

Abstract
Bacterial culture broth extracts have been the starting point for the development of numerous therapeutics. However, only a small fraction of bacterial biosynthetic diversity is accessible using this strategy. Here, we apply a discovery approach that bypasses the culturing step entirely by bioinformatically predicting small molecule structures from the primary sequences of the biosynthetic gene clusters. These structures are then chemically synthesized to give synthetic-bioinformatic natural products (syn-BNPs). Using this approach, we screened syn-BNPs inspired by nonribosomal peptide synthetases against microbial pathogens, and discovered an antibiotic for which no resistance could be identified and an antifungal agent with activity against diverse fungal pathogens.
Synthetic chemists have long focused on the synthesis of small molecules resembling those isolated from nature as a means of generating bioactive compounds (Figure 1a). In fact, the majority of antimicrobials in clinical use today are either natural products (NPs) or synthetic small molecules inspired by NPs.1 Unfortunately, traditional NP discovery methods are limited by the fact that the majority of environmental bacteria remain refractory to culture2 and that only a fraction of the chemical encoding capacity of most cultured bacteria is accessed in laboratory fermentation studies.3 We see an emerging opportunity for using synthetic chemistry to access new bioactive molecules, driven by the increasing cost effectiveness of high-throughput DNA sequencing and the rapidly improving bioinformatic algorithms for predicting the chemical output of bacterial secondary metabolite biosynthetic gene clusters (BGCs) using DNA sequence alone. In the approach outlined here, chemical structures are predicted bioinformatically from the primary sequence data and are then produced by chemical synthesis to generate syn-BNPs (Figure 1b).4 By applying a syn-BNP approach to sequenced nonribosomal peptide synthetase (NRPS) BGCs, we have identified a broad-spectrum antibiotic and an antifungal agent.
Figure 1.
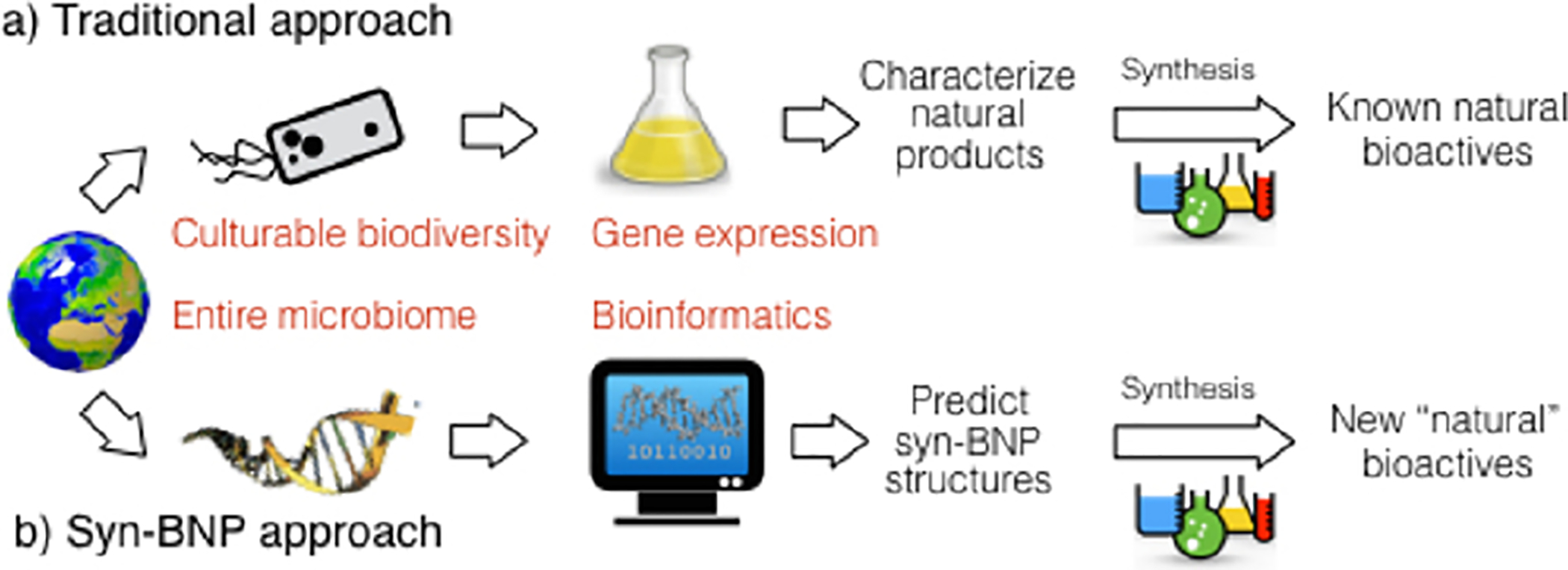
Comparison of the scope and limitation of traditional NP discovery methods (a) and the syn-BNP approach (b).
