

Abstract
Poly‐L‐lysine is one of the biocompatible polymers having amino and carboxyl groups in its structure. This attractive feature of poly‐L‐lysine makes it very convenient for bioactive molecule attachment. This study details the preparation of poly‐L‐lysine‐based pencil graphite electrodes (PLL/PGEs) and use of the coated electrodes for direct ultrasensitive DNA hybridization detection. In the first part of this study, poly‐L‐lysine coated electrodes were prepared using L‐lysine as the monomer by cyclic voltammetry (CV) with different cyclic scans. The effect of these cyclic scans during the electropolymerization was investigated. Coated electrodes were characterized by cyclic voltammetry, electrochemical impedance spectroscopy (EIS) and scanning electron microscopy (SEM). Then, one‐pot preparation of poly‐L‐lysine composites with graphene (GN) and multi‐walled carbon nanotubes (MWCNTs) onto the pencil graphite electrodes were achieved. Electrochemical responses of these 3 electrodes were compared. After all, electrochemical DNA hybridization was performed using the poly‐L‐lysine‐based electrodes prepared at optimum polymerization condition. The PLL/PGE coated electrode presented a good linear response in the target concentration range of 1.0×10−13 to 1.0×10−6 with a detection limit of 2.25×10−14 using differential pulse voltammetry as the detection method. We believe that poly‐L‐lysine‐based surfaces will be useful for further clinical applications.
Keywords: L-lysine; electrochemical polymerization; poly-L-lysine, DNA detection

1. Introduction
Deoxyribonucleic acid (DNA) is one of the most important molecules of our life process. It is the responsible molecule for carrying the genetic information in cells. The detection of specific DNA sequences, DNA damage and DNA‐anticancer drug interactions have been the hot topics of all times since they are directly related to human health 1, 2, 3, 4. They have found many applications in medicine, pharmacy, molecular biology, environmental monitoring and food safety. In particular, detection of specific DNA sequences is great importance for the diagnosis of diseases, genomic sequencing and pathogen identification 5, 6, 7, 8. There are several ways of detecting DNA and monitoring DNA‐related studies, such as polymerase chain reaction (PCR)‐based, spectroscopy‐based or biosensor‐based 9, 10, 11, 12, 13. Among these techniques, DNA biosensors constitute an important place due to their advantage of recognizing base pairs efficiently in shorter time values compared to laborious classical methods. Biosensors that use electrochemical transducers have been used commonly in order to construct sensitive, selective, inexpensive, rapid, simple and miniaturized biosensing platforms 14, 15, 16, 17, 18, 19.
Various approaches can be used for electrochemical DNA hybridization detection using direct electrochemistry of DNA or a redox reporter, amplifications with nanoparticles and polymer coated electrodes. In this context, Prof. Joseph Wang's group have made significant contributions to the electrochemical DNA biosensors. For example, Wang et al. have used electrochemical stripping detection of colloidal gold tag for DNA hybridization at single‐use thick film carbon electrodes 20. In their study, magnetic bead‐linked oligonucleotide probe and target hybridization have been followed by binding of streptavidin‐coated metal nanoparticles to the captured DNA and dissolution of gold. Another remarkable study has been performed using a ternary monolayer involving hexanedithiol (HDT) co‐immobilized with a thiolated capture probe on gold surfaces by Campuzano et al. 21. 6‐mercapto‐1‐hexanol (MCH) has been used as the diluent of the ternary monolayer. This ternary monolayer has presented a quantification of target DNA down to 7 pM and 17 pM in untreated serum and urine samples, respectively. Then, Kuralay et al. have used this ternary monolayer approach at screen‐printed gold electrodes (Au/SPEs) 22. A quantification of target DNA down to 25 pM and 100 pM in untreated serum and urine samples have been obtained, respectively.
Electroactive polymers (or electropolymerized films) can be effectively used in various areas, such as nanotechnology and biotechnology. They have attracted great attention in DNA sensing because of their availability of forming roboust, uniform and biocompatible surfaces having charges or their own functional groups which make them very convenient for biomolecule immobilization 23, 24, 25, 26. Besides these advantages, many of them have good conductivities, porous structures and high electroactive surface areas. Their easy preparation is commonly rely on the electrochemical oxidation of their monomers in the presence of a dopant molecule 27, 28, 29, 30. Gu et al. have fabricated polyaniline/polyacrylate (PANI/PAA)‐modified boron‐doped diamond electrode for electrochemical impedance sensing of DNA hydridization 31. The electrode has presented a detection limit of 2×10−8 M. Another DNA biosensor with polyethylene glycolylated (PEGylated) PANI has been constructed by Hui et al. 32. The biosensor has exhibited a response to breast cancer susceptibility gene (BRCA1) with a linear range from 0.01 pM to 1 nM. Prof. Joseph Wang's group have electropolymerized polypyrrole (PPy) in the presence of nucleic acid probes as the dopant molecules. The probes have served as the sole counter anion during the electropolymerization and maintained their hybridization activity 33. Jiang and Wang have also demonstrated a genoelectronic route for discriminating between short oligonucleotides and chromosomal DNA. This route has been depended on the electropolymeric growth of PPy. The response of system has found proportional to the concentration of the oligonucleotide dopant 34. Peptide modified poly(3,4‐ethylenedioxythiophene) (PEDOT) coated glassy carbon electrode (GCE) has been used for BRCA1 detection and a detection limit of 0.03 fM has been found. The biosensor has exhibited a satisfying accuracy and low fouling in 5 % (v/v) human plasma 35.
