effect of interventions, eg, trials with high risk of bias may significantly overestimate intervention effects (Schulz 1995; Moher 1998; Kjaergard 2001; Wood 2008). As all of the included trials were of high risk of bias, we cannot exclude the possibility that the beneficial effects of UDCA on liver biochemistry may be due to bias or random errors (Wetterslev 2008; Brok 2009; Thorlund 2009). However, comparable observations in patients with primary biliary cirrhosis (Gong 2008) and viral hepatitis (Chen 2007) may support the positive effects of UDCA on some liver biochemical variables.
We also cannot exclude the possibility that the identified trials did not correctly assess the potential beneficial effects of bile acids on hard outcomes in patients with PSC. First, the treatment duration in the included trials may have been too short; the median duration of UDCA treatment in the included trials was three years. This is a relatively short duration, considering the slow progress of PSC, which commonly takes decades to cause portal hypertension and eventual premature death from liver failure (LaRusso 1999). Second, the apparent lack of clinical benefit of UDCA may also be due to patient selection. Most of the patients in the eight trials had an advanced histological stage of PSC with substantial fibrosis. Thus, the disease may have been too advanced in many of the included patients to achieve a positive response from medical therapy. However, the majority of patients with PSC do not present with early disease. Third, the sample size of the included trials ranged from 14 to 219 patients. Further, the sample size calculation in three trials (Beuers 1992; Lindor 1997; Stiehl 1994) was based on biochemical variables, such as serum transaminases, rather than on hard outcomes (death or liver transplantation). This may explain our failure to detect a significant effect of UDCA on clinically important outcome measures. Due to the difficulty in identifying many patients with PSC, multicentre randomised trials are required to examine the effects of UDCA on clinically important outcomes.
PSC is a chronic cholestatic hepato‐biliary disease in which progressive obliterative fibrosis of the intrahepatic and extrahepatic bile ducts leads to biliary cirrhosis, portal hypertension, and liver failure. The purpose of the trials assessing UDCA for PSC has not been to evaluate whether this bile acid could reverse the decompensated or terminal stage of the disease, but rather if UDCA could slow progression towards the more advanced stages. It has also been a main goal to study the effect of UDCA on liver histology and cholangiography. We were not able to identify any significant effect of UDCA on these outcome measures. We also performed sensitivity analyses to examine whether this failure may be due to insufficient doses of UDCA or too short a duration of treatment and follow‐up, but could not find any significant difference either. We have also been unable to find evidence that the improvement of biochemical variables can predict a decrease of death or stop the progress of the disease. Only one trial (Mitchell 2001) reported a significant reduction in the progression of cholangiographic appearances and liver fibrosis as assessed by disease staging under treatment with high dose UDCA (20 mg/kg body weight/day) for two years. However, two newly included trials (Olsson 2005; Lindor 2009) used an even higher dosage of UDCA throughout a longer period of time, but without reporting positive changes in the progression of the mentioned parameters. It is possible that absorption of UDCA is decreased in patients with PSC due to impaired alkalinisation of bile by the diseased biliary epithelium (Paumgartner 2002; Paumgartner 2004) and that higher doses of UDCA could have beneficial effects. However, evidence for this is still very poor (Beuers 2009).
Kim et al (Kim 2000) have developed a prognostic model for PSC patients based on 405 patients, followed up for a median of 36 months. Age; presence of ascites, hepatomegaly, splenomegaly, and variceal bleeding; haemoglobin level, platelet count, and prothrombin time; and serum levels of aspartate aminotransferase, albumin, and total bilirubin were significantly (P < 0.01) associated with patient survival (Kim 2000). However, these observations do not prove that the effect of UDCA on surrogate outcome measures such as bilirubin and liver enzyme activities translates into a more favourable prognosis. In clinical practice, some physicians still base therapeutic decisions on non‐validated surrogate outcomes such as the biochemical variables mentioned above (Gluud 2007). Two systematic reviews on the effect of bile acids in primary biliary cirrhosis (Gong 2008) and viral hepatitis (Chen 2007) reported significant improvement in liver biochemistry, but not on hard outcome measures such as mortality or liver transplantation. The recent meta‐analysis by Shi 2009 also reports a statistically significant beneficial effect of UDCA on liver biochemistry in patients with PSC, but without significant effect on mortality, symptoms, and need for liver transplantation. One could argue whether more trials should be performed to eventually confirm a beneficial effect of UDCA on clinically important outcomes in PSC. However, we have to state that current results and conclusions are based mostly on trials with a rather small number of patients and a high risk of bias, and that some of the trials are published only as abstracts. Another important aspect that perhaps should be more investigated is the quality of life in PSC patients undergoing UDCA treatment. Only one trial included in our review reported on this aspect, without finding any statistically significant difference in various physical and mental components of patients' quality of life.
The mechanisms for the beneficial effects of UDCA on PSC include the protection of cholangiocytes against the cytotoxicity of hydrophobic bile acids, stimulation of hepato‐biliary secretion, and protection of hepatocytes against bile acid‐induced apoptosis (Paumgartner 2004). Phospholipids in bile can protect cholangiocytes against membrane damage induced by hydrophobic bile acids. Administration of UDCA is also helpful in rendering bile more hydrophilic and less cytotoxic (Van Nieuwkerk 1996; Perez 2009). The protection of UDCA may also result from stimulation of hepato‐biliary secretion (Beuers 1993; Beuers 1996; Beuers 2001; Paumgartner 2004). Furthermore, recent data from basic research demonstrate that bile acids including UDCA appear to affect both death receptors (Caspase 8/10) and cell survival cascades (UDCA‐epidermal growth factor‐mitogen activated protein kinase) (Guicciardi 2002; Qiao 2002; Paumgartner 2004). The beneficial effects of UDCA on liver biochemical variables suggest that in patients with PSC (the present review), primary biliary cirrhosis (Gong 2008), and viral hepatitis (Chen 2007) the stimulation of the cell survival cascades by UDCA seems to overpower its concomitant stimulation of the cell death receptors. However, beneficially affecting cell death of, for example, hepatocytes may not be important enough to arrest or slow the progression of a disease such as PSC, which primarily affects the biliary tract. Similar or alternative disease mechanisms may be at play in primary biliary cirrhosis and viral hepatitis. Therefore, clinicians should base their clinical practice on solid research evidence rather than on evidence from animal studies or the opinion of experts. Furthermore, when UDCA induced mitogen‐activated protein kinase signalling (for survival cascades) was abolished in rodent hepatocytes, UDCA induced apoptosis was enhanced (Qiao 2002). At present we know too little about the effects of the abolishment of mitogen‐activated protein kinase in humans. The mechanisms mentioned above may explain the observed improvement of liver biochemistry but this does not provide an impetus to treat PSC patients with UDCA. Evidence of beneficial effects of UDCA on clinically meaningful outcomes is needed to recommend UDCA for PSC patients.
Long‐term UDCA administration in patients with PSC was associated with a very low incidence of adverse events in the eight included trials; these findings confirm previous observations in patients with primary biliary cirrhosis and viral hepatitis (Chen 2007; Gong 2008). The major adverse event reported in these trials was diarrhoea. In the treatment of PSC, it was unclear whether patients with inflammatory bowel disease, a frequently associated condition, would tolerate UDCA treatment as well as patients without inflammatory bowel disease. In the Beuers 1992 and Stiehl 1994 trials, patients treated with UDCA experienced severe diarrhoea without signs of inflammation. This seemed to have been related to UDCA because the diarrhoea stopped promptly following the termination of treatment or lowering the dosage of UDCA. In the Mitchell 2001 trial, although the patients received high‐dose UDCA treatment, no adverse events, such as diarrhoea, were reported. In summary, the evidence collected in this review does not show any convincing beneficial effects of UDCA on death, liver complications, liver histology, or cholangiographic deterioration in patients with PSC. Treatment of PSC patients with UDCA needs to be evaluated in appropriately powered randomised trials of low risk of bias.
Authors' conclusions
Implications for practice.
There is insufficient evidence either to support or to refute treatment with bile acids for patients with PSC.
Implications for research.
A need for adequately powered and long‐term conducted randomised clinical trials to support or change the current results and knowledge of the effects of UDCA for PSC still exist. Since UDCA seems to be well‐tolerated, the effects of high‐dose UDCA (eg, greater than 20 mg/kg body weight/day) compared with placebo intervention are worth exploring. We were not able to find trials evaluating other types of bile acids in the treatment of patients with PSC, so these investigations could also be worth performing. The relative rarity of PSC necessitates multicentre and likely multi‐national co‐operation.
What's new
| Date | Event | Description |
|---|---|---|
| 7 October 2010 | New search has been performed | Two new trials are included. |
| 7 October 2010 | New citation required but conclusions have not changed | This review is an updated version of a review first published in Issue 2, 2003 of The Cochrane Library. |
Notes
Changes to the protocol While working on this review we found some shortcomings in the protocol. We amended these shortcomings as described below. None of the amendments were data driven.
