

Abstract
Neurotropic coronavirus infection of mice results in acute encephalomyelitis followed by viral persistence. Whereas cellular immunity controls acute infection, humoral immunity regulates central nervous system (CNS) persistence. Maintenance of serum Ab was correlated with tissue distribution of virus‐specific Ab‐secreting cells (ASC). Although virus‐specific ASC declined in cervical lymph node and spleen after infectious virus clearance, virus‐specific serum Ab was sustained at steady levels, with a delay in neutralizing Ab. Virus‐specific ASC within the CNS peaked rapidly 1 wk after control of infectious virus and were retained throughout chronic infection, consistent with intrathecal Ab synthesis. Surprisingly, frequencies of ASC in the BM remained low and only increased gradually. Nevertheless, virus‐specific ASC induced by peripheral infection localized to both spleen and BM. The data suggest that CNS infection provides strong stimuli to recruit ASC into the inflamed tissue through sustained up‐regulation of the CXCR3 ligands CXCL9 and CXCL10. Irrespective of Ag deprivation, CNS retention of ASC coincided with elevated BAFF expression and ongoing differentiation of class II+ to class II–CD138+CD19+ plasmablasts. These results confirm the CNS as a major ASC‐supporting environment, even after resolution of viral infection and in the absence of chronic ongoing inflammation.
Keywords: Antibody, Central nervous system, Coronavirus
Abbreviations:
- ASC:
Ab‐secreting cell
- BBB:
blood brain barrier
- CLN:
cervical lymph nodes
- CNS:
central nervous system
- CSF:
cerebral spinal fluid
- JHMV:
JHM strain of mouse hepatitis virus
- MS:
multiple sclerosis
- p.i.:
post‐infection
Introduction
The preeminent goal of the immune system is to eliminate pathogens and establish immunological memory 1. Both T cells and Ab participate in eliminating a variety of pathogens; however, sustained serum Ab is an important criteria for many vaccination strategies, as they provide the first line of defense against re‐infection 2. Upon Ag encounter in regional lymph nodes, B cells undergo clonal expansion in extrafollicular foci and within germinal centers 3, 4. Rapidly activated B cells secrete low‐affinity Ab but can undergo isotype switching and limited BCR hypermutation as they differentiate into plasmablasts 3. In the milieu of accessory cells and cytokines, germinal center B cells undergo affinity maturation and ultimately differentiate into both Ab‐secreting cells (ASC) and memory B cells. As Ag is depleted, ASC and memory B cells are detected with increasing frequency in BM 2, where both stromal cells and other resident cells provide soluble as well as contact‐dependent survival signals, including CXCL12 and BAFF 5. Ab secretion by terminally differentiated plasma cells is independent of both Ag and T cell regulation 2, 6. Long‐lived ASC in BM and spleen maintain serum Ab, thus providing protective immunity to re‐infection, sometimes for the life time of the host 2. Upon re‐infection memory B cells rapidly expand and differentiate into plasmablasts, constituting a crucial source to increase pathogen‐specific Ab and replenish ASC 4, 6.
In contrast to Ag encountered in the periphery, the regulation of B cell activation by Ag sequestered within the central nervous system (CNS) is less clear. The absence of dedicated lymphatic drainage and the presence of the blood brain barrier (BBB) limits Ag transport from the CNS into secondary lymphoid tissue as well as trafficking of both cells and macromolecules into the CNS. In addition, CNS resident cells do not express MHC molecules 7–9; therefore, in the absence of inflammation, there is little possibility for endogenous Ag presentation, consistent with the inability to initiate immune responses within the CNS 10. Nevertheless, Ag deposition into the CNS, under conditions in which the BBB integrity remains relatively intact, results in a robust local humoral immune response 11. In situ ASC differentiation at the site of Ag deposition is preceded by peripheral activation, as implied by the early detection of Ag‐specific ASC in cervical lymph nodes (CLN) and spleen and the necessity of CLN for intrathecal Ab production 11. These data suggest that an Ag depot within the CNS is recognized as non‐self and that functions associated with peripheral B cell differentiation are sustained within the CNS. A variety of CNS inflammatory diseases support trafficking and preferential retention of ASC within the CNS. During virus‐induced encephalitis, B cells as well as virus‐specific ASC and ASC of unknown specificities are found in the inflamed CNS 12–16. Indirect evidence for intrathecal Ab synthesis includes oligoclonal Ab bands with restricted specificities in the cerebral spinal fluid (CSF) of patients with subacute sclerosing panencephalitis (SSPE) and multiple sclerosis (MS) 14, 17, 18. Increased CSF to serum Ab ratios detected following a variety of experimental CNS viral infections and measles virus‐induced encephalitis in humans further support local Ab production 15, 19, 20. Similarly, during acute MS the BBB is compromised, potentially allowing ASC recruitment 21, 22. Only a small portion of the oligoclonal bands in MS CSF are specific for a defined Ag 18, 23, 24, while the oligoclonal Ab bands in the CSF of SSPE patients are specific for measles virus 19, 20. These data suggest that B cells are recruited into the CNS during acute peripheral infections with neurological involvement as well as during loss of BBB integrity associated with autoimmunity. They also imply that the CNS provides a milieu capable of differentiating and maintaining ASC. Although chemokines and survival factors that influence preferential localization of ASC to the BM have recently been defined 5, 6, 15, relatively little is known about mechanisms sustaining ASC within the CNS.
Prolonged maintenance of anti‐viral intrathecal Ab synthesis is a common trait following control of many acute viral CNS infections 15, 16, 25, 26. Infection of the murine CNS with the neurotropic JHM strain of mouse hepatitis virus (JHMV) results in an acute CNS inflammatory response containing all cell types associated with anti‐viral immunity, including B cells 12, 27. Despite rapid recruitment of B cells into the CNS following JHMV infection, serum Ab is detected only after the majority of infectious virus has been eliminated 16. The concept that humoral immunity is redundant during acute infection is supported by unimpaired initial clearance of infectious virus from the CNS of mice lacking the ability to secrete Ab 28, 29 and the absence of anti‐viral ASC in the CNS of WT mice until after virus clearance 16. However, a local Ab response within the CNS is required to control viral persistence 29.
