

Summary
Immunity that controls parasitemia and inflammation during Plasmodium falciparum (Pf) malaria can be acquired with repeated infections. A limited understanding of this complex immune response impedes the development of vaccines and adjunctive therapies. We conducted a prospective systems biology study of children who differed in their ability to control parasitemia and fever following Pf infection. By integrating whole-blood transcriptomics, flow-cytometric analysis, and plasma cytokine and antibody profiles, we demonstrate that a pre-infection signature of B cell enrichment; upregulation of T-helper type 1 (Th1) and Th2 cell-associated pathways, including interferon responses; and p53 activation associated with control of malarial fever and coordinated with Pf-specific IgG and Fc receptor activation to control parasitemia. Our hypothesis-generating approach identified host molecules that may contribute to differential clinical outcomes during Pf infection. As a proof of concept, we have shown that enhanced p53 expression in monocytes attenuated Plasmodium-induced inflammation and predicted protection from fever.
Introduction
The parasite Plasmodium falciparum (Pf) causes approximately 219 million cases of malaria and 435,000 deaths annually (World Health Organization, 2018). In humans, Plasmodium infection begins in the liver as a clinically silent process that progresses to blood-stage infection, during which the merozoite form of the parasite invades and replicates within erythrocytes. In non-immune individuals, the cyclical rupture of infected erythrocytes and release of daughter merozoites can rapidly progress to an ‘uncomplicated’ febrile illness or to severe, life-threatening syndromes. In areas of intense transmission, children who survive the first five years of life have typically acquired immunity to severe malaria; however, they remain susceptible to repeated bouts of non-life-threatening febrile malaria into adolescence as they gradually acquire more complete immunity (Langhorne et al., 2008). Yet, even after decades of repeated P. falciparum infections, sterile immunity that prevents blood-stage infection appears to be rarely acquired (Tran et al., 2013).
Although Pf-specific IgG is known to play a key role in controlling malarial fever and parasitemia (Cohen et al., 1961), less is known about cellular immunity to malaria in humans. Pf-elicited interferon-gamma (IFN-γ) production by γδ and CD4+ T cells has been associated with clinical immunity to malaria in humans (D’Ombrain et al., 2008; Reece et al., 2004); however, IFN-γ can induce pathology if not regulated by mediators such as interleukin-10 (IL-10) from type 1 regulatory T (Tr1) cells (Portugal et al., 2014). More recently, the Vδ2+ subset of γδ T cells was implicated in both malaria pathogenesis (Jagannathan et al., 2014; Stanisic et al., 2014) and protection from subsequent Pf infection (Jagannathan et al., 2017). Thus, the mechanisms regulating the balance between immunity and immunopathology during malaria remain unclear. We recently profiled whole-blood transcriptomes by RNA-seq in adults before and during febrile malaria and found that relative to malaria-experienced individuals, malaria-naive individuals had increased activation of pro-inflammatory pathways despite lower parasitemia (Tran et al., 2016), providing evidence for modulation of inflammatory responses during malaria. To further elucidate the mechanisms underlying host control of parasitemia and Pf-induced inflammation, we applied systems biology approaches to a prospective study of Malian children who differed in their ability to control fever and parasitemia after documented Pf infection. By integrating whole-blood transcriptomics with flow cytometric analysis of peripheral blood cells and plasma cytokine and Pf-specific antibody profiles, we demonstrate that a pre-infection signature of B cell enrichment; upregulation of T helper type 1 (Th1) and Th2 pathways, including IFN responses; and p53 activation associates with control of malarial fever, coordinating with Pf-specific IgG and Fc receptor activation to control parasitemia. Our hypothesis-generating approach identified host molecules that may be responsible for differential clinical outcomes after Pf infection. As a proof of concept, we demonstrate that enhanced p53 expression in monocytes attenuates Plasmodium-induced inflammation and predicts protection from fever.
Results
Prospective surveillance defines three classes of malaria susceptibility
In a prospective-cohort study of 695 subjects in a Malian village, we detected 1424 malaria episodes (fever ≥37.5°C and >2500 asexual Pf parasites/μl) through passive and active clinical surveillance over three years. We focused on children aged 6–11 years, the age during which malaria immunity begins to be acquired in this region (Tran et al., 2013), and excluded those with the malaria-protective HbS allele. We identified children who were Pf-PCR negative before the six-month malaria season and determined their time to first Pf infection during the ensuing malaria season by retrospective PCR analysis of dried blood spots that had been collected bi-weekly (Figure S1A). We identified three distinct outcomes to Pf infection during the first malaria season: 1) infection without progression to fever or other malaria symptoms during the entire malaria season (Immune, n=20), 2) infection with a delay of 2–14 days until progression to fever (Delayed Fever, n=34), and 3) infection with concurrent fever (Early Fever, n=26) (Figure 1A). For these 80 children, we performed a systems analysis using blood collected before, during, and after infection (Figure S1B). Demographic characteristics between classes were similar except that Immune children were ~1.5 years older despite our pre-defined age restrictions and were more likely to be infected with the helminth Schistosoma haematobium (Table S1). Independent of age, malaria episodes were less common in the Immune class relative to the other two classes over three years of continuous clinical surveillance (Figure 1B), suggesting durable clinical immunity to malaria. The difference in time to fever in children who progressed to fever was not due to stochastic sampling, as demonstrated by a significant difference in infection-to-fever intervals with no overlap between classes (Figure 1C). Clinical immunity to malaria is typically associated with control of parasitemia and increased Pf-specific IgG (Crompton et al., 2014). Indeed, Pf parasite densities at the first PCR-positive time point were similar if the child was afebrile, irrespective of class (Immune and Delayed Fever), with significantly lower densities relative to densities during febrile malaria episodes (Figure 1D). Before infection, Immune children showed increased magnitude and breadth of Pf-specific IgG relative to Early Fever children (Figures 1E and 1F), consistent with the critical role of IgG in malaria immunity (Cohen et al., 1961), with enhanced reactivity of blood-stage-specific IgG3 (Figures S2A and S2B), a malaria-protective subclass that potently activates complement and triggers NK cell-mediated, antibody-dependent cellular cytotoxicity (Arora et al., 2018; Boyle et al., 2015; Hart et al., 2019). Thus, the Immune class controlled both fever and parasitemia; the Delayed Fever class initially controlled fever and parasitemia but progressed to fever and higher parasitemia; while the Early Fever class did not control fever or parasitemia.
Figure 1. Prospective time-to-event data defined three clinically distinct classes of malaria susceptibility.
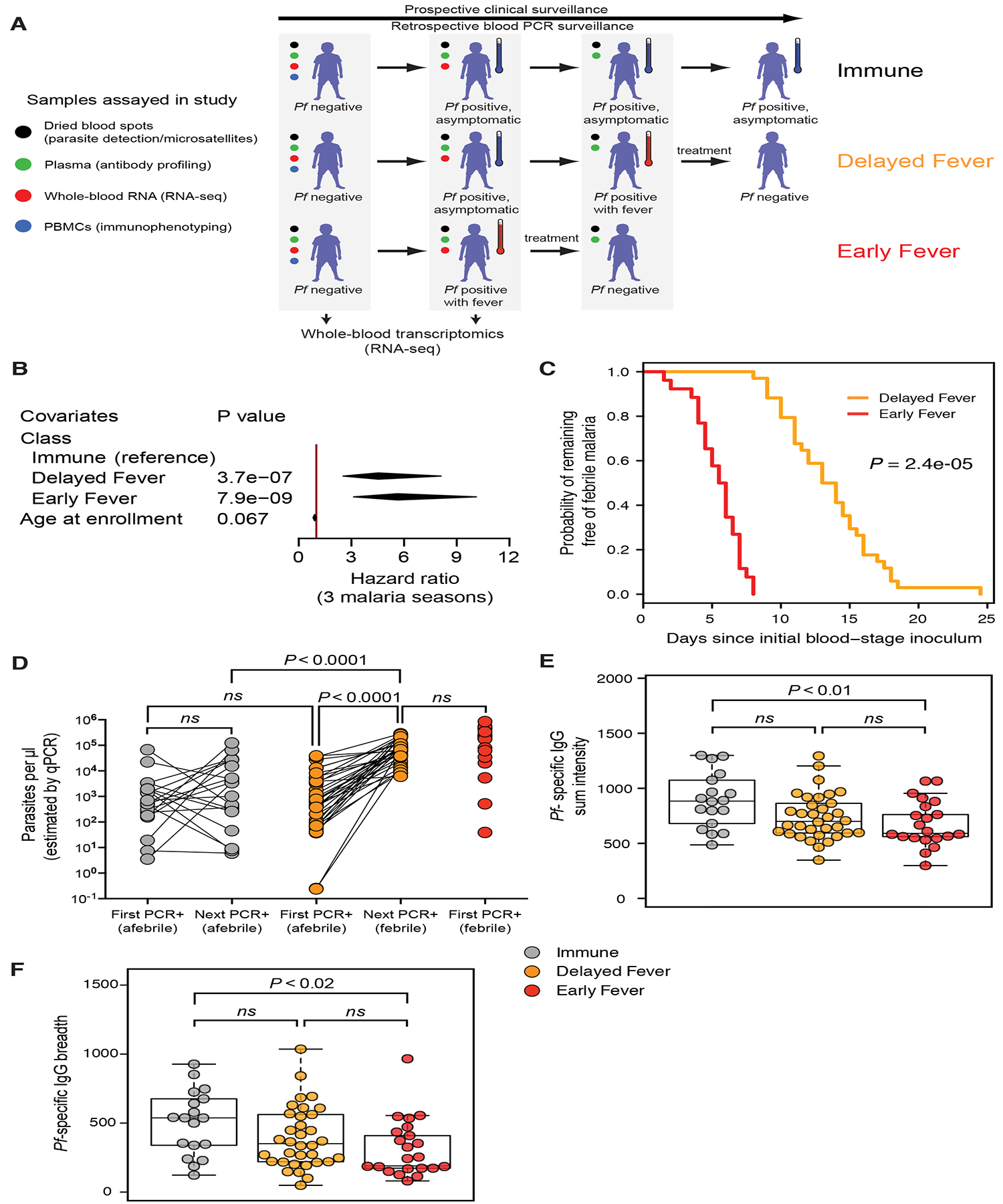
(A) Study design. Weekly clinical surveillance identified children with febrile malaria during one malaria season. Blood smears were performed at symptomatic visits, and anti-malarials administered if positive for parasitemia. Pf-specific PCR of dried blood spots collected at bi-weekly surveillance visits and at self-referred symptomatic visits were performed retrospectively to determine the first visit at which a child was infected with Pf irrespective of symptoms. Children classified as “Immune” (n=20) were infected but never developed malarial symptoms and thus never received anti-malarials. Asymptomatic children at the time of first infection who subsequently developed febrile malaria were classified as “Delayed Fever” (n=34). Children who were febrile at the time of first infection were classified as “Early Fever” (n=26). Please see also Figure S1 and Table S1. (B) Multiple malaria episodes over three consecutive malaria seasons were analyzed using the Andersen–Gill extension of the Cox regression model with class and age as co-variates. Diamonds represent 95% confidence intervals. (C) Kaplan-Meier curve showing probability of remaining free of febrile malaria since the first blood-stage inoculum (midpoint between last negative Pf PCR and first positive Pf PCR). P value determined by asymptotic log-rank test of left-censored data. (D) qPCR-estimated Pf densities at first infection and subsequent visit (within 2 weeks) in blood. Since the Early Fever class received anti-malarials at the first infection, qPCR was not performed for the subsequent visit. Significance was determined using a nested ANOVA with Tukey’s honest significant difference adjustment. (E, F) Pf-specific IgG responses prior to the first PCR-positive infection by class. Plasma was probed against a protein array containing 1087 Pf antigens. Shown are fluorescence intensities summed for reactive Pf antigens within a sample (E) or the number of Pf antigens reactive above background (F). Beeswarm plots are overlaid on boxplots indicating quartiles (boxes) and range (whiskers). Shown are Bonferroni-adjusted P values from Wilcoxon tests. Please see also Figure S2.
A pre-infection transcriptional signature associates with immunity to febrile malaria
We performed RNA-seq of whole blood collected from children at their uninfected baseline to determine a transcriptional signature predictive of immunity to febrile malaria. Principal components analysis (PCA) using normalized expression data for the most variably expressed genes across all uninfected samples demonstrated minimal separation of Immune and Delayed Fever from Early Fever (Figure 2A). To assess broad transcriptional differences, we deconvoluted whole-blood, gene expression data using a leukocyte gene signature matrix (Newman et al., 2015) and observed increased B cell signatures in the Immune class (Figure 2B).
Figure 2. Whole-blood transcriptomic analysis before Pf infection reveals differences in BCR signaling and Th1-Th2 cell balance among classes of malaria risk.
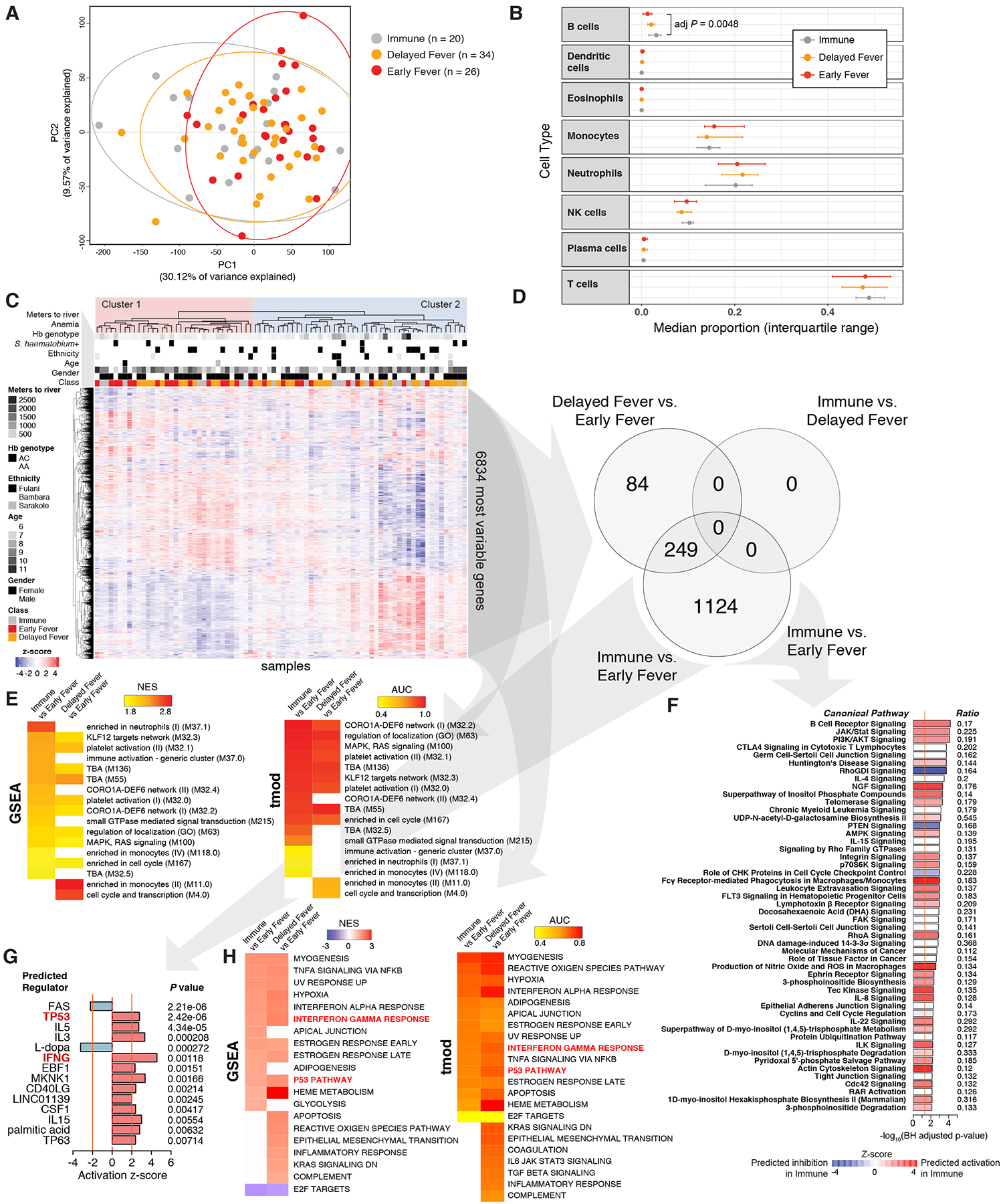
(A) Principal components analysis of pre-infection whole-blood RNA-seq data across all subjects. Ellipses represent 95% confidence intervals. (B) Median proportions of cell types determined by deconvoluting gene expression using a leukocyte gene signatures matrix (CIBERSORT). (C) Unsupervised hierarchical clustering heat map for the top 50% most variably expressed genes (6,834 genes) using the complete method and Pearson correlation distance. Baseline presence of indicated characteristics are shown in black. For age and meters, darker grayscale intensities represent increasing quintiles. (D) Venn diagram showing the number of differentially expressed genes (DEGs) at a false discovery rate (FDR) <5% with no fold-change cut-off. Please also see Supplemental Dataset 1. (E) Blood transcription modules found to be differentially enriched between classes across three testing methods (FDR<5% across all methods). Please also see Table S2. Only gene set enrichment analysis (GSEA) and tmod results are shown. (F) Significant canonical pathways using DEGs with FDR<5% (no fold-change cut-off) for the Immune vs. Early Fever comparison. Ratio indicates the proportion of DEGs in the dataset overrepresented in the pathway. (G) Top upstream regulators using DEGs with FDR<5% (no fold-change cut-off) for the Immune vs. Early Fever comparison. Only predicted regulators with a z-score > |2| and P < 0.05 are shown. (H) Hallmark genesets from MSigDB v6.2 found to be differentially enriched between classes in both GSEA and tmod (FDR<5% across both methods).