General style We have used fewer abbreviations in order to make the review easier to read.
Types of studies To document adverse events, we considered study types other than randomised clinical trials because rare adverse events are seldom captured in small trials. Accordingly, we sought information on rare adverse events from large cohort studies as well as previously published meta‐analyses and systematic reviews.
Outcome measures We changed the outcomes as follows: 5. Radiological response: number of patients with radiological progression or no improvement. into 5. Radiological response: number of patients with radiological deterioration.
and
6. Histological response: number of patients with histological progression or no improvement. into 6. Histological response: number of patients with histological deterioration.
Acknowledgements
We thank all the patients and investigators who were involved in the clinical trials mentioned in this review. We thank Wendong Chen for drafting the previous version of the review. We thank the staff of The Cochrane Hepato‐Biliary Group Editorial Team, especially Dimitrinka Nikolova, for their contribution and excellent collaboration.
Appendices
Appendix 1. Search strategies
| Data Base | Time span | Search Strategy |
| The Cochrane Hepato‐Biliary Group Controlled Trials Register | October 2010. | ('primary sclerosing cholangitis' OR PSC) AND ('bil* acid*' OR 'lithocolic acid*' OR LCA OR 'chenodeoxycholic acid*' OR CDCA OR 'ursodeoxycholic acid*' OR UDCA OR 'deoxycholic acid*' OR DCA OR 'dehydrocholic acid*' OR DHCA OR 'tauro‐ursodeoxycholic acid*' OR TDCA) |
| The Cochrane Central Register of Controlled Trials in The Cochrane Library | Issue 4, 2010 | #1 MeSH descriptor Cholangitis, Sclerosingexplode all trees #2 primary sclerosing cholangitis or PSC #3 (#1 OR #2) #4 MeSH descriptor Bile Acids and Saltsexplode all trees #5 bil* acid* or lithocolic acid* or LCA or chenodeoxycholic acid* or CDCA or ursodeoxycholic acid* or UDCA or deoxycholic acid* or DCA or dehydrocholic acid* or DHCA or tauro‐ursodeoxycholic acid* or TDCA #6 (#4 OR #5) #7 (#3 AND #6) |
| MEDLINE (Ovid SP) | 1950 to October 2010. | 1. exp Cholangitis, Sclerosing/ 2. (primary sclerosing cholangitis or PSC).mp. [mp=title, original title, abstract, name of substance word, subject heading word, unique identifier] 3. 1 or 2 4. exp "Bile Acids and Salts"/ 5. (bil* acid* or lithocolic acid* or LCA or chenodeoxycholic acid* or CDCA or ursodeoxycholic acid* or UDCA or deoxycholic acid* or DCA or dehydrocholic acid* or DHCA or tauro‐ursodeoxycholic acid* or TDCA).mp. [mp=title, original title, abstract, name of substance word, subject heading word, unique identifier] 6. 4 or 5 7. 3 and 6 8. (random* or blind* or placebo* or meta‐analysis).mp. [mp=title, original title, abstract, name of substance word, subject heading word, unique identifier] 9. 7 and 8 |
| EMBASE (Ovid SP) | 1980 to October 2010. | 1. exp primary sclerosing cholangitis/ 2. (primary sclerosing cholangitis or PSC).mp. [mp=title, abstract, subject headings, heading word, drug trade name, original title, device manufacturer, drug manufacturer name] 3. 1 or 2 4. exp bile acid/ 5. (bil* acid* or lithocolic acid* or LCA or chenodeoxycholic acid* or CDCA or ursodeoxycholic acid* or UDCA or deoxycholic acid* or DCA or dehydrocholic acid* or DHCA or tauro‐ursodeoxycholic acid* or TDCA).mp. [mp=title, abstract, subject headings, heading word, drug trade name, original title, device manufacturer, drug manufacturer name] 6. 4 or 5 7. 3 and 6 8. (random* or blind* or placebo* or meta‐analysis).mp. [mp=title, abstract, subject headings, heading word, drug trade name, original title, device manufacturer, drug manufacturer name] 9. 7 and 8 |
| Science Citation Index Expanded (http://apps.isiknowledge.com) | 1900 to October 2010. | # 5 #4 AND #3 # 4 TS=(random* or blind* or placebo* or meta‐analysis) # 3 #2 AND #1 # 2 TS=(bil* acid* or lithocolic acid* or LCA or chenodeoxycholic acid* or CDCA or ursodeoxycholic acid* or UDCA or deoxycholic acid* or DCA or dehydrocholic acid* or DHCA or tauro‐ursodeoxycholic acid* or TDCA) # 1 TS=(primary sclerosing cholangitis or PSC) |
Data and analyses
Comparison 1. UDCA versus control (placebo or no treatment).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Mortality at the end of treatment | 8 | 592 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.00 [0.46, 2.20] |
| 2 Number of treatment failures at the end of treatment | 4 | 500 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.22 [0.91, 1.64] |
| 3 Adverse events | 6 | 402 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.10 [0.76, 1.60] |
| 4 Quality of life ‐ physical component | 1 | 219 | Mean Difference (IV, Fixed, 95% CI) | ‐0.80 [‐3.19, 1.59] |
| 5 Quality of life ‐ mental component | 1 | 219 | Mean Difference (IV, Fixed, 95% CI) | ‐0.20 [‐2.69, 2.29] |
| 6 Number of patients with liver histological deterioration at the end of treatment | 4 | 185 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.89 [0.45, 1.74] |
| 7 Histological inflammatory score at the end of treatment | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 8 Number of patients with cholangiographic deterioration at the end of treatment | 3 | 86 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.6 [0.23, 1.57] |
| 9 Number of patients with worsening clinical symptoms (fatigue and/or pruritus) at the end of treatment | 3 | 151 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.99 [0.35, 2.80] |
| 10 Serum bilirubin level (µmol/l) at the end of treatment | 3 | 108 | Mean Difference (IV, Fixed, 95% CI) | ‐14.64 [‐18.70, ‐10.58] |
| 11 Serum alkaline phosphatases activity (IU/L) at the end of treatment | 4 | 120 | Mean Difference (IV, Fixed, 95% CI) | ‐506.24 [‐582.93, ‐429.55] |
| 12 Serum aspartate aminotransferase activity (IU/L) at the end of treatment | 2 | 88 | Mean Difference (IV, Fixed, 95% CI) | ‐46.44 [‐77.33, ‐15.55] |
| 13 Serum gamma‐glutamyltranspeptidase activity (IU/L) at the end of treatment | 2 | 42 | Mean Difference (IV, Fixed, 95% CI) | ‐259.69 [‐314.83, ‐204.55] |
| 14 Serum albumin concentration (g/l) at the end of treatment | 3 | 108 | Mean Difference (IV, Fixed, 95% CI) | ‐0.20 [‐1.91, 1.50] |
| 15 Cost effectiveness (No data in the trials) | 0 | 0 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 2. Sensitivity analyses.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Mortality ‐ Duration of the treatment with UDCA | 8 | 592 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.00 [0.46, 2.20] |
| 1.1 Short treatment duration (less than 24 months) | 2 | 34 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.43 [0.02, 9.00] |
| 1.2 Long treatment duration (24 months or more) | 6 | 558 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.08 [0.48, 2.44] |
| 2 Mortality ‐ Dose of UDCA in the treatment | 8 | 592 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.00 [0.46, 2.20] |
| 2.1 Low dose of UDCA (less than 13 mg/kg body weight /day) | 3 | 78 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 2.2 High dose of UDCA (13 mg/kg body weight/day or more) | 5 | 514 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.00 [0.46, 2.20] |
| 3 Mortality ‐ Different duration of the treatment with different dose of UDCA | 8 | 592 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.00 [0.46, 2.20] |
| 3.1 Short treatment duration with low dose of UDCA | 1 | 20 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 3.2 Short treatment duration with high dose of UDCA | 1 | 14 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.43 [0.02, 9.00] |
| 3.3 Long treatment duration with low dose of UDCA | 2 | 58 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 3.4 Long treatment duration with high dose of UDCA | 4 | 500 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.08 [0.48, 2.44] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Beuers 1992.
| Methods | Study design: a prospective, randomised, double‐blind, placebo‐controlled trial. | |
| Participants | Country: Germany.