Virus‐specific ASC were measured to gain an understanding of organ‐specific distribution following an inflammatory response confined to the CNS. Similar to virus‐specific T cells 30, ASC were detected within the spleen and CLN prior to the CNS 16. Stable levels of anti‐viral serum Ab suggest ASC in the BM as a primary source throughout persistence. However, virus‐specific ASC were preferentially retained within the CNS and were barely detectable in BM throughout persistent infection of the CNS by JHMV, in contrast to preferential accumulation in BM observed for heterologous peripheral infections 2, 31–33. High levels of virus‐induced CXCL9 and CXCL10 mRNA within the CNS implicate CXCR3 ligands as mediators of ASC recruitment. Furthermore, BAFF mRNA up‐regulation preceded ASC accumulation and remained detectable throughout ASC persistence. These data suggest that virus‐induced CNS inflammation creates an environment that sustains local ASC, preenpting accumulation in the BM as a homeostatic maintenance site.
Results
Ab response following CNS viral infection
Activation of virus‐specific immune effectors occurs predominantly in CLN following CNS infection with JHMV 30. To ensure that an infection confined to the CNS induces continued Ab production similar to peripheral Ag encounter 2, 31, sera were examined for anti‐viral Ab. Virus‐specific IgM was initially detected at day 7 post‐infection (p.i.), while anti‐viral IgG was first detected slightly delayed at day 10 p.i. (Fig. 1A). Both reached maximum levels prior to the peak of neutralizing Ab and remained elevated (Fig. 1). Neutralizing Ab was initially detected at day 10 p.i. (Fig. 1B), consistent with previous results 16, increased further until day 45 p.i. and then remained stable until day 90 p.i. (Fig. 1B). The kinetics as well as retention of both neutralizing and isotype‐specific Ab in serum were similar in infected BALB/c (H‐2d) and C57BL/6 (H‐2b) mice (data not shown), demonstrating the absence of a strain‐dependent difference in the mouse haplotypes most commonly used to examine mouse hepatitis virus pathogenesis. Neutralizing as well as IgG and IgM serum Ab are thus sustained at constant levels following control of virus replication in the CNS.
Figure 1.
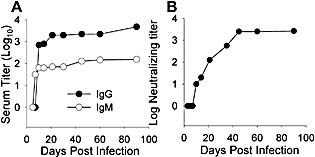
Kinetics of serum and neutralizing Ab responses. (A) Virus‐specific IgG (solid circle) and IgM (open circle) and neutralizing Ab (B) in sera of infected mice are shown. Data are representative of pooled sera from three to five mice per time point from at least three experiments.
Peripheral ASC retention
Following resolution of acute systemic and tissue‐specific infections not involving the CNS, spleen and BM are the major sites harboring the ASC that maintain serum Ab levels 4, 6. Sustained detection of serum JHMV‐specific IgM and IgG after clearance of infectious virus from the CNS thus suggested continued Ig secretion by virus‐specific ASC in BM or spleen. To examine potential contribution(s) of lymphoid organ‐derived ASC in maintaining serum Ab levels following CNS infection, JHMV‐specific ASC frequencies were examined longitudinally in spleen and BM. As ASC frequencies decline rapidly in CLN after day 15 p.i. 16, maintenance in this compartment was not pursued. ASC frequencies in spleen were already low at day 21 p.i., with similar frequencies of virus‐specific IgM and IgG ASC (Fig. 2A), consistent with the decline after day 10 p.i. 16. Splenic IgG ASC further declined gradually with time p.i. but remained detectable. In contrast, the complete disappearance of IgM ASC by day 45 p.i. suggests distinct trafficking, differentiation and/or survival of IgM versus IgG ASC (Fig. 2A). IgG‐secreting ASC in BM were barely detectable at day 21 p.i., whereas IgM ASC were not detected until day 45 p.i. (Fig. 2B). The frequency of ASC in BM increased very slowly with time, implying ongoing recruitment and/or continued differentiation of memory B cells or plasmablasts. However, even at day 90 p.i., only a minimal frequency of ASC was detected in BM. These data suggest that only a small population of virus‐specific ASC in peripheral organs contribute to sustained serum anti‐viral Ab levels.
Figure 2.
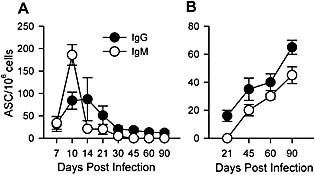
JHMV‐specific IgG and IgM ASC in peripheral lymphoid organs. Frequencies of JHMV‐specific IgG (solid circle) and IgM (open circle) ASC in spleen (A) and BM (B) measured by ELISPOT. Data represent the average of three to five separate experiments per time point ± SEM.
CNS ASC retention
Previous data revealed that virus‐specific ASC frequencies peak within the CNS by day 21 p.i. and persist to at least day 30 p.i. at much higher levels than in secondary lymphoid organs 16. The prolonged presence of ASC within the CNS following clearance of infectious virus contrasts with the rapid ASC decline in spleens and lungs following resolution of LCMV and Sendai virus infections 2, 31. To determine the fate of ASC during persistence, virus‐specific ASC frequencies within the CNS were monitored longitudinally. Both IgG and IgM virus‐specific ASC in the CNS declined steadily after the peak at day 21 p.i. (Fig. 3). IgG ASC in the CNS declined to approximately 50% by day 60 p.i., and a further 50% loss occurred between days 60 and 90 after infection. The peak frequency of virus‐specific IgM ASC was significantly reduced compared to IgG ASC and dropped by ∼50% between days 21 and 45 after infection. However, in contrast to IgG ASC, the frequency of virus‐specific IgM ASC remained relatively constant between days 45 and 90 p.i. (Fig. 3). Overall, the frequency of virus‐specific ASC in the CNS at day 90 p.i. exceeded frequencies in peripheral organs (Fig. 2).
Figure 3.
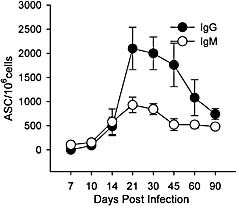
JHMV‐specific IgG and IgM ASC within the CNS. Frequencies of JHMV‐specific IgG (solid circle) and IgM (open circle) ASC in CNS measured by ELISPOT. Data represent the average of two to five separate experiments per time point ± SEM.
To characterize the distribution of ASC within the CNS, CD138 was used as a marker for pre‐plasma and terminally differentiated plasma cells, with the caveat that this approach does not specifically identify virus‐specific ASC. CD138+ cells were present in both gray and white matter, with a significant number in areas of demyelination in both spinal cord (Fig. 4A) and brain (data not shown) at day 35 after infection. The respective areas were confirmed as demyelinated by staining adjacent serial sections with luxol fast blue (data not shown). At day 90 p.i., viral Ag had been cleared from the CNS, and demyelination was significantly ameliorated (data not shown); however, CD138+ cells were still present in the CNS (Fig. 4B, C). Substantial numbers of both IgG and IgM ASC were also detected at day 90 after infection. At 90 days p.i., CD138+, IgG+ and IgM+ cells were widely distributed in both gray and white matter within the brain parenchyma and in perivascular areas within the spinal cords (Fig. 4D–F). These data contrast with an ultrastructural examination of the CNS of mice approximately 1 year p.i. with a distinct variant of JHMV, which suggested preferential retention of plasma cells in the meninges 34. Retention of ASC within the CNS parenchyma, associated with intrathecal Ab production, is consistent with an increased CSF to serum ratio of anti‐viral Ab observed following JHMV infection in both mice and rats 35–37.