Unsupervised hierarchical clustering of expression data from the most variable genes revealed two main clusters: 16 of 26 Early Fever children grouped within Cluster 1 and 36 of 54 Immune or Delayed Fever children grouped within Cluster 2 (P=0.029 by Fisher’s exact test; Figure 2C). Neither cluster clearly associated with gender, HbAC genotype, ethnicity, distance to river (mosquito breeding sites), anemia, or S. haematobium infection (Figure 2C). Given the age differences between classes (Table S1), we used an age-adjusted model to determine differential gene expression (Supplemental Dataset 1). Differentially expressed genes (DEGs) were identified between Immune and Early Fever (1373 DEGs) and between Delayed Fever and Early Fever (333 DEGs) but not between Immune and Delayed Fever at a 5% false discovery rate (FDR) (Figure 2D).
To identify differences in immunologically relevant molecular signatures and pathways between classes, we applied results from the differential gene expression analyses to three enrichment methods using blood transcription modules (BTMs) (Li et al., 2014) as gene sets: gene set enrichment analysis (GSEA) (Sergushichev, 2016; Subramanian et al., 2005), Camera (Wu and Smyth, 2012), and tmod (Weiner 3rd and Domaszewska, 2016), as well as Ingenuity Pathways Analysis (IPA). Mutually significant BTMs that were upregulated in the Immune versus Early Fever across all enrichment methods (FDR<5%) included platelet activation, targets for the transcription factor Kruppel Like Factor 12 (KLF12); gene networks for an actin regulator important for T cell migration and homeostasis CORO1A (Foger et al., 2006) and a guanine nucleotide exchange factor critical for both Th1 and Th2 cell responses DEF6 (Becart et al., 2007; Tanaka et al., 2003); MAPK-RAS signaling; neutrophil enrichment; cell cycle enrichment; and small GTPase-mediated signal transduction (Figure 2E; Table S2). IPA showed increased signaling through the B cell receptor (BCR), the canonical IFN signaling pathway JAK-STAT, and the PI3K-AKT pathways in Immune versus Early Fever (Figure 2F). Significantly different BTMs between Delayed Fever and Early Fever included monocyte enrichment; platelet activation; KLF12 targets network; cell cycle and transcription; and CORO1A-DEF6 network modules (Figure 2E). There were no differentially expressed canonical pathways between Delayed Fever and Early Fever by IPA (FDR<5%).
Upstream regulator analysis predicted increased activation of the cell cycle regulator p53 (encoded by TP53) in Immune or Delayed Fever relative to Early Fever (Figures 2G and S3). With regard to adaptive immunity, several proteins related to T helper cell subsets were predicted to be activated in Immune or Delayed Fever relative to Early Fever, including the T helper 1 (Th1) cell cytokine IFNγ (Figures 2G and S3); the Th1 cell transcription factor STAT4 (Figure S3); the Th2 cell-associated cytokines IL-5 (Figures 2G and S3), and IL-13 (Figure S3); and the co-stimulatory and T follicular helper (Tfh) cell protein CD40 ligand (CD40LG) (Figures 2G and S3). Notably, the p53 pathway and the IFNγ response were also identified as coherently upregulated processes in Immune or Delayed Fever relative to Early Fever by GSEA and tmod using “hallmark” gene sets (MSigDB v6.2; Figure 2H) (Liberzon et al., 2015). To validate predicted upstream regulators that are transcription factors (TF), we performed self-contained gene set testing for each two-way comparison using a compendium of human TF networks derived from experimentally determined genome-wide maps of in vivo DNaseI footprints, limiting the analysis to blood cells (Neph et al., 2012). Binding targets of HIF1A, EBF1, SPI1, TP53, and RELA were differentially expressed between either Immune or Delayed Fever versus Early Fever (Supplemental Dataset 2).
To assess if differential cell type enrichment contributed to the observed differences in activation of functional pathways, we compared cell lineage marker expression by RNA-seq and flow cytometry between classes. Relative to Early Fever, the Immune class exhibited age-independent increases in gene and protein expression of the B cell marker CD19 (Figures 3A–B). No class differences were observed in B cell subsets (Figure 3C) or with lineage markers for monocytes, neutrophils, NK cells, or T cells (Figures S4A and S4B). Taken together, the data suggest that differential B cell enrichment or activation; platelet and monocyte activity; T helper subset differentiation; and p53 pathway activation prior to infection may play a role in modulating clinical outcomes during subsequent Pf infection.
Figure 3. Flow cytometric analyses of PBMCs before infection reveals B cell enrichment and Th2 cell bias in Immune children.
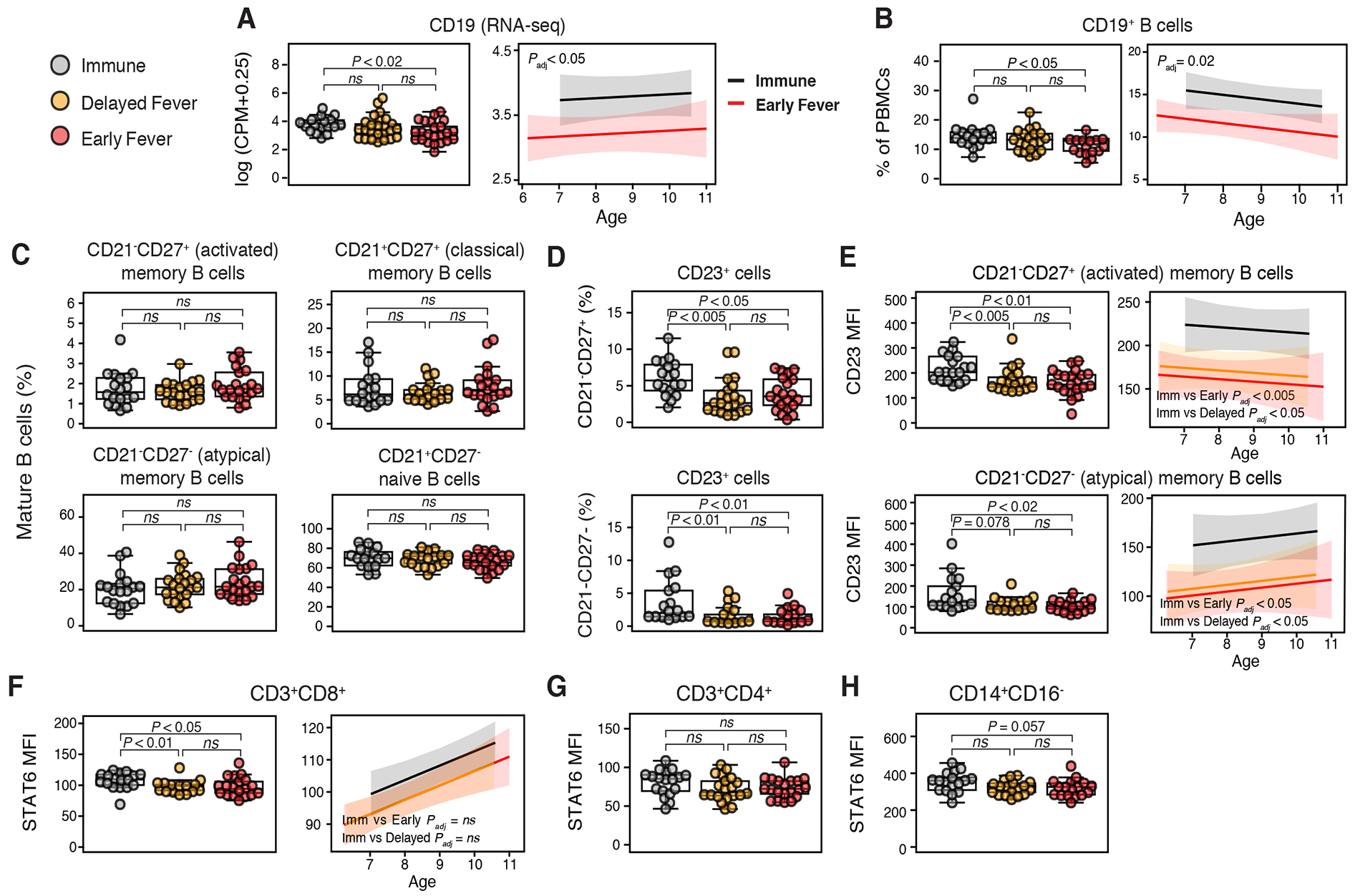
Differences between (A) CD19 expression by whole-blood RNA-seq counts per million reads (CPM) and (B) CD19+ B cell percentages by class (left panel) and between Immune and Early Fever with adjustment for age (right panel). (C) % B cell subsets among CD19+CD10− mature B cells: activated MBCs (CD21−CD27+), classical memory B cells (MBCs) (CD21+CD27+), atypical MBCs (CD21−CD27−), and naïve B cells (CD21+CD27−). (D) % CD23+ (FcεRII) cells among activated and atypical MBCs. (E) Mean fluorescence intensity (MFI) of CD23 on activated and atypical MBCs and relationship between CD23 MFI and age. P values are for class comparisons of CD23 MFI with adjustments for age. STAT6 MFI in (F) CD8+ T cells (left panel), (G) CD4+ T cells, and (H) CD14+CD16− monocytes. For (F), the right panel shows the relationship between STAT6 and age for CD8+ T cells by class. For beeswarm plots, overlaid boxplots indicate quartiles (boxes) and range (whiskers); significance determined by Mann-Whitney test. For regression lines in panels A, B, E, and F, P values are for class comparisons of flow cytometry (response) variable with age as a covariate. Shown are 95% confidence bands. P values for all panels were Bonferroni adjusted. Please see also Figure S4.
CD23 expression on activated and atypical memory B cells associates with subsequent control of Pf-induced fever and parasitemia
Given differential enrichment of the Th1 and Th2 cell-related DEF6 network (Figure 2E) and increased activation by IFNγ and IL-5 (Figures 2G) in the Immune class, we focused our flow cytometric analysis on Th1 and Th2 cell-related proteins using PBMCs collected at the pre-infection baseline for a subset of 60 children. Relative to the other two classes, the Immune class demonstrated an age-independent increase in expression of the IL-4-inducible, low-affinity IgE receptor CD23 in CD21−CD27+ (activated) and CD21−CD27− (atypical) memory B cells (Figures 3D and 3E). This finding was accompanied by an age-dependent increase in STAT6 expression in CD8+ T cells from Immune children (Figure 3F), but no class differences in CD4+ T cells or CD14+CD16− monocytes (Figures 3G and 3H), suggesting these peripheral cell types are unlikely to provide the IL-4 needed for CD23 upregulation. No class differences in expression of phosphorylated STAT6, the Th2 cell transcription factor GATA3, or the Th1 cell transcription factor T-bet were detected in T cell subsets (not shown). To further assess for Th2 cell bias, we measured cytokines, total IgE, and Pf-specific IgE in plasma collected at the uninfected baseline but observed no class differences (data not shown).
Acute Pf infection induces a subclinical interferon-related, pro-inflammatory signature in Immune children
To determine which host genes and immune pathways are important for control of malaria fever and parasitemia, we used paired analysis to assess whole-blood transcriptional changes by RNA-seq during the first PCR-confirmed Pf infection of the season relative to each child’s uninfected baseline (Δ). Transcriptional differences among the most variably expressed genes distinguished febrile malaria (Δ Early Fever) from asymptomatic infections by PCA and unsupervised hierarchical clustering, but could not distinguish asymptomatic infections that remained afebrile (Δ Immune) from those that progressed to fever (Δ Delayed Fever) (Figures 4A and 4B). Clinical class, rather than host factors or co-infections, best explained separation of Cluster 1 (44 of 46 Immune or Delayed Fever children) and Cluster 2 (21 of 25 Early Fever children; P=5.0×10−12 by Fisher’s exact test; Figure 4B).
Figure 4. Differential gene expression can be detected during asymptomatic P. falciparum infection in Immune but not Delayed Fever children.