In the present work, we describe the preparation of poly‐L‐lysine coated pencil graphite electrodes (PLL/PGEs) and use of them for ultrasensitive DNA hybridization detection. PLL is an attractive surface for biosensing applications due to its biocompability and positively/negatively charged groups 36, 37. For example, Shan et al. have been used graphene sheets functionalized with poly‐L‐lysine for the detection of hydrogen peroxide (H2O2) 38. There have been few attempts on DNA sensing based on PLL. Jiang et al. have been presented an electrochemical biosensor with poly‐L‐lysine/single walled carbon nanotubes for EIS detection of PAT gene fragment and PCR amplification of NOS gene 39. Carboxylic group‐functionalized single‐walled carbon nanotubes (SWNTs) have been dropped onto the carbon paste electrode (CPE) and then PLL films have been electropolymerized by cyclic voltammetry (CV). The detection limit has been 3.1×10−13 M using electrochemical impedance spectroscopy (EIS). Another DNA biosensor based on functionalized graphene oxide and poly‐L‐lysine modified glassy carbon electrode (GCE) has been constructed by Sun et al. for the detection of tlh gene sequence related to vibrio parahaemolyticus 40. PLL films have been electropolymerized by cyclic voltammetry (CV) and functionalized graphene modification has been performed subsequently by dropping onto the coated electrode. Hybridization event has been monitored with methylene blue. The detection limit has been found as 1.69×10−13 M. A genesensor to detect severe acute respiratory syndrome (SARS) virus has been prepared on poly‐L‐ysine modified screen‐printed carbon electrodes (SPCEs) 41. Enzyme catalyzed hydrolysis of the 3‐indoxyl phosphate (3‐IP) to indigo has been monitored by cyclic voltammetry. In the second part, the target has been labeled using an Au (I) complex. The detection limits have been found as 8 pM and 0.5 nM, respectively. In our study, we present a simple protocol based on the electropolymerization of L‐lysine onto disposable pencil graphite electrodes (PLL/PGEs) by cyclic voltammetry. PLL/PGEs were prepared using various cyclic scans (5, 10 and 15 cycles) in order to investigate the effect of polymeric film thickness on the electrochemical response of the coated electrode. The response of the coated surface was also compared with the response of the uncoated electrode for double‐stranded DNA (dsDNA) detection. Improved results were obtained with the coated one. Thus, we used the coated electrode in DNA hybridization sensing in 1 : 1 diluted serum samples. According to our knowledge, this is the first to fabricate PLL coated pencil graphite surfaces and to present their application as DNA biosensor in serum samples. The hybridization step didn't require any indicator step and oxidation signals of guanine (G) was used, directly. The prepared electrode presented a sensitive and selective response towards label‐free DNA detection. Besides this, herein we fabricated poly‐L‐lysine/graphene (PLL/GN) and poly‐L‐lysine/multi‐walled carbon nanotubes (PLL/MWCNTs) nanocomposites coated PGEs. The electroactive polymer/nanomaterials‐based nanocomposites coated surfaces have attracted great attention for different applications 4. This is the first to demonstrate one‐pot, rapid, robust and easy fabrication of polyamino acids/nanomaterials composites coated disposable surfaces in the literature. Electrochemical polymerization was performed with L‐lysine monomer in the presence of graphene or multi‐walled carbon nanotubes onto the PGEs. Electrochemical characterization was performed and also the response of the coated electrodes for DNA biosensing was compared.
2. Experimental
2.1. Reagents and Instrumentation
L‐Lysine (98 %), multi‐walled carbon nanotubes, potassium ferricyanide (K3[Fe(CN)6]), potassium ferrocyanide (K4[Fe(CN)6]), sodium chloride (NaCl), potassium chloride (KCl) and human serum (from human male AB plasma) were obtained from Sigma‐Aldrich. Graphene powder (99.9 %) was from Aldrich. Other chemicals were in analytical reagent grade and they were supplied from Sigma and Merck. All the experiments were carried out at room temperature.
Double‐stranded DNA (dsDNA) were purchased from Serva (Germany). Oligonucleotides (ODNs) used in the experiments were purchased from TIB MOLBIOL (Germany). Their sequences are listed below:
- Thiol linked probe:
5′‐SH‐AATACCACATCATCCATATA
- Target:
5′‐TATATGGATGATGTGGTATT
- Mismatch (MM):
5′‐TATGTGGATGATGTGGTATT
- Noncomplementary (NC):
5′‐AATACCTGTATTCCTCGCCTGTC
All DNA and ODN solutions were prepared with ultra pure water. Then, stock solutions of dsDNA (1000 mg L−1) and ODNs were prepared with Tris‐EDTA buffer (10 mM Tris‐HCl, 1 mM EDTA, TE, pH 8.0). More diluted solutions of dsDNA (100 mg L−1) and probe DNA (50 mM) were prepared with 50 mM acetate buffer containing 20 mM NaCl (ABS, pH 4.8). Target, MM and NC ODNs were prepared in diluted serum sample (diluted with acetate buffer containing 20 mM NaCl in 1 : 1 ratio).