Figure 4.
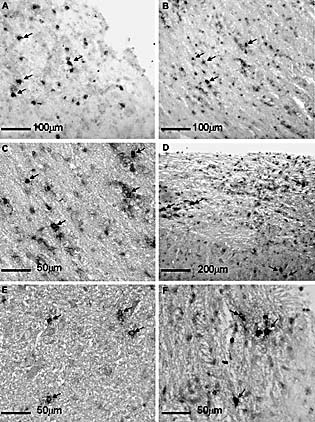
Plasma cell retention in the CNS following JHMV infection. Distribution of CD138+ plasma cells in spinal cord at day 35 p.i (A) and day 90 p.i. (B, C) as detected by immunohistochemistry. IgG+ cells (D, E) and IgM+ cells (F) in spinal cord at day 90 p.i. as detected by immunohistochemistry.
Organ‐specific ASC distribution
ASC frequency analysis reflects enrichment of distinct populations but does not recapitulate total numbers per organ. Data derived from experiments depicted in Fig. 2 and 3 were converted to ASC per tissue to account for overall higher numbers of lymphocytes in spleen and BM compared to the CNS. Vastly higher numbers of ASC were detected within the CNS compared to BM and spleen at day 21 p.i., irrespective of isotype (Fig. 5A, B), suggesting preferential trafficking of pre‐plasma cells into the CNS and/or more rapid local differentiation 11. On a total tissue basis, IgG ASC numbers progressively declined within the CNS, remained relatively constant in spleen and slowly increased in BM, eventually reaching numbers equivalent to spleen at days 40–60 p.i. (Fig. 5A). Slightly different dynamics were observed between days 60 and 90 p.i., when numbers continued to decline in spleen and CNS but increased in BM (Fig. 5A, B). The continued decline of CNS IgG ASC by day 90 p.i. eventually resulted in an equal distribution of total IgG ASC in CNS and BM (Fig. 5A). Although IgM ASC were not detected in BM at day 21 p.i. (Fig. 5B), they ultimately increased, reaching numbers similar to the CNS by day 90 p.i. (Fig. 5B). As terminally differentiated ASC are sessile 4, 5, these dynamics presumably reflect progressive loss of survival factors within the CNS but continued differentiation of ASC within the BM, rather than redistribution from the CNS to the BM. However, the vastly greater virus‐specific ASC frequencies and numbers per organ imply enhanced recruitment/survival factors within the CNS compared to peripheral lymphoid compartments following the resolution of encephalitis (Fig. 2, 4, 5). Consistent with enhanced recruitment/survival within the CNS, increased virus‐specific IgG plaque diameter with time p.i. suggests ASC maturation 38. In contrast, the diameter of virus‐specific plaques derived from BM remained relatively constant throughout infection (data not shown).
Figure 5.
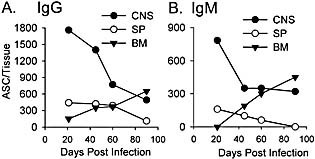
Total JHMV‐specific IgG (A) and IgM (B) ASC in CNS, spleen (SP) and BM. Numbers derived from frequencies were converted to total cell numbers in each tissue.
To further explore differentiation of ASC within the CNS, infiltrating cells were analyzed phenotypically for expression of plasmablast markers. ASC are typically characterized by down‐regulation of the B cell lineage markers B220 and CD19 but expression of CD138 4. Plasma cells furthermore lose class II expression through transcriptional repression of the class II transcriptional activator CIITA 4. Flow cytometric analysis of CD45hi brain infiltrates indicated a prominent increase of CD138+ cells between days 14 and 21 p.i. (Fig. 6A), consistent with detection of virus‐specific ASC by ELISPOT. CD138+ cells were B220– but retained CD19 expression (data not shown). Although their percentage was low, CD138+ cells increased in frequency by day 70 p.i. (Fig. 6A). Furthermore, whereas the majority of CD138+ cells expressed class II early during persistence, class II– cells dominated later. CD138+class II– cells were even more abundant in spinal cords, the predominant site of persistence and demyelination 39, by day 70 p.i. (Fig. 6B). Surprisingly, CD138+ cells retained CD19 expression, suggesting incomplete differentiation. Nevertheless, decreasing class II surface expression on CD138+ pre‐plasma cells supports ongoing differentiation for at least a 2‐month period. These data suggest that plasmablasts are recruited into the CNS at early stages of differentiation from secondary lymphoid organs.
Figure 6.
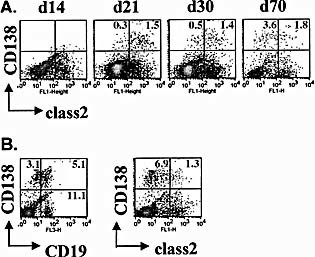
Recruitment kinetics of CD138+CD19+ B cells and differentiation during viral persistence. CNS mononuclear cells isolated from JHMV‐infected mice were stained for CD45, CD19, CD138 and class II expression and analyzed by 4‐color flow cytometry at the indicated time points after infection. (A) Density plots depicting CD138 and class II expression on CD45hi cells from brains throughout infection (gated on CD45hi cells). (B) Density plots gated on CD45hi cells from spinal cords depicting CD138 expression together with CD19 (left) and class II (right) at day 70 after infection. Numbers indicate percentages of cells in the respective quadrants.
Migration of plasmablasts into inflamed tissues frequently correlates with CXCL10 expression 40–42. Transient responsiveness to the CXCR3 ligands CXCL9, 10 and 11 has also been shown to mediate plasmablast migration in vitro 40. JHMV infection induces CNS expression of both CXCL9 and CXCL10 within 3 days p.i. 43, potentially supporting recruitment of ASC. During CNS autoimmune disease, ASC accumulation is associated with expression of CXCL13, a ligand involved in B cell migration toward follicular DC, and expression of BAFF, a B cell and ASC survival factor 42, 44, 45. Expression of mRNA for the chemokines CXCL9, CXCL10 and CXCL13 as well as BAFF was thus analyzed during acute replication and the persistent phase (Table 1). CXCL9 and CXCL10 mRNA were most prominent between days 8 and 14 p.i. and decreased substantially following clearance of infectious virus, confirming previous results 43. The lack of CXCL13 mRNA expression implies the absence of follicular DC involvement and local lymphoid follicle formation 44. BAFF mRNA levels peaked delayed compared to the CXCR3 ligands but preceded peak ASC accumulation (Table 1). Although BAFF mRNA decreased slowly after day 21 p.i., levels were still elevated 5‐fold over naive levels 75 days after infection. Thus, maximum BAFF mRNA levels preceded peak accumulation of virus‐specific ASC and declined with similar kinetics as ASC during viral persistence. These data suggest that CXCL9 and CXCL10 function as recruitment cytokines and that BAFF constitutes a potential survival factor for ASC within the brain.