Bioinformatic analysis of microbial sequence data indicates that nonribosomal peptides (NRPs) are likely the most commonly encoded secondary metabolites across bacterial taxa.5 The importance of this structurally diverse class of NPs to antimicrobial discovery is highlighted by their prevalence among recently reported antibacterial and antifungal agents.4,6 Multiple, increasingly accurate bioinformatics algorithms have been developed to predict small molecule structures encoded by NRPS BGCs based solely on the primary sequence of the NRP megasynthetases.7 The development of these algorithms coupled with the versatility of solid-phase peptide synthesis (SPPS) makes NRPs an appealing test case for a syn-BNP approach (Figure 2a).
Figure 2.

(a) Overview of the syn-BNP approach. (b) Results of primary screening against ESKAPE pathogens and Candida albicans.(c) Summary of primary (zone of growth inhibition) and secondary (resynthesis and validation) screening data. Here we report in detail on antibiotic 1.69 (1) and an antifungal agent 3.23 (2).
To identify BGCs from which to generate syn-BNPs for antimicrobial screening, complete bacterial genomes were obtained from the NCBI database and the NRPS BGCs were bioinformatically culled from these data. Hybrid BGCs and those with less than 5 modules were removed, as short NRPs are often highly modified and therefore not easily accessible using SPPS alone. Within the remaining data set, we focused on NRPS BGCs that contained either starter condensation domains or thioesterase domains and excluded those that appear to diverge from the classic collinear rule. The algorithms we used for predicting the peptide sequences represent three generations of NPRS prediction software (NRPSPred2, Stachelhaus, Minowa).7a–c NRPSPred2 is the most sophisticated algorithm, and we adhered to its predictions whenever possible. If it failed to predict a specific peptide, we consulted predictions from the other algorithms.
Based on predictions from 280 NRPS BGCs, 288 peptides (3 × 96-well plates) were produced by SPPS using Fmoc chemistry (Table S1). Syn-BNPs were made in a 96-well microtiter plate format using an automated peptide synthesizer. Depending on the bioinformatic prediction, the N-terminus of each peptide was either left as a free amine or derivatized with acetic acid, a long chain fatty acid, or benzoic acid. Syn-BNPs were released by hydrolysis using NaOH, neutralized with acetic acid, and directly screened against target microbes (Figure 2a).
All syn-BNPs were screened for bioactivity against the ESKAPE pathogens (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, Enterobacter cloacae) and the fungal pathogen Candida albicans. ESKAPE pathogens cause the majority of hospital-acquired infections in the US,8 whereas C. albicans is the fourth leading cause of hospital bloodstream infections9 and a leading cause of oral and genital fungal infections.10 Syn-BNPs that resulted in a clear zone of growth inhibition on a lawn of any pathogen were considered hits (Figure 2b). Twelve primary hits were resynthesized, purified, and reassayed (Figure 2c, Tables S2 and S3). Six syn-BNP primary hits were validated as having activity (MIC ≤ 16 μg/mL) against at least one pathogen.
The most active syn-BNPs were inspired by BGCs present in bacteria from taxa that have not been a focus of NP discovery programs. None of the bacteria from which active syn-BNPs were predicted has been previously reported to produce a NP that resembles the syn-BNP. These are not surprising observations as very few bacterial taxa have been extensively explored in traditional NP discovery programs and most BGCs remain silent in laboratory culture conditions. Based on potency, spectrum of activity and therapeutic index we selected the antibacterial syn-BNP 1 and the antifungal syn-BNP 2 for detailed analysis (Table 1, Figures 3, 4, and 5).