Electrochemical studies were carried out with AUTOLAB‐PGSTAT 204 system supported with a NOVA software package (Metrohm, The Netherlands). A disposable pencil graphite as a working electrode (PGE), an Ag/AgCl as a reference electrode (BASi, USA) and a Pt wire (BASi, USA) as a counter electrode were used. The connector of the PGE (0.5 mm HB Tombow) was a Tombow pencil. Surface morphologies of the electrodes were examined using scanning electron microscopy (SEM) with Hitachi SU 1510 (Japan).
2.2. Electropolymerization Step
Preparation of electropolymerization solutions: 10 mM L‐lysine solution was prepared in 50 mM pH 7.4 phosphate buffer containing 0.1 M NaCl. The solution was prepared freshly and deoxygenated with nitrogen gas (99.99 %, BOS, Turkey) before each experiment. For the experiments that were carried out to fabricate poly‐L‐lysine/graphene coated PGE (PLL/GN/PGE), 10 mM L‐lysine and 1 mg mL−1 graphene were prepared in 50 mM pH 7.4 phosphate buffer containing 0.1 M NaCl. For the experiments that were carried out to fabricate poly‐L‐lysine/multi‐walled carbon nanotubes coated PGE (PLL/MWCNTs/PGE), 10 mM L‐lysine and 1 mg mL−1 multi‐walled carbon nanotubes were prepared in 50 mM pH 7.4 phosphate buffer containing 0.1 M NaCl. The solutions were prepared freshly and deoxygenated with nitrogen gas (99.99 %, BOS, Turkey) before each experiment.
Modification of pencil graphite electrode (PGE) with poly‐L‐lysine (PLL): The coated electrode was prepared with cyclic voltammetry (CV) with various cyclic scans (5, 10 and 15 cycles) at a potential range of −0.50 V to +1.80 V vs. Ag/AgCl at a scan rate of 100 mV s−1. Same procedure was used for the fabrication of (PLL/GN/PGE) and PLL/MWCNTs/PGE.
2.3. Electrochemical Characterization of Poly‐L‐lysine Coated Pencil Graphite Electrodes (PLL/PGEs)
Cyclic voltammetric characterization was performed at a potential range of −0.30 V to 0.60 V vs. Ag/AgCl at a scan rate of 100 mV s−1 in 0.1 M KCl containing 5 mM Fe(CN)6 3−/4−. Electrochemical impedance spectroscopy (EIS) studies were recorded at +0.40 V under an AC amplitude of 10 mV and a frequency range from 10−2 to 105 Hz.
2.4. Oligonucleotide (ODN) Immobilization onto the Pencil Graphite Electrode
Probe DNA immobilization was carried out by dipping the pencil graphite electrode (PGE) into the probe DNA solution (50 mM) for 30 min. For DNA hybridization or selectivity experiments (with NC or MM), probe DNA immobilized PLL/PGEs were dipped into the target, NC or MM solutions for 15 min. The concentrations for these solutions were 1 mM.
Differential pulse voltammetric responses of the ODNs immobilized PLL/PGEs were recorded between +0.60 V to +1.40 V vs. Ag/AgCl at a step potential of 50 mV and a scan rate of 10 mV s−1.
3. Results and Discussion
In the first part of this study, we aim to prepare poly‐L‐lysine coated disposable pencil graphite electrodes (PLL/PGEs) and investigate the electrochemical behaviors of these PLL/PGEs. PLL coated surfaces have attracted great attention due to their positively charged amino groups (−NH2) and negatively charged carboxyl groups (−COOH), versatility and ease of electrochemical polymerization 39, 40, 41. In addition, Poly‐L‐lysine/graphene (PLL/GN) and poly‐L‐lysine/multi‐walled carbon nanotubes (PLL/MWCNTs) nanocomposites coated PGEs were prepared in one‐pot synthesis and introduced for the first time in the literature. Electrochemical polymerization was performed with L‐lysine monomer in the presence of graphene or multi‐walled carbon nanotubes onto the PGEs. Electrochemical responses of the coated electrodes were compared. In the second part, the use of the poly‐L‐lysine‐based surfaces were examined as a DNA biosensor. The schematic presentation of the work is given as Figure 1.
Figure 1.

Schematic presentation of the work.
Different polymeric surfaces with various cyclic scans were prepared in order to fabricate poly‐L‐lysine coated electrodes. Figure 2A to C present the electropolymerization curves for poly‐L‐lysine prepared using cyclic voltammetry technique with 5, 10 and 15 cycles, respectively. In Figure 2D, all polymerization curves were overlaid. As seen from these figures, electropolymerization was accomplished in the same current scale with various cyclic scans presenting the unique and reproducible fabrication of poly‐L‐lysine onto the disposable graphite surfaces.