Publication language: English. Inclusion criteria: ‐ diagnosis of PSC by endoscopic retrograde cholangiography, hepatobiliary histological appearance, and a cholestatic serum enzyme pattern in the absence of evidence of secondary sclerosing cholangitis, hepatobiliary malignancies, or other viral, metabolic, or autoimmune liver disease; ‐ ALP level at least 1.5 times the normal value. Exclusion criteria ‐ pregnancy; ‐ therapy of PSC within the last three months with UDCA, azathioprine, chlorambucil, colchicine, cyclosporine, methotrexate, D‐penicillamine, or corticosteroids; ‐ serum bilirubin level > 15 mg/dl; ‐ other liver diseases in addition to PSC. Participants: ‐ UDCA group (n = 6) Mean age (years): 29 (range 17 to 46); Ratio of sex (M/F): 6/0; Concurrent IBD no.: 5/6 (83%); Symptoms: fatigue: 3/6 (50%); ascites: 1/6 (17%). ‐ Placebo group (n = 8) Mean age (years): 46 (range 24 to 62); Ratio of sex (M/F): 5/3; Concurrent IBD no.: 5/8 (63%); Symptoms: fatigue: 5/8 (63%); RUQ pain: 1/8 (12.5%); pruritus: 2/8 (25%); jaundice: 1/8 (12.5%) |
|
| Interventions | UDCA group:
‐ Dose: 13 to 15 mg/kg body weight/day in two divided doses.
‐ Route: orally.
‐ Duration: one year. Placebo group: ‐ identical‐appearing capsules administered in the same quantity and manner. |
|
| Outcomes | Biochemical changes at the end of treatment; Clinical signs and symptoms at the end of treatment; Liver histology at the end of treatment; and adverse events. | |
| Notes | Letter was sent to the authors in August 2010. A reply with additional information was received shortly after. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Adequate sequence generation? | Low risk | A computer‐generated block randomisation was used. |
| Allocation concealment? | Unclear risk | Method of allocation concealment not described. |
| Blinding? All outcomes | Low risk | Identical appearing placebo in 250 mg capsules were used. |
| Incomplete outcome data addressed? | Low risk | Follow‐up time: 1 year. Numbers and reasons for withdrawal were stated. |
| Free of selective reporting? | Low risk | Study outcomes clearly pre‐specified and data reported. |
| Sample size calculation | Low risk | Stated and used. |
| Intention‐to‐treat analysis | High risk | Not stated and not used. According to the principal author contacted during August 2010, two patients from the control group were excluded from data analysis. |
de Maria 1996.
| Methods | Study design: a randomised controlled trial. | |
| Participants | Country: United States of America .
Publication language: English. Inclusion criteria PSC documented by endoscopic cholangiography, liver biopsy, and a battery of clinical, biochemical, and serologic parameters for PSC. Exclusion criteria not stated. Participants (1) UDCA group (n = 20) Mean age (years +/‐ standard deviation): 32.0 +/‐ 5.1; Ratio of sex (M/F): 14/6; Concurrent IBD no. : 9 (45%). (2) Control group (n = 20) Mean age (years +/‐ standard deviation): 31.2 +/‐ 5.0; Ratio of sex (M/F): 14/6; Concurrent IBD no. : 8 (40%). |
|
| Interventions | UDCA group:
Dose: 300 mg twice a day.
Route: orally.
Duration: two years. Control group: no treatment. |
|
| Outcomes | Liver histological changes at the end of treatment; Endoscopic cholangiography changes at the end of treatment; Biochemical variables at the end of treatment; and clinical symptoms changes at the end of treatment. | |
| Notes | Letter was sent to the authors in September 2010. No reply was received. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Adequate sequence generation? | Unclear risk | No description of sequence generation method given. Quote: "A total of 59 subjects...were randomised sequentially to 1 of the 3 groups in 1:1:1 block design." |
| Allocation concealment? | Unclear risk | No information provided. |
| Blinding? All outcomes | Unclear risk | No information provided. |
| Incomplete outcome data addressed? | Low risk | Follow‐up time: 2 years. No withdrawals occurred. |
| Free of selective reporting? | Unclear risk | Study outcomes were not pre‐specified. |
| Sample size calculation | High risk | Not stated and not used. |
| Intention‐to‐treat analysis | Low risk | Not stated but used. |
Lindor 1997.
| Methods | Study design: a multicenter, randomised, double‐blind, placebo‐controlled trial. | |
| Participants | Country: United States of America .
Publication language: English. Inclusion criteria ‐ a chronic cholestatic liver disease of at least six months duration; ‐ a serum ALP level at least 1.5 times the upper normal limit; ‐ retrograde, operative, or percutaneous cholangiographic findings of intrahepatic or extrahepatic biliary‐duct obstruction, beading, or narrowing consistent with primary sclerosing cholangitis; ‐ a liver biopsy in the previous three months with compatible findings. Exclusion criteria (1) treatment with UDCA, colchicine, corticosteroids, cyclosporine, methotrexate, or penicillamine in the preceding three months; (2) anticipated need for liver transplantation within one year on the basis of the Mayo survival model; (3) recurrent variceal haemorrhage, spontaneous uncontrolled encephalopathy, or ascites resistant to diuretic agents; (4) pregnancy; (5) an age of less than 18 years or more than 70 years; (6) features suggestive of coexisting liver diseases, including primary biliary cirrhosis, chronic alcoholic liver disease, autoimmune hepatitis, chronic hepatitis B or C, or cholangiocarcinoma; (7) a history of intraductal stones or biliary tract operations apart from cholecystectomy; (8) recurrent ascending cholangitis requiring hospitalisation more than twice a year. Participants (1) UDCA group (n = 53) Mean age (years +/‐ standard deviation): 41.7 +/‐ 1.8; Ratio of sex (M/F): 32/21; Concurrent IBD no. : 41 (77%). (2) Placebo group (n = 52) Mean age (years +/‐ standard deviation): 43.8 +/‐ 1.6; Ratio of sex (M/F): 29/23; Concurrent IBD no. : 44 (85%). |
|
| Interventions | UDCA group:
Dose: 13 to 15 mg/kg body weight/day in four divided doses.
Route: orally.
Duration: two years. Placebo group: identical‐appearing capsules administered in the same quantity and manner. |
|
| Outcomes | Number of treatment failure (withdrawal from study, liver transplantation, worsening of symptoms, varices, ascites, encephalopathy, histologic progression, and death) at the end of treatment; Serum bilirubin, ALP, AST, and albumin level at the end of treatment; and adverse events. | |
| Notes | Letter was sent to the authors in August 2010. A reply with no additional information was received shortly after. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Adequate sequence generation? | Low risk | Computer‐generated, blocked randomisation. |
| Allocation concealment? | Unclear risk | Method of allocation concealment not described. |
| Blinding? All outcomes | Low risk | Placebo was given in identical tablets and in the same way as the interventional drug, and patients, physicians, nurses and study coordinators were blinded to the administration of both. |
| Incomplete outcome data addressed? | High risk | The number and reasons for withdrawal of patients were not stated in the report, although a table shows a withdrawal of 13 patients from the UDCA‐group and 9 patients from the control group with no explanations given. Median follow‐up time was 2.2 years. |
| Free of selective reporting? | Unclear risk | Not enough information provided. Study outcomes are not clearly stated and all data are not shown. |
| Sample size calculation | Low risk | Stated and used. |
| Intention‐to‐treat analysis | High risk | Stated but not used. |
Lindor 2009.
| Methods | Study design: a long‐term, randomised, double‐blind controlled trial. | |
| Participants | Country: United States of America.
Publication language: English. Inclusion criteria: ‐ chronic cholestatic disease of at least 6 months duration; ‐ serum alkaline phosphatase at least 1½ times the upper limits of normal; ‐ retrograde, operative, percutaneous, or magnetic resonance cholangiography demonstrating intrahepatic and/or extrahepatic biliary duct obstruction, beading or narrowing consistent with PSC within 1 year of the study entry; ‐ liver biopsy in the previous 1 year that was available for review and compatible with the diagnosis of PSC. Exclusion criteria: ‐ coexistent conditions such as preexisting advanced malignancies or severe cardiopulmonary disease that would limit their life expectancy to less than 2 years; ‐ inability to provide consent; ‐ treatment with UDCA, pentoxifylline, corticosteroids, cyclosporine, colchicine, azathioprine, methotrexate, D‐penicillamine, budesonide, nicotine, pirfenidone, or tacrolimus in the 3 months prior to study entry; ‐ inflammatory bowel disease patients requiring specific treatment in the preceding 3 months except for maintenance therapy with 5‐ASA compound; ‐ anticipated need for liver transplantation within 2 years (expected survival of <80% at 2 years based on Mayo risk score); ‐ recurrent variceal bleeding, spontaneous uncontrolled encephalopathy, INR >1.5 uncorrected by vitamin K, or resistant ascites that suggested an anticipated survival of less than 1 year; ‐ pregnancy or lactation; ‐ age less than 18 years or greater than 75 years; ‐ findings highly suggestive of liver disease of other etiology ‐ previous intraductal stones or operations on the biliary tree, other than cholecystectomy ‐ recurrent ascending cholangitis requiring hospitalisation occurring more than two times per year. Participants: ‐ UDCA group (n = 76): Mean age (years): 47.9 (range 20.5 to 75.6); Ratio of sex (M/F): 38/38; Duration of disease (years): 1.3 (range 0.1 to 13.4); Concurrent IBD no. : 55 (72%). ‐ Placebo group (n = 74): Mean age (years): 45.3 (range 17.9 to 73.6); Ratio of sex (M/F): 48/26; Duration of disease (years): 1.0 (range 0.0 to 49.5); Concurrent IBD no. : 61 (%). |
|
| Interventions | UDCA group:
‐ Dose: 28 to 30 mg/kg body weight/day in divided doses given with meals and a bedtime snack.