Table 1.
| Syn-BNP | Ef | Sa | Kp | Ab | Pa | Ec | Ca | HT-29 |
|---|---|---|---|---|---|---|---|---|
| 1 | 4 | 16 | > | 16 | > | > | 128 | 64 |
| 2 | > | > | > | 64 | > | > | 4 | > |
Minimal inhibitory concentration (MIC) values are in μg/mL units. The “>” symbol denotes MIC > 128 μg/mL (highest concentration tested).
Abbreviations: E. faecium (Ef), S. aureus (Sa), K. pneumoniae (Kp), A. baumannii (Ab), P. aeruginosa (Pa), E. cloacae (Ec), C. albicans (Ca); HT-29 is a colon cancer cell line used as a surrogate to assess cytotoxicity.
Figure 3.
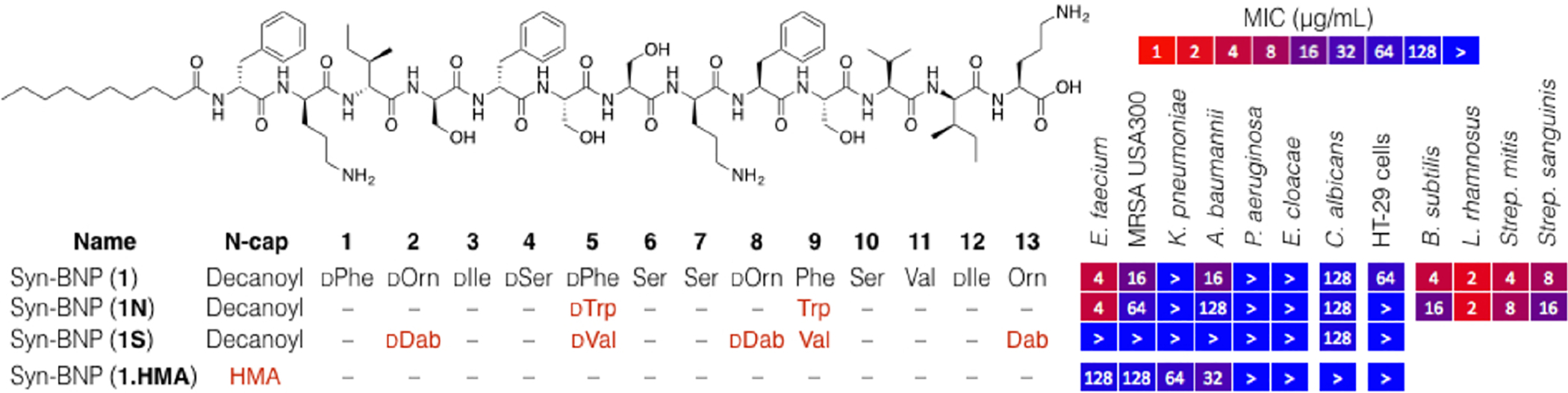
SAR and activity studies of syn-BNP 1. We focused on the synthesis of bioinformatic congeners as well as an exploration of the N-terminal modification. “>” denotes MIC > 128 μg/mL (highest concentration tested)