Table 1.
Kinetics of chemokines and BAFF gene expression in the CNS following JHMV infection
|
Day p.i. |
CXCL9a) |
CXCL10a) |
CXCL13a) |
BAFFa) |
|---|---|---|---|---|
|
(Naive) |
2.9±0.1 |
2.9±0.2 |
0.5±0.2 |
53±13 |
|
8 |
4157±635 |
28 640±3854 |
2.1±1.4 |
1149±390 |
|
14 |
3802±973 |
35 621±658 |
1.6±1.3 |
2235±1225 |
|
21 |
508±80 |
2182±1197 |
1.6±1.2 |
1511±670 |
|
35 |
511±98 |
1165±623 |
1.1±0.8 |
815±169 |
|
75 |
123±49 |
409±195 |
0.5±0.2 |
313±43 |
a) Relative levels were normalized to ubiquitin mRNA expression
CNS versus peripheral ASC
To analyze whether the preferential localization of ASC within the CNS compared to BM is unique to virus‐induced encephalitis, virus‐specific ASC distribution was analyzed following a relatively benign self‐limiting peripheral JHMV infection. Frequencies as well as total numbers of virus‐specific IgG and IgM ASC were combined to facilitate comparison of CNS‐restricted (Fig. 7A) and transient peripheral (Fig. 7B) viral infection. In contrast to the low frequencies of total virus‐specific ASC in spleen and BM following CNS infection, peripheral infection resulted in substantially increased frequencies in the spleen as well as BM (Fig. 7B). No ASC were detected in the CNS following peripheral infection, suggesting that in contrast to activated T cells 46, activated B cells do not traffic into the CNS. Differences in ASC distribution were even more dramatic at the level of total number per tissue. Contrasting with the CNS infection (Fig. 7C), the vast majority of virus‐specific ASC localized to the spleen following peripheral infection (Fig. 7D). Although total numbers of virus‐specific ASC in BM of peripherally infected mice were lower than in spleen (Fig. 7D), ASC numbers in BM still exceeded those in the CNS of infected mice (Fig. 7B), suggesting no inherent defect in trafficking to BM. The overall paucity of peripheral ASC following CNS infection is consistent with limited viral Ag in lymphoid tissues and rapid recruitment into the CNS. Furthermore, enhanced virus‐specific ASC elicited by peripheral viral infection presumably reflect higher viral Ag load. Nevertheless, the preferential retention of ASC in spleen following a peripheral infection contrasts with the distribution of ASC following immunization with inert Ag and either systemic or respiratory viral infections 2, 31, 47.
Figure 7.
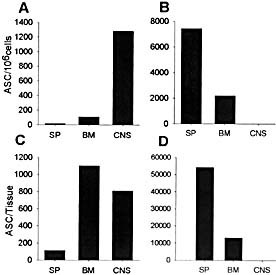
Comparison of ASC frequencies and total numbers per tissue following CNS and peripheral infection. Frequencies and total numbers of virus‐specific ASC per tissue 90 days following CNS (A, C) or peripheral (B, D) JHMV infection are shown (SP: spleen). Frequencies and total numbers represent a combination of IgG and IgM ASC. Data are representative of three separate experiments.
Discussion
Humoral immune responses are essential for resolution of many acute viral infections, especially those caused by cytopathic viruses 48. Furthermore, persisting serum Ab induced by previous infection or vaccination provides a strong defense against re‐infection 2. Long‐lived BM ASC are thought to provide a major source of persisting serum Ab following a variety of acute systemic viral infections 2, 31, 49. However, it has long been evident that the CNS also provides a preferred ASC survival niche 13, 15, 17. Studies of experimental CNS infections in animal models, systemic viral infections in humans resulting in encephalitis as well as CNS autoimmune disease, especially MS, all imply intrathecal Ab production 13, 15, 17–20, 44, 50. However, B cell recruitment and CNS survival signals following virus‐induced inflammation are largely unexplored.
The present data demonstrate that a neurotropic coronavirus infection limited to the CNS results in preferential recruitment and long‐term ASC retention, despite clearance of infectious virus. Although the site of ASC differentiation is not resolved, kinetic parameters suggest initial activation in the periphery. Virus‐specific ASC are detected in the lymphoid tissues prior to the CNS 16, similar to findings for T cells 30. Consistent with their derivation from secondary lymphoid organ germinal centers, virus‐specific ASC in the CNS emerge subsequent to germinal center reactions, which peak ∼12 days p.i. 4, 6, and are sustained after viral Ag and inflammation have subsided. Assessment of plasmablast recruitment via their CD138+ phenotype revealed kinetics similar to those demonstrated by ELISPOT. Retained expression of CD19 and class II indicates migration at an early differentiation state. The subsequent relative increase in CD138+ cells and gradual loss of class II expression during the persistent state suggest ongoing local differentiation within the CNS and/or preferential survival of a more differentiated phenotype 21. The former notion is supported by a substantially increased proportion of class II–CD138+ cells in spinal cords compared to brains at day 70 p.i. as well as increased spot size of ASC derived from brains at late compared to early times p.i. (data not shown).