Figure 2.
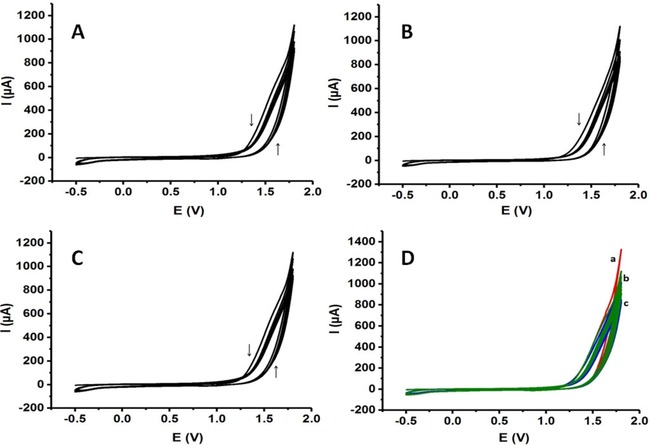
Electropolymerization curves of poly‐L‐lysine obtained with cyclic voltammetry: (A) 5 cyclic scans, (B) 10 cyclic scans, (C) 15 cyclic scans, (D) 3 conditions together (Scan rate: 100 mV s−1).
Electrochemical characterization of the coated surfaces are important to emphasize the advantage of the coated surfaces compared to uncoated electrodes in means of improved electrochemical properties such as electroactive surface areas and reversibility. Electrochemical behaviors of the PLL coated electrodes first characterized with cyclic voltammetry in 0.1 M KCl solution containing 5 mM Fe(CN)6 3−/4. Figure 3A‐a to d illustrate the cyclic voltammograms of PLL/PGE prepared with 5 cycles, PLL/PGE prepared with 10 cycles, PLL/PGE prepared with 15 cycles and uncoated PGE, respectively.
Figure 3.
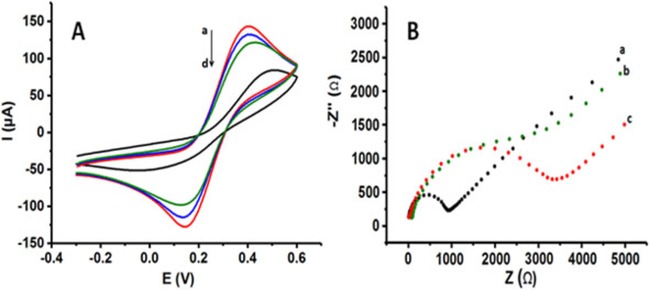
(A) CVs of a) PLL/PGE prepared with 5 cyclic scans, b) PLL/PGE prepared with 10 cyclic scans, c) PLL/PGE prepared with 15 cyclic scans, d) uncoated PGE, (B) Nyquist diagrams of a) PLL/PGE prepared with 5 cyclic scans, b) PLL/PGE prepared with 10 cyclic scans, c) uncoated PGE in 0.1 M KCl solution containing 5 mM Fe(CN)6 3−/4 redox probe (Scan rate for CV: 100 mV s−1, Frequency range for EIS: 10−2 to 105 Hz at +0.40 V).
PLL coated electrodes exhibited an enhanced electrochemical properties compared to the uncoated one. In each polymerization condition, higher oxidation and reduction peaks for Fe(CN)6 3−/4− redox probe were observed. In addition, more reversible peaks were obtained. Well‐defined peaks due to the oxidation and reduction of redox probe were obtained with the coating of graphite surface with PLL. SI Table 1 summarizes the anodic/cathodic peak potentials (Epa/Epc), the anodic/cathodic peak currents (Ipa/Ipc). Best electrochemical response was achieved with thinner PLL layer onto the PGE. We evaluate the increasing polymerization cyclic scans as the indicator of the polymeric film thickness 42. Thus, with the increasing polymeric film thickness, reversibility and the oxidation/reduction peak currents of redox probe were decreased proportionally. However, this decrease was not a dramatic one. Our results proved the advantage of integration of PLL layer with pencil graphite surface. 3.5‐fold increase in the oxidation of the redox probe and 4.5‐fold increase in the reduction of the redox probe were found with the coated electrode prepared with 5 cycles of electropolymerization compared to the uncoated electrode.
Electrochemical impedance spectroscopy was also used for the characterization and comparison of the surfaces. Figure 3B‐a to c show the impedance spectra of PLL/PGE prepared with 5 cyclic scans (Figure 3B‐a), PLL/PGE prepared with 10 cyclic scans (Figure 3B‐b) and uncoated PGE (Figure 3B‐c) in 0.1 M KCl containing 5 mM Fe(CN)6 3−/4− redox probe. Charge‐transfer resistance (Rct) values (the diameter of the semicircle in the Nyquist plot) were found as 1077 Ω, 2790 Ω and 3470 Ω, respectively. With the PLL/PGE prepared with 5 cyclic scans, the electron transfer was faster than the other electrodes. The PLL/PGE prepared with 10 cycles also gave better Rct value than the uncoated electrode. These results were in a good agreement with the results obtained with CV proving the electropolymerized layer onto the PGE. Polymeric layer faciliates electron transfer between the solution and the electrode interface.