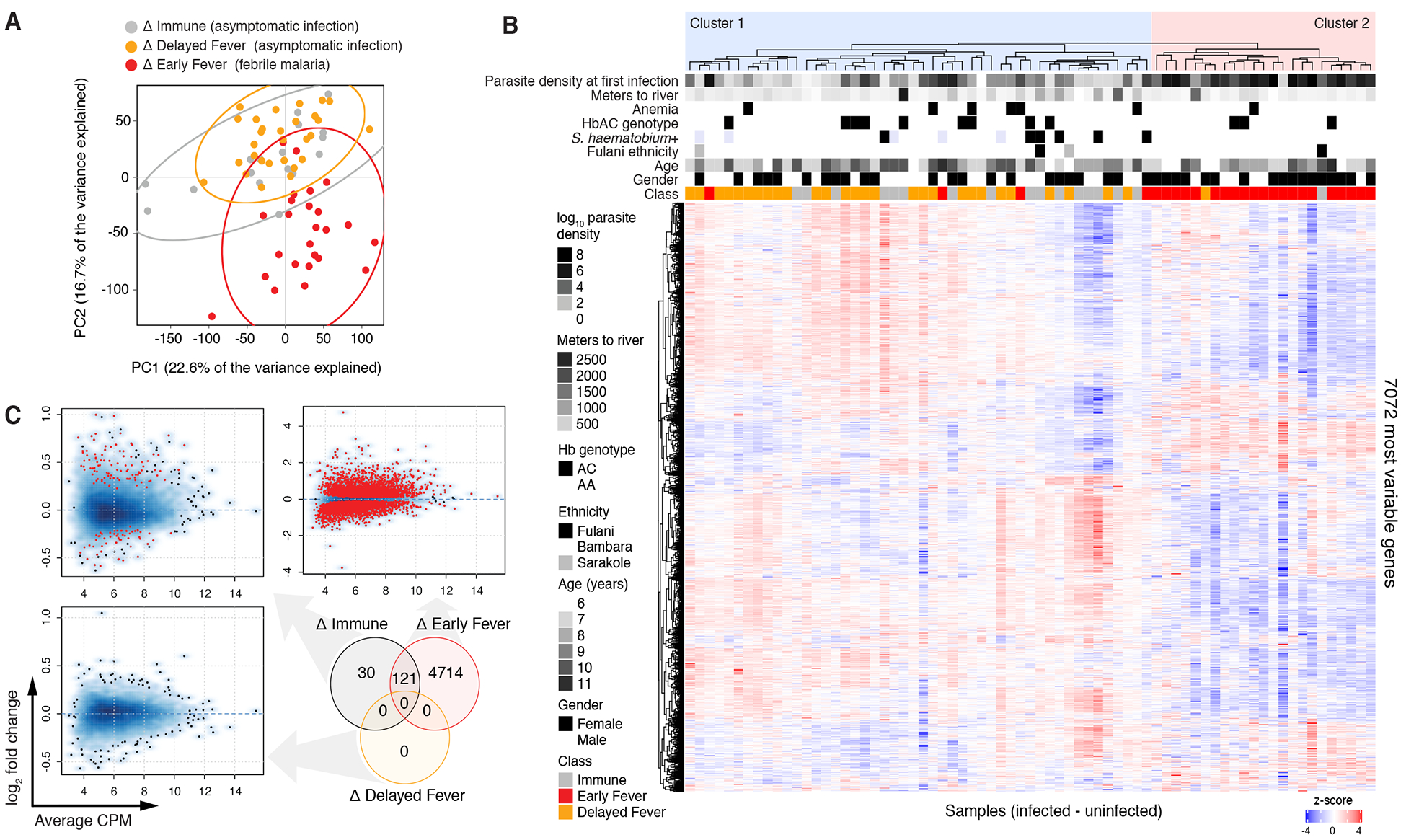
Paired analysis of infected (I) vs. uninfected (U) samples is indicated by Δ. (A) Principal components analysis of paired RNA-seq samples (infected – uninfected baseline for each subject) with 95% confidence intervals (ellipses). (B) Unsupervised hierarchical clustering heat map for the top 50% most variably expressed genes (7,072 genes) using the complete method and Pearson correlation distance. (C) Differential gene expression for paired infected vs. uninfected baseline samples within each clinical class. For smear density plots, DEGs with FDR<5% are shown as red points. Venn diagram shows the number of differentially expressed genes (DEGs) at a false discovery rate (FDR) <5% with no fold-change cut-off. ns = not significant. Please see also Supplemental Datasets 3 and 4 as well as Figure S6.
Pf infection induced the greatest number of transcriptional changes during febrile malaria (Δ Early Fever; 4835 DEGs at 5% FDR; Figure 4C; Supplemental Dataset 3). Enrichment analysis using BTMs revealed upregulation of monocyte; activated dendritic cell; cell cycle and transcription; Type I IFN; toll-like receptor (TLR) and inflammatory signaling signatures; but downregulation of lymphocyte signatures during febrile malaria (Figure 5A). By IPA, in Δ Early Fever we observed enrichment of DEGs involved in innate and adaptive immune pathways, including nuclear factor of activated T cells (NFAT) signaling, CD28 signaling in T helper cells, BCR signaling, Fcγ receptor-mediated phagocytosis in macrophages and monocytes, phosphoinositide 3-kinase signaling in B cells, and T cell receptor signaling (Figure S5A), with predicted activation of interferon-related genes (IFNG, IRF7, IFNA2, IFNL1, IRF1, IFNB1, IRF3) as well as genes related to adaptive responses (IL15, CD40LG, IL5) and innate inflammation (TNF, TLR4) (Figure S5B). Overall, Δ Early Fever exhibited a robust interferon-driven, pro-inflammatory signature that included both innate and adaptive immune components, which is consistent with previous blood-transcriptomic studies of susceptible individuals during febrile malaria (Griffiths et al., 2005; Idaghdour et al., 2012; Lee et al., 2018; Nallandhighal et al., 2018; Ockenhouse et al., 2006; Tran et al., 2012; Yamagishi et al., 2014).
Figure 5. P. falciparum infection in Immune children induces expression of genes related to protein synthesis, activation of monocytes, and inflammation.
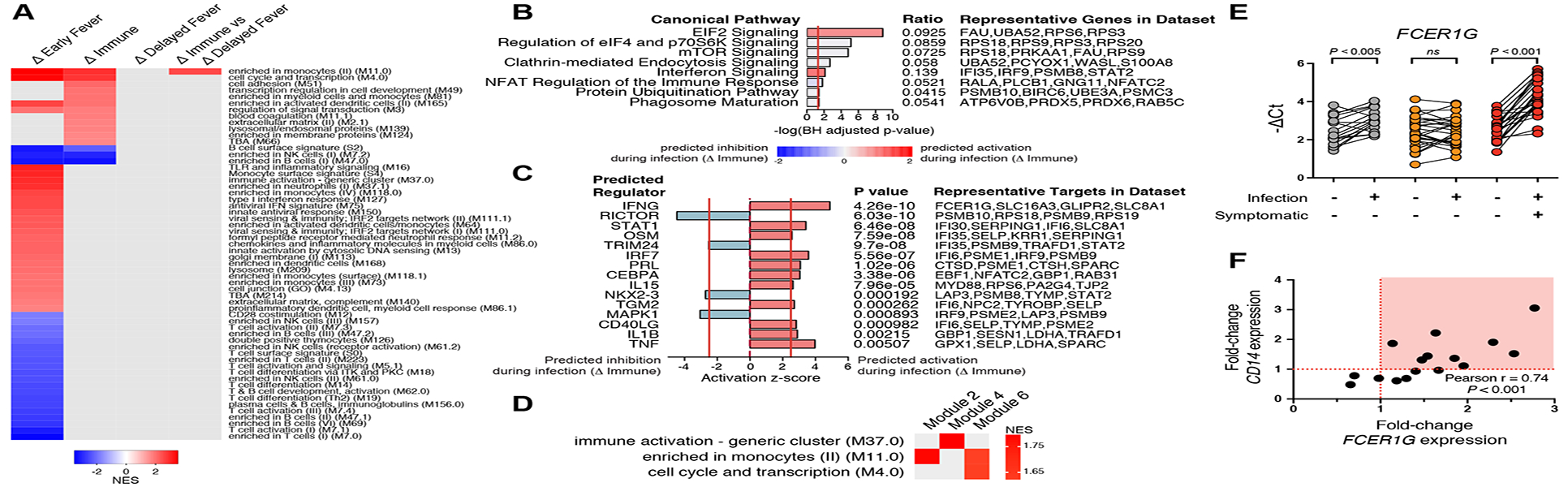
Paired analysis of infected (I) vs. uninfected (U) samples is indicated by Δ. (A) Blood transcription modules (BTM) found to be differentially enriched between classes across three methods (FDR<5% for all methods). Only gene set enrichment analysis (GSEA) results are shown. (B) Significant Pf-induced canonical pathways and (C) upstream regulators using DEGs with FDR<5% (no FC cut-off) for the ΔImmune comparison. (D) BTM enrichment of co-expression network modules which differed between Δ Immune and Δ Delayed Fever classes using weighted-gene correlation network analysis with Δ log2 counts per million expression values as input. Only significant BTMs (adjusted P values <0.05) are shown. (E) Confirmation of class differences in whole-blood FCER1G expression before and during asymptomatic infection by qPCR. Significance was determined by paired t test. Shown are Bonferroni-adjusted P values. (F) Relationship between Pf-induced changes in expression of CD14 and FCER1G by qPCR in 16 Immune children with available samples. Samples with increased expression of both FCER1G and CD14 are highlighted in the red quadrant.
Among asymptomatically infected children, gene-level transcriptional activity was detected in Δ Immune (151 DEGs at 5% FDR; Figure 4C), but, notably, not in Δ Delayed Fever (0 DEGs at 5% FDR; Figure 4C). By GSEA, asymptomatic infections within Δ Immune were characterized by upregulated enrichment of modules related to monocytes and activated dendritic cells (DC); cell cycle, transcription, and cell adhesion; lysosomal and endosomal proteins; and blood coagulation; but downregulated enrichment in B and NK cell signatures (Figure 5A). Similarly, IPA showed overrepresentation of pathways important to translational control and cell growth (eukaryotic initiation factor 2 [eIF2], p70S6K, mTOR signaling) and immune response (interferon signaling, NFAT, phagosome maturation; Figure 5B), with predicted activation by the pro-inflammatory genes IFNG, IRF7, IL1B, and TNF (Figure 5C). In contrast to Δ Immune, asymptomatic infections within Δ Delayed Fever did not induce differential enrichment of any transcriptional modules (Figure 5A). Direct comparison of Pf-induced transcriptional changes within Δ Immune against those within Δ Delayed Fever by GSEA revealed a monocyte enrichment signature (M11.0) as the only significantly different BTM between the two asymptomatic classes (Figure 5A). To assess class differences in data-derived, co-expression network modules induced by incident Pf parasitemia, we applied weighted-gene co-expression network analysis (WGCNA) to the Δ expression data. WGCNA constructs biologically relevant modules based on hierarchical clustering of co-expression networks determined via pairwise correlations between gene expression profiles (Langfelder and Horvath, 2008). Network modules that were different between the two asymptomatic classes showed upregulated enrichment in the M11.0 monocyte signature in Δ Immune relative to Δ Delayed Fever (Figure 5D). At the gene level, DEGs for the Δ Immune vs. Δ Delayed Fever comparison included HAUS4, CCDC66, FCER1G, and NUP54 (FDR<5%, Supplemental Dataset 3). Differential expression of FCER1G, which encodes the common γ chain of the Fc receptor, was confirmed by qPCR (Figure 5E). In Δ Immune, Pf-induced increases in whole-blood FCER1G expression correlated with increases in the expression of the monocyte lineage marker CD14, (Figure 5F), suggesting that monocytosis may have contributed to enhanced FCER1G expression during subclinical Pf infection. Taken together, the Pf-induced transcriptomic analyses provide evidence that monocytes are important for control of parasitemia and fever.
We investigated other potential causes for differential host responses to Pf infection, including the presence of intestinal helminths (Supplemental Dataset 4) and blood-borne viruses (Figures S6A and S6B) at the pre-infection baseline, as well as the genetic diversity of Pf isolates obtained from the first infection (Figures S6C and S6D) but did not find significant class differences.
Baseline Pf-specific IgG and CD23 expression on B cells correlate with lower parasitemia and delayed fever, respectively
To identify potential modulators of the Pf-induced inflammatory response and parasitemia, we determined pairwise correlations between baseline biological variables, including relevant gene expression data, and measures related to host response to the first Pf infection (Figure 6A) and mapped the significant relationships (adjusted P<0.01; Figure 6B). Pre-infection Pf-specific IgG negatively correlated with parasite density at the first detected infection, consistent with the known anti-parasite effects of IgG (Cohen et al., 1961). Pf-specific IgG at the first infection positively correlated with age, pre-infection Pf-specific IgG, and baseline percentage of p53+ T cells. Age positively correlated with baseline expression of STAT6 in CD8+ T cells and CD14+ monocytes. At the level of whole-blood gene expression, baseline expression of TP53, STAT6, CD19, and CD23 positively correlated with each other and negatively correlated with STAT4. Baseline STAT6 expression negatively correlated with the resting CD4+ T cell memory signature. Days from baseline to first febrile malaria episode negatively correlated with in vivo parasite growth rate and positively correlated with CD23 expression on atypical and activated memory B cells. Baseline variables, including p53-related parameters, did not correlate with changes in expression (Δ) of any inflammatory genes except for the baseline CD8+ T cell signature, which positively correlated with ΔIFNG.
Figure 6. Correlations between immunological, clinical, parasitological, and select transcriptomic variables.
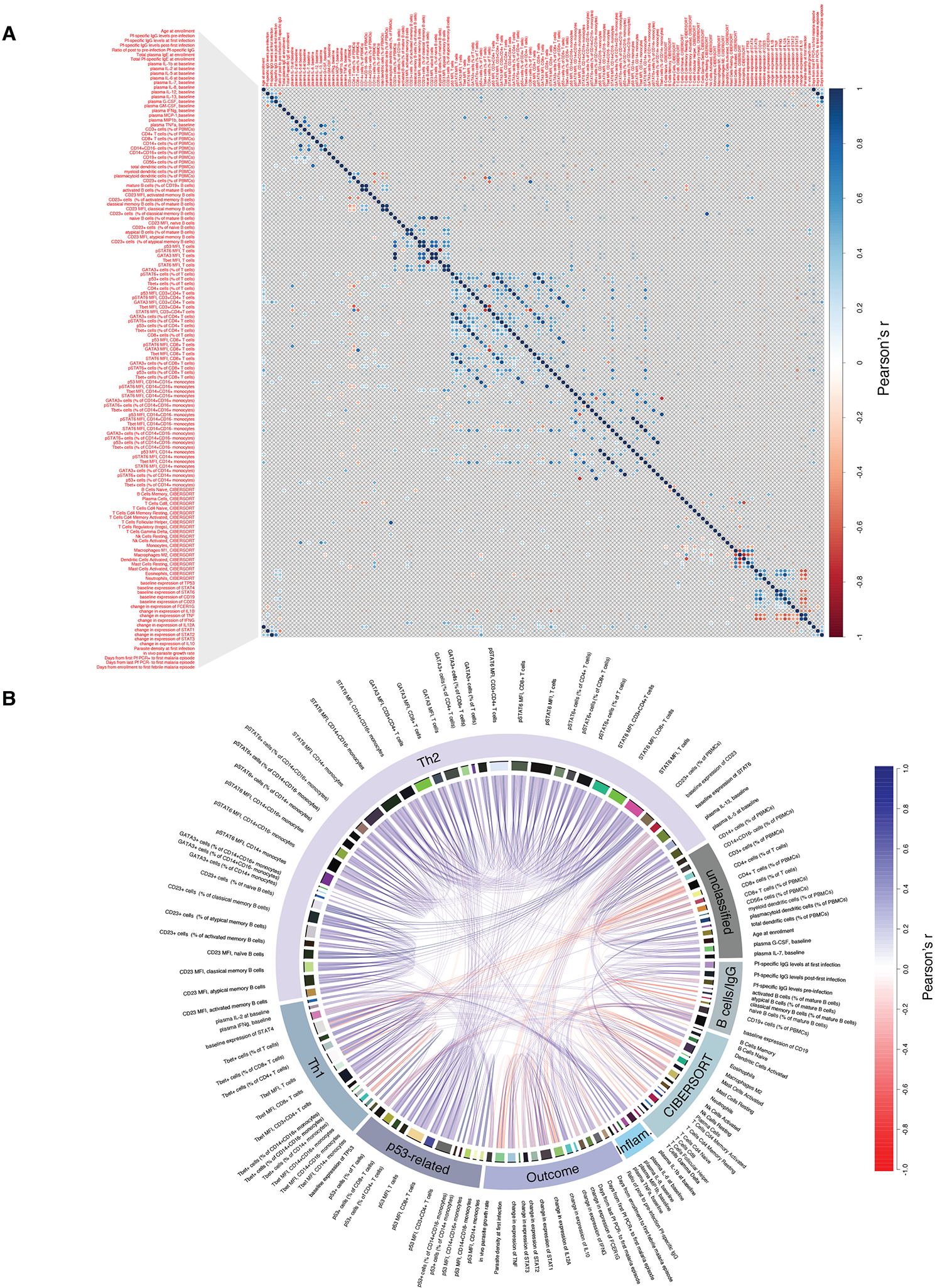
(A) Correlation plot for pair-wise comparisons between immunological, clinical, parasitological, and select transcriptomic variables. Comparisons with Benjamini-Hochberg-adjusted and unadjusted P<0.01 are shown above and below the diagonal, respectively. (B) Chord diagram showing relationships between significant correlations with Benjamini-Hochberg-adjusted P<0.01. Inflam. = pro-inflammatory.