‐ Route: orally.
‐ Duration: five years. Placebo group: ‐ identical placebo. |
|
| Outcomes | Primary outcome measures: Death or transplantation; Development of cirrhosis, varices, cholangiocarcinoma. | |
| Notes | Letter was sent to the authors in August 2010. A reply with no additional information was received shortly after. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Adequate sequence generation? | Low risk | Computer‐generated sequence. |
| Allocation concealment? | Unclear risk | The method for allocation not described. |
| Blinding? All outcomes | Low risk | Identical‐appearing placebo. |
| Incomplete outcome data addressed? | Low risk | Number and reasons for patients withdrawal from study were reported. |
| Free of selective reporting? | Low risk | Study outcomes clearly pre‐specified and data reported. |
| Sample size calculation | Low risk | Stated and used. |
| Intention‐to‐treat analysis | Low risk | Stated and used. |
Lo 1992.
| Methods | Study design: a double‐blind placebo controlled trial. | |
| Participants | Country: United Kingdom.
Publication language: English. Inclusion criteria ‐ not stated. Exclusion criteria ‐ not stated. Participants ‐ UDCA group (n = 8) ‐ placebo group (n = 10) (in total = 11 males; mean age: 47 years, range 23 to 58; 11 patients with ulcerative colitis). |
|
| Interventions | UDCA group:
Dose: 10 mg/kg body weight/day.
Route: orally.
Duration: two years. Placebo group: identical‐appearing capsules administered in the same quantity and manner. |
|
| Outcomes | Liver histological changes at the end of treatment; Endoscopic cholangiography changes at the end of treatment; Biochemical parameters at the end of treatment; Clinical symptom changes at the end of treatment; and adverse events. | |
| Notes | Abstract. Letter was sent to the authors in September 2010. No reply was received. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Adequate sequence generation? | Unclear risk | Method of sequence generation was not described. |
| Allocation concealment? | Unclear risk | No information provided. |
| Blinding? All outcomes | Unclear risk | No information provided. |
| Incomplete outcome data addressed? | Low risk | Follow‐up time: 2 years. Quote: "Four patients were withdrawn from the study; one in the UDCA group due to development of colonic carcinoma and 3 in the placebo group because of clinical deterioration or self‐withdrawal." |
| Free of selective reporting? | Unclear risk | Outcomes were not pre‐specified. |
| Sample size calculation | High risk | Not stated and not performed. |
| Intention‐to‐treat analysis | High risk | Not stated and probably not performed. |
Mitchell 2001.
| Methods | Study design: a double‐blind, randomised trial. | |
| Participants | Country: Germany.
Publication language: English. Inclusion criteria ‐ satisfying the cholangiographic criteria for the diagnosis of PSC; ‐ all patients had a liver biopsy with histological features compatible with the diagnosis and stable liver biochemical tests for three months before entry with a cholestatic enzyme pattern. Exclusion criteria ‐ age less than 18 or greater than 80 years; ‐ treatment with UDCA in the preceding year; ‐ previous bile‐duct surgery; ‐ dominant extrahepatic or bile duct stricture; ‐ previous choledocholithiasis; ‐ recurrent ascending cholangitis; ‐ previous history of variceal haemorrhage; ‐ decompensated liver disease; ‐ cholangiocarcinoma; ‐ active inflammatory bowel disease; ‐ and any features of a coexisting liver disease or an overlap syndrome. Participants ‐ UDCA group (n = 13) Mean age (years): 52 (range 22 to 79); Ratio of sex (M/F): 9/4; Concurrent IBD no. : 11 (84%); Symptoms: 6/13. ‐ Placebo group (n = 13) Mean age (years): 52 (range 28 to 74); Ratio of sex (M/F): 10/3; Concurrent IBD no. : 10 (70%); Symptoms: 5/13. |
|
| Interventions | UDCA group:
‐ Dose: 20 mg/kg body weight/day in two divided doses.
‐ Route: orally.
‐ Duration: two years. Placebo group: ‐ identical‐appearing capsules administered in the same quantity and manner. |
|
| Outcomes | Liver histological changes at the end of treatment; Endoscopic cholangiography changes at the end of treatment; Biochemical parameters at the end of treatment; Clinical symptoms changes at the end of treatment; and any adverse events. | |
| Notes | Letter was sent to the authors in September 2010. No reply was received. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Adequate sequence generation? | Unclear risk | No information provided. |
| Allocation concealment? | Unclear risk | No information provided. |
| Blinding? All outcomes | Low risk | Quote: "...an identical‐appearing capsule administered in the same quantity and manner." |
| Incomplete outcome data addressed? | High risk | Follow‐up time: 2 years. Reasons for withdrawal are clearly stated for two patients. Two patients were not considered as withdrawn from study, but were not included in the analysis. One patient was not included in the analysis and no reasons were reported. |
| Free of selective reporting? | Low risk | Outcomes pre‐specified and required data reported. |
| Sample size calculation | High risk | Not stated and not used. |
| Intention‐to‐treat analysis | High risk | Stated but not used. |
Olsson 2005.
| Methods | Study design: a 5‐year multicenter, randomised, placebo‐controlled trial | |
| Participants | Country: Sweden, Norway, Denmark.
Publication language: English. Inclusion criteria: ‐ diagnosis of PSC based on cholangiography with conventional radiological criteria; ‐ age 18 ‐ 70 years; ‐ body weight ≤ 115 kg; ‐ expected survival > 1 year. Exclusion criteria: ‐ earlier treatment with UDCA; ‐ planned pregnancy within the forthcoming 5 years; ‐ alcohol abuse and other forms of abuse; ‐ positive tests for hepatitis B surface antigen or anti‐hepatitis C virus; ‐ another concomitant cause of liver disease. Participants: ‐ UDCA group (n = 97): Mean age (years +/‐ SD): 43.6 +/‐ 12.7; Ratio of sex (M/F): 70/26; Mean body weight (kg +/‐ SD): 75.8 +/‐ 13.2; Concurrent IBD no. : 81 (84%); IBD treatment: 43 (44%); Prevalence of symptoms before start: pruritus: 33 (34%), RUQ abdominal pain: 32 (33%), fever: 18 (19%), jaundice: 24 (25%), ascites: 2 (2%). ‐ Placebo group (n = 101): Mean age (years +/‐ SD): 43.1 +/‐ 11.2; Ratio of sex (M/F): 69/32; Mean body weight (kg +/‐ SD): 74.5 +/‐ 12.9; Concurrent IBD no. : 87 (86%); IBD treatment: 43 (43%); Prevalence of symptoms before start: pruritus: 38 (38%), RUQ abdominal pain: 29 (29%), fever: 16 (16%), jaundice: 27 (27%), ascites: 0 (0%). |
|
| Interventions | UDCA group:
‐ Dose: 17‐23 mg/kg body weight/day in two divided doses.
‐ Route: orally.
‐ Duration: five years. Placebo group: ‐ identical 250‐mg gelatin capsules containing microcrystalline cellulose. |
|
| Outcomes | Primary outcomes:
‐ death or liver transplantation; Secondary outcomes: ‐ changes in the frequency of PSC‐related symptoms; ‐ changes in self‐estimated quality of life; ‐ changes in liver laboratory tests; Tertiary outcomes: ‐ effect of UDCA on intestinal symptoms in patients with concomitant IBD. |
|
| Notes | Letter was sent to the authors in August 2010. A reply with additional information was received shortly after. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Adequate sequence generation? | Unclear risk | Method of sequence generation not described. Quote: "...randomised by a hospital pharmacist into blocks of 4 patients." |
| Allocation concealment? | Unclear risk | Method of allocation concealment not described. Quote: "The trial code was kept at the pharmacies in the hospitals." |
| Blinding? All outcomes | Low risk | Quote:"...placebo in identical 250‐mg gelatin capsules containing microcrystalline cellulose..." |
| Incomplete outcome data addressed? | Low risk | Number and reasons for withdrawal were reported. |
| Free of selective reporting? | Unclear risk | Study outcomes were clearly pre‐specified, but not all actual data were reported. |
| Sample size calculation | Low risk | Stated and used. |
| Intention‐to‐treat analysis | High risk | Not stated and not used. |
Stiehl 1994.
| Methods | Study design: a 3‐year pilot study with a placebo‐controlled study period. | |
| Participants | Country: Germany.