Figure 4.
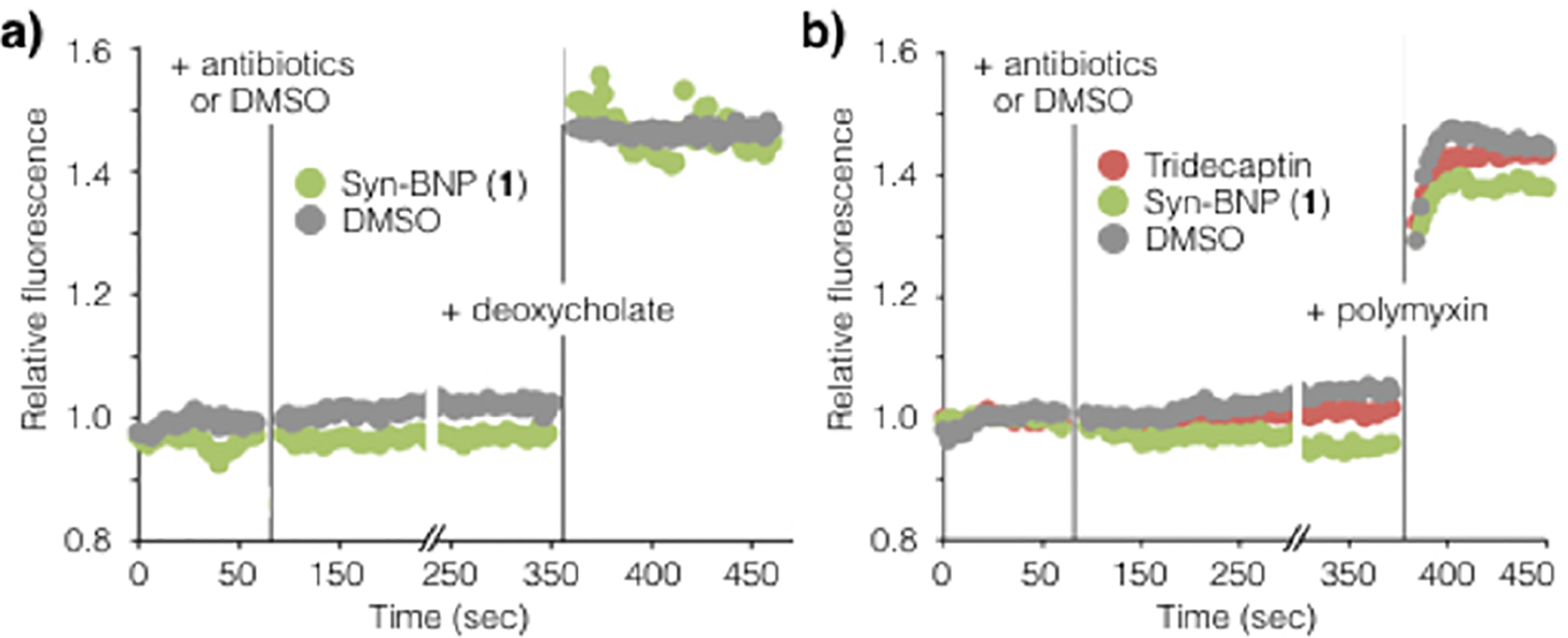
Membrane depolarization performed on E. faecium (a) and A. baumannii (b) indicate that membrane integrity is maintained in the presence of 1N. Deoxycholate and polymyxin were used as the positive control, respectively. Addition of sodium deoxycholate to E. faecium or polymyxin to A. baumannii resulted in rapidly enhanced DiBAC4 fluorescence.
Figure 5.
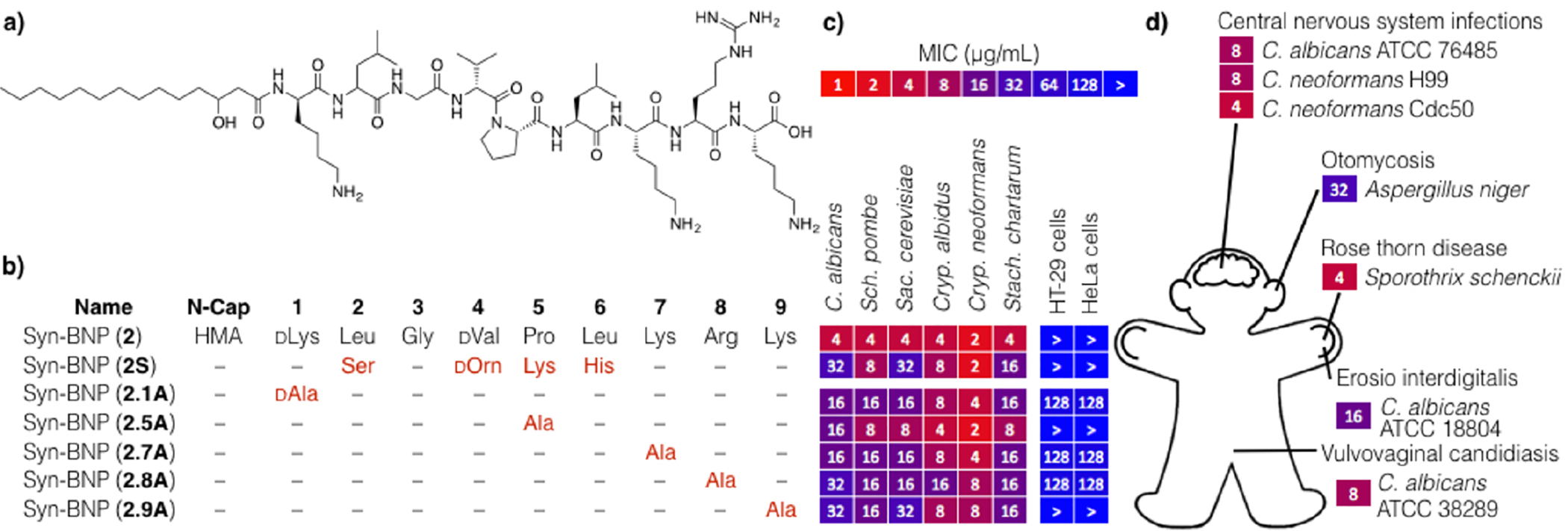
(a) Structure and (b) SAR studies of syn-BNPs 2. (c) MICs of various structural analogs of syn-BNP 2. (d) Syn-BNP 2 proves effective against a number of clinically relevant pathogens that cause hand, ear, as well as central nervous system infections.