Surface characterization of PLL/PGEs and uncoated PGE was carried out with scanning electron microscopy (SEM). Figure 4A presents the SEM images of uncoated (bare) PGE (Figure 4A) at magnitudes of 50 mm, 10 mm and 5 mm. The graphite layers were clearly seen in these images in parallel to the literature 43, 44. SEM images of PLL/PGE prepared with 5 cyclic scans (Figure 4B) and PLL/PGE prepared with 10 cyclic scans (Figure 4C) were also given at the same magnitudes. It is clear from the images that after the modification of the electrode surfaces with PLL, the morphology of the new surfaces changed. The polymeric film formation on the graphite layers of the bare electrode was obvious in each cases compared to the uncoated PGE.
Figure 4.

SEM images of (A) uncoated (bare) electrode, (B) PLL/PGE prepared with 5 cyclic scans, (C) PLL/PGE prepared with 10 cyclic scans (Magnitudes: 50 mm, 10 mm and 5 mm).
The fabrication of electroactive polymer/nanomaterials composites on various electrode materials have attracted great attention for different applications. In addition, their one‐pot synthesis onto electrodes are among very important topics 4, 46. After the optimization studies for PLL/PGEs, we demonstrated the first example of one‐pot, rapid, robust and easy fabrication of polyamino acid/nanomaterials composites coated disposable surfaces in the literature. Electrochemical polymerization was performed with L‐lysine monomer in the presence of graphene (GN) or multi‐walled carbon nanotubes (MWCNTs) onto the PGEs using 5 cyclic scans. Figure 5a‐d present the electrochemical behaviors of PLL/GN/PGE, PLL/MWCNTs/PGE, PLL/PGE and bare PGE in 0.1 M KCl solution containing 5 mM Fe(CN)6 3−/4, respectively. As seen from these cyclic voltammetric behaviors all coated electrodes exhibited very good electrochemical responses compared to the bare electrode. In addition, SI Table 2 summarizes the anodic/cathodic peak potentials (Epa/Epc) and the anodic/cathodic peak currents (Ipa/Ipc). According to these results, the reversibility of the electrodes were better in the presence of graphene or multi‐walled carbon nanotubes as expected. The best performance was obtained with the PLL/GN/PGE. The results emphasized the catalytical effect of nanomaterials on the electrode material. However, the peak currents which was related to the effective areas were almost the same for 3 electrodes 4.
Figure 5.
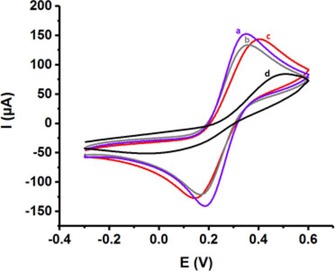
(A) CVs of a) PLL/GN/PGE, b) PLL/MWCNTs/PGE, c) PLL/PGE, d) uncoated PGE in 0.1 M KCl solution containing 5 mM Fe(CN)6 3−/4 redox probe (Scan rate: 100 mV s−1).
After the electropolymerization and characterization studies, we present the application of PLL‐based pencil graphite electrodes for DNA biosensing. The outer layer of the electrode serves as a positively/negatively charged surface in order to bind DNA electrostatically. The electropolymerization condition was kept as 5 cycles of polymerization since the PLL coated electrode showed the best electrochemical response at this working condition. Firstly, the advantage of the coated electrodes was presented by using them in 100 mg L−1 dsDNA solution. In Figure 6A‐a to d, differential pulse voltammograms (DPVs) of PLL/GN/PGE, PLL/MWCNTs/PGE, PPL/PGE and uncoated PGE in dsDNA solution prepared in acetate buffer (pH 4.8) were overlaid. As seen from these DPVs, electroactive 4 DNA bases (guanine (G), adenine (A), thymine (T), cytosine (C), respectively) were able to be differentiated in a good seperation in the same current scale by using PLL/GN/PGE, PLL/MWCNTs/PGE and PLL/PGE. PLL/GN/PGE, PLL/MWCNTs/PGE and PLL/PGE exhibited almost the same behavior. The separations of adenine (A) and thymine (T) were slightly more sensitive by using the PLL/GN/PGE and there was also a catalytic effect in parallel to our cyclic voltammetric results with the PLL/GN/PGE and PLL/MWCNTs/PGE. However, there was no clear seperation of the DNA bases using uncoated PGE and the peak currents were in a lower current values compared to the ones obtained with PLL‐based electrodes. These results signified the use of PLL‐based surfaces for DNA biosensing. Then, the usability of PLL/PGE electrode was presented in human serum which was diluted with acetate buffer (pH 4.8) in 1 : 1 ratio. The experiments for %100 serum sample for the differentiation of DNA bases was also performed. However, the sensitivity was poor in these results. Figure 6B‐a illustrates the DPV of PLL/PGE in 100 mg L−1 dsDNA solution prepared in acetate buffer (pH 4.8) and Figure 6B‐b illustrates the DPV of PLL/PGE in 100 mg L−1 dsDNA solution prepared in serum. The results were in a good aggrement to prove the usability of PLL/PGE in real samples. Thus, the PLL/PGEs can be good candidates for further clinical applications.