Publication language: English. Inclusion criteria ‐ a typical endoscopic retrograde cholangiographic investigation, which showed multiple stenoses; ‐ a serum alkaline phosphatases level at least twice the normal range; ‐ a negative antimitochondrial antibody; ‐ and a liver biopsy compatible with the diagnosis of PSC. Exclusion criteria ‐ decompensation of cirrhosis; ‐ a history of neoplastic disease; ‐ and/or coexisting hepatic disease. Participants ‐ UDCA group (n = 10) Mean age (years): 36 (range 18 to 58); Ratio of sex (M/F): 8/2; Concurrent IBD no. : 9 (90%); Ratio of intra‐ and extra‐hepatic in ERCP: 10:9. ‐ Placebo group (n = 10) Mean age (years): 41 (range 22 to 57); Ratio of sex (M/F): 7/3; Concurrent IBD no. : 8 (80%); Ratio of intrahepatic and extrahepatic PSC in ERCP: 10:9. |
|
| Interventions | UDCA group:
‐ Dose: 750 mg/day.
‐ Route: orally.
‐ Duration: three months. Placebo group: ‐ identical‐appearing capsules administered in the same quantity and manner. |
|
| Outcomes | Liver histological changes at the end of treatment; Endoscopic cholangiography changes at the end of treatment; Biochemical parameters at the end of treatment; and clinical symptom changes at the end of treatment. | |
| Notes | Letter was sent to the authors in August 2010. A reply with additional information was received shortly after. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Adequate sequence generation? | Low risk | A computerised randomisation list was used. |
| Allocation concealment? | Unclear risk | No information provided. |
| Blinding? All outcomes | Unclear risk | Not adequately described. |
| Incomplete outcome data addressed? | Low risk | We contacted the principal author during August 2010 and he stated that no dropouts occurred during the study period. |
| Free of selective reporting? | Unclear risk | Study outcomes were not pre‐specified. |
| Sample size calculation | Low risk | Stated and used. The trial attained the calculated sample size. |
| Intention‐to‐treat analysis | High risk | Not stated and not used. |
ALP: alkaline phosphatases. AST: aspartate aminotransferase. ERCP: endoscopic retrograde cholangiopancreatography. IBD: inflammatory bowel disease. PSC: primary sclerosing cholangitis. UDCA: ursodeoxycholic acid. RUQ: right upper quadrant. INR: international normalized ratio.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Charatcharoenwitthaya '08 | A longitudinal, cohort study performed to determine the clinical features of small‐duct PSC, as well as to determine the influence of IBD and the use of UDCA on the clinical course of the livers disease. Forty‐two patients with small‐duct PSC were followed for up to 24.9 years. UDCA treatment of 13 to 15 mg/kg body weight/day for an average of 40 months showed biochemical improvement (P < 0.001) in UDCA‐treated patients, while no statistically significant change occurred in untreated patients. However, UDCA therapy did not show a statistically significant effect on disease progression. IBD had no impact on survival or transplantation in small‐duct PSC patients. |
| Chazouilleres 1990 | A case series study that prospectively evaluated the effects of UDCA (dose range 750 to 1250 mg/day) in 15 patients with PSC. A clinical and biochemical evaluation was carried out at 3 and 6 months after treatment initiation. Six months of UDCA treatment resulted in a statistically significant improvement of clinical and biochemical variables. |
| Garioud 2006 | A prospective, multicentre observational study which included 174 patients with PSC of at least four years duration. Study objective was to describe the outcome of PSC under treatment with low dose UDCA. The observed survival was compared to the predicted survival by the revised Mayo model. During the study period 10 patients died and 28 were transplanted. Observed survival and predicted survival were similar. |
| Gilger 2000 | A case series study including ten children with primary sclerosing cholangitis. Data were retrospectively analysed to evaluate the effect of UDCA in a mean dose of 17 mg/kg body weight/day (range from 9 to 37 mg/kg body weight/day). One patient was not included in the analysis because of loss to follow‐up. There were no adverse events from the medication recorded in any of the patients. Results showed a significant reduction in activities of serum ALP, ALT, AST, and GGT after 20‐month treatment period. |
| Harnois 2001 | A case series study including thirty patients with PSC treated with high‐dose UDCA (25 to 30 mg/kg body weight/day) for one year. Changes in the Mayo risk score at 1 year of treatment and projected survival at 4 years were analysed in comparison with a placebo group of patient (n = 52) and a group of patients (n = 53) treated with low‐dose UDCA (13 to 15 mg/kg body weight/day). A marked improvement in serum activities of ALP, AST, and the concentration of albumin and total bilirubin occurred at end of treatment. |
| O'Brien 1991 | A case series study to evaluate the effect of UDCA treatment (10 mg/kg body weight/day) on liver enzyme activity, bilirubin, cholesterol and bile acids levels, and symptoms in patients with PSC. Twelve patients with persistently elevated pretreatment ALP and GGT activities were observed for a median of 37 months. Significant reductions in serum total cholesterol levels and enzyme activities during treatment were found. Improvements continued after two years of treatment in ten patients. |
| Okolicsanyi 2003 | A multicentre retrospective study to evaluate the efficacy of low‐dose UDCA treatment in PSC patients. Data of 86 patients from eight different centres were analysed. Sixty‐nine were treated with UDCA (8 to 13 mg/kg body weight/day) and seventeen were treated symptomatically and served as controls. Treatment effect was determined by symptom analysis and standard liver function tests. Results showed a statistically significant improvement of liver function tests and there was no amelioration of symptoms including fatigue, jaundice and body weight loss. |
| Rudolph 1991 | A case series study of fifteen patients on UDCA therapy (8 to 10 mg/kg body weight/day) followed for two years or longer to assess the effect of UDCA administration on survival in patients with PSC. After two years, one patient died due to bile duct carcinoma, and one liver transplantation was necessary. According to the Mayo risk factors the predicted risk of death did not increase. |
| Schonfeld 1996 | A case series study of eleven patients who received 10 mg/kg body weight/day of UDCA for three to six years to assess the effect of UDCA on liver function. UDCA significantly reduced serum activities of ALP, GGT, AST, and ALT. Parameters of synthetic liver function remained constant during the observation period. Quantitative liver function tests showed little change during the treatment. |
| van de Meeberg 1996 | A case series study that included PSC and PBC patients designed to evaluate the efficacy of a single or multiple dose regimen on liver enzyme activity and serum and biliary bile salts composition. Twenty‐seven patients (19 PSC, 8 PBC) received 10 mg/kg body weight/day of UDCA in a single dose at bed time or in three divided doses with meals over three months. Liver biochemical variables improved equally in both groups. |
| van Hoogstraten 1998 | A multicentre randomised trial assessing the effects of 10 mg/kg body weight/day of UDCA, given in either single or multiple daily doses, on symptoms, serum liver tests, cholangiographic and histological findings, and treatment failure rates. Forty‐eight patients with PSC were enrolled. After two years treatment, no beneficial effects of UDCA on symptoms, liver histology, or mortality were observed. Serum biochemical variables including ALP, GGT, and AST improved significantly in both groups. |
| van Hoogstraten 2000 | An eight‐week randomised double blind pilot study assessing the effects of 9 mg of budesonide (n = 6) versus 3 mg of budesonide (n = 6) or 10 mg of prednisone (n = 6) in patients who had been treated with 12 mg/kg body weight/day of UDCA for at least five months without achieving biochemical remission. Pruritus decreased significantly more in the prednisone group, compared to both the 3 mg and 9 mg budesonide groups. ALP decreased in the prednisone group whereas serum bilirubin, GGT, AST, and ALT did not change significantly. |
| van Milligen 1999 | A case series study evaluating the effect of UDCA treatment in patients with PSC by assessing biochemical variables, immunological markers and inflammation indices. Seventeen PSC patients were enrolled for one year of treatment with UDCA (dose range 12 to 15 mg/kg body weight/day). Treatment with UDCA was associated with significant improvements in serum biochemical liver tests, immunoglobulin levels, and blood coagulation factors. |
| van Thiel 1992 | A randomised clinical trial comparing UDCA with colchicine or no treatment. It was not possible to ascertain from the abstract, which is the only published information on the trial, which treatment the participants of the control group received. A total of 16 control group patients and 32 UDCA treated patients with PSC were studied for a mean of 18.1 months, standard deviation 1.0. UDCA was administered at a dose of 300 mg twice a day. Patients in the control group received either colchicine (0.6 mg twice a day) or no treatment. Patients in the UDCA group experienced a significant reduction in serum bilirubin level. No difference in ALT, AST, ALP, or albumin levels was observed between the two groups and there was no percent change in these parameters compared to baseline. |
ALP = alkaline phosphatases. ALT = alanine aminotransferase. AST= aspartate aminotransferase. GGT = gamma glutamyltransferase. PBC = primary biliary cirrhosis. PSC = primary sclerosing cholangitis. UDCA = ursodeoxycholic acid.