Syn-BNP 1 is an N-acylated 13-mer linear peptide containing seven D-amino acids and three nonproteinogenic residues. It is active against Gram-positive bacteria but not against the common fungal pathogen C. albicans at the highest concentration tested (MIC > 128 μg/mL), and is only mildly cytotoxic (MIC 64 μg/mL) against the HT-29 cancer cell line. Among the ESKAPE pathogens it is most active against E. faecium (MIC 4 μg/mL) but also shows activity against the Gram-negative pathogen A. baumannii.
Syn-BNP 1 was predicted from a BGC found in the genome of Paenibacillus mucilaginosus K02, a silicate solubilizing Gram-positive environmental bacterium that is used in China as a growth-promoting additive in fertilizer.11 It resembles the tridecaptins, a family of linear 13-mer peptides isolated from cultures of Paenibacillus polymyxa E681.12 The BGC associated with 1 is related to gene clusters that encode the tridecaptins; however, bioinformatic analysis of the megasynthetases supports the production of distinct metabolites by these clusters (Figure S1). These antibiotics share 5 of 13 residues and the N-terminal fatty acids. Interestingly, syn-BNP 1 acts broadly against Gram-positive bacteria with only limited activity against Gram-negative bacteria whereas tridecaptin A1 shows the opposite spectrum of activity (Figure 3 and Table S3).13
Our structure–activity relationship (SAR) study of 1 focused on the synthesis of bioinformatically predicted congeners as well as an exploration of the N-terminal modification (Figure 3). The originally synthesized peptide (syn-BNP 1) was more active than any other analog we synthesized. Changing the fatty acid from C10 to C14:(3-OH) (3-hydroxymyristic acid, HMA) abrogated antibacterial activity completely (syn-BNP 1.HMA).
To explore the mode of action of this syn-BNP, we initially attempted to identify E. faecium resistant mutants. Interestingly, we were unable to raise resistance using either direct plating on concentrations of 1N just above its MIC or by serial passage of cultures into increasingly higher concentrations of 1N. As many antibiotics for which resistance does not arise function through disruption of the membrane, we tested 1N for membrane depolarization activity using the dye DiBAC4 (bis(1,3-dibutylbarbituric acid) trimethine oxonol), which shows increased fluorescence upon entering a depolarized cell.14 Based on fluorescence change in a DiBAC4 assay, 1 does not induce membrane depolarization in either E. faecium or A. baumannii (Figure 4). The structurally related antibiotic tridecaptin, which induces antibiosis through lipid II binding, does not depolarize the membrane in this assay either. In fact, lipid II binding antibiotics in general (e.g., teixobactin, vancomycin, tridecaptin) do not depolarize bacterial mem-branes and have very low (to nonexistent) resistance rates.6b,13,15
Syn-BNP 2 is an N-acylated nonapeptide with antifungal activity that at the highest concentration tested (128 μg/mL) did not kill HT-29 or HeLa cells, two cancer cell lines used to assess cytotoxicity (Figure 5a). It is active against diverse, clinically relevant fungal pathogens, including clinical isolates of vaginal and central nervous system infections (Figure 5d). Syn-BNP 2 was predicted from a BGC found in the genome of Xenorhabdus nematophila.16 X. nematophila is a Gram-negative entomopathogen that lives in symbiosis with Steinernema carpocapsae, an insect-parasitizing nematode that is used as an insect pests biological control agent. The BGC from which syn-BNP 2 was predicted contains only one close relative within sequenced genomes (Figure S2),17 and no structurally similar NPs have been reported.