A role of CNS‐localized persisting viral Ag in driving ASC differentiation and retention remains elusive. Intrathecal Ab secretion is associated with a variety of CNS infectious and autoimmune diseases and involves responses to self Ag as well as persisting microbial Ag 14, 51–53. Although a small frequency of oligoclonal Ab in the CSF of MS patients are specific for Ag associated with viral infections acquired as young adults, the majority have as‐yet‐undefined specificities 18. These data suggest that ASC can be retained within the human CNS for prolonged periods in the apparent absence of microbial Ag. However, a confounding issue is the role of self Ag in potentially propagating novel ASC subpopulations during chronic inflammation 23, 44. During JHMV infection, virus‐specific T cells decline rapidly as viral replication is controlled and Ag diminishes; nevertheless, T cell retention is prolonged in the continued presence of viral Ag/RNA 39. In contrast, virus‐specific ASC only reach peak levels after viral Ag is reduced to barely detectable levels and infectious virus is below detection limits. Their frequencies and total numbers subsequently also decline, albeit at a much reduced rate compared to T cells. These data imply distinct and largely Ag‐independent ASC survival signals. Up‐regulation of BAFF mRNA prior to peak ASC accumulation and a gradual concomitant decrease in BAFF mRNA levels and ASC after day 21 p.i. support BAFF as a likely candidate molecule required for ASC survival in the CNS 42, 44, 45. Furthermore, the inability to detect CXCL13 weakens the concept of a potential role for local follicle formation in supporting de novo ASC generation, in contrast to involvement of this mechanism during chronic autoimmune disease 44. In addition, ASC of multiple isotypes are retained within the CNS for prolonged periods following CNS JHMV infection 16. Retention of IgM ASC in the CNS, persisting serum IgM and gradual accumulation in BM suggest a potential role for IgM in control of JHMV persistence, similar to other viral infections 48. Irrespective of unique isotype patterns induced by distinct CNS infections, all isotypes can be sustained within the CNS 13. Retention thus appears dependent upon the host's response to the virus itself and not a generalized response to CNS inflammation.
Transient ASC detection within spleen is a common denominator in infections confined to the CNS as well as systemic, respiratory and intestinal infections 2, 13, 15, 31. However, in contrast to systemic infections, in which a decline in splenic ASC numbers is counterbalanced by an increase in BM ASC, few ASC ever appear in the BM following JHMV infection. Preferential CNS retention, concomitant with poor BM accumulation, appears unique compared to localized infections of lung or intestinal tract 31, 33 and may reflect the primary role of intrathecal Ab in controlling viral persistence 13, 15, 16. A prominent intrathecal Ab response to JHMV infection is supported by increased CFS to serum ratios of virus‐specific antibodies in rats and mice 36. In extreme cases, some animals only exhibited viral Ab in CSF and not in the corresponding serum 36. Extremely limited ASC localization to BM suggests that ASC are diverted from a default trafficking pathway into the BM characteristic of peripheral infections 2. Our data suggest that strong induction of both CXCL9 and CXCL10 within the CNS during acute infection mediates recruitment of CXCR3‐expressing plasmablasts. Continued expression of these chemokines as virus is cleared may sustain ongoing recruitment as ASC continue to emerge from lymphoid organs. The elevated level of BAFF mRNA within the CNS supports induction of factors necessary for prolonged ASC survival 54. Furthermore, in contrast to ASC apoptosis within inflammatory lesions following virus clearance from peripheral tissues, few apoptotic cells are detected in the CNS 55, and ASC are retained in apparent preference to BM.
Together these data support the concept that the CNS provides a milieu for sustained ASC viability and Ab secretion. The results are consistent with intrathecal Ab synthesis in a variety of human pathological conditions as well as rodent models 11, 13, 15, 18–20. Importantly, the paucity of ASC in BM suggests that differentiating ASC are diverted and migrate toward virus‐induced chemokines within the CNS. CXCL9 and CXCL10, early chemokines induced by JHMV 43, are maintained during peak ASC recruitment and support a role for CXCR3 ligands in ASC recruitment. CXCL12 and BAFF, which are constitutively expressed at high levels in BM, may be candidates facilitating ASC retention in the CNS 6. The recent discovery of BAFF up‐regulation in CNS plaques associated with MS and in mice with experimental autoimmune encephalomyelitis 44, 45 provides a mechanism for the promotion of ASC survival during CNS autoimmune disease. Similarly, following CNS infection, BAFF was up‐regulated concurrently with viral clearance, and mRNA expression was maintained within the CNS throughout persistence. These data suggest that maintenance of ASC during chronic viral infections associated with limited ongoing inflammation may involve components similar to chronic inflammatory diseases, albeit with limited contribution of viral Ag.
Materials and methods
Mice and virus
Male 6‐wk‐old C57BL/6 and BALB/c mice, sero‐negative for mouse hepatitis virus, were obtained from the National Cancer Institute (Frederick, MD). Mice were maintained in a specific pathogen‐free environment and infected within 1 wk of arrival. CNS infection was induced by intracerebral inoculation of the neutralizing mAb‐selected 2.2v‐1 variant of JHMV 56. Mice were injected in the left hemisphere with 30 μL containing 250 PFU JHMV diluted in endotoxin‐free Dulbecco's PBS. For peripheral infections mice were injected i.p. with 1 × 106 PFU JHMV as previously described 55. All procedures were performed in compliance with Keck School of Medicine Institutional Animal Care and Use Committee approved protocols.
Serum and neutralizing Ab
Serum anti‐JHMV Ab levels were determined by ELISA as previously described 16. Briefly, 2‐fold serum dilutions (pooled from three to five individuals per time point) were adsorbed overnight at 4°C onto 96‐well plates coated with a serum‐free supernatant derived from JHMV‐infected DBT cells (6 × 103 PFU/well) diluted in 0.1 M sodium phosphate buffer (pH 9). Bound Ab was detected with biotinylated goat anti‐mouse IgG and IgM (Jackson ImmunoResearch Laboratories, West Grove, PA), avidin peroxidase and 1 mg/mL 2,2′‐azinobis‐(3‐ethylbenzothiazoline‐6‐sulphonic acid) (ABTS; Roche Diagnostics Corp., Indianapolis, IN) in PBS containing H2O2. Optical densities were determined at 405 nm in a Microplate Autoreader (Bio‐Tek Instruments, Winooski, VT) and titers calculated as reciprocals of the highest dilution that exceeded three standard deviations over the mean negative control. Neutralizing titers were determined as previously described using sera pooled from three to five individuals per time point 16. Briefly, heat‐inactivated sera (56°C for 30 min) were incubated with 200 PFU JHMV in 96‐well plates for 90 min at 37°C. Following addition of DBT cells (9 × 104 cells/well), cultures were incubated for 48 h at 37°C. Neutralization titers represent the highest dilution that prevented virus‐induced cytopathic effects.