Differences between protocol and review
We assessed the risk of bias in included trials for two more components than it was stated in the review protocol ‐ incomplete outcome data and selective outcome measure report. Quasi‐randomised, observational, and case‐control studies were included for the report of adverse events.
Contributions of authors
GP and VG independently performed searches of relevant databases. GP validated the search, selected trials for inclusion, contacted the authors of trials, performed data extraction and data analysis, and drafted the systematic review. CG participated in the conduct of the protocol as well as the first version of this review (New Reference). VG revised the review update. CG and DS revised the review update, solved discrepancies of data extraction, validated data analysis, and provided consultation.
Sources of support
Internal sources
The Copenhagen Trial Unit, Denmark.
External sources
Danish Medical Research Council Grant on Getting Research into Practice (GRIP), Denmark.
Declarations of interest
None known.
New search for studies and content updated (no change to conclusions)
References
References to studies included in this review
Beuers 1992 {published data only}
- Beuers U, Spengler U, Kruis W, Aydemir U, Heldwein W, Weinzierl M, et al. Effect of ursodeoxycholic acid in primary sclerosing cholangitis: a controlled trial. Hepatology 1991;14(4 pt 2):64A. [DOI] [PubMed] [Google Scholar]
- Beuers U, Spengler U, Kruis W, Aydemir U, Wiebecke B, Heldwein W, et al. Ursodeoxycholic acid for treatment of primary sclerosing cholangitis: a placebo‐controlled trial. Hepatology 1992;16(3):707‐14. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
de Maria 1996 {published data only}
- Maria N, Colantoni A, Rosenbloom E, Thiel DH. Ursodeoxycholic acid does not improve the clinical course of primary sclerosing cholangitis over a 2‐year period. Hepato‐Gastroenterology 1996;43(12):1472‐9. [MEDLINE: ] [PubMed] [Google Scholar]
Lindor 1997 {published data only}
- Lindor KD, for the Mayo Primary Sclerosing Cholangitis‐Ursodeoxycholic Acid Study Group. Ursodiol for primary sclerosing cholangitis. The New England Journal of Medicine 1997;336(10):691‐5. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Lindor 2009 {published data only}
- Lindor KD, Enders FB, Schmoll JA, Hoskin TL, Jorgensen RA, Petz JL, et al. Randomized, double‐blind controlled trial of high‐dose ursodeoxycholic acid (UDCA) for primary sclerosing cholangitis (PSC). Hepatology 2008;48(4 (Suppl)):378A. [Google Scholar]
- Lindor KD, Kowdley KV, Luketic VAC, Harrison ME, McCashland T, Befeler AS, et al. High‐dose ursodeoxycholic acid for the treatment of primary sclerosing cholangitis. Hepatology 2009;50(3):808‐14. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Lo 1992 {published data only}
- Lo SK, Herrmann R, Chapman RW, Fleming KA, Shearman J, Cusick P, et al. Ursodeoxycholic acid in primary sclerosing cholangitis: a double‐blind placebo controlled trial [AASLD abstract]. Hepatology 1992;16(2 Pt 2):92A. [Google Scholar]
Mitchell 2001 {published data only}
- Bansi D, Christie J, Fleming K, Chapman R. High‐dose ursodeoxycholic acid in primary sclerosing cholangitis: a randomized double‐blind, placebo‐controlled trial. Gastroenterology 1996;110:1146A. [DOI] [PubMed] [Google Scholar]
- Mitchell SA, Bansi D, Hunt N, Christie J, Fleming K, Chapman R. High‐dose ursodeoxycholic acid (UDCA) in primary sclerosing cholangitis (PSC): results after two years of a randomised double‐blind, placebo‐controlled trial. Gut 1997;40(Suppl 1):A29. [Google Scholar]
- Mitchell SA, Bansi DS, Hunt N, Bergmann KV, Fleming KA, Chapman RW. A preliminary trial of high‐dose ursodeoxycholic acid in primary sclerosing cholangitis. Gastroenterology 2001;121(4):900‐7. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Olsson 2005 {published data only}
- Olsson R, Boberg KM, Muckadell OS, Lindgren S, Hultcrantz R, Folvik G, et al. High‐dose ursodeoxycholic acid in primary sclerosing cholangitis: A 5‐year multicenter, randomized, controlled study. Gastroenterology 2005;129(5):1464‐72. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Stiehl 1994 {published data only}
- Stiehl A, Raedsch R, Rudolph G, Theilmann L. Treatment of primary sclerosing cholangitis with ursodeoxycholic acid: first results of a controlled study. Hepatology 1989;10(4):602. [Google Scholar]
- Stiehl A, Walker S, Stiehl L, Rudolph G, Hofmann WJ, Theilmann L. Effect of ursodeoxycholic acid on liver and bile duct disease in primary sclerosing cholangitis. A 3‐year pilot study with a placebo‐controlled study period. Journal of Hepatology 1994;20(1):57‐64. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Charatcharoenwitthaya '08 {published data only}
- Charatcharoenwitthaya P, Angulo P, Enders FB, Lindor KD. Impact of inflammatory bowel disease and ursodeoxycholic acid therapy on small‐duct primary sclerosing cholangitis. Hepatology 2008;47(1):133‐42. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Chazouilleres 1990 {published data only}
- Chazouilleres O, Poupon R, Capron JP, Metman EH, Dhumeaux D, Amouretti M, et al. Ursodeoxycholic acid for primary sclerosing cholangitis. Journal of Hepatology 1990;11(1):120‐3. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Garioud 2006 {published data only}
- Garioud A, Seksik P, Chretien Y, Corpechot C, Poupon R, Poupon RE, et al. Four‐year outcome of primary sclerosing cholangitis (PSC) under low‐dose ursodeoxycholic acid (UDCA): a French prospective study. Journal of Hepatology 2006;44(Suppl 2):S234‐S235. [DOI: 10.1016/S0168-8278(06)80632-X] [DOI] [Google Scholar]
Gilger 2000 {published data only}
- Gilger MA, Gann ME, Opekun AR, Gleason WA Jr. Efficacy of ursodeoxycholic acid in the treatment of primary sclerosing cholangitis in children. Journal of Pediatric Gastroenterology and Nutrition 2000;31(2):136‐41. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Harnois 2001 {published data only}
- Harnois DM, Angulo P, Jorgensen RA, Larusso NF, Lindor KD. High‐dose ursodeoxycholic acid as a therapy for patients with primary sclerosing cholangitis. American Journal of Gastroenterology 2001;96(5):1558‐62. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
O'Brien 1991 {published data only}
- O'Brien CB, Senior JR, Arora‐Mirchandani R, Batta AK, Salen G. Ursodeoxycholic acid for the treatment of primary sclerosing cholangitis: a 30‐month pilot study. Hepatology 1991;14(5):838‐47. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Okolicsanyi 2003 {published data only}
- Okolicsanyi L, Groppo M, Floreani A, Morselli‐Labate AM, Rusticali AG, Battocchia A, et al. Treatment of primary sclerosing cholangitis with low‐dose ursodeoxycholic acid: results of a retrospective Italian multicentre survey. Digestive and Liver Disease 2003;35(5):325‐31. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Rudolph 1991 {published data only}
- Rudolph G, Walker S, Theilmann L, Folsch U, Bircher J, Stiehl A. The effect of ursodeoxycholic acid on survival in patients with primary sclerosing cholangitis [EASL abstract]. Journal of Hepatology 1991;13(Suppl 2):S165. [Google Scholar]
Schonfeld 1996 {published data only}
- Schonfeld J, Zotz R, Mahl M, Beste M, Breuer N, Goebell H. Primary sclerosing cholangitis: conventional and quantitative liver function tests during long‐term therapy with ursodeoxycholic acid [Primar sklerosierende cholangitis: konventionelle und quantitative leberfunktionstests unter einer mehrjahrigen therapie mit ursodesoxycholsaure]. Zeitschrift fur Gastroenterologie 1996;34(2):123‐7. [MEDLINE: ] [PubMed] [Google Scholar]
van de Meeberg 1996 {published data only}