A random forests model yields the best predictors of malaria protection
To determine which variables best predicted protection from febrile malaria, we tested three types of machine learning models (i.e. random forests, K-nearest neighbors and elastic net) for classifying children as Immune, Delayed Fever, or Early Fever using age and 7778 biological features tested prior to infection, including Pf-antigen-specific IgG reactivity by protein array, IgG subclass reactivity against blood-stage antigens, Pf-specific and total IgE, plasma cytokines, flow cytometric data, baseline RNA-seq data, and change in expression of inflammation-related genes as input (refer to STAR Methods). Random forests models resulted in the highest predictive accuracies (62–71%) in all 63 runs of the data mining process. Top features appearing in the random forests model at >100-folds cross validation among 63 runs of K = 6-fold cross validation were pre-infection baseline expression of IFNAR2, the pseudogene ANKRD36BP1, LILRA6, CLCC1, ALPL, MFSD11 and CACNA1E as well as IgG reactivity to the Pf array antigens PFK9, GEXP5 and gammaGCS (Supplemental Dataset 5).
Increased p53 attenuates Plasmodium-induced inflammatory responses
Finally, as a ‘proof of concept’ we sought to validate specific hypotheses generated by our unbiased systems-based analysis. Given the unexpected finding of pre-infection p53 activation in Immune and Delayed Fever relative to Early Fever (Figures 2G and 2H), we further investigated the role of p53 in malaria. Pre-infection expression of TP53 was increased in both Immune and Delayed Fever relative to Early Fever by both RNA-seq and qPCR (Figures 7A and 7B). We corroborated the upstream regulator results (Figure 2G) by showing that a significant proportion of targets known to be activated by p53 (Fischer, 2017) were overexpressed in both Immune and Delayed Fever children relative to Early Fever at the pre-infection baseline by self-contained gene set testing (Figure 7C).
Figure 7. TP53 expression is associated with regulation of host inflammatory response but not control of parasite growth.
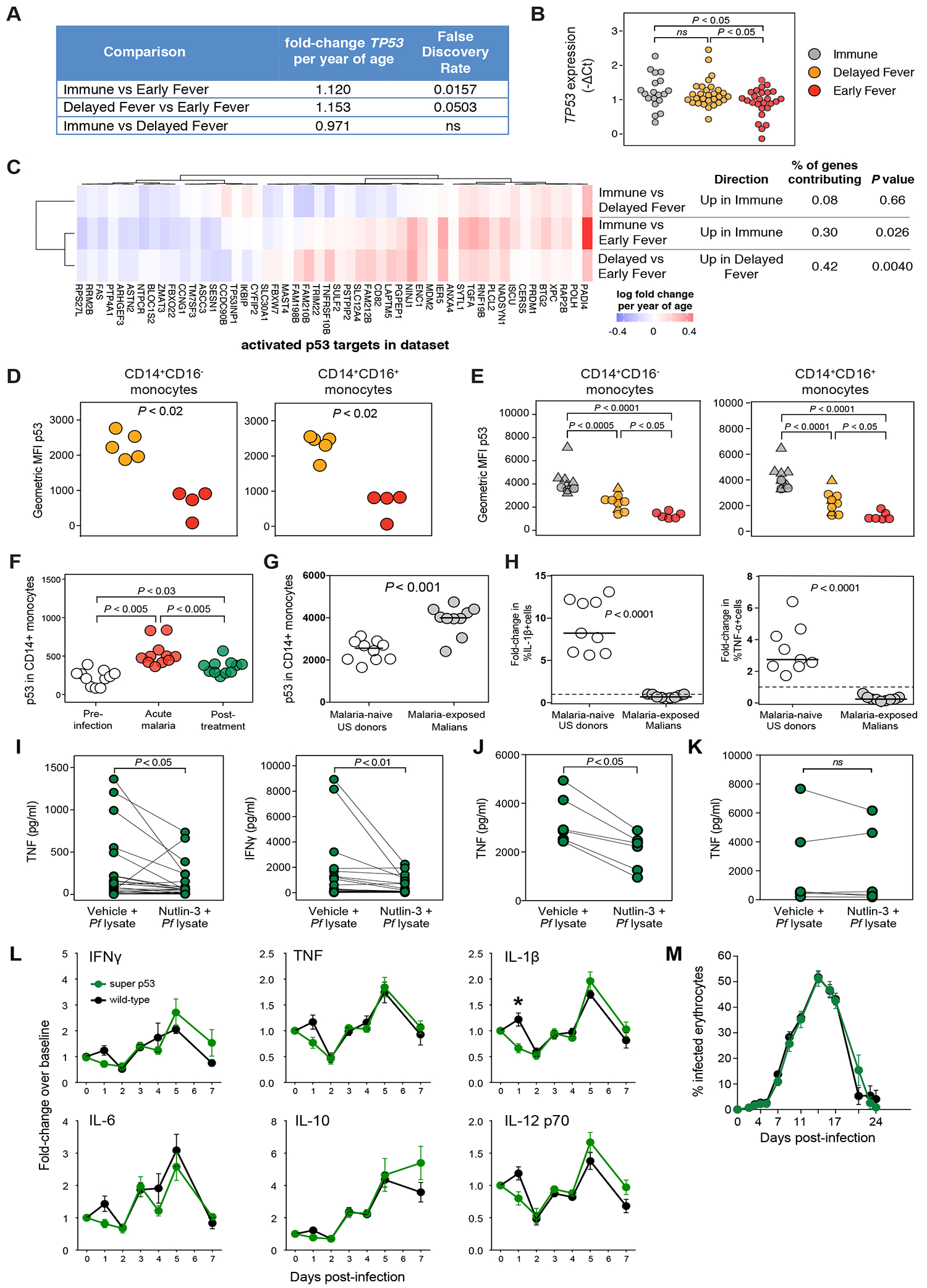
(A) Differential gene expression analysis for TP53 at the uninfected baseline. (B) TP53 expression in whole blood by qPCR before Pf infection. (C) Rotation self-contained gene set testing using a set of targets known to be activated by p53 for all comparisons at the uninfected baseline. Class differences in p53 expression determined by flow cytometry in peripheral CD14+ monocytes obtained at (D) the uninfected baseline (n=9) and (E) in May 2012 (n=26) using the anti-p53 monoclonal antibody clone PAb240. Triangles represent Pf-infected individuals. (F) Longitudinal p53 expression in CD14+ monocytes before, during acute malaria, and 7 days after treatment (n=11 subjects). (G) p53 expression in CD14+ monocytes in malaria-naïve US donors and malaria-exposed Malian donors using PAb240. (H) Change in % of IL-1β+ or TNF+ cells by intracellular staining after 12h stimulation of isolated CD14+ monocytes with Pf-infected red blood cell lysate (Pf lysate). Plasma concentrations of indicated cytokines in culture supernatants of (I) PBMCs (n=25 pairs); (J) CD14+ monocytes (n=6 pairs); and (K) CD14− non-monocytes (n=6 pairs) after 12 h stimulation with Pf lysate with and without the p53 stabilizer nutlin-3. (L) Changes in plasma concentrations of the indicated cytokines and (M) parasitemia in super p53 mice, which have an extra copy of Trp53, and wild-type littermates infected with Plasmodium yoelii 17XNL. Data are represented as mean ± standard error of the mean. Results are summarized data from five experiments that included 31 super-p53 mice and 33 wild-type mice. Significance differences between groups were determined by ANOVA with post-hoc Tukey’s HSD (B, E, F), Mann-Whitney (D, G, H), paired Wilcoxon tests (I, J, K), or t tests with Holm-Sidak-adjusted P values (L, M). ns = not significant, *P < 0.05.
Despite differences in TP53 gene expression, we initially did not find significant correlations between intracellular p53 and measures of clinical response to Pf infection nor class differences in p53 in B cell, T cell, or monocyte subsets for 60 children at their uninfected baseline by flow cytometry using the anti-p53 monoclonal antibody clone DO-1. We re-assessed p53 cellular expression with the anti-p53 monoclonal antibody clone PAb240, which we found to be more sensitive than DO-1 at detecting differences in p53 (data not shown), in PBMCs collected at the uninfected baseline (May 2011) from the remaining Delayed Fever and Early Fever children and found increased p53 expression in CD14+CD16+ and CD14+CD16− monocytes among Delayed Fever relative to Early Fever (Figure 7D). These findings were confirmed in cells collected prior to the next malaria season (May 2012), which showed that p53 expression in CD14+CD16− and CD14+CD16+ monocytes increased with increasing clinical immunity to malaria (Figure 7E). Although p53 expression appears to be highest in children with asymptomatic Pf infections, uninfected Immune children demonstrated higher p53 than uninfected children in both the Delayed Fever and Early Fever classes (Figure 7E). Next, we investigated the effect of acute Pf infection on p53 in peripheral blood monocytes and found that p53 increased during acute febrile malaria and remained above the pre-infection baseline seven days post-treatment (Figure 7F), implicating prior malaria exposure as an explanation for increased p53 observed in the malaria-protected classes. In support of this, malaria-exposed Malian adults demonstrated higher p53 in monocytes than malaria-naïve US donors (Figure 7G).
Previous studies have shown that the absence of p53 promotes the induction of pro-inflammatory genes via a NF-κB-dependent pathway, resulting in fatal pathogen-induced inflammation in mice (Madenspacher et al., 2013), whereas overexpression of p53 can blunt LPS-mediated pro-inflammatory responses in vitro (Komarova et al., 2005). Therefore, we investigated the potential role of p53 in modulating Plasmodium-induced inflammation. We found Pf-stimulated monocytes from malaria-exposed Malian adults produced decreased intracellular IL-1β and TNF in vitro relative to monocytes from malaria-naïve US donors (Figure 7H). Pre-treatment of PBMCs from malaria-naïve US donors with the p53 pathway activator nutlin-3 abrogated the production of TNF and IFNγ by PBMCs in response to Pf stimulation (Figure 7I). The reduction in Pf-induced TNF production with nutlin-3 treatment was replicated in purified CD14+ (Figure 7J) but not CD14– cells (Figure 7K), suggesting that the TNF effect is specific and intrinsic to monocytes. Furthermore, blood-stage infection with the non-lethal murine strain Plasmodium yoelii 17XNL induced the pro-inflammatory cytokines IFNγ, TNF, IL-1β, IL-6, and IL-12 at day 1 post-infection in wild-type mice but not in mice expressing one extra copy of the gene encoding p53; however, only IL-1β was significantly different after multiple-testing correction (Figure 7L). These early differences in pro-inflammatory cytokines were not sustained and did not impact parasitemia (Figures 7L and 7M). These data taken together suggest that increased p53 dampens Plasmodium-induced inflammation without impacting parasitemia.
Discussion
Previous studies that employed blood transcriptional profiling during Pf infections compared malaria-susceptible cases to uninfected controls (Griffiths et al., 2005; Idaghdour et al., 2012; Nallandhighal et al., 2019; Ockenhouse et al., 2006; Yamagishi et al., 2014). None of these studies investigated immune responses to Pf infections in individuals protected from malaria symptoms. Here, we prospectively evaluated differential responses to the first PCR-detectable Pf infection of the malaria season in children who either remained asymptomatic or progressed to febrile malaria, allowing comparison of immune responses between protected and susceptible children. By using each child as his or her own healthy, pre-infection control, we limited inter-individual variability and enhanced our ability to detect infection-driven differences between classes.
We prospectively captured the host immune response before and during incident, PCR-detected Pf infections in children with three levels of immunity to malaria. The most protected children (Immune) remained asymptomatic and afebrile after Pf infection and were able to control parasite growth, while the least protected children (Early Fever) were symptomatic with fever and high parasitemia at the time of incident infection. An intermediate phenotype comprised children who were asymptomatic with low parasitemia at the time of incident infection, but then progressed to fever and high parasitemia within two weeks (Delayed Fever). These clinical phenotypes provide evidence for the acquisition of partial immunity in malaria-experienced children in which clinical “tolerance” to blood-stage infection (anti-disease immunity) can be acquired independently of the ability to inhibit parasite growth (anti-parasite immunity).
The immunological mechanisms contributing to anti-disease immunity and/or anti-parasite immunity can be inferred by comparing immunological signatures among the three phenotypes before and during incident infection. We confirmed the importance of IgG in malaria protection (Cohen et al., 1961) by showing increased Pf-specific IgG, particularly of the cytophilic subclass IgG3, and increased peripheral B cells prior to infection in the Immune class. Notably, independent of humoral immunity, the Immune class demonstrated increased expression of genes involved in multiple immune pathways (e.g. monocyte and platelet function, T cell homeostasis and Th1-Th2 cell responses [CORO1A-DEF6], and targets for IFNγ and IL-5).
Despite evidence of dual Th1 and Th2 cell activation at the level of the whole-blood transcriptome, our analysis of Th1 and Th2 cellular proteins revealed only increases in Th2 cell-related proteins, with increased CD23 on activated and atypical memory B cells and STAT6 in CD8+ T cells in Immune children, but no differences in CD4+ T cells. Although the role of Th2 cell immune responses in malaria protection is not completely understood, prior studies generally support a model by which Th2-polarized CD4+ T cells enhance the production of Pf-specific antibodies during blood-stage infection (Perez-Mazliah and Langhorne, 2014), particularly PD-1+CXCR3−CXCR5+CD4+ Tfh cells, which appear to be efficient enhancers of B cell help in malaria-exposed children (Obeng-Adjei et al., 2015).
What could be the etiology of immune activation in the Immune class at the pre-infection baseline? We previously reported that co-infection with S. haematobium decreases the risk of febrile malaria in this population (Doumbo et al., 2014), raising the possibility of immunomodulation by helminth infection. Due to mass administration of anti-helminthic drugs, only a few children in our study were infected with helminths at baseline. Even so, six of the seven children infected with S. haematobium were in the Immune class (n=20). It remains possible that low-density S. haematobium infections undetectable by microscopy or sustained immunomodulatory effects despite recent clearance of helminth infections contribute to the Th2 cell-like signature in malaria-protected children.
Baseline immune activation might also be due to more recent or prior cumulative malaria exposure given the slightly older age of the Immune class. Indeed, a recent whole-blood transcriptomic analysis revealed that children who previously experienced high numbers of malaria episodes demonstrated greater immune activation with upregulation of genes involved in interferon and B cell receptor signaling (Bediako et al., 2019), a signature that resembles that observed in the Immune class. We also previously showed that recent Pf malaria episodes result in increased activation of innate and adaptive pathways in unstimulated peripheral immune cells during convalescence, when symptoms have resolved, and cell composition has normalized to baseline (Portugal et al., 2014).
In the current study, we detected interferon and pro-inflammatory signatures among asymptomatic first Pf infections that never progressed to fever (Immune), but not in asymptomatic first infections that would later progress to fever (Delayed Fever). In contrast to Delayed Fever, the Immune class responded to asymptomatic infection in a manner that suggested a coordinated response marked by activation of innate and adaptive immune cells combined with trafficking of these cells in and out of the peripheral blood: enhanced interferon signaling and activation by pro-inflammatory cytokines, enrichment of monocytes and activated dendritic cells, and decreased B lymphocytes in the peripheral blood. FCER1G, which encodes the common γ chain of the Fc receptor and is induced by IFN-γ (Scholl and Geha, 1993), was differentially induced in the Immune but not the Delayed Fever class during acute Pf infection. Reduction in B cell modules may be explained by trafficking of these cells to lymphoid organs.