Figure 6.

DPVs of (A) a) PPL/GN/PGE, b) PLL/MWCNTs/PGE, c) PLL/PGE, d) uncoated PGE in 100 mg L−1 dsDNA solution prepared in 50 mM acetate buffer (pH 4.8), (B) a) PLL/PGE in 100 mg L−1 dsDNA solution prepared in 50 mM acetate buffer (pH 4.8), b) PLL/PGE in 100 mg L−1 dsDNA solution prepared in human serum diluted with 50 mM acetate buffer (in 1 : 1 ratio) (Step potential: 50 mV, Scan rate: 10 mV s−1).
In the last part of the study, DNA hybridization was investigated with PLL/PGEs and PLL/GN/PGEs. The effect of probe DNA immobilization time was examined by immersing the PLL/PGEs into the probe DNA solution for 15 min (SI Figure 1a), 30 min (SI Figure 1b) and 60 min (SI Figure 1c). Adenine oxidation peak was used to monitor the changes using differential pulse voltammetry. As seen from these results, the oxidation peak gave the best response at 30 min of probe immobilization. Thus, 30 min probe DNA immobilization time was used in further studies.
DNA hybridization studies were performed by incubating the probe DNA immobilized PLL/PGEs and PLL/GN/PGEs with different concentrations of target DNA prepared in diluted serum samples for 15 min. Guanine oxidation peak was used to monitor the hybridization in this case. Figure 7A presents the DPVs (from a–n) related to the response of the electrode for target DNA concentration. The response of the PLL/PGE in diluted serum was also added in Figure 7A (black line). The calibration graph was given as Figure 7B. This novel PLL/PGE presented a good linear response to target DNA in the concentration range of 1.0×10−13 to 1.0×10−6 M (R2=0.9930) (n=3). The detection limit was calculated as 2.25×10−14 M. This value was a good one compared to the literature, in particular to the ones that could be only used in hybridization buffer 31, 32, 40, 41, 47, 48. The comparison of the performance of the electrode with various polymer coated electrodes was given in SI Table 3.
Figure 7.

(A) DPVs for guanine oxidation peaks after hybridization of probe with different concentrations of target (at PLL/PGE): 1.0×10−13 M (a), 5.0×10−13 M (b), 1.0×10−12 M (c), 5.0×10−12 M (d), 1.0×10−11 M (e), 5.0×10−11 M (f), 1.0×10−10 M (g), 5.0×10−10 M (h), 1.0×10−9 M (i), 5.0×10−9 M (j), 1.0×10−8 M (k), 5.0×10−8 M (l), 2.5×10−7 M (m), 1.0×10−6 M (n) (black line: DPV of PLL/PGE in diluted serum), (B) Calibration graph (n=3), (C) DPVs of a) probe DNA immobilized PLL/PGE, b) probe DNA immobilized PLL/PGE after 15 min of target DNA interaction, c) probe DNA immobilized PLL/PGE after 15 min of NC DNA interaction, d) probe DNA immobilized PLL/PGE after 15 min of target/MM DNA (in 1 : 1 ratio) interaction.
Selectivity of the biosensor was also tested using noncomplemantary (NC) DNA and mismatch (MM) DNA. Figure 7C illustrates the DPVs of probe DNA immobilized PLL/PGE (Figure 7C‐a), probe DNA immobilized PLL/PGE after 15 min of target DNA interaction (Figure 7C‐b), probe DNA immobilized PLL/PGE after 15 min of NC DNA interaction (Figure 7D‐c), probe DNA immobilized PLL/PGE after 15 min of target/MM DNA (in 1 : 1 ratio) interaction (Figure 7C‐d). As seen from these results, the biosensor is selective to target DNA and there was no response in the case of NC DNA. The reproducility of the PLL/PGEs were investigated using 1.0×10−13 M target DNA. The RSD was found as 5.6 % (n=5) giving a promising result for routine clinical applications. The average guanine oxidation peak current value was 1.40±0.06 mA obtained with the probe DNA immobilized PLL/PGE after 15 min of target DNA interaction (Figure 7C‐b) and it was 1.60±0.06 with probe DNA immobilized PLL/PGE after 15 min of target/MM DNA (in 1 : 1 ratio) interaction (Figure 7C‐d) (n=3).