Isolation of mononuclear cells and flow cytometry
Groups of four to six JHMV‐infected mice were perfused with PBS at various times after infection. Single‐cell suspensions from spleen or BM were depleted of red blood cells by Tris‐based ammonium chloride treatment and resuspended in RPMI medium containing 25 mM Hepes (pH 7.2) supplemented with 10% FCS. CNS mononuclear cells were isolated as previously described 16. Briefly, brains and spinal cords were mechanically dissociated and the homogenate adjusted to 30% Percoll (Amersham Pharmacia Biotech, Piscataway, NJ). A 70% Percoll underlay was added prior to centrifugation at 800 × g for 25 min at 4°C. Mononuclear cells were recovered from the 30%/70% interface, diluted in serum‐free RPMI medium containing 25 mM Hepes, collected by centrifugation and washed three times. For ELISPOT analysis cells were resuspended in RPMI containing 25 mM Hepes and 10% FCS. For flow cytometry, cells were blocked with 10% mouse sera and rat anti‐mouse CD16/32 (2.4G2; BD PharMingen, San Diego, CA) prior to staining with labeled mAb specific for CD45 (30‐F11), CD19 (1D3), B220 (RA3–6B2), CD138 (281–2) and MHC class II (2G9). All mAb were obtained from BD PharMingen. Cells were analyzed with a FACSCalibur flow cytometer (Becton Dickinson, Mountain View, CA) using Cellquest Pro software. Forward and side scatter signals were used to establish a gate excluding dead cells and tissue debris, and at least 50 000 events were analyzed.
Ab‐secreting cells (ASC)
Virus‐specific ASC were detected by ELISPOT as previously described 16. Briefly, Millipore Multiscreen 96‐well plates (Bedford, MA) were coated by overnight incubation at 4°C with supernatant collected from JHMV‐infected DBT cells propagated under serum‐free conditions (∼5 × 105 PFU/well). Control wells were coated with virus‐free medium. Mononuclear cells isolated from spleen, BM and CNS were added at various dilutions and incubated for 4 h at 37°C. Single‐cell spots were visualized by sequential addition of biotinylated goat anti‐mouse IgG or IgM (Jackson ImmunoResearch Laboratories), avidin peroxidase (Sigma, St. Louis, MO) and 3,3′‐Diaminobenzidine substrate (Sigma). Color was developed at room temperature for 5–10 min. The wells were then washed with water, air dried and the spots counted using a dissecting microscope. ELISPOT images were taken by using Zeiss KS Elispot imaging system.
Histology
Mice were perfused with PBS, and brains and spinal cords were removed and snap frozen. Distribution of IgG and IgM ASC was determined by staining acetone‐fixed sections with biotinylated goat anti‐mouse IgG or IgM (Jackson ImmunoResearch Laboratories). Plasma cell distribution was examined using anti‐CD138 (BD PharMingen). Slides were counterstained with hematoxylin and scored in a blinded manner.
PCR Analysis
RNA was extracted from half brains of individual mice (three per group) by homogenization in guanidine isothiocyanate. Samples were sheared prior to centrifugation in 5.7 M cesium chloride (100 000 × g for 18 h) and treated with DNAase I (Roche) to avoid DNA contamination. AMV reverse transcriptase (Promega, Madison, WI) was used for reverse transcription. Real‐time PCR was performed with SYBR Green PCR reagents (Applied Biosystems, Foster City, CA) using the DNA Engine OPTICON (MJ Research, Waltham, MA) with the following primers: BAFF, forward 5′‐CAGGAACAGACGCGCTTTC‐3′ and reverse 5′‐GTTGAGAATGGCGGCATCC‐3′; CXCL13, forward 5′‐CATAGATCGGATTCAAGTTACGCC‐3′ and reverse 5′‐TCTTGGTCCAGATCACAACTTCA‐3′; CXCL10, forward 5′GACGGTCCGCTGCAACTG‐3′ and reverse 5′‐GCTTCCCTATGGCCCTCATT‐3′; CXCL9, forward 5′‐TGCACGATGCTCCTGCA‐3′ and reverse 5′‐AGGTCTTTGAGGGATTTGTAGTGG‐3′; ubiquitin, forward 5′‐TGGCTATTAATTATTCGGTCTGCAT‐3′ and reverse 5′‐GCAAGTGGCTAGAGTGCAGAGTAA‐3′. Levels were normalized relative to ubiquitin mRNA and converted to a linearized value using the formula of 1.8 – Ct gene x) × 105.
Acknowledgements
This work was supported by National Institutes of Health Grants AI 47249 and NS 18146. The authors wish to express their gratitude to Wenqiang Wei, Emmanuel Dimacali and Ernesto Barron for excellent technical assistance with the immunohistochemistry, real‐time PCR and photography.
Footnotes
WILEY‐VCH
WILEY‐VCH
WILEY‐VCH
WILEY‐VCH
WILEY‐VCH
WILEY‐VCH
WILEY‐VCH
References
- 1. Gray, D., A role for antigen in the maintenance of immunological memory. Nat. Rev. Immunol. 2002. 2: 60–65. [DOI] [PubMed] [Google Scholar]
- 2. Slifka, M. K. and Ahmed, R., Long‐lived plasma cells: a mechanism for maintaining persistent antibody production. Curr. Opin. Immunol. 1998. 10: 252–258. [DOI] [PubMed] [Google Scholar]
- 3. Manser, T., Textbook germinal centers? J. Immunol. 2004. 172: 3369–3375. [DOI] [PubMed] [Google Scholar]
- 4. Shapiro‐Shelef, M. and Calame, K., Regulation of plasma‐cell development. Nat. Rev. Immunol. 2005. 5: 230–242. [DOI] [PubMed] [Google Scholar]
- 5. Kunkel, E. J. and Butcher, E. C., Plasma‐cell homing. Nat. Rev. Immunol. 2003. 3: 822–829. [DOI] [PubMed] [Google Scholar]
- 6. Manz, R. A., Hauser, A. E., Hiepe, F. and Radbruch, A., Maintenance of serum antibody levels. Annu. Rev. Immunol. 2005. 23: 367–386. [DOI] [PubMed] [Google Scholar]
- 7. Hickey, W. F., Lassmann, H. and Cross, A. H., Lymphocyte entry and the initiation of inflammation in the central nervous system. In Keane, R. W. and Hickey, W. F. (Eds.) Immunology of the nervous system. Oxford University Press 1997, pp 200–225. [Google Scholar]
- 8. Sedgwick, J. C. and Hickey, W. F., Antigen presentation in the central nervous system. In Kearney, J. and Hickey, W. F. (Eds.) Immunology of the nervous system. Oxford University Press 1997, pp 364–418. [Google Scholar]
- 9. Barker, C. F. and Billingham, R. E., Immunologically privileged sites. Adv. Immunol. 1977. 25: 1–54. [PubMed] [Google Scholar]
- 10. Fabry, Z., Raine, C. S. and Hart, M. N., Nervous tissue as an immune compartment: the dialect of the immune response in the CNS. Immunol. Today 1994. 15: 218–224. [DOI] [PubMed] [Google Scholar]