In the case of 2, the NRPSPred2 and Minowa algorithms predicted the same syn-BNP structure. The Stachelhaus algorithm predicted a related peptide that turned out to be less active (2S). Our SAR study thus focused on a partial alanine scan (Figure 5b,c). We found that replacing any of the anionic residues or the proline with alanine reduced the antifungal activity by 2–8-fold.
We did not observe a change in MIC in the presence of either ergosterol or sorbitol, suggesting that 2 does not function by disruption of ergosterol biology or the cell wall, two of the most common antifungal mechanisms of action. The molecular target of this syn-BNP remains to be determined.
Cultures of X. nematophila have been extensively studied for NPs and 2 does not resemble NPs characterized in those studies.18 P. mucilaginosus was not available to us, and therefore we could not determine if it produces a NP related to 1. This highlights two key benefits of a syn-BNP approach: (1) it can provide access to molecules inspired by BGCs that are not accessible using traditional methods, and (2) it is not necessary to have a cultured organism, only its genomic sequence data, for the NPs it encodes to serve as inspiration for chemical synthesis studies.
Until now, the translation of biosynthetic instructions into chemical entities has only been possible using biological processes. Even with this modest initial application of a syn-BNP screening approach, we identified molecules with biomedically relevant activities (e.g., antibiosis and antifungal activity). Moreover, syn-BNP approach allowed us to produce hundreds of naturally inspired compounds in parallel, unlike other genome mining methods, which have so far failed to provide access to large numbers of small molecules.
Bacterial (meta)genome sequence data will continue to grow in volume for the foreseeable future, providing access to instructions for an ever increasing number of NPs. We began our syn-BNP studies using NRPS BGCs because of the advanced nature of the algorithms for predicting NRP structures from sequence data7a–c and the ease with which peptide structures can be generated by SPPS. With the development of improved bioinformatic algorithms for converting diverse BGCs into small molecule structure predictions and the incorporation of more sophisticated chemical and chemo-enzymatic synthesis methods to produce more complex syn-BNPs, we postulate syn-BNPs libraries represent a new resource for identifying new bioactive small molecules.
Supplementary Material
ACKNOWLEDGMENTS
We thank the Fischetti (MRSA, K. pneumonia, A. baumannii), Hang (E. faecium), Kapoor (S. pombe, S. cerevisiae), and Tomasz (MRSA) laboratories for strains.
Funding
This work was supported by NIH U19AI109713.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b11861.
Bioinformatic, experimental details, and full author lists of references 16 and 17 (PDF)
Data tables (XLSX)
The authors declare no competing financial interest.
REFERENCES
- (1).Newman DJ; Cragg GM J. Nat. Prod 2016, 79, 629. [DOI] [PubMed] [Google Scholar]
- (2).(a) Handelsman J; Rondon MR; Brady SF; Clardy J; Goodman RM Chem. Biol 1998, 5, R245. [DOI] [PubMed] [Google Scholar]; (b) Rappe MS; Giovannoni SJ Annu. Rev. Microbiol 2003, 57, 369. [DOI] [PubMed] [Google Scholar]
- (3).(a) Piel J Annu. Rev. Microbiol 2011, 65, 431. [DOI] [PubMed] [Google Scholar]; (b) Charlop-Powers Z; Milshteyn A; Brady SF Curr. Opin. Microbiol 2014, 19,70. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Brady, S. F. Nat. Protoc 2007, 2, 1297. [DOI] [PubMed] [Google Scholar]
- (4).Chu J; Vila-Farres X; Inoyama D; Ternei M; Cohen LJ; Gordon EA; Reddy BVB; Charlop-Powers Z; Zebroski HA; Gallardo-Macias R; Jaskowski M; Satish S; Park S; Perlin DS; Freundlich JS; Brady SF Nat. Chem. Biol 2016, 12, 1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).(a) Doroghazi JR; Albright JC; Goering AW; Ju KS; Haines RR; Tchalukov KA; Labeda DP; Kelleher NL; Metcalf WW Nat. Chem. Biol 2014, 10, 963. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Charlop-Powers Z; Owen JG; Reddy BVB; Ternei M; Guimaraes DO; de Frias UA; Pupo MT; Seepe P; Feng ZY; Brady SF eLife 2015, 4, e05048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).(a) Zipperer A; Konnerth MC; Laux C; Berscheid A; Janek D; Weidenmaier C; Burian M; Schilling NA; Slavetinsky C; Marschal M; Willmann M; Kalbacher H; Schittek B; Brotz-Oesterhelt H; Grond S; Peschel A; Krismer B Nature 2016, 535, 511. [DOI] [PubMed] [Google Scholar]; (b) Ling LL; Schneider T; Peoples AJ; Spoering AL; Engels I; Conlon BP; Mueller A; Schaberle TF; Hughes DE; Epstein S; Jones M; Lazarides L; Steadman VA; Cohen DR; Felix CR; Fetterman KA; Millett WP; Nitti AG; Zullo AM; Chen C; Lewis K Nature 2015, 517, 455. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) De Lucca AJ; Walsh TJ Antimicrob. Agents Chemother 1999, 43, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).(a) Stachelhaus T; Mootz HD; Marahiel MA Chem. Biol 1999, 6, 493. [DOI] [PubMed] [Google Scholar]; (b) Minowa Y; Araki M; Kanehisa MJ Mol. Biol 2007, 368, 1500. [DOI] [PubMed] [Google Scholar]; (c) Rottig M; Medema MH; Blin K; Weber T; Rausch C; Kohlbacher O Nucleic Acids Res 2011, 39, W362. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Blin K; Medema MH; Kazempour D; Fischbach MA; Breitling R; Takano E; Weber T Nucleic Acids Res 2013, 41, W204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Pendleton JN; Gorman SP; Gilmore BF Expert Rev. Anti-Infect. Ther 2013, 11, 297. [DOI] [PubMed] [Google Scholar]
- (9).(a) Wisplinghoff H; Bischoff T; Tallent SM; Seifert H; Wenzel RP; Edmond MB Clin. Infect. Dis 2004, 39, 309. [DOI] [PubMed] [Google Scholar]; (b) Wisplinghoff H; Ebbers J; Geurtz L; Stefanik D; Major Y; Edmond MB; Wenzel RP; Seifert H Int. J. Antimicrob. Agents 2014, 43, 78. [DOI] [PubMed] [Google Scholar]
- (10).Jabra-Rizk MA; Kong EF; Tsui C; Nguyen MH; Clancy CJ; Fidel PL Jr.; Noverr M Infect. Immun 2016, 84, 2724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).(a) Liu W; Xu X; Wu X; Yang Q; Luo Y; Christie P Environ. Geochem. Health 2006, 28, 133. [DOI] [PubMed] [Google Scholar]; (b) Hu XF; Gao YY; Fang QL; Wu JG; Chen JS J. Nucl. Agric. Sci 2008, 22, 420. [Google Scholar]
- (12).(a) Shoji J; Hinoo H; Sakazaki R; Kato T; Wakisaka Y; Mayama M; Matsuura S; Miwa HJ Antibiot. 1978, 31, 646. [DOI] [PubMed] [Google Scholar]; (b) Lohans CT; van Belkum MJ; Cochrane SA; Huang Z; Sit CS; McMullen LM; Vederas JC ChemBioChem 2014, 15, 243. [DOI] [PubMed] [Google Scholar]
- (13).Cochrane SA; Findlay B; Bakhtiary A; Acedo JZ; Rodriguez-Lopez EM; Mercier P; Vederas JC Proc. Natl. Acad. Sci. U. S. A 2016, 113, 11561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Clementi EA; Marks LR; Duffey ME; Hakansson AP J. Biol. Chem 2012, 287, 27168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Breukink E; de Kruijff B Nat. Rev. Drug Discovery 2006, 5, 321. [DOI] [PubMed] [Google Scholar]
- (16).Chaston JM; Suen G; Tucker SL; Andersen AW; Bhasin A; Bode E; Bode HB; Brachmann AO; Cowles CE; Cowles KN; et al. PLoS One 2011, 6, e27909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Duchaud E; Rusniok C; Frangeul L; Buchrieser C; Givaudan A; Taourit S; Bocs S; Boursaux-Eude C; Chandler M; Charles J-F; et al. Nat. Biotechnol 2003, 21, 1307. [DOI] [PubMed] [Google Scholar]
- (18).Bode HB Curr. Opin. Chem. Biol 2009, 13, 224. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.