Then, the novel PLL/GN/PGEs were used for DNA hybridization in order to compare the PLL‐based disposable pencil graphite electrodes. Figure 8A presents the DPVs (from a–g) related to the response of the electrode for target DNA concentration. The calibration graph was given in Figure 8B. This novel PLL/GN/PGE also presented a good linear response to target DNA in the concentration range of 1.0×10−13 to 1.0×10−6 M (R2=0.9970) (n=3). The detection limit was calculated as 1.30×10−14 M. This value was slightly better than the one we obtained with PLL/PGE. Selectivity of the biosensor was tested in the same way (SI Figure 2). The biosensor presented selectivity to its target and other unwanted components such as NC and MM sequences. The reproducibility of the PLL/GN/PGEs were investigated using 1.0×10−13 M target DNA. The RSD was found as 4.8 % (n=5) giving a promising result for routine clinical applications.
Figure 8.

(A) DPVs for guanine oxidation peaks after hybridization of probe with different concentrations of target (at PLL/GN/PGE): 1.0×10−13 M (a), 1.0×10−12 M (b), 5.0×10−12 M (c), 5.0×10−11 M (d), 1.0×10−10 M (e), 5.0×10−8 M (f), 1.0×10−6 M (g), (B) Calibration graph (n=3),
4. Conclusions
In the current work, poly‐L‐lysine‐based disposable pencil graphite surfaces were fabricated by cyclic voltammetry in a fast, low‐cost and practical way. The electrode prepared with 5 cyclic scans gave the best electrochemical response in terms of reversibility and higher oxidation/reduction peak currents for the redox probe (Fe(CN)6 3−/4−). Furthermore, for the first time in the literature the fabrication of one‐pot synthesis of polyamino acid/nanomaterials coated electrodes was achieved. Cyclic voltammetric behaviors of all PLL‐based electrodes exhibited very good electrochemical responses compared to the bare electrode. The results also emphasized the catalytical effect of nanomaterials on the electrode material. The PLL‐based surfaces exhibited a good discrimination of electroactive DNA bases in a high sensitivity, while uncoated PGE gave poor results. In the last part of the study, DNA hybridization was performed with PLL/PGE and PLL/GN/PGE. The electrodes presented a good linear response, high sensitivity and selectivity and good reproducibility for target DNA prepared in diluted serum samples (50 % serum). The hybridization step didn't require any indicator step in the study and the prepared electrodes presented a sensitive and selective response towards label‐free DNA detection. Therefore, disposable PLL‐based PGEs can be used for further routine clinical applications.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
F. Kuralay acknowledges Turkish Academy of Sciences (TÜBA) as an associate member and TÜBA‐GEBİP Program.
F. Kuralay, N. Dükar, Y. Bayramlı, Electroanalysis 2018, 30, 1556.
References
- 1. Wang J., Anal. Chim. Acta 2002, 469, 63. [Google Scholar]
- 2. Agüí L., Yáñez-Sedeño P., Pingarrón J. M., Anal. Chim. Acta 2008, 622, 11. [DOI] [PubMed] [Google Scholar]
- 3. Palecek E., Fojta M., Tomschik M., Wang J., Biosens. Bioelectron. 1998, 13, 621. [DOI] [PubMed] [Google Scholar]
- 4. Kuralay F., Erdem A., Analyst 2015, 140, 2876. [DOI] [PubMed] [Google Scholar]
- 5. Qaddare S. H., Salim A., Biosens. Bioelectron. 2017, 89, 773. [DOI] [PubMed] [Google Scholar]
- 6. Palecek E., Electroanalysis 1996, 8, 7. [Google Scholar]
- 7. Wang J., Nucleic Acid Res. 2000, 28, 3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Llopis X., Perez B., Del Valle M., Alegret S., Tr. Anal. Chem. 2005, 24, 826. [Google Scholar]
- 9. Miotke L., Lau B. T., Rumma R. T., Ji H. P., Anal. Chem. 2014, 86, 2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Liu B., Bazan G. C., Chem. Mater. 2004, 16, 4467. [Google Scholar]
- 11. Cao Y. C., Jin R., Mirkin C. A., Science 2002, 297, 1536. [DOI] [PubMed] [Google Scholar]
- 12. Kuralay F., Erdem A., Abacı S., Özyörük H., Yıldız A., Anal. Chim. Acta 2009, 643, 83. [DOI] [PubMed] [Google Scholar]
- 13. Erdem A., Ozsoz M., Electroanalysis 2002, 14, 965. [Google Scholar]