- 11. Knopf, P. M., Harling‐Berg, C. J., Cserr, H. F., Basu, D., Sirulnick, E. J., Nolan, S. C., Park, J. T. et al., Antigen‐dependent intrathecal antibody synthesis in the normal rat brain: tissue entry and local retention of antigen‐specific B cells. J. Immunol. 1998. 161: 692–701. [PubMed] [Google Scholar]
- 12. Williamson, J. S., Sykes, K. C. and Stohlman, S. A., Characterization of brain‐infiltrating mononuclear cells during infection with mouse hepatitis virus strain JHM. J. Neuroimmunol. 1991. 32: 199–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tyor, W. R. and Griffin, D. E., Virus specificity and isotype expression of intraparenchymal antibody‐secreting cells during Sindbis virus encephalitis in mice. J. Neuroimmunol. 1993. 48: 37–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Vandvik, B., Norrby, E., Nordal, H. J. and Degre, M., Oligoclonal measles virus‐specific IgG antibodies isolated from cerebrospinal fluids, brain extracts, and sera from patients with subacute sclerosing panencephalitis and multiple sclerosis. Scand. J. Immunol. 1976. 5: 979–992. [DOI] [PubMed] [Google Scholar]
- 15. Tyor, W. R., Wesselingh, S., Levine, B., Griffin, D. E. and Irani, D. N., Long term intraparenchymal Ig secretion after acute viral encephalitis in mice. J. Immunol. 1992. 149: 4016–4020. [PubMed] [Google Scholar]
- 16. Tschen, S. I., Bergmann, C. C., Ramakrishna, C., Morales, S., Atkinson, R. and Stohlman, S. A., Recruitment kinetics and composition of antibody‐secreting cells within the central nervous system following viral encephalomyelitis. J. Immunol. 2002. 168: 2922–2929. [DOI] [PubMed] [Google Scholar]
- 17. Persson, M. A., Laurenzi, M. A. and Vranjesevic, D., Extended repertoire of specific antibodies in CSF of patients with subacute sclerosing panencephalitis compared to those with multiple sclerosis: anti‐bacterial antibodies are also increased. J. Neuroimmunol. 1989. 22: 135–142. [DOI] [PubMed] [Google Scholar]
- 18. Cross, A. H., Trotter, J. L. and Lyons, J.‐A., B cells and antibodies in CNS demyelinating disease. J. Neuroimmunol. 2001. 112: 1–14. [DOI] [PubMed] [Google Scholar]
- 19. Mehta, P., Kane, A. and Thormar, H., Quantitation of measles virus‐specific immunoglobulins in serum, CSF, and brain extract from patients with subactue sclerosing panencephalitis. J. Immunol. 1977. 118: 2254–2261. [PubMed] [Google Scholar]
- 20. Pohl‐Koppe, A., Kaiser, R., Meulen, V. T. and Liebert, U. G., Antibody reactivity to individual structural proteins of measles virus in the CSF of SSPE and MS patients. Clin. Diagn. Virol. 1995. 4: 135–147. [DOI] [PubMed] [Google Scholar]
- 21. Corcione, A., Casazza, S., Ferretti, E., Giunti, D., Zappia, E., Pistorio, A., Gambini, C. et al., Recapitulation of B cell differentiation in the central nervous system of patients with multiple sclerosis. Proc. Natl. Acad. Sci. USA 2004. 101: 11064–11069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ritchie, A. M., Gilden, D. H., Williamson, R. A., Burgoon, M. P., Yu, X., Helm, K., Corboy, J. R. and Owens, G. P., Comparative analysis of the CD19+ and CD138+ cell antibody repertoires in the cerebrospinal fluid of patients with multiple sclerosis. J. Immunol. 2004. 173: 649–656. [DOI] [PubMed] [Google Scholar]
- 23. Gerritse, K., Deen, C., Fasbender, M., Ravid, R., Boersma, W. and Claassen, E., The involvement of specific anti myelin basic protein antibody‐forming cells in multiple sclerosis immunopathology. J. Neuroimmunol. 1994. 49: 153–159. [DOI] [PubMed] [Google Scholar]
- 24. Genain, C. P., Cannella, B., Hauser, S. L. and Raine, C. S., Identification of autoantibodies associated with myelin damage in multiple sclerosis. Nat. Med. 1999. 5: 170–175. [DOI] [PubMed] [Google Scholar]
- 25. Mokhtarian, F., Huan, C. M., Roman, C. and Raine, C. S., Semliki Forest virus‐induced demyelination and remyelination‐‐involvement of B cells and anti‐myelin antibodies. J. Neuroimmunol. 2003. 137: 19–31. [DOI] [PubMed] [Google Scholar]
- 26. Diamond, M. S., Shrestha, B., Marri, A., Mahan, D. and Engle, M., B cells and antibody play critical roles in the immediate defense of disseminated infection by West Nile encephalitis virus. J. Virol. 2003. 77: 2578–2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Marten, N. W., Stohlman, S. A. and Bergmann, C. C., MHV infection of the CNS: mechanisms of immune‐mediated control. Viral Immunol. 2001. 14: 1–18. [DOI] [PubMed] [Google Scholar]
- 28. Lin, M. T., Hinton, D. R., Marten, N. W., Bergmann, C. C. and Stohlman, S. A., Antibody prevents virus reactivation within the central nervous system. J. Immunol. 1999. 162: 7358–7368. [PubMed] [Google Scholar]
- 29. Ramakrishna, C., Bergmann, C. C., Atkinson, R. and Stohlman, S. A., Control of central nervous system viral persistence by neutralizing antibody. J. Virol. 2003. 77: 4670–4678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Marten, N. W., Stohlman, S. A., Zhou, J. and Bergmann, C. C., Kinetics of virus‐specific CD8+ ‐T‐cell expansion and trafficking following central nervous system infection. J. Virol. 2003. 77: 2775–2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sangster, M., Hyland, L., Sealy, R. and Coleclough, C., Distinctive kinetics of the antibody‐forming cell response to Sendai virus infection of mice in different anatomical compartments. Virology 1995. 207: 287–291. [DOI] [PubMed] [Google Scholar]
- 32. Sangster, M. Y., Topham, D. J., D'Costa, S., Cardin, R. D., Marion, T. N., Myers, L. K. and Doherty, P. C., Analysis of the virus‐specific and nonspecific B cell response to a persistent B‐lymphotropic gammaherpesvirus. J. Immunol. 2000. 164: 1820–1828. [DOI] [PubMed] [Google Scholar]