Thus, children in the Immune class appear to tolerate the Pf-induced IFNγ and pro-inflammatory response, benefiting from the possible parasite-killing effect of this response without suffering from pathological inflammation. Although a baseline Th2 cell-bias is implicated as a possible modulator of pro-inflammatory response to Pf infection in our study, other pathways may also control the host inflammatory response to provide malaria tolerance. In other disease models, p53 has been shown to modulate the inflammatory response to infection or autoimmunity (Dijsselbloem et al., 2007; Liu et al., 2009; Madenspacher et al., 2013; Zheng et al., 2005). Although studies in mice show that p53 controls Plasmodium liver stage infection (Kaushansky et al., 2013) and suppresses Plasmodium-induced lymphoma (Robbiani et al., 2015), the role of p53 during blood-stage infection is unknown. Here, we have shown that Immune children have increased TP53 expression and p53-pathway activation at their pre-infection baseline. We also showed that increased p53 levels can blunt Plasmodium-induced inflammatory responses in human immune cells and a mouse model of malaria, suggesting that p53 upregulation may be a mechanism by which tolerance to blood-stage infection is achieved in malaria-immune populations. Although this anti-inflammatory effect neither aids in the control of blood-stage infection nor appears to be sufficiently robust to reduce fever in the face of rising parasitemia, systemic increases in p53 may also benefit the host by reducing the burden of subsequent liver stage Pf infections (Kaushansky et al., 2013).
In this study, we demonstrated that malaria-protected children are markedly different from malaria-susceptible children at the transcriptomic and cellular levels prior to Pf infection and in response to acute Pf infection. Differences in age and Pf-specific Ig reactivity suggest that previous exposure to malaria, and possibly other pathogens or commensals, plays a role in these findings. It is also possible that host genetic factors contribute to such wide-ranging immunological differences. Although large genome-wide association studies have examined host genetic susceptibility to severe malaria (Lee et al., 2013; Malaria Genomic Epidemiology et al., 2015; Timmann et al., 2012), formal studies addressing genetic susceptibility to uncomplicated malaria are still needed. Here, it is notable that among the top class-predicting features in the machine-learning analysis was baseline expression of LILRA6, a gene encoding for an activating leukocyte immunoglobulin-like receptor (LILRA) that exhibits copy number variation and is restricted to monocytes (Bashirova et al., 2014). LILRAs can control immune responses by associating with Fc receptor γ chain containing immunoreceptor tyrosine-based activation motifs. LILRA6 copy number variation could provide a genetic explanation for the enhanced control of inflammation during Pf infection observed in the Immune class. Further studies are needed to validate and determine the biological importance of this finding.
Taken together, our analysis provides a model of naturally acquired clinical immunity to malaria. In this model, a pre-infection, activated immune state consisting of monocyte and platelet activation, interferon signaling, T helper activation, and B cell enrichment, combined with greater Pf-specific IgG reactivity, identifies individuals who are primed to respond favorably to future Pf infection by controlling Pf-inducible inflammatory symptoms and parasitemia. When new Pf infections trigger interferon-driven pro-inflammatory responses in these individuals, they remain asymptomatic but respond appropriately by increasing Fc receptors through enrichment or activation of innate cells, thus facilitating the phagocytosis of Pf-infected erythrocytes opsonized by pre-existing Pf-specific IgG. In contrast, enhanced p53 pathways, differential cell cycle regulation, and a Th2-biased environment identify individuals capable of controlling Pf-induced inflammation, but only at lower parasitemia. New blood-stage infections in these individuals are initially asymptomatic but then progress to higher parasitemia and malarial fever in the absence of the activated immune state and high Pf-specific IgG reactivity prior to infection.
The current study addressed molecular signatures associated with protection from febrile malaria using analysis of whole-blood transcriptomes, which limits the ability to detect signatures within specific cell populations. Future studies that examine signatures within immune cell subsets using single-cell technology may identify additional signatures relevant to malaria immunity. Nevertheless, this prospective systems-immunology analysis of a pediatric population may provide researchers with a useful resource for testing hypotheses regarding host molecules and pathways that may contribute to differential clinical outcomes during acute infections in children.
STAR Methods
Lead contact and Materials Availability
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Tuan M. Tran (tuantran@iu.edu).
Experimental Model and Subject Details
Human Studies
The Ethics Committee of the Faculty of Medicine, Pharmacy and Dentistry at the University of Sciences, Technique and Technology of Bamako, and the Institutional Review Board of the National Institute of Allergy and Infectious Diseases, National Institutes of Health approved this study. The study site and study population have been previously described (Doumbo et al., 2014; Tran et al., 2013). Briefly, the study was conducted in the village of Kalifabougou, Mali, where Pf malaria transmission is intense and seasonal, occurring from June through December (Tran et al., 2013). In May 2011, we enrolled 695 healthy children and adults, aged 3 months to 25 years, into a longitudinal observational cohort study to investigate malaria immunity in which bi-weekly active malaria surveillance was conducted with interval week home check-ups and passive surveillance by self-referral. Baseline hemoglobin values measured by a HemoCue analyzer were used to determine anemia status based on World Health Organization criteria (http://www.who.int/vmnis/indicators/haemoglobin.pdf). Exclusion criteria at enrollment included a hemoglobin level <7 g/dL, axillary temperature ≥37.5°C, acute systemic illness, underlying chronic disease, use of antimalarial or immunosuppressive medications in the past 30 days, or pregnancy. Malaria episodes were defined as parasitemia of ≥2500 parasites/μL, an axillary temperature of ≥37.5°C within 24 hours, and no other cause of fever discernible by physical exam. We collected whole-blood for RNA (200 μl whole blood in 400 μl of Tempus solution [Applied Biosystems]), dried blood spots (DBS) on filter paper (Protein Saver 903, Whatman), cell pellets, and plasma from healthy, uninfected children at enrollment (before the 6-month malaria season), during bi-weekly scheduled visits, and at their first malaria episode of the ensuing season (Figure S1A). Additional samples collected at enrollment included PBMCs isolated by Ficoll density gradient for subsequent flow cytometric analysis and urine and stool for determination of parasitic co-infections. After transport from the field site, RNA, blood pellets, plasma, and stool were stored at −80C, and PBMCs were stored in liquid nitrogen until further analysis. Point-of-care blood smears were performed for individuals symptomatic at any clinic visit, with anti-malarials given for any Plasmodium positive smears per the Malian national guidelines. First Pf infections were detected retrospectively by PCR of longitudinally collected DBS as previously described (Tran et al., 2013). First malaria episodes were determined from the clinical visit data. From a subset of children aged 6–11 years (the age range during which the acquisition of clinical immunity to uncomplicated malaria begins in areas of intense malaria transmission), we selected 1) children whose clinically silent, retrospectively detected Pf infections never progressed to fever and were not treated with anti-malarials (Immune, n=20); 2) children whose Pf infections progressed to fever within 2–14 days (Delayed Fever, n=34); and 3) children who were febrile at the time of Pf infection (Early Fever, n=26) for further analysis using systems biology approaches.
Mouse studies
Animal experiments were approved by the National Institute of Allergy and Infectious DiseasesAnimal Care and Use Committee (NIAID ACUC). The NIAID ACUC approved the Animal Study Proposal identification number LIG-1E which adheres to the regulations of the Animal Welfare Regulations (AWAR) and Public Health Service (PHS) Policy on Humane Care and Use of Laboratory Animals. Super p53 C57BL/6 mice, originally described by Manuel Serrano (Garcia-Cao et al., 2002), were bred at LIG using male breeders (generously provided by David G. Kirsch, Duke University). Super p53 C57BL/6 mice and littermate C57B6 wild-type controls were infected intraperitoneally with 1×106 P. yoelii 17XNL GFP parasites (Ono et al., 2007) that were previously passaged through a donor C57BL/6 mouse. Venous blood was obtained for plasma and determination of parasitemia starting on Day 1 through Day 24 post infection. Plasma cytokine levels were determined by Luminex at a plasma dilution of 1:4 using a Bioplex 7-plex mouse cytokine kit (BioRad) per the manufacturer’s instructions. Only the six inflammatory cytokines from this panel were reported. Parasitemia was determined on a FACScalibur flow cytometer (BD Biosciences) by determining the percentage of GFP-positive erythrocytes relative to total erythrocytes.
Method details
Blood PCR for determination of parasite density and parasite genotyping
Genomic DNA (gDNA) was extracted from DBS using the QIAamp DNA Mini Kit (Qiagen, Valencia, CA) for determination of Pf parasite density by quantitative real-time PCR (qPCR) (Tran et al., 2014) and six-loci microsatellite genotyping using established protocols (Shaukat et al., 2012). For analysis of genetic diversity from microsatellite data, only the dominant peak for each locus was considered. Pairwise fixation indexes with bootstrapped confidence internals were calculated with the diveRsity package in R (Keenan et al., 2013).
Screening for helminth co-infections
Screening for helminths was performed as previously described (Doumbo et al., 2014). Briefly, filtered urine and stool prepared by Kato-Katz were assessed by microscopy for Schistosoma haematobium and Schistosoma mansoni ova, respectively (Katz et al., 1972), and soil transmitted helminths were detected in stool by multi-parallel qPCR (Easton et al., 2016). qPCR-detection of the bloodborne filarial nematode Mansonella perstans was performed on gDNA extracted from two 3-mm DBS punches using an established protocol (Drame et al., 2016).
Generation of RNA-seq data
RNA processing and sequencing has been previously described (Tran et al., 2016). Briefly, total RNA was extracted from whole blood and depleted of ribosomal and globin RNA prior to amplification using the ScriptSeq Complete Gold Kit (Illumina, San Diego, CA). Directional RNA-seq libraries were prepared using the ScriptSeq v2 kit (Epicentre, Madison, WI). Sequencing of 2×100 bp paired-end reads was performed on a HiSeq 2000 using V3 reagents (Illumina). The Illumina sequences were trimmed of bases with a Phred quality score <15 and any contaminating adapters used in cDNA and sequencing library preparation. Only paired-end reads which survived trimming and were ≥ 60 bases in length were mapped to the human (GRCh37, version 17, Ensembl 72) and P. falciparum 3D7 (version 3 annotation, Dec 2012) genomes in parallel using TopHat2 (Kim et al., 2013). Transcript abundance was determined by Cufflinks (Trapnell et al., 2012).
Verification of human RNA-seq data by qPCR
For subjects who had sufficient RNA remaining after RNA-seq library preparation, verification of expression for select genes was performed by qPCR. Globin- and rRNA-depleted RNA was reverse transcribed into cDNA using the SuperScript VILO cDNA Synthesis Kit (Thermo Fisher Scientific, Grand Island, NY) and then purified by QIAquick 96 PCR Purification kit (Qiagen). Constitutively expressed housekeeping gene candidates were ranked by expression level and low coefficient of variation in the normalized RNA-seq data. PUM1 was chosen as an internal reference control for all qPCR reactions. Three differentially expressed genes (CD14, FCER1G, and TP53) were selected for verification of RNA-seq data. Probe and primer sets for CD14, FCER1G, and PUM1 were designed using Primer Express, version 3.0 software (Thermo Fisher Scientific, Grand Island, NY) and purchased from Biosearch Technologies (Petaluma, CA). For TP53 (and PUM1 reference), TaqMan gene expression assays were also procured from Thermo Fisher Scientific. To minimize pipetting errors and optimize consistency in volumes of cDNA template and master mix, RT-qPCR reactions were set-up in triplicate in 384-well plates using the Biomek NXP Laboratory Automation Workstation (Beckman Coulter, Fullerton, CA). PCR amplification efficiency was determined for each gene in singleplex and then in multiplex reactions using serial dilutions of a DNA standard. Amplification efficiencies ranged from 86%−100%. Multiplex reactions for the simultaneous amplification of each target gene and the PUM1 reference gene were performed in a 20 μl volume composed of 5 μl of cDNA and 1X EXPRESS qPCR Supermix plus 1X TaqMan gene expression assay or 0.4 μM each primer and 0.120 μM fluorescently tagged probe. Thermo-cycling conditions were as follows: 50°C for 2 min, 95°C for 2 min, followed by 45 amplification cycles of 95°C for 15 s, and 60°C for 1 min. The 2-ΔΔCt method was used to determine quantitative relative expression values.
Differential gene expression analysis
Gene expression analysis of RNA-seq read counts mapping to the human genome was performed using the edgeR package (Robinson et al., 2010). Analyses, including filtering and normalization, were performed separately for: 1) uninfected samples (U); 2) uninfected and infected samples paired by subject for children (I-U). After filtering sex-specific genes (male-specific gene on the Y chromosome and genes that escape X-inactivation) and genes with low expression across all samples, trimmed mean of M values (TMM) normalization (Robinson and Oshlack, 2010) was applied to the top 50% most variable genes. Expression data for these genes were converted to log count-per-million (logCPM) for data visualization with PCA plots and unsupervised hierarchical clustering heatmaps. For the paired analysis, logCPMI – logCPMU (ΔlogCPM) values were used for each subject pair for data visualization. Given the age differences between the classes (Table S1), for the uninfected analysis, differential gene expression (DGE) analysis was performed using the glmLRT function as 2-way comparisons between the three classes using the following model matrix formula:
Here, age is a continuous variable, Batch1 is the extraction batch, and Batch2 is the sequencing batch.
For the paired analysis, we wanted to both compare the effect of infection within individuals as well as the differences between classes while taken into account the pairing of samples by subject. We used the following model matrix formula:
Age was not included as a covariate in the paired analysis given that age differences were accounted for by the subject effect. DGE analysis was performed with the glmLRT function for the following within class comparisons, set up as contrasts:
Between class comparisons were also performed, with the following being the most informative:
Functional analysis of transcriptomic data
Deconvolution of RNA-seq expression data to assess the relative proportions of immune cells was performed with CIBERSORT (Newman et al., 2015) using count data for each sample and the LM22 signature genes file. Proportions for cell subsets within each cell lineage were summed, and the sums for 8 most abundant cell lineages were reported. Differential expression of functional pathways was assessed by:
Determining mutually significant (FDR<5%) enrichment across different enrichment methods using blood transcription modules (Li et al., 2014) or the “hallmark” MSigDB collection (v6.2) (Liberzon et al., 2015) as gene sets: Correlation Adjusted MEan RAnk (Camera) competitive gene set testing (Wu and Smyth, 2012); “fast” geneset enrichment analysis (GSEA; (Sergushichev, 2016; Subramanian et al., 2005); and tmod (Weiner 3rd and Domaszewska, 2016). Each analysis used the same model matrix formula and contrasts as for the differential gene expression analysis above. GSEA was performed on differentially expressed genes (FDR <0.25) ranked by decreasing log2fold-change per year of age (uninfected analysis) or log2fold-change (paired analysis). For Camera analysis, intergene correlation was set at 0.01. For tmod, the U test was employed on gene ranks derived from the differential gene expression analyses above.
Ingenuity Pathways Analysis (Build 478438M, version 44691306) using differential expressed genes with a FDR<5% as determined from the edgeR differential gene expression analysis above. Upstream regulators selected for visualization in figures were limited to the following Molecule Types: “transcription regulator”, “chemical - endogenous mammalian”, “other”, “cytokine”, “kinase”, “growth factor”, “transport”, “transmembrane receptor”, “translation regulator”, “enzyme”, and “group”.
For the weighted gene co-expression network analysis (Langfelder and Horvath, 2008), co-expression network was constructed using Δ log counts per million (cpm) expression data, defined as log cpm at first Pf infection minus log cpm at the uninfected baseline in a paired manner for 71 subjects for whom expression data at both time points were available. The adjacency matrix was calculated using a beta of 4, which was determined using a scale-free topology fit index curve. Modules were identified using average linkage hierarchical clustering of dissimilarity measure derived from the adjacency matrix followed by adaptive branch pruning of the clustering dendogram with a minimum cluster size of 30. Co-expression modules were related to class differences by calculating the significance measure for each gene using Welch’s t test, with effect size being mean expression of the Immune class minus the expression of the Delayed Fever class. Genes within each co-expression network module were ranked by effect size, and the functional significance of co-expression network modules was determined by fast GSEA the pre-ranked gene list using BTMs as gene sets.