- Meeberg PC, Wolfhagen FH, Berge‐Henegouwen GP, Salemans JM, Tangerman A, Buuren HR, et al. Single or multiple dose ursodeoxycholic acid for cholestatic liver disease: biliary enrichment and biochemical response. Journal of Hepatology 1996;25(6):887‐94. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
van Hoogstraten 1998 {published data only}
- Hoogstraten HJ, Wolfhagen FH, Meeberg PC, Kuiper H, Nix GA, Becx MC, et al. Ursodeoxycholic acid therapy for primary sclerosing cholangitis: results of a 2‐year randomized controlled trial to evaluate single versus multiple daily doses. Journal of Hepatology 1998;29(3):417‐23. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
van Hoogstraten 2000 {published data only}
- Hoogstraten HJ, Vleggaar FP, Boland GJ, Steenbergen W, Griffioen P, Hop WC, et al. Budesonide or prednisone in combination with ursodeoxycholic acid in primary sclerosing cholangitis: a randomized double‐blind pilot study. Belgian‐Dutch PSC Study Group. American Journal of Gastroenterology 2000;95(8):2015‐22. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
van Milligen 1999 {published data only}
- Milligen de Wit AW, Kuiper H, Camoglio L, Bracht J, Jones EA, Tytgat GN, et al. Does ursodeoxycholic acid mediate immunomodulatory and anti‐inflammatory effects in patients with primary sclerosing cholangitis?. European Journal of Gastroenterology and Hepatology 1999;11(2):129‐36. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
van Thiel 1992 {published data only}
- Thiel DH, Wright HI, Gavaler JS. Ursodeoxycholic acid (UDCA) therapy for primary sclerosing cholangitis (PSC): preliminary report of a randomised controlled trial [AASLD abstract]. Hepatology 1992;16(2 Pt 2):62A. [Google Scholar]
Additional references
Angulo 1999
- Angulo P, Lindor KD. Primary sclerosing cholangitis. Hepatology 1999;30(1):325‐32. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Beuers 1993
- Beuers U, Nathanson MH, Isales CM, Boyer JL. Tauroursodeoxycholic acid stimulates hepatocellular exocytosis and mobilizes extracellular Ca++ mechanisms defective in cholestasis. Journal of Clinical Investigation 1993;92(6):2984‐93. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Beuers 1996
- Beuers U, Throckmorton DC, Anderson MS, Isales CM, Thasler W, Kullak‐Ublick GA. Tauroursodeoxycholic acid activates protein kinase C in isolated rat hepatocytes. Gastroenterology 1996;110(5):1553‐63. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Beuers 2001
- Beuers U, Bilzer M, Chittattu A, Kullak‐Ublick GA, Keppler D, Paumgartner G, et al. Tauroursodeoxycholic acid inserts the apical conjugate export pump, Mrp2, into canalicular membranes and stimulates organic anion secretion by protein kinase C‐dependent mechanisms in cholestatic rat liver. Hepatology 2001;33(5):1206‐16. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Beuers 2009
- Beuers U, Kullak‐Ublick GA, Pusl T, Rauws ER, Rust C. Medical treatment of primary sclerosing cholangitis: a role for novel bile acids and other (post‐) transcriptional modulators?. Clinical Reviews in Allergy and Immunology 2009;36(1):52‐61. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Brok 2009
- Brok J, Thorlund K, Wetterslev J, Gluud C. Apparently conclusive meta‐analysis may be inconclusive ‐ Trial sequential analysis adjustment of random error risk due to repetitive testing of accumulating data in apparently conclusive neonatal meta‐analysis. International Journal of Epidemiology 2009;38(1):298‐303. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Burak 2003
- Burak KW, Angulo P, Lindor KD. Is there a role for liver biopsy in primary sclerosing cholangitis?. American Journal of Gastroenterology 2003;98(5):1155‐8. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Calmus 1990
- Calmus Y, Gane P, Rouger P, Poupon R. Hepatic expression of class I and class II major histocompatibility complex molecules in primary biliary cirrhosis: effect of ursodeoxycholic acid. Hepatology 1990;11(1):12‐5. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Card 2008
- Card TR, Solaymani‐Dodaran M, West J. Incidence and mortality of primary sclerosing cholangitis in the UK: a population‐based cohort study. Journal of Hepatology 2008;48(6):939‐44. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Chen 1984
- Chen LY, Goldberg HI. Sclerosing cholangitis: broad spectrum of radiographic features. Gastrointestinal Radiolology 1984;9(1):39‐47. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Chen 2007
- Chen W, Liu J, Gluud C. Bile acids for viral hepatitis. Cochrane Database of Systematic Reviews 2007, Issue 4. [DOI: 10.1002/14651858.CD003181.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Crosignani 1996
- Crosignani A, Battezzati PM, Setchell KDR, Invernizzi P, Covini G, Zuin M, et al. Tauroursodeoxycholic acid for treatment of primary biliary cirrhosis. A dose‐response study. Digestive Diseases and Sciences 1996;41(4):809‐15. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Cullen 2005
- Cullen SN, Chapman RW. Review article: current management of primary sclerosing cholangitis. Alimentary Pharmacology and Therapeutics 2005;21(8):933‐48. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Demets 1987
- Demets DL. Methods for combining randomized clinical trials: strengths and limitations. Statistics in Medicine 1987;6(3):341‐50. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
DerSimonian 1986
- DerSimonian R, Laird N. Meta‐analysis in clinical trials. Controlled Clinical Trials 1986;7(3):177‐88. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Donaldson 1991
- Donaldson PT, Farrant JM, Wilkinson ML, Hayllar K, Portmann BC, Williams R. Dual association of HLA DR2 and DR3 with primary sclerosing cholangitis. Hepatology 1991;13(1):129‐33. [MEDLINE: ] [PubMed] [Google Scholar]
Egger 1997
- Egger M, Davey Smith G, Schneider M, Minder C. Bias in meta‐analysis detected by a simple graphical test. BMJ (Clinical Research Ed.) 1997;315(7109):629‐34. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Gluud 2001
- Gluud C. Alcoholic hepatitis: no glucocorticosteroids?. In: Leuschner U, James OFW, Dancygier H editor(s). Steatohepatitis (NASH and ASH). Falk Symposium 121. Lancaster: Kluwer Academic Publisher, 2001:322‐42. [Google Scholar]
Gluud 2007
- Gluud C, Brok J, Gong Y, Koretz RL. Hepatology may have problems with putative surrogate outcome measures. Journal of Hepatology 2007;46(4):734‐42. [DOI] [PubMed] [Google Scholar]
Gluud 2010
- Gluud C, Nikolova D, Klingenberg SL, Alexakis N, Als‐Nielsen B, Colli A, et al. Cochrane Hepato‐Biliary Group. About The Cochrane Collaboration (Cochrane Review Groups (CRGs)). 2010, Issue 9. Art. No.: LIVER.
Gong 2008
- Gong Y, Huang ZB, Christensen E, Gluud C. Ursodeoxycholic acid for primary biliary cirrhosis. Cochrane Database of Systematic Reviews 2008, Issue 3. [DOI: 10.1002/14651858.CD000551.pub2] [DOI] [PubMed] [Google Scholar]
Gordon 2008
- Gordon FD. Primary sclerosing cholangitis. Surgical Clinics of North America 2008;88(6):1385‐407. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Guicciardi 2002
- Guicciardi M, Gores GJ. Ursodeoxycholic acid cytoprotection: dancing with death receptors and survival pathways. Hepatology 2002;35(4):971‐3. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Higgins 2002
- Higgins JPT, Thompson SG. Quantifying heterogeneity in a meta‐analysis. Statistics in Medicine 2002;21(11):1539‐58. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Higgins 2009
- Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.0.2 [updated September 2009]. The Cochrane Colloboration, 2009. Available from www.cochrane‐handbook.org.
ICH‐GCP 1997
- International Conference on Harmonisation Expert Working Group. International conference on harmonisation of technical requirements for registration of pharmaceuticals for human use. ICH harmonised tripartite guideline. Guideline for good clinical practice1997 CFR & ICH Guidelines. Vol. 1, PA 19063‐2043, USA: Barnett International/PAREXEL, 1997. [Google Scholar]
Jaeschke 2002
- Jaeschke H, Gores GJ, Cederbaum AI, Hinson JA, Pessayre D, Lemasters JJ. Mechanisms of hepatotoxicity. Toxicological Sciences 2002;65(2):166‐76. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Kaplan 2007
- Kaplan GG, Laupland KB, Butzner D, Urbanski SJ, Lee SS. The burden of large and small duct primary sclerosing cholangitis in adults and children: a population‐based analysis. American Journal of Gastroenterology 2007;102(5):1042‐9. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Kim 2000
- Kim WR, Therneau TM, Wiesner RH, Poterucha JJ, Benson JT, Malinchoc M, et al. A revised natural history model for primary sclerosing cholangitis. Mayo Clinic Proceedings 2000;75(7):688‐94. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Kjaergard 2001
- Kjaergard LL, Villumsen J, Gluud C. Reported methodological quality and discrepancies between large and small randomized trials in meta‐analyses. Annals of Internal Medicine 2001;135(11):982‐9. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
LaRusso 1999
- LaRusso NF, Wiesner RH, Ludwig J. Sclerosing cholangitis. In: Bircher J, Benhamou JP, McIntyre N, Rizzetto M, Rodes J editor(s). Oxford Textbook of Clinical Hepatology. 2. Vol. II, Oxford: Oxford University Press, 1999:1121‐30. [Google Scholar]
Lee 1995
- Lee YM, Kaplan MM. Primary sclerosing cholangitis. New England Journal of Medicine 1995;332(14):924‐33. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Linder 2001
- Linder S, Soderlund C. Endoscopic therapy in primary sclerosing cholangitis: outcome of treatment and risk of cancer. Hepatogastroenterology 2001;48(38):387‐92. [MEDLINE: ] [PubMed] [Google Scholar]
Lindor 1987
- Lindor KD, Wiesner RH, LaRusso NF. Recent advances in the management of primary sclerosing cholangitis. Seminars in Liver Disease 1987;7(4):322‐7. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Ludwig 1981
- Ludwig J, Barham SS, LaRusso NF, Elveback LR, Wiesner RH, McCall JT. Morphologic features of chronic hepatitis associated with primary sclerosing cholangitis and chronic ulcerative colitis. Hepatology 1981;1(6):632‐40. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Ludwig 1986
- Ludwig J, LaRusso NF, Wiesner RH. Primary sclerosing cholangitis. In: Peters RL, Craig JR editor(s). Liver Pathology‐Contemporary Issues in Surgical Pathology. New York: Churchill Livingstone, 1986:193‐213. [Google Scholar]
Ludwig 1991
- Ludwig J. Small‐duct primary sclerosing cholangitis. Seminars in Liver Disease 1991;11(1):11‐7. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Meagher 2007
- Meagher S, Yusoff I, Kennedy W, Martel M, Adam V, Barkun A. The roles of magnetic resonance and endoscopic retrograde cholangiopancreatography (MRCP and ERCP) in the diagnosis of patients with suspected sclerosing cholangitis: a cost‐effectiveness analysis. Endoscopy 2007;39(3):222‐8. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Michaels 2008
- Michaels A, Levy C. Endoscopic and surgical management of primary sclerosing cholangitis. Medscape Journal of Medicine 2008;10(10):242. [MEDLINE: ] [PMC free article] [PubMed] [Google Scholar]
Moher 1998
- Moher D, Pham B, Jones A, Cook DJ, Jadad AR, Moher M, et al. Does quality of reports of randomised trials affect estimates of intervention efficacy reported in meta‐analyses. Lancet 1998;352(9128):609‐13. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Olerup 1995
- Olerup O, Olsson R, Hultcrantz R, Broome U. HLA‐DR and HLA‐DQ are not markers for rapid disease progression in primary sclerosing cholangitis. Gastroenterology 1995;108(3):937‐40. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Palmer 1972
- Palmer RH. Bile acids, liver injury and liver disease. Archives of Internal Medicine 1972;130(4):606‐17. [MEDLINE: ] [PubMed] [Google Scholar]
Paumgartner 2002
- Paumgartner G, Beuers U. Ursodeoxycholiic acid in cholestatic liver disease: mechanisms of action and therapeutic use revisited. Hepatology 2002;36(3):525‐31. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Paumgartner 2004
- Paumgartner G, Beuers U. Mechanisms of action and therapeutic efficacy of ursodeoxycholic acid in cholestatic liver disease. Clinics in Liver Disease 2004;8(1):67‐81. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Perez 2009
- Perez MJ, Briz O. Bile‐acid‐induced cell injury and protection. World Journal of Gastroenterology 2009;15(14):1677‐89. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Podda 1996
- Podda M, Crosignani A, Battezzati PM, Quagliuolo M, Valsania C, Invernizzi P, et al. Tauroursodeoxycholic acid for the treatment of chronic hepatitis: a dose‐response study (IASL Abstract). Hepatology 1996;23(1):I‐70. [MEDLINE: ] [Google Scholar]
Qiao 2002
- Qiao L, Yacoub A, Studer E, Gupta S, Pei XY, Grant S, et al. Inhibition of the MAPK and PI3K pathways enhances UDCA‐induced apoptosis in primary rodent hepatocytes. Hepatology 2002;35(4):779‐89. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
RevMan 2008 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version Version 5.0. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2008.
Royle 2003
- Royle P, Milne R. Literature searching for randomized controlled trials used in Cochrane reviews: rapid versus exhaustive searches. International Journal of Technology Assessment in Health Care 2003;19(4):591‐603. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Schulz 1995
- Schulz KF, Chalmers I, Hayes R, Altman D. Empirical evidence of bias. JAMA 1995;273(5):408‐12. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Shi 2009
- Shi J, Li Z, Zeng X, Lin Y, Xie WF. Ursodeoxycholic acid in primary sclerosing cholangitis: meta‐analysis of randomized controlled trials. Hepatology Research 2009;39(9):865‐73. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Sholmerich 1984
- Sholmerich J, Becher MS, Schmidt KH, Schubert R, Kremer B, Felhaus S, et al. Influence of hydroxylation and conjugation of bile salts on their membrane damaging properties. Hepatology 1984;4(4):661‐7. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Silveira 2008
- Silveira MG, Lindor KD. Primary sclerosing cholangitis. Canadian Journal of Gastroenterology 2008;22(8):689‐98. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Stiehl 1996
- Stiehl A. Ursodeoxycholic acid in the treatment of primary sclerosing cholangitis. The Italian Journal of Gastroenterology 1996;28(3):178‐80. [MEDLINE: ] [PubMed] [Google Scholar]
Stiehl 1997
- Stiehl A, Rudolph G, Sauer P, Benz C, Stremmel W, Walker S, et al. Efficacy of ursodeoxycholic acid treatment and endoscopic dilation of major duct stenoses in primary sclerosing cholangitis. An 8‐year prospective study. Journal of Hepatology 1997;26(3):560‐6. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Talwalkar 2004
- Talwalkar JA, Angulo P, Johnson CD, Petersen BT, Lindor KD. Cost‐minimization analysis of MRC versus ERCP for the diagnosis of primary sclerosing cholangitis. Hepatology 2004;40(1):39‐45. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Talwalkar 2005
- Talwalkar JA, Lindor KD. Primary sclerosing cholangitis. Inflammatory Bowel Diseases 2005;11(1):62‐72. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Thorlund 2009
- Thorlund K, Devereaux PJ, Wetterslev J, Guyatt G, Ioannidis JP, Thabane L, et al. Can trial sequential monitoring boundaries reduce spurious inferences from meta‐analysis?. International Journal of Epidemiology 2009;38(1):276‐86. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Tischendorf 2008
- Tischendorf JJ, Geier A, Trautwein C. Current diagnosis and management of primary sclerosing cholangitis. Liver Transplantation 2008;14(6):735‐46. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Van Nieuwkerk 1996
- Nieuwkerk CM, Elferink RP, Groen AK, Ottenhoff R, Tytgat GN, Dingemans KP, et al. Effects of Ursodeoxycholate and cholate feeding on liver disease in FVB mice with a disrupted mdr2 P‐glycoprotein gene. Gastroenterology 1996;111(1):165‐71. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Wee 1985a
- Wee A, Ludwig J. Pericholangitis in chronic ulcerative colitis: primary sclerosing cholangitis of the small bile ducts?. Annals of Internal Medicine 1985;102(5):581‐7. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Wee 1985b
- Wee A, Ludwig J, Coffey RJ Jr, LaRusso NF, Wiesner RH. Hepatobiliary carcinoma associated with primary sclerosing cholangitis and chronic ulcerative colitis. Human Pathology 1985;16(7):719‐26. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Wetterslev 2008
- Wetterslev J, Thorlund K, Brok J, Gluud C. Trial sequential analysis may establish when firm evidence is reached in cumulative meta‐analysis. Journal of Clinical Epidemiology 2008;61(1):64‐75. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Wood 2008
- Wood L, Egger M, Gluud LL, Schulz KF, Juni P, Altman DG, et al. Empirical evidence of bias in treatment effect estimates in controlled trials with different interventions and outcomes: meta‐epidemiological study. BMJ (Clinical Research Ed.) 2008;336(7644):601‐5. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Yoshikawa 1998
- Yoshikawa M, Matsui Y, Kawamoto H, Toyohara M, Matsumura K, Yamao J, et al. Intragastric administration of ursodeoxycholic acid suppresses immunoglobulin secretion by lymphocytes from liver, but not from peripheral blood, spleen and Peyer's patches in mice. International Journal of Immunopharmacology 1998;20(1‐3):29‐38. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
Chen 2003
- Chen W, Gluud C. Bile acids for primary sclerosing cholangitis. Cochrane Database of Systematic Reviews 2003, Issue 2. [DOI: 10.1002/14651858.CD003626] [DOI] [PubMed] [Google Scholar]