- 33. Youngman, K. R., Franco, M. A., Kuklin, N. A., Rott, L. S., Butcher, E. C. and Greenberg, H. B., Correlation of tissue distribution, developmental phenotype, and intestinal homing receptor expression of antigen‐specific B cells during the murine anti‐rotavirus immune response. J. Immunol. 2002. 168: 2173–2181. [DOI] [PubMed] [Google Scholar]
- 34. Erlich, S. S., Fleming, J. O., Stohlman, S. A. and Weiner, L. P., Experimental neuropathology of chronic demyelination induced by a JHM virus variant (DS). Arch. Neurol. 1987. 44: 839–842. [DOI] [PubMed] [Google Scholar]
- 35. Fleming, J. O., Ting, J. Y., Stohlman, S. A. and Weiner, L. P., Improvements in obtaining and characterizing mouse cerebrospinal fluid. Application to mouse hepatitis virus‐induced encephalomyelitis. J. Neuroimmunol. 1983. 4: 129–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dorries, R., Watanabe, R., Wege, H. and ter Meulen, V., Analysis of the intrathecal humoral immune response in Brown Norway (BN) rats, infected with the murine coronavirus JHM. J. Neuroimmunol. 1987. 14: 305–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dorries, R., Watanabe, R., Wege, H. and ter Meulen, V., Murine coronavirus‐induced encephalomyelitis in rats: analysis of immunoglobulins and virus‐specific antibodies in serum and cerebrospinal fluid. J. Neuroimmunol. 1986. 12: 131–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Holt, P. G., Sedgwick, J. D., Stewart, G. A., O'Leary, C. and Krska, K., ELISA plaque assay for the detection of antibody secreting cells: observations on the nature of the solid phase and on variations in plaque diameter. J. Immunol. Methods 1984. 74: 1–7. [DOI] [PubMed] [Google Scholar]
- 39. Marten, N. W., Stohlman, S. A. and Bergmann, C. C., Role of viral persistence in retaining CD8(+) T cells within the central nervous system. J. Virol. 2000. 74: 7903–7910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hauser, A. E., Debes, G. F., Arce, S., Cassese, G., Hamann, A., Radbruch, A. and Manz, R. A., Chemotactic responsiveness toward ligands for CXCR3 and CXCR4 is regulated on plasma blasts during the time course of a memory immune response. J. Immunol. 2002. 169: 1277–1282. [DOI] [PubMed] [Google Scholar]
- 41. Mackay, F., Schneider, P., Rennert, P. and Browning, J., BAFF AND APRIL: a tutorial on B cell survival. Annu. Rev. Immunol. 2003. 21: 231–264. [DOI] [PubMed] [Google Scholar]
- 42. Uccelli, A., Aloisi, F. and Pistoia, V., Unveiling the enigma of the CNS as a B‐cell fostering environment. Trends Immunol. 2005. 26: 254–259. [DOI] [PubMed] [Google Scholar]
- 43. Lane, T. E., Asensio, V. C., Yu, N., Paoletti, A. D., Campbell, I. L. and Buchmeier, M. J., Dynamic regulation of α‐ and β‐ chemokine expression in the central nervous system during mouse hepatitis virus‐induced demyelinating disease. J. Immunol. 1998. 160: 970–978. [PubMed] [Google Scholar]
- 44. Serafini, B., Rosicarelli, B., Magliozzi, R., Stigliano, E. and Aloisi, F., Detection of ectopic B‐cell follicles with germinal centers in the meninges of patients with secondary progressive multiple sclerosis. Brain Pathol. 2004. 14: 164–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Krumbholz, M., Theil, D., Derfuss, T., Rosenwald, A., Schrader, F., Monoranu, C. M., Kalled, S. L. et al., BAFF is produced by astrocytes and up‐regulated in multiple sclerosis lesions and primary central nervous system lymphoma. J. Exp. Med. 2005. 201: 195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chen, A., Khanna, N., Stohlman, S. A. and Bergmann, C. C., Bystander T cells recruited during viral induced encephalomyelitis accumulate transiently and do not exert effector function. J. Virol. 2005. 79: 4700–4708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Manz, R. A., Thiel, A. and Radbruch, A., Lifetime of plasma cells in the bone marrow. Nature 1997. 388: 133–134. [DOI] [PubMed] [Google Scholar]
- 48. Zinkernagel, R. M., Immunology taught by viruses. Science 1996. 271: 173–178. [DOI] [PubMed] [Google Scholar]
- 49. Slifka, M. K. and Ahmed, R., Long‐term humoral immunity against viruses: revisiting the issue of plasma cell longevity. Trends Microbiol. 1996. 4: 394–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Villar, L. M., Sadaba, M. C., Roldan, E., Masjuan, J., Gonzalez‐Porque, P., Villarrubia, N., Espino, M. et al., Intrathecal synthesis of oligoclonal IgM against myelin lipids predicts an aggressive disease course in MS. J. Clin. Invest. 2005. 115: 187–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Pedersen, N. S., Kam‐Hansen, S., Link, H. and Mavra, M., Specificity of immunoglobulins synthesized within the central nervous system in neurosyphilis. Acta Pathol. Microbiol. Immunol. Scand. [C] 1982. 90: 97–104. [DOI] [PubMed] [Google Scholar]
- 52. Sindic, C. J., Monteyne, P. and Laterre, E. C., The intrathecal synthesis of virus‐specific oligoclonal IgG in multiple sclerosis. J. Neuroimmunol. 1994. 54: 75–80. [DOI] [PubMed] [Google Scholar]
- 53. Magliozzi, R., Columba‐Cabezas, S., Serafini, B. and Aloisi, F., Intracerebral expression of CXCL13 and BAFF is accompanied by formation of lymphoid follicle‐like structures in the meninges of mice with relapsing experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2004. 148: 11–23. [DOI] [PubMed] [Google Scholar]
- 54. Avery, D. T., Kalled, S. L., Ellyard, J. I., Ambrose, C., Bixler, S. A., Thien, M., Brink, R. et al., BAFF selectively enhances the survival of plasmablasts generated from human memory B cells. J. Clin. Invest. 2003. 112: 286–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Stohlman, S. A., Bergmann, C. C., Lin, M. T., Cua, D. J. and Hinton, D. R., CTL effector function within the central nervous system requires CD4+ T cells. J. Immunol. 1998. 160: 2896–2904. [PubMed] [Google Scholar]
- 56. Fleming, J. O., Trousdale, M. D., el‐Zaatari, F. A., Stohlman, S. A. and Weiner, L. P., Pathogenicity of antigenic variants of murine coronavirus JHM selected with monoclonal antibodies. J. Virol. 1986. 58: 869–875. [DOI] [PMC free article] [PubMed] [Google Scholar]