Identification of viral sequences in Illumina reads
The National Center for Biotechnology Information (NCBI) viral database was downloaded from the NCBI ftp site (ftp://ftp.ncbi.nlm.nih.gov/genomes/Viruses/) and masked against low complexity regions and tandem repeats with RepeatMasker (http://www.repeatmasker.org) (Smit et al., 2013–2015) to reduce spurious hits. Illumina sequencing reads were mapped against the human reference assembly hg19 (downloaded from ftp://ftp.ensembl.org/pub/release-86/fasta/homo_sapiens/dna/) with CLC Genomics Workbench v9. Unmapped reads were then mapped to the masked NCBI viral database with bowtie2 (Langmead and Salzberg, 2012) at high stringency. Discrimination between hits targeting specific low complexity regions on viral sequences and those spanning entire viral genomes was performed with in-house perl scripts.
Pf-specific antibody profiling by protein microarray
When plasma samples were available for a subject, we determined IgG reactivities by probing plasma samples collected at the pre-Pf infection (n = 77), infection (n = 80), and post-infection (n = 76) timepoints against a protein microarray containing 1,087 Pf antigens as previously described (Obeng-Adjei et al., 2015). Protein microarray data were log2-transformed, background subtracted and robust linear model normalized, then each sample was evaluated to determine the magnitude (i.e. sum of all antibody reaction intensities) and breadth (i.e. count of unique antibodies with intensities higher than background) (Obeng-Adjei et al., 2015). Post-infection to pre-infection IgG ratios were determined for 73 individuals.
Pf-specific IgG subclass profiling by Luminex
Plasma samples were run in a customized Luminex assay to quantify the relative concentration of immunoglobulin istoypes. Carboxylated magplex-microspheres (Luminex) were coupled to antigen by covalent NHS-ester linkages via EDC and NHS (Thermo Scientific) as previously described (Brown et al., 2012), except with magnetic bead washing, rather than centrifugation. Antigens included Pf CSP (3D7), MSP142, and AMA1(FVO) from David Narum (Laboratory of Malaria Immunology and Vaccinology, NIAID, NIH), and RH5 from Gavin Wright (Wellcome Trust Sanger Institute). Assays were performed following a previously described protocol (Brown et al., 2012). Briefly, antigen-coated microspheres (approximately 500 beads per antigen, per well) were added to a 384 well plate Luminex plates (Greiner Bio-One)-Microplate. Each serum sample (diluted 1:100 in 1xPBS) was added to two replicate wells within a 384-well plate and incubated with beads for 2 hrs, with shaking at 900 RPM at RT. The microspheres were washed by magnetic bead separation and then incubated with IgG1-, IgG2-, IgG3-, IgG4-, IgA- or IgM secondary PE-conjugated antibody (Southern Biotech) for 1 hr, with shaking at 900 RPM at RT. The beads were then washed and read on a Bio-Plex 200 System (Bio-rad). Results are reported as the average median fluorescent intensity of two replicates. Given the focus of this study on blood-stage immunity, analysis was limited to reactivity data for IgG1, IgG2, IgG3, and IgG4 against the blood-stage antigens MSP142, AMA1, and RH5.
Total IgE and Pf-specific IgE determination
For 79 individuals, total IgE was determined using the Bio-Plex Pro human IgE isotyping assay (Bio-Rad) at a 1:500 plasma dilution using the manufacturer’s recommended protocol. Pf-specific IgE was determined at a plasma dilution of 1:50 using lysates of mature stages of magnetically separated Pf-infected RBCs (iRBC) of the laboratory strain 3D7 (Portugal et al., 2014) or lysates of uninfected RBCs. Maxisorp 96-well plates (Nunc) were coated with iRBC or uRBC lysates overnight at 4°C. After washing with PBS/0.1% Tween-20 (4 cycles) and PBS (1 cycle), wells were blocked with PBS/0.1% Tween-20/10% dry milk (blocking buffer) for 1h at room temperature followed by another 5 cycles of washing as before. Wells were incubated with human plasma diluted 1:50 overnight at room temperature followed by 5 cycles of washing. Wells were incubated with anti-human IgE diluted 1:5000 in blocking buffer for 1h at room temperature followed by 5 cycles of washing. Detection was carried out using SureBlue TMB Microwell Peroxidase Substrate Kit (KPL) per the manufacturer’s instructions.
Flow cytometry and immunophenotyping
Initial immunophenotyping and intracellular cytokine staining of PBMCs collected at enrollment (uninfected baseline) were performed on a subset of 60 children (Immune = 18, Delayed Fever = 21, and Early Fever = 21) in two separate batches with a similar distribution of classes. PBMCs were thawed, washed in PBS with 4% fetal bovine serum (FACS buffer), blocked with human Fc Blocker (BD Biosciences) for 20 min, stained with a cell viability stain in PBS for 30 min followed by staining with B cell, T cell, and monocyte cell surface markers in FACS buffer for 30 min at 4°C (antibodies listed in Key Resources Table). Cells were fixed and permeabilized using the FoxP3/transcription factor intracellular staining kit (eBioscience) prior to staining with intracellular antibodies for 30 min (antibodies listed in Key Resources Table). A minimum of 100,000 gated cells were acquired on an LSR II flow cytometer (BD). Dead cells were excluded from the analysis. Flow cytometric analysis was done using FlowJo 10 software (TreeStar). p53 staining was also performed using the anti-p53 monoclonal antibody clone PAb240 on PBMCs of 9 additional children (Delayed Fever = 5, and Early Fever = 4).
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-human CD10 Brilliant Violet 510 clone HI10A | BioLegend | Cat#312219; RRID: AB_2561722 |
| anti-human CD10 Brilliant Violet 605 clone HI10A | BD Biosciences | Cat#562978; RRID: AB_2737929 |
| anti-human CD14 Brilliant Violet 785 clone M5E2 | BioLegend | Cat#301840; RRID: AB_2563425 |
| anti-human CD14 PE clone M5E2 | BD Biosciences | Cat#555398; RRID: AB_395799 |
| anti-human CD14 PE-Cy7 clone M5E2 | BD Biosciences | Cat#557742; RRID: AB_396848 |
| anti-human CD16 Brilliant Violet 605 clone 3G8 | BioLegend | Cat#302040; RRID: AB_2562990 |
| anti-human CD19 Brilliant Violet 785 clone HIB19 | BioLegend | Cat#302240; RRID: AB_2563442 |
| anti-human CD19 Brilliant Violet Violet 480 clone SJ25C1 | BD Biosciences | Cat#566103; RRID: AB_2739505 |
| anti-human CD21 APC clone HB5 | Ebioscience/Thermo Fisher Scientific | Cat#17-0219-42; RRID: AB_1582217 |
| anti-human CD21 PE clone HB5 | Ebioscience/Thermo Fisher Scientific | Cat#12-0219-42; RRID: AB_1548734 |
| anti-human CD23 Brilliant Violet Violet 711 clone EBVCS-5 | BD Biosciences | Cat#743430; RRID: AB_2741503 |
| anti-human CD23 PE-Cy7 clone EBVCS-5 | BioLegend | Cat#338516; RRID: AB_2278308 |
| anti-human CD27 Brilliant Violet 421 clone M-T271 | BioLegend | Cat#356418; RRID: AB_2562599 |
| anti-human CD27 Brilliant Violet 650 clone L128 | BD Biosciences | Cat#563228; RRID: AB_2744352 |
| anti-human CD3 Horizon V500 clone UCHT1 | BD Biosciences | Cat#561416; RRID: AB_10612021 |
| anti-human CD4 PE-CF594 clone RPA-T4 | BD Biosciences | Cat#562281; RRID: AB_11154597 |
| anti-human CD4 PerCP clone RPA-T4 | BioLegend | Cat#300527; RRID: AB_893327 |
| anti-human CD4 PerCP/Cy5.5 clone RPA-T4 | BioLegend | Cat#300530; RRID: AB_893322 |
| anti-human CD56 Brilliant Violet 711 clone 5.1H11 | BioLegend | Cat#362541; RRID: AB_2565919 |
| anti-human CD56 Brilliant Violet 650 clone NCAM16.2 | BD Biosciences | Cat#564057; RRID: AB_2738568 |
| anti-human CD8 PE-Cy7 clone RPA-T8 | BD Biosciences | Cat#557746; RRID: AB_396852 |
| anti-human CD8a Brilliant Violet 570 clone RPA-T8 | BioLegend | Cat#301038; RRID: AB_2563213 |
| anti-human GATA3 PE-Cy7 clone L50–823 | BD Biosciences | Cat#560405; RRID: AB_1645544 |
| anti-human p53 Alexa Fluor 647 clone DO-1 | SantaCruz | Cat#sc-126 AF647 |
| anti-human p53 Alexa Fluor 647 clone PAb 240 | Novus Biologicals | Cat#NB200–103AF647 |
| anti-human phospho-STAT6 PE-conjugated Ab clone Y641 | R&D Systems | Cat#IC3717P; RRID: AB_2255459 |
| anti-human STAT6 PerCP-Cy5.5 clone 23/Stat6 | BD Biosciences | Cat#564149; RRID: AB_2738622 |
| anti-human T-bet PE-CF594 clone O4–46 | BD Biosciences | Cat#562467; RRID: AB_2737621 |
| Biological Samples | ||
| human peripheral blood mononuclear cells | Kalifabougou cohort of malaria immunity | https://clinicaltrials.gov/ct2/show/NCT01322581 |
| human plasma | Kalifabougou cohort of malaria immunity | https://clinicaltrials.gov/ct2/show/NCT01322581 |
| human whole-blood genomic DNA | Kalifabougou cohort of malaria immunity | https://clinicaltrials.gov/ct2/show/NCT01322581 |
| human whole-blood RNA | Kalifabougou cohort of malaria immunity | https://clinicaltrials.gov/ct2/show/NCT01322581 |
| human stool genomic DNA | Kalifabougou cohort of malaria immunity | https://clinicaltrials.gov/ct2/show/NCT01322581 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Nutlin-3 | Sigma | Cat#N-6287 |
| Insolution™ Nutlin-3 Racemic | EMD Millipore | Cat#444151 |
| Ficoll-Paque™ Premium Sterile Solution | GE Healthcare | Cat#17-5442-02 |
| Critical Commercial Assays | ||
| Bio-Plex Pro Human Cytokine 17-plex Assay | Bio-Rad | Cat#M5000031YV |
| Bio-Plex Pro Human Cytokine Screening Panel 7-plex Express Assay | Cat#17005101 | |
| Bio-Plex Pro Human IgE Isotyping Assay | Bio-Rad | Cat#171A3102M |
| Bio-Plex Pro Mouse Cytokine Th1 Panel | Bio-Rad | Cat#L6000004C6 |
| EasySep Human Monocyte Isolation Kit | Stemcell Technologies | Cat#19359 |
| EasySep Human CD14 Positive Selection Kit II | Stemcell Technologies | Cat#17858 |
| HiSeq 2000 paired-end Sequencing Kit, V3 | Illumina | Cat#SY-401–1001 |
| Pf1000 Antigen protein array | University of California Irvine | Probed on 05/07/2014 Pf1000 chip printed at UCI, Omnigrid 100, 16 April 2014 Chips used 1–41 PNG reference sera used |
| ScriptSeq Complete Gold Kit (Blood) | Illumina | Cat#BGGB1324 |
| ScriptSeq™ v2 RNA-Seq Library Preparation Kit | Epicentre (Illumina) | Cat#SSV21124 |
| SuperScript® VILO cDNA Synthesis Kit | Invitrogen | Cat#11754050 |
| Deposited Data | ||
| Resource website for the Systems Immunology of Malaria publication | this paper | https://www.immport.org/shared/study/SDY1172 |
| RNA-seq data | this paper | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE52166 |
| Experimental Models: Organisms/Strains | ||
| Plasmodium falciparum 3D7 | MR4 | Cat#MRA-102G |
| Plasmodium falciparum HB3 | MR4 | Cat#MRA-155G |
| Plasmodium yoelii 17XNL GFP | MR4 | Cat#MRA-817 |
| super p53 C57BL/6 mice | García-Cao et al., 2002 | David G Kirsch (Duke University) |
| wild-type C57BL/6 mice (littermate controls) | This paper | |
| Oligonucleotides | ||
| P. falciparum microsatellite analysis | ||
| ARA2–3(F): GTACATATGAATCACCAA | Shaukat et al., 2012 | N/A |
| ARA2-F: GAATAAACAAAGTATTGCT (FAM) | Shaukat et al., 2012 | N/A |
| ARA2-R: GCTTTGAGTATTATTAATA | Shaukat et al., 2012 | N/A |
| PFPK2–3R: CCTCAGACTGAAATGCAT | Shaukat et al., 2012 | N/A |
| PFPK2-F: CTTTCATCGATACTACGA | Shaukat et al., 2012 | N/A |
| PFPK2-R: AAAGAAGGAACAAGCAGA (HEX) | Shaukat et al., 2012 | N/A |
| Poly α−3(IR): GAAATTATAACTCTACCA (FAM) | Shaukat et al., 2012 | N/A |
| Poly α-F: AAAATATAGACGAACAGA | Shaukat et al., 2012 | N/A |
| Poly α-R: ATCAGATAATTGTTGGTA | Shaukat et al., 2012 | N/A |
| TA40 For: AAGGGATTGCTGCAAGGT | Shaukat et al., 2012 | N/A |
| TA40 Rev-1: GAAATTGGCACCACCACA | Shaukat et al., 2012 | N/A |
| TA40 Rev-2: CATCAATAAAATCACTACTA (PET) | Shaukat et al., 2012 | N/A |
| TA81–3F: GAAGAAATAAGGGAAGGT | Shaukat et al., 2012 | N/A |
| TA81-F: TGGACAAATGGGAAAGGATA (PET) | Shaukat et al., 2012 | N/A |
| TA81-R: TTTCACACAACACAGGATT | Shaukat et al., 2012 | N/A |
| TA87–3F: ATGGGTTAAATGAGGTACA | Shaukat et al., 2012 | N/A |
| TA87-F: AATGGCAACACCATTCAAC (HEX) | Shaukat et al., 2012 | N/A |
| TA87-R: ACATGTTCATATTACTCAC | Shaukat et al., 2012 | N/A |
| P. falciparum parasite density | ||
| rFAL1: 5′ TTAAACTGGTTTGGGAAAACCAAATATATT 3′ | Tran et al., 2014 | N/A |
| rFAL2: 5′ ACACAATGAACTCAATCATGACTACCCGTC 3′ | Tran et al., 2014 | N/A |
| rPLU5: 5′ CCTGTTGTTGCCTTAAACTTC 3′ | Tran et al., 2014 | N/A |
| rPLU6: 5′ TTAAAATTGTTGCAGTTAAAACG 3′ | Tran et al., 2014 | N/A |
| human GAPDH F2: 5′ CGACCACTTTGTCAAGCTCA 3′ | Tran et al., 2014 | N/A |
| human GAPDH R2: 5′ GGTGGTCCAGGGGTCTTACT 3′ | Tran et al., 2014 | N/A |
| Human RT qPCR targets | ||
| CD14 forward TCAGAGGTTCGGAAGACTTATCG | This paper | N/A |
| CD14 probe FAM-CAGGACGCGCGCTCCATGG-BHQ1 | This paper | N/A |
| CD14 reverse TCATCGTCCAGCTCACAAGGT | This paper | N/A |
| FCER1G forward AGTGCGAAAGGCAGCTATAACC | This paper | N/A |
| FCER1G probe FAM_CGTAAGTCTCCTGGTTCCTGGTGCTCA-BHQ1 | This paper | N/A |
| FCER1G reverse TACTGTGGTGGTTTCTCATGCTTC | This paper | N/A |
| PUM1 forward GGAGTCTACCCTGCCAGTCT | This paper | N/A |
| PUM1 probe CalFluorGold540-ACGGAGAACCTGCTGCTGTCCTTGCT-BHQ1 | This paper | N/A |
| PUM1 reverse GACGTTGGCTGGCTCCTC | This paper | N/A |
| PUM1 Taqman primer/probe set | Thermo Fisher Scientific | Cat#Hs00472881_m1 (VIC) |
| TP53 Taqman primer/probe set | Thermo Fisher Scientific | Cat#Hs01034249_m1 (FAM) |
| Other pathogen detection | ||
| Ancylostoma duodenale FWD Primer (5’−3’) GTATTTCACTCATATGATCGAGTGTTC | Easton et al., 2016 | N/A |
| Ancylostoma duodenale Probe 5’-TGACAGTGTGTCATACTGTGGAAA-3’ | Easton et al., 2016 | N/A |
| Ancylostoma duodenale REV Primer (5’−3’) GTTTGAATTTGAGGTATTTCGACCA | Easton et al., 2016 | N/A |
| Ascaris lumbricoides FWD Primer (5’−3’) GTAATAGCAGTCGGCGGTTTCTT | Easton et al., 2016 | N/A |
| Ascaris lumbricoides Probe 5’-TTGGCGGACAATTGCATGCGAT-3’ | Easton et al., 2016 | N/A |
| Ascaris lumbricoides REV Primer (5’−3’) GCCCAACATGCCACCTATTC | Easton et al., 2016 | N/A |
| Mansonella perstans FWD Primer (5’−3’) CGTTGATATTTGAATGCACGACAATG | Drame et al., 2016 | N/A |
| Mansonella perstans Probe 5’-CCGACGTGCACACCAT-3’ | Drame et al., 2016 | N/A |
| Mansonella perstans REV Primer (5’−3’) GGCCGAAACATTCAATTACCTCAA | Drame et al., 2016 | N/A |
| Necator americanus FWD Primer (5’−3’) CCAGAATCGCCACAAATTGTAT | Easton et al., 2016 | N/A |
| Necator americanus Probe 5’-CCCGATTTGAGCTGAATTGTCAAA-3’ | Easton et al., 2016 | N/A |
| Necator americanus REV Primer (5’−3’) GGGTTTGAGGCTTATCATAAAGAA | Easton et al., 2016 | N/A |
| Strongyloides stercoralis FWD Primer (5’−3’) CGCTCCAGAATTAGTTCCAGTT | Easton et al., 2016 | N/A |
| Strongyloides stercoralis Probe 5’-ACAGTCTCCAGTTCACTCCAGAAGAGT-3’ | Easton et al., 2016 | N/A |
| Strongyloides stercoralis REV Primer (5’−3’) GCAGCTTAGTCGAAAGCATAGA | Easton et al., 2016 | N/A |
| Trichuris trichiura FWD Primer (5’−3’) GGCGTAGAGGAGCGATTT | Easton et al., 2016 | N/A |
| Trichuris trichiura Probe 5’-TTTGCGGGCGAGAACGGAAATATT-3’ | Easton et al., 2016 | N/A |
| Trichuris trichiura REV Primer (5’−3’) TACTACCCATCACACATTAGCC | Easton et al., 2016 | N/A |
| Software and Algorithms | ||
| bowtie2 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| caret R package | Kuhn, 2008 | https://CRAN.R-project.org/package=caret |
| circlize R package | Gu et al., 2014 | http://zuguang.de/circlize_book/book/ |
| corrplot R package | https://CRAN.R-project.org/package=corrplot | |
| Cufflinks | Trapnell et al., 2012 | http://cole-trapnell-lab.github.io/cufflinks/ |
| diveRsity R package | Keenan et al., 2013 | https://cran.r-project.org/web/packages/diveRsity/index.html |
| edgeR R package | Robinson et al., 2012 | https://bioconductor.org/packages/release/bioc/html/edgeR.html |
| fsgea package | Sergushichev, 2016 | https://bioconductor.org/packages/release/bioc/html/fgsea.html |
| glmnet R package | Friedman et al., 2010 | https://cran.r-project.org/web/packages/glmnet/index.html |
| impute R package | Troyanskaya et al., 2001 | http://bioconductor.org/packages/release/bioc/html/impute.html |
| R version 3.5 | https://www.r-project.org | |
| RepeatMasker | http://repeatmasker.org | |
| survminer R package | https://CRAN.R-project.org/package=survminer | |
| tmod package | Weiner and Domaszewska, 2016 | https://CRAN.R-project.org/package=tmod |
| TopHat2 | Kim et al., 2013 | http://ccb.jhu.edu/software/tophat |
| trimmed mean of M values (TMM) normalization | Robinson and Oshlack, 2010 | https://bioconductor.org/packages/release/bioc/html/edgeR.html |
| WGCNA package | Langfelder and Horvath (2008) | https://cran.r-project.org/web/packages/WGCNA/index.html |
| Other | ||
Cell culture and stimulation experiments
PBMCs were cultured in complete RPMI (RPMI 1640 plus 10% fetal calf serum, 1% penicillin/streptomycin, 2-mercaptoethanol) in flat-bottom 96-well plates, at 37°C in a 5% CO2 atmosphere. 5×105 PBMCs were pre-treated with nutlin-3 (20uM) or DMSO control for 12 h prior to stimulation with infected red blood cell (iRBCs) or uninfected RBC lysate at a ratio of 3 RBCs per PBMC for another 12 h without change of media. Following stimulation, cells were centrifuged, and supernatants were recovered and frozen at −80°C for cytokine analysis. Levels of IFN-γ and TNF were determined from thawed supernatants (neat) using the Bio-plex human cytokine kit (Bio-Rad) and a Luminex 200 device (Bio-Rad) per the manufacturer’s protocol. Final concentrations were calculated from the mean fluorescence intensity and expressed in pg/mL using standard curves with known concentrations of each cytokine.
Monocyte isolation, culture and stimulation
For flow cytometric studies including intracellular cytokine staining, monocytes were isolated from the blood of healthy, malaria-naïve United States (US) donors at the National Institutes of Health Blood Bank using a two-step procedure beginning with automated leukopheresis followed by counterflow elutriation (Stroncek et al., 2014). Elutriated monocytes were further purified using the EasySep Monocyte isolation kit (StemCell Technologies). Monocytes from Malian patient samples were isolated from cryoperserved PBMCs using the same monocyte isolation kit after thawing. Monocytes were confirmed to be >96% purity by flow cytometry using anti-human CD14 monoclonal antibodies. Cells were cultured in RPMI medium 1640 supplemented with penicillin, streptomycin, 2 mM glutamine, and 10% heat-inactivated pooled AB human serum. Monocytes were plated in either 24 or 6 well culture plates, kept at incubator for 2 hrs of rest and stimulated with iRBC lysate at a ratio of 1:10 (monocyte:Pf lysate) for 18 hrs. eBioscience™ Protein Transport Inhibitor Cocktail was added along with the iRBC and kept throughout the stimulation period. Cells were collected after stimulation and stained for surface markers and intracellular cytokines as described in “Cell culture stimulation.”
To measure secreted cytokines in supernatants of stimulated monocytes, CD14+ monocytes were isolated using the EasySep Human CD14 Positive Selection Kit II (Stemcell Technologies) from PBMCs obtained by Ficoll gradient density centrifugation of blood collected from healthy, malaria-naïve US donors. Monocytes were confirmed to be >98% pure by flow cytometry. Purified CD14+ or CD14− cells were plated in 96-well tissue culture plates and rested for 2 h prior to stimulation as described above for PBMCs in the Cell culture stimulation experiments section.
Multiplex cytokine analysis of human plasma
For 79 individuals whose plasma samples were available at enrollment, we performed multiplex cytokine analysis using the Bio-Plex Pro human cytokine 17-plex assay (Bio-Rad) and a Luminex 200 device (Bio-Rad) at a plasma dilution of 1:4 per the manufacturer’s instructions.
Data Mining
Data from 80 individuals were used to determine if the three classes of acquired immunity to Pf (Immune, Delayed Fever, or Early Fever) could be predicted using a combination of 6,822 RNA-seq features, 1072 protein microarray features, 107 FACS features, 24 Luminex features, 14 multiplex cytokine analysis features plus total and Pf-specific IgE features (8,927 total features). Prior to data mining, missing data was imputed using K-Nearest Neighbors using the impute R package (Troyanskaya et al., 2001). Data from the various experiments were also normalized by subtracting the mean of each feature and dividing by its standard deviation (i.e. Xnew = X – mean(X) / sd(X)). Three basic types of classification models with were tested using K = 6-fold cross-validation: K-Nearest Neighbors (KNN), random forests (RF) and elastic net (EN) models (Friedman et al., 2010). At each fold of the cross-validation, random forests feature selection from the caret R package library (Kuhn, 2008) was used to reduce the number of features prior to model training, selecting the maximum number of features identified by either the caret package itself or a score calculated using the ranked values of each feature number’s accuracy, accuracy SD, Cohen’s kappa and kappa SD. KNN models were trained for K = 1, 3, 5 and 7 neighbors. RF models were trained on 12 parameter combinations, with number of trees = 250, 500, 750 or 1000 and node = 1, 2 or 3. Elastic net models were trained using 30 different combinations of their alpha and lambda parameters, specifically 35 pairwise combinations of alpha = 0, 0.25, 0.5, 0.75, 1 with lambda = 0.05, 0.04, 0.03, 0.02, 0.01, and 0. Elastic net models with alpha = 0 are also known as ridge regression models, while models with alpha = 1 are lasso regression models. Performance of each classifier was evaluated using the mean and standard deviation of its accuracy and its Cohen’s kappa coefficient, with the overall performance of the model evaluated using
Scores increase when Mean Accuracy is greater than 50%, but scores are penalized when Mean Accuracy is below 50%. Similarly, scores increase when Kappa increases, when Accuracy SD decreases and Kappa SD decreases. All four of these criteria are desirable qualities, equally weighted in the scoring system. The top scoring models were identified, and coefficients from the top 3 elastic net or random forest models were identified to determine which features (i.e. genes, antibodies, etc.) were most important in classifying the samples. This data mining process was repeated over 63 “runs”, each with different randomly generated subsets of samples assigned to the K = 6 folds. We had planned for more than 100 runs, but were limited by computation time and many runs failed due to long computation times, possibly because the randomly generated folds may have yielded very small or zero sample sizes for one malaria class (Immune, Delayed or Early). Among these 63 runs, we searched for features that appeared in the most cross validation folds among the 63 runs and K = 6 folds to identify the best predictive features. Features appearing in > 100 cross validation folds appeared to be very high-quality predictors.
Quantification and Statistical Analysis
Details of the statistical tests applied to datasets shown in Figures can be found in the above methods and in corresponding Figure legends. All data points and “n” values reflect biological replicates. Statistical analyses were performed in R (version 3.5.1) or GraphPad prism 7.0b. Plots used in this manuscript were generated using the following R packages: ggplot2 (general plots); survminer (time-to-event analyses), corrplot (correlation matrix), and circlize (Gu et al., 2014).
Data and Code Availability
Raw data and analytical tools are available on ImmPort (https://www.immport.org), a NIAID-funded repository focusing on immunological data sets, under study accession number SDY1172. Sequencing data is available on the Gene Expression Omnibus (GEO) database under the accession number GSE52166. Data presented in this systems analysis can be explored via interactive apps that are accessible at https://malariasystems.org.
Supplementary Material
Supplemental Dataset 4. Molecular and microscopic evaluations for parasitic co-infections in urine, stool, and blood at the P. falciparum-negative baseline (study enrollment). Related to Figure 4.
Supplemental Dataset 2. Rotational gene set testing (roast) using human transcription factor networks derived from experimentally determined genome-wide maps of in vivo DNaseI footprints (Neph et al., 2012). Related to Figures 2 and S3. Analysis was performed for each two-way comparison and limited to blood-cell types.
Supplemental Dataset 5. Top coefficients using the random forests class prediction model. Related to STAR Methods (Data Mining).
Supplemental Dataset 3. Differential gene expression analysis during the first P. falciparum infection of the malaria season. Related to Figure 4. Δ (delta) = paired comparison between first P. falciparum infection of the malaria season vs. uninfected baseline.
Supplemental Dataset 1. Differential gene expression analysis at the uninfected baseline prior to the malaria season. Related to Figure 2.
Acknowledgements
We thank the residents of Kalifabougou, Mali for participating in this study. This project was supported with federal funds from the National Institute of Allergy and Infectious Diseases (NIAID), National Institutes of Health, Department of Health and Human Services under contract number HHSN272200900007C, award number U19AI110819, and the Division of Intramural Research. T.M.T. was also supported by K08AI125682 (NIAID) and the Doris Duke Charitable Foundation Clinical Scientist Development Award. Protein microarray experiments were funded by NIAID grants U19AI089686 and R01AI095916 (P.L.F.). Super p53 mice were kindly provided by David G. Kirsch (Duke University). Recombinant Pf antigens were kindly provided by David L. Narum (Laboratory of Malaria Immunology and Vaccinology, NIAID, NIH) and Gavin J. Wright (Wellcome Trust Sanger Institute).
Footnotes
Declaration of Interests
P.L.F. has an equity interest in Antigen Discovery, Inc., which is developing products related to the protein microarray platform used in this study. The University of California reviewed and approved in accordance with its conflict of interest policies.
References
- Arora G, Hart GT, Manzella-Lapeira J, Doritchamou JY, Narum DL, Thomas LM, Brzostowski J, Rajagopalan S, Doumbo OK, Traore B, et al. (2018). NK cells inhibit Plasmodium falciparum growth in red blood cells via antibody-dependent cellular cytotoxicity. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bashirova AA, Apps R, Vince N, Mochalova Y, Yu XG, and Carrington M (2014). Diversity of the human LILRB3/A6 locus encoding a myeloid inhibitory and activating receptor pair. Immunogenetics 66, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becart S, Charvet C, Canonigo Balancio AJ, De Trez C, Tanaka Y, Duan W, Ware C, Croft M, and Altman A (2007). SLAT regulates Th1 and Th2 inflammatory responses by controlling Ca2+/NFAT signaling. The Journal of clinical investigation 117, 2164–2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bediako Y, Adams R, Reid AJ, Valletta JJ, Ndungu FM, Sodenkamp J, Mwacharo J, Ngoi JM, Kimani D, Kai O, et al. (2019). Repeated clinical malaria episodes are associated with modification of the immune system in children. BMC medicine 17, 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle MJ, Reiling L, Feng G, Langer C, Osier FH, Aspeling-Jones H, Cheng YS, Stubbs J, Tetteh KK, Conway DJ, et al. (2015). Human antibodies fix complement to inhibit Plasmodium falciparum invasion of erythrocytes and are associated with protection against malaria. Immunity 42, 580–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown EP, Licht AF, Dugast AS, Choi I, Bailey-Kellogg C, Alter G, and Ackerman ME (2012). High-throughput, multiplexed IgG subclassing of antigen-specific antibodies from clinical samples. J Immunol Methods 386, 117–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen S, Mc GI, and Carrington S (1961). Gamma-globulin and acquired immunity to human malaria. Nature 192, 733–737. [DOI] [PubMed] [Google Scholar]
- Crompton PD, Moebius J, Portugal S, Waisberg M, Hart G, Garver LS, Miller LH, Barillas-Mury C, and Pierce SK (2014). Malaria immunity in man and mosquito: insights into unsolved mysteries of a deadly infectious disease. Annual review of immunology 32, 157–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Ombrain MC, Robinson LJ, Stanisic DI, Taraika J, Bernard N, Michon P, Mueller I, and Schofield L (2008). Association of early interferon-gamma production with immunity to clinical malaria: a longitudinal study among Papua New Guinean children. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America 47, 1380–1387. [DOI] [PubMed] [Google Scholar]
- Dijsselbloem N, Goriely S, Albarani V, Gerlo S, Francoz S, Marine JC, Goldman M, Haegeman G, and Vanden Berghe W (2007). A critical role for p53 in the control of NF-kappaB-dependent gene expression in TLR4-stimulated dendritic cells exposed to Genistein. Journal of immunology 178, 5048–5057. [DOI] [PubMed] [Google Scholar]
- Doumbo S, Tran TM, Sangala J, Li S, Doumtabe D, Kone Y, Traore A, Bathily A, Sogoba N, Coulibaly ME, et al. (2014). Co-infection of long-term carriers of Plasmodium falciparum with Schistosoma haematobium enhances protection from febrile malaria: a prospective cohort study in Mali. PLoS neglected tropical diseases 8, e3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drame PM, Montavon C, Pion SD, Kubofcik J, Fay MP, and Nutman TB (2016). Molecular Epidemiology of Blood-Borne Human Parasites in a Loa loa-, Mansonella perstans-, and Plasmodium falciparum-Endemic Region of Cameroon. The American journal of tropical medicine and hygiene 94, 1301–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Easton AV, Oliveira RG, O’Connell EM, Kepha S, Mwandawiro CS, Njenga SM, Kihara JH, Mwatele C, Odiere MR, Brooker SJ, et al. (2016). Multi-parallel qPCR provides increased sensitivity and diagnostic breadth for gastrointestinal parasites of humans: field-based inferences on the impact of mass deworming. Parasites & vectors 9, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer M (2017). Census and evaluation of p53 target genes. Oncogene 36, 3943–3956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foger N, Rangell L, Danilenko DM, and Chan AC (2006). Requirement for coronin 1 in T lymphocyte trafficking and cellular homeostasis. Science 313, 839–842. [DOI] [PubMed] [Google Scholar]
- Friedman J, Hastie T, and Tibshirani R (2010). Regularization Paths for Generalized Linear Models via Coordinate Descent. J Stat Softw 33, 1–22. [PMC free article] [PubMed] [Google Scholar]
- Garcia-Cao I, Garcia-Cao M, Martin-Caballero J, Criado LM, Klatt P, Flores JM, Weill JC, Blasco MA, and Serrano M (2002). “Super p53” mice exhibit enhanced DNA damage response, are tumor resistant and age normally. EMBO J 21, 6225–6235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths MJ, Shafi MJ, Popper SJ, Hemingway CA, Kortok MM, Wathen A, Rockett KA, Mott R, Levin M, Newton CR, et al. (2005). Genomewide analysis of the host response to malaria in Kenyan children. The Journal of infectious diseases 191, 1599–1611. [DOI] [PubMed] [Google Scholar]
- Gu Z, Gu L, Eils R, Schlesner M, and Brors B (2014). circlize Implements and enhances circular visualization in R. Bioinformatics 30, 2811–2812. [DOI] [PubMed] [Google Scholar]
- Hart GT, Tran TM, Theorell J, Schlums H, Arora G, Rajagopalan S, Sangala ADJ, Welsh KJ, Traore B, Pierce SK, et al. (2019). Adaptive NK cells in people exposed to Plasmodium falciparum correlate with protection from malaria. The Journal of experimental medicine. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Idaghdour Y, Quinlan J, Goulet JP, Berghout J, Gbeha E, Bruat V, de Malliard T, Grenier JC, Gomez S, Gros P, et al. (2012). Evidence for additive and interaction effects of host genotype and infection in malaria. Proceedings of the National Academy of Sciences of the United States of America 109, 16786–16793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagannathan P, Kim CC, Greenhouse B, Nankya F, Bowen K, Eccles-James I, Muhindo MK, Arinaitwe E, Tappero JW, Kamya MR, et al. (2014). Loss and dysfunction of Vdelta2(+) gammadelta T cells are associated with clinical tolerance to malaria. Science translational medicine 6, 251ra117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagannathan P, Lutwama F, Boyle MJ, Nankya F, Farrington LA, McIntyre TI, Bowen K, Naluwu K, Nalubega M, Musinguzi K, et al. (2017). Vdelta2+ T cell response to malaria correlates with protection from infection but is attenuated with repeated exposure. Sci Rep 7, 11487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz N, Chaves A, and Pellegrino J (1972). A simple device for quantitative stool thick-smear technique in Schistosomiasis mansoni. Revista do Instituto de Medicina Tropical de Sao Paulo 14, 397–400. [PubMed] [Google Scholar]
- Kaushansky A, Ye AS, Austin LS, Mikolajczak SA, Vaughan AM, Camargo N, Metzger PG, Douglass AN, MacBeath G, and Kappe SH (2013). Suppression of host p53 is critical for Plasmodium liver-stage infection. Cell reports 3, 630–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keenan K, McGinnity P, Cross TF, Crozier WW, and Prodöhl PA (2013). diveRsity: An R package for the estimation and exploration of population genetics parameters and their associated errors. Methods in Ecology and Evolution 4, 782–788. [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, and Salzberg SL (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komarova EA, Krivokrysenko V, Wang K, Neznanov N, Chernov MV, Komarov PG, Brennan ML, Golovkina TV, Rokhlin OW, Kuprash DV, et al. (2005). p53 is a suppressor of inflammatory response in mice. FASEB J 19, 1030–1032. [DOI] [PubMed] [Google Scholar]
- Kuhn M (2008). Building Predictive Models in R Using the caret Package. J Stat Softw 28, 1–26.27774042 [Google Scholar]
- Langfelder P, and Horvath S (2008). WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9, 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langhorne J, Ndungu FM, Sponaas AM, and Marsh K (2008). Immunity to malaria: more questions than answers. Nature immunology 9, 725–732. [DOI] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HJ, Georgiadou A, Walther M, Nwakanma D, Stewart LB, Levin M, Otto TD, Conway DJ, Coin LJ, and Cunnington AJ (2018). Integrated pathogen load and dual transcriptome analysis of systemic host-pathogen interactions in severe malaria. Science translational medicine 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JC, Espeli M, Anderson CA, Linterman MA, Pocock JM, Williams NJ, Roberts R, Viatte S, Fu B, Peshu N, et al. (2013). Human SNP links differential outcomes in inflammatory and infectious disease to a FOXO3-regulated pathway. Cell 155, 57–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Rouphael N, Duraisingham S, Romero-Steiner S, Presnell S, Davis C, Schmidt DS, Johnson SE, Milton A, Rajam G, et al. (2014). Molecular signatures of antibody responses derived from a systems biology study of five human vaccines. Nature immunology 15, 195–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, and Tamayo P (2015). The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst 1, 417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, Park YJ, Tsuruta Y, Lorne E, and Abraham E (2009). p53 Attenuates lipopolysaccharide-induced NF-kappaB activation and acute lung injury. Journal of immunology 182, 5063–5071. [DOI] [PubMed] [Google Scholar]
- Madenspacher JH, Azzam KM, Gowdy KM, Malcolm KC, Nick JA, Dixon D, Aloor JJ, Draper DW, Guardiola JJ, Shatz M, et al. (2013). p53 Integrates host defense and cell fate during bacterial pneumonia. The Journal of experimental medicine 210, 891–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malaria Genomic Epidemiology N, Band G, Rockett KA, Spencer CC, and Kwiatkowski DP (2015). A novel locus of resistance to severe malaria in a region of ancient balancing selection. Nature 526, 253–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nallandhighal S, Park GS, Ho YY, Opoka RO, John CC, and Tran TM (2018). Whole-blood transcriptional signatures composed of erythropoietic and Nrf2-regulated genes differ between cerebral malaria and severe malarial anemia. The Journal of infectious diseases. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nallandhighal S, Park GS, Ho YY, Opoka RO, John CC, and Tran TM (2019). Whole-Blood Transcriptional Signatures Composed of Erythropoietic and NRF2-Regulated Genes Differ Between Cerebral Malaria and Severe Malarial Anemia. The Journal of infectious diseases 219, 154–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neph S, Stergachis AB, Reynolds A, Sandstrom R, Borenstein E, and Stamatoyannopoulos JA (2012). Circuitry and dynamics of human transcription factor regulatory networks. Cell 150, 1274–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, Hoang CD, Diehn M, and Alizadeh AA (2015). Robust enumeration of cell subsets from tissue expression profiles. Nat Methods 12, 453–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obeng-Adjei N, Portugal S, Tran TM, Yazew TB, Skinner J, Li S, Jain A, Felgner PL, Doumbo OK, Kayentao K, et al. (2015). Circulating Th1-Cell-type Tfh Cells that Exhibit Impaired B Cell Help Are Preferentially Activated during Acute Malaria in Children. Cell reports 13, 425–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ockenhouse CF, Hu WC, Kester KE, Cummings JF, Stewart A, Heppner DG, Jedlicka AE, Scott AL, Wolfe ND, Vahey M, and Burke DS (2006). Common and divergent immune response signaling pathways discovered in peripheral blood mononuclear cell gene expression patterns in presymptomatic and clinically apparent malaria. Infection and immunity 74, 5561–5573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono T, Tadakuma T, and Rodriguez A (2007). Plasmodium yoelii yoelii 17XNL constitutively expressing GFP throughout the life cycle. Experimental parasitology 115, 310–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Mazliah D, and Langhorne J (2014). CD4 T-cell subsets in malaria: TH1/TH2 revisited. Front Immunol 5, 671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portugal S, Moebius J, Skinner J, Doumbo S, Doumtabe D, Kone Y, Dia S, Kanakabandi K, Sturdevant DE, Virtaneva K, et al. (2014). Exposure-dependent control of malaria-induced inflammation in children. PLoS pathogens 10, e1004079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reece WH, Pinder M, Gothard PK, Milligan P, Bojang K, Doherty T, Plebanski M, Akinwunmi P, Everaere S, Watkins KR, et al. (2004). A CD4(+) T-cell immune response to a conserved epitope in the circumsporozoite protein correlates with protection from natural Plasmodium falciparum infection and disease. Nature medicine 10, 406–410. [DOI] [PubMed] [Google Scholar]
- Robbiani DF, Deroubaix S, Feldhahn N, Oliveira TY, Callen E, Wang Q, Jankovic M, Silva IT, Rommel PC, Bosque D, et al. (2015). Plasmodium Infection Promotes Genomic Instability and AID-Dependent B Cell Lymphoma. Cell 162, 727–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, and Oshlack A (2010). A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol 11, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholl PR, and Geha RS (1993). Physical association between the high-affinity IgG receptor (Fc gamma RI) and the gamma subunit of the high-affinity IgE receptor (Fc epsilon RI gamma). Proceedings of the National Academy of Sciences of the United States of America 90, 8847–8850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sergushichev A (2016). An algorithm for fast preranked gene set enrichment analysis using cumulative statistic calculation. bioRxiv. [Google Scholar]
- Shaukat AM, Gilliams EA, Kenefic LJ, Laurens MB, Dzinjalamala FK, Nyirenda OM, Thesing PC, Jacob CG, Molyneux ME, Taylor TE, et al. (2012). Clinical manifestations of new versus recrudescent malaria infections following anti-malarial drug treatment. Malaria journal 11, 207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit AFA, Hubley R, and Green P (2013–2015). RepeatMasker Open-4.0. [Google Scholar]
- Stanisic DI, Cutts J, Eriksson E, Fowkes FJ, Rosanas-Urgell A, Siba P, Laman M, Davis TM, Manning L, Mueller I, and Schofield L (2014). gammadelta T cells and CD14+ monocytes are predominant cellular sources of cytokines and chemokines associated with severe malaria. The Journal of infectious diseases 210, 295–305. [DOI] [PubMed] [Google Scholar]
- Stroncek DF, Fellowes V, Pham C, Khuu H, Fowler DH, Wood LV, and Sabatino M (2014). Counter-flow elutriation of clinical peripheral blood mononuclear cell concentrates for the production of dendritic and T cell therapies. J Transl Med 12, 241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y, Bi K, Kitamura R, Hong S, Altman Y, Matsumoto A, Tabata H, Lebedeva S, Bushway PJ, and Altman A (2003). SWAP-70-like adapter of T cells, an adapter protein that regulates early TCR-initiated signaling in Th2 lineage cells. Immunity 18, 403–414. [DOI] [PubMed] [Google Scholar]
- Timmann C, Thye T, Vens M, Evans J, May J, Ehmen C, Sievertsen J, Muntau B, Ruge G, Loag W, et al. (2012). Genome-wide association study indicates two novel resistance loci for severe malaria. Nature 489, 443–446. [DOI] [PubMed] [Google Scholar]
- Tran TM, Aghili A, Li S, Ongoiba A, Kayentao K, Doumbo S, Traore B, and Crompton PD (2014). A nested real-time PCR assay for the quantification of Plasmodium falciparum DNA extracted from dried blood spots. Malaria journal 13, 393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran TM, Jones MB, Ongoiba A, Bijker EM, Schats R, Venepally P, Skinner J, Doumbo S, Quinten E, Visser LG, et al. (2016). Transcriptomic evidence for modulation of host inflammatory responses during febrile Plasmodium falciparum malaria. Sci Rep 6, 31291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran TM, Li S, Doumbo S, Doumtabe D, Huang CY, Dia S, Bathily A, Sangala J, Kone Y, Traore A, et al. (2013). An intensive longitudinal cohort study of Malian children and adults reveals no evidence of acquired immunity to Plasmodium falciparum infection. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America 57, 40–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran TM, Samal B, Kirkness E, and Crompton PD (2012). Systems immunology of human malaria. Trends in parasitology 28, 248–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, and Pachter L (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7, 562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troyanskaya O, Cantor M, Sherlock G, Brown P, Hastie T, Tibshirani R, Botstein D, and Altman RB (2001). Missing value estimation methods for DNA microarrays. Bioinformatics 17, 520–525. [DOI] [PubMed] [Google Scholar]
- Weiner J 3rd, and Domaszewska T (2016). tmod: an R package for general and multivariate enrichment analysis. PeerJ Preprints 4, e2420v2421. [Google Scholar]
- World Health Organization (2018). World Malaria Report 2018 (World Health Organization). [Google Scholar]
- Wu D, and Smyth GK (2012). Camera: a competitive gene set test accounting for intergene correlation. Nucleic Acids Res 40, e133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagishi J, Natori A, Tolba ME, Mongan AE, Sugimoto C, Katayama T, Kawashima S, Makalowski W, Maeda R, Eshita Y, et al. (2014). Interactive transcriptome analysis of malaria patients and infecting Plasmodium falciparum. Genome research 24, 1433–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng SJ, Lamhamedi-Cherradi SE, Wang P, Xu L, and Chen YH (2005). Tumor suppressor p53 inhibits autoimmune inflammation and macrophage function. Diabetes 54, 1423–1428. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Dataset 4. Molecular and microscopic evaluations for parasitic co-infections in urine, stool, and blood at the P. falciparum-negative baseline (study enrollment). Related to Figure 4.
Supplemental Dataset 2. Rotational gene set testing (roast) using human transcription factor networks derived from experimentally determined genome-wide maps of in vivo DNaseI footprints (Neph et al., 2012). Related to Figures 2 and S3. Analysis was performed for each two-way comparison and limited to blood-cell types.
Supplemental Dataset 5. Top coefficients using the random forests class prediction model. Related to STAR Methods (Data Mining).
Supplemental Dataset 3. Differential gene expression analysis during the first P. falciparum infection of the malaria season. Related to Figure 4. Δ (delta) = paired comparison between first P. falciparum infection of the malaria season vs. uninfected baseline.
Supplemental Dataset 1. Differential gene expression analysis at the uninfected baseline prior to the malaria season. Related to Figure 2.
Data Availability Statement
Raw data and analytical tools are available on ImmPort (https://www.immport.org), a NIAID-funded repository focusing on immunological data sets, under study accession number SDY1172. Sequencing data is available on the Gene Expression Omnibus (GEO) database under the accession number GSE52166. Data presented in this systems analysis can be explored via interactive apps that are accessible at https://malariasystems.org.