- 14. Campuzano S., Pedrero M., García J. L., García E., García P., Pingarrón J. M., Anal. Bioanal. Chem. 2011, 399, 2413. [DOI] [PubMed] [Google Scholar]
- 15. Drummond T. G., Hill M. G., Barton J. K., Nature Biotechnol. 2003, 21, 1192. [DOI] [PubMed] [Google Scholar]
- 16. Widaningrum T., Widyastuti E., Pratiwi F. W., Faidoh Fatimah A. I., Rijiravanich P., Somasundrum M., Surareungchai W., Talanta 2017, 167, 14. [DOI] [PubMed] [Google Scholar]
- 17. Gooding J., Electroanalysis 2002, 14, 1149. [Google Scholar]
- 18. Zhou M., Guo S., ChemCatChem 2015, 7, 744. [Google Scholar]
- 19. Bo X., Zhou M., Guo L., Biosens. Bioelectron. 2017, 89, 167. [DOI] [PubMed] [Google Scholar]
- 20. Wang J., Xu D., Kawde A.-N., Polsky R., Anal. Chem. 2001, 73, 5576. [DOI] [PubMed] [Google Scholar]
- 21. Campuzano S., Kuralay F., Lobo-Castañón M. Jesús, Bartošík M., Vyavahare K., Paleček E., Haake D. A., Wang J., Biosens. Bioelectron. 2011, 26, 3577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kuralay F., Campuzano S., Haake D. A., Wang J., Talanta 2011, 85, 1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lee T.-Y., Shim Y.-B., Anal. Chem. 2001, 73, 5629. [DOI] [PubMed] [Google Scholar]
- 24. Evtugyn G., Hianik T., Tr. Anal. Chem. 2016, 79, 168. [Google Scholar]
- 25. Kuralay F., Erdem A., Abacı S., Özyörük H., Yıldız A., Electrochem. Commun. 2009, 11, 1242. [Google Scholar]
- 26. Gerard M., Chaubey A., Malhotra B. D., Biosens. Bioelectron. 2002, 17, 345. [DOI] [PubMed] [Google Scholar]
- 27. Reisberg S., Piro B., Noël V., Pham M. C., Anal. Chem. 2005, 77, 3351. [DOI] [PubMed] [Google Scholar]
- 28. Thompson L. A., Kowalik J., Josowicz M., Janata J., J. Am. Chem. Soc. 2003, 125, 324. [DOI] [PubMed] [Google Scholar]
- 29. Spain E., Kojima R., Kaner R. B., Wallace G. G., O'Grady J., Lacey K., Barry T., Keyes T. E., Forster R. J., Biosens. Bioelectron. 2011, 26, 2613. [DOI] [PubMed] [Google Scholar]
- 30. Zhu B., Travas-Sejdic J., Analyst 2018, 7, 687. [DOI] [PubMed] [Google Scholar]
- 31. Gu H., Su X. di, Loh K. Ping, J. Phys. Chem. B 2005, 109, 13611. [DOI] [PubMed] [Google Scholar]
- 32. Hui N., Sun X., Niu S., Luo X., ACS Appl. Mater. Interfaces 2017, 9, 2914. [DOI] [PubMed] [Google Scholar]
- 33. Wang J., Jiang M., Fortes A., Mukherjee B., Anal. Chim. Acta 1999, 402, 7. [Google Scholar]
- 34. Jiang M., Wang J., J. Electroanal. Chem. 2001, 500, 584. [Google Scholar]
- 35. Wang G., Han R., Su X., Li Y., Xu G., Luo X., Biosens. Bioelectron. 2017, 92, 396. [DOI] [PubMed] [Google Scholar]
- 36. Jalit Y., Rodríguez M. C., Rubianes M. D., Bollo S., Rivas G. A., Electroanalysis 2008, 20, 1623. [Google Scholar]
- 37. Gasnier A., Gonzalez-Dominguez J. M., Anson-Casaos A., Hernandez-Ferrer J., Pedano M. L., Rubianes M. D., Martinez M. T., Rivas G., Electroanalysis 2014, 26, 1676. [Google Scholar]
- 38. Shan C., Yang H., Han D., Zhang Q., Ivaska A., Niu L., Langmuir 2009, 25, 12030. [DOI] [PubMed] [Google Scholar]
- 39. Jiang C., Yang T., Jiao K., Gao H., Electrochim. Acta 2008, 53, 2917. [Google Scholar]
- 40. Sun W., Zhang Y., Ju X., Li G., Gao H., Sun Z., Anal. Chim. Acta 2012, 752, 39. [DOI] [PubMed] [Google Scholar]
- 41. Diaz-Gonzalez M., Escosura-Muniz A. De la, Gonzalez-Garcia M. B., Costa-Garcia A., Biosens. Bioelectron. 2008, 23, 1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kuralay F., Özyörük H., Yıldız A., Sens. Actuators B 2005, 109, 194. [Google Scholar]
- 43. Erdem A., Congur G., Electroanal. 2018, 30, 3067. [Google Scholar]
- 44. Kuralay F., Demirci S., Kiristi M., Oksuz L., Oksuz A. U., Coll. Surfaces B: Biointerfaces 2014, 123, 825. [DOI] [PubMed] [Google Scholar]
- 45. Zhou M., Zhai Y., Dong S., Anal. Chem. 2009, 81, 5603. [DOI] [PubMed] [Google Scholar]
- 46. Muti M., Kuralay F., Erdem A., Coll. Surfaces B: Biointerfaces 2012, 91, 77. [DOI] [PubMed] [Google Scholar]
- 47. Du M., Yang T., Li X., Jiao K., Talanta 2012, 88, 439. [DOI] [PubMed] [Google Scholar]
- 48. Zhou N., Yang T., Jiang C., Du M., Jiao K., Talanta 2009, 77, 1021. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary