

Abstract
Although CD8 T cells are key players in neuroinflammation, little is known about their trafficking cues into the central nervous system (CNS). We used a murine model of CNS autoimmunity to define the molecules involved in cytotoxic CD8 T‐cell migration into the CNS. Using a panel of mAbs, we here show that the α4β1‐integrin is essential for CD8 T‐cell interaction with CNS endothelium. We also investigated which α4β1‐integrin ligands expressed by endothelial cells are implicated. The blockade of VCAM‐1 did not protect against autoimmune encephalomyelitis, and only partly decreased the CD8+ T‐cell infiltration into the CNS. In addition, inhibition of junctional adhesion molecule‐B expressed by CNS endothelial cells also decreases CD8 T‐cell infiltration. CD8 T cells may use additional and possibly unidentified adhesion molecules to gain access to the CNS.
Keywords: α4β1‐Integrin, CD8 T cell, Junctional adhesion molecule‐B, Migration, Neuroinflammation
Introduction
The central nervous system (CNS) is considered as a unique immune‐privileged environment allowing a basal immune surveillance under physiological conditions, and restraining potentially deleterious inflammatory reactions in disease states 1, 2. A tight control of the trafficking of immune cells toward the CNS is a major parameter contributing to this immune‐privileged status 3. Indeed, circulating immune cells have to cross protective barriers using a complex multistep cascade that involves distinct trafficking cues both at the surface of the CNS endothelial cells and of immune cells 4.
The current knowledge regarding T‐cell migration into the CNS derives mainly from CD4 T cells whereas little is known for CD8 T cells. Under inflammatory conditions, either due to autoimmune responses or infection, the inflamed blood‐brain barrier (BBB) endothelium upregulates the expression of adhesion molecules (selectins and cell adhesion molecules of the immunoglobulin superfamily). Several surface molecules expressed by CD4 T cells, such as P‐selectin glycoprotein ligand‐1, αLβ2, α4β1, CD6, or α6β1, regulate sequential steps for transmigration that include tethering, rolling, capture, firm adhesion, and finally diapedesis of blood‐borne circulating T cells across the endothelial layer 4. Antigen recognition in the subarachnoidal spaces then allow T cells to migrate through the glia limitans superficialis into the CNS parenchyma, leading to clinical disease 5. In this scenario, a pivotal role for the interaction between the α4β1‐integrin heterodimer expressed by activated T helper 1 CD4 cells, and vascular cell adhesion protein 1 (VCAM‐1), its main ligand on BBB endothelial cells, has been demonstrated 6, 7, 8, 9, 10. However, as Th17 CD4 cells were shown to infiltrate the brain in the absence of α4‐integrin in an αLβ2‐dependent manner 10, 11, α4‐independent routes to the CNS exist for other crucial T‐cell subsets.
Although cumulative evidence points to a key role for CD8 T cells in several inflammatory CNS disorders such as MS, autoimmune encephalitis, or immune reconstitution inflammatory syndrome (IRIS) affecting the CNS 12, 13, 14, little is known about trafficking of CD8 T cells into the CNS. Interaction between P‐selectin glycoprotein ligand‐1 and P‐selectin contributes to the recruitment of human CD8 T cells in brain vessels 15. CD8 T‐cell migration is not affected by blocking interactions between αLβ2/ICAM‐1, ALCAM/CD6, PECAM‐1/PECAM‐1, or CCL2/CCR2, while all of them were previously shown to partake in the recruitment of other immune cell populations to the CNS 16, 17. The conflicting results about neutralization of α4‐integrin in EAE and in coronavirus‐induced encephalitis, restricting or not CD8 T‐cell infiltration of the CNS, suggest that these cells may use alternative and possibly unidentified adhesion molecules to gain access to the CNS, raising the potential for selective control of their trafficking into the brain 10, 17, 18.
Using a murine model of CNS autoimmunity, the aim of our study was to define the role of α4‐integrins in the CD8 T‐cell migration across the BBB during neuroinflammation, and to specify which of the heterodimers containing the α4‐integrin subunit, namely α4β1 and α4β7, is implicated.
Results
α4β1‐Integrin blockade protects CamK‐HA mice from developing CD8 T‐cell‐mediated encephalomyelitis
CamK‐HA mice selectively express the Haemagglutinin (HA) of the Influenza virus in neurons under the control of the calmodulin kinase promoter. These mice, but not control littermates, exhibited severe encephalomyelitis characterized by weight loss, neurological signs, and death in 40–50% of recipients after the adoptive transfer of 5 × 106 in vitro generated cytotoxic HA‐specific CD8 T cells. Diabetes insipidus caused by the CD8‐mediated destruction of arginine‐vasopressin hypothalamic neurons was a prominent clinical manifestation in the surviving CamK‐HA recipients 19. Endogenous CD4 and CD8 T cells also infiltrated the CNS of CamK‐HA mice developing encephalomyelitis (Supporting Information Fig. 1A–B). However, because CD4 T cell‐depleted recipients underwent weight loss and diabetes insipidus, endogenous CD4 T cells are not required for disease development (Supporting Information Fig. 1C–D).
To delineate in this model the role of α4‐integrin in CD8 T‐cell migration to the CNS, we first showed that in vitro differentiated cytotoxic HA‐specific CD8 T cells express both α4‐ and β7‐integrin subunits, and the α4β7‐integrin heterodimer (Fig. 1A). Because there is no mAb against the α4β1‐integrin heterodimer, we used an in vitro binding assay to indirectly show that in vitro differentiated cytotoxic HA‐specific CD8 T cells express the α4β1‐integrin heterodimer. While HA‐specific CD8 T cells untreated or treated with a control IgG bind to recombinant ICAM‐1 and VCAM‐1, an anti‐α4‐integrin mAb completely inhibited binding to VCAM‐1 (one‐way ANOVA, p < 0.00001), but did not affect binding to ICAM‐1 (Fig. 1B). As VCAM‐1 is the main ligand of the α4β1‐integrin heterodimer, this suggests that HA‐specific CD8 T cells express the functional α4β1‐integrin heterodimer.
Figure 1.
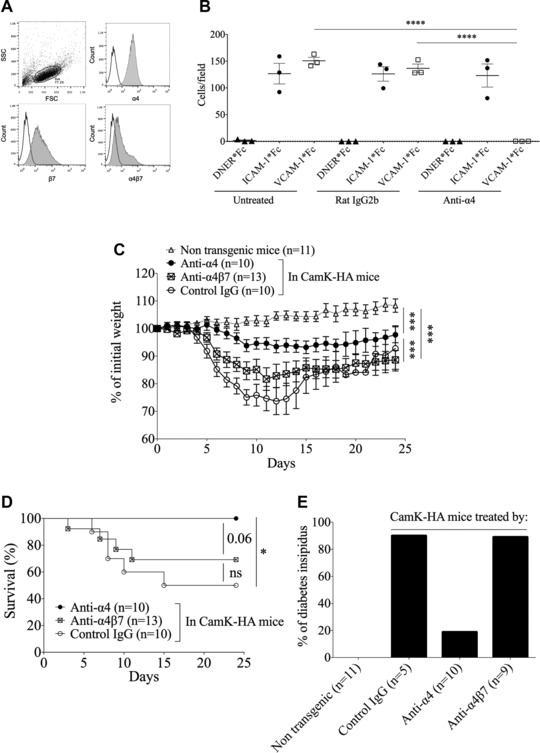
α4β1‐integrin blockade protects CamK‐HA mice from developing CD8 T‐cell‐mediated encephalomyelitis. (A) FACS analysis of α4‐integrin subunit, β7‐integrin subunit, and α4β7‐integrin heterodimer expression on in vitro differentiated cytotoxic HA‐specific CD8 T cells (one experiment representative of three experiments of in vitro differentiation of HA‐specific cytotoxic CD8 T cells).(B) In vitro binding assay of cytotoxic HA‐specific CD8 T cells to recombinant ICAM‐1 or VCAM‐1 IgG and a control protein (DNER‐IgG), after pretreatment with isotype control rat IgG2b or anti‐α4‐integrin‐IgG. Pooled data from three independent experiments, each point representing the mean of triplicates. One‐way ANOVA ****p < 0.00001. (C and D) Adoptive transfer at day 0 of 5 × 106 cytotoxic HA‐specific CD8 T cells in nontransgenic mice or in CamK‐HA mice treated at day 0, 4, and 8 by an anti‐α4, an anti‐α4β7, or a control IgG mAb. Impact on the weight (C) and on survival (D) of the recipient mice is shown. (C) One‐way ANOVA ***p < 0.0001. (D) Log‐rank (Mantel–Cox) test *p < 0.01. (E) Incidence of diabetes insipidus among survivors at day 24 after the adoptive transfer of 5 × 106 cytotoxic HA‐specific CD8 T cells in nontransgenic mice or in CamK‐HA mice treated at day 0, 4, and 8 by an anti‐α4, an anti‐α4β7, or a control IgG mAb. Diabetes insipidus is defined using dipsticks by a urine‐specific gravity ≤ 1010 on three consecutive days. (C), (D), and (E) are pooled data from three independent experiments, each involving three to five mice per group. Data represent mean ± SEM.
We then assessed whether treatment of CamK‐HA mice with anti‐α4‐integrin mAb could protect against CD8 T‐cell‐mediated encephalomyelitis. Therefore, we treated CamK‐HA recipients with an anti‐α4‐integrin or a control IgG at day 0, 4, and 8 after the adoptive transfer of 5 × 106 cytotoxic HA‐specific CD8 T cells. Compared to recipients receiving the control IgG, CamK‐HA recipients treated by anti‐α4‐integrin mAb lost less weight (one‐way ANOVA, p < 0.0001) and showed no death (0/10 versus 5/10, Log‐rank test, p = 0.006). In addition, survivors experienced less diabetes insipidus (2/10 versus 4/5) (Fig. 1C–E). Since α4‐integrin is a subunit shared by both α4β1‐ and α4β7‐integrin heterodimers, we then investigated which one is involved in CD8 T‐cell migration into the CNS. Currently no mAb is available that specifically recognizes the α4β1‐integrin heterodimer, while the DATK32 mAb recognizes and blocks specifically the α4β7‐integrin heterodimer 7. We therefore treated CamK‐HA recipients with this anti‐α4β7‐integrin mAb. Recipients underwent the same clinical phenotype as mice receiving the control mAb. Indirectly, this strongly suggests that the α4β1‐integrin is the essential heterodimer for activated CD8 T‐cell migration into the CNS in our model. However, α4‐integrin blockade did not fully abrogate the neurological signs induced by HA‐specific CD8 T‐cell transfer in CamK‐HA mice as significant weight loss developed when compared to control littermates (Fig. 1C), and central diabetes insipidus was detected in 20% of treated recipients.
Blockade of the α4β1‐integrin reduces CD8 T‐cell infiltration and microglial activation in the CNS
We next investigated the impact of α4β1‐integrin blockade on CNS inflammatory infiltrate composition and tissue damage. Eight days after the adoptive transfer of 5 × 106 cytotoxic HA‐specific CD8 T cells, CamK‐HA recipients treated with an anti‐α4‐integrin mAb or a control IgG were sacrificed and their CNS‐infiltrating cells were analyzed by flow cytometry. A drastic reduction in the number of CD45high Thy1.2+ T cells infiltrating the CNS was observed in recipients treated with the anti‐α4‐integrin mAb (1333 ± 263 versus 35 452 ± 5748 cells/CNS, respectively, Mann–Whitney test, p < 0.0001; Fig. 2A–B). Ex vivo analysis of splenocytes showed that the anti‐α4‐integrin mAb did not deplete CD8 T cells in the periphery, and even promoted the accumulation of transferred cells in the secondary lymphoid organs (Supporting Information Fig. 2A–B). Moreover, ex vivo analyses and restimulation with the cognate HA peptide showed that the anti‐α4‐integrin mAb also did not affect activation of transferred CD8 T cells collected from spleen and lymph nodes (Supporting Information Fig. 2C–F). In the CNS, we observed a decrease in microglial activation, evaluated by MHC class II expression on CD45int CD11b+ cells, in recipients treated by the anti‐α4‐integrin mAb (MFI for MHC class II 589 ± 127 versus 6825 ± 1172, Mann–Whitney test, p = 0.0002; Fig. 2C–D). Histological analysis also corroborated the marked reduction in the parenchymal CD8 T‐cell infiltration in recipients treated by the anti‐α4‐integrin mAb‐treated animals (Mann–Whitney test, p = 0.0002; Fig. 2E–G), while some meningeal CD8 T cells could still be detected. Antagonization of α4β1‐integrin, therefore inhibits CD8 T‐cell infiltration and microglial activation in the CNS parenchyma of recipient CamK‐HA mice.
Figure 2.
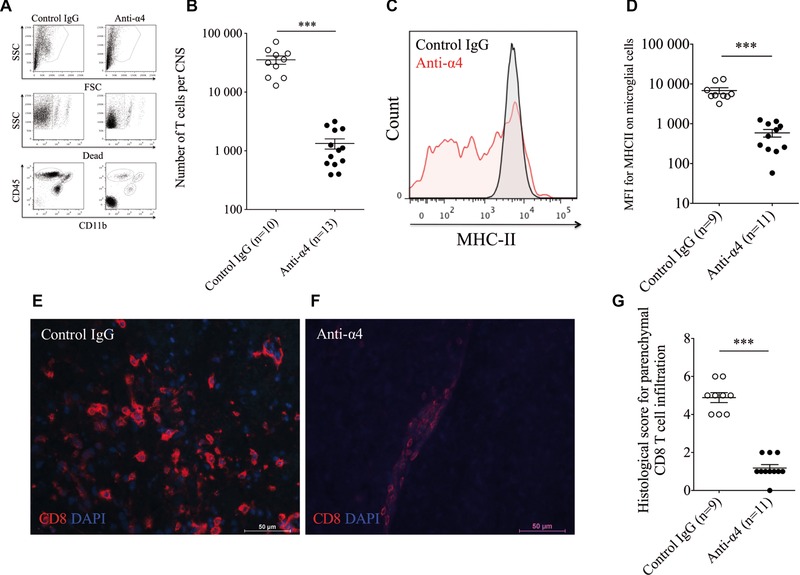
Blockade of the α4β1‐integrin reduces T‐cell infiltration and microglial activation in the CNS. (A–D) FACS analysis of CNS‐infiltrating cells at day 8 after adoptive transfer of 5 × 106 cytotoxic HA‐specific CD8 T cells in CamK‐HA mice treated at day 0 and 4 by an anti‐α4 or a control IgG mAb. (A) Illustration of the gating strategy. (B) Absolute number of CD45high Thy1.2+ T cells infiltrating the CNS. Each dot shows an individual animal. Mann–Whitney test ***p < 0.0001. (C) Overlay of MHC class II expression on CD45int CD11b+ cells in mice treated by a control IgG (black line), or anti‐α4 mAb (red line). One representative out of three independent experiments is shown. (D) The MFI of MHC class II expression on CD45int CD11b+ microglial cells is shown. Mann–Whitney test ***p = 0.0002. Each dot shows an individual animal. (B and D) are pooled data from three independent experiments, each involving three to five mice per group. Data represent mean ± SEM. (E–G) Brain CD8 T‐cell infiltration at day 8 after adoptive transfer of 5 × 106 cytotoxic HA‐specific CD8 T cells in CamK‐HA mice treated at day 0 and 4 by a control IgG (E) or an anti‐α4‐integrin mAb (F; bar = 50 μm). (G) Histological scoring for parenchymal CD8 T‐cell infiltration. Pool of three independent experiments, each involving three to five mice per group. Mann–Whitney test ***p = 0.0002. Data represent mean ± SEM.
VCAM‐1 blockade retains the clinical phenotype and only partly decreases CD8 T cell CNS infiltration
To further investigate how α4β1‐integrin contributes to CD8 T cell trafficking to the CNS, we investigated which ligand expressed on brain microvascular endothelial cells may interact with α4β1‐integrin. First, we blocked in vivo VCAM‐1, the major ligand for α4β1‐integrin 20, using two distinct mAbs. As both mAbs provided identical results (Fig. 3A), the two groups were pooled in Figure 3B and C. CamK‐HA recipients treated by anti‐VCAM‐1 mAb lost more weight (one‐way ANOVA, p < 0.0001) and developed diabetes insipidus more frequently (9/11 versus 1/6, chi‐square test with Yates’ correction, p = 0.036) compared to mice treated by the anti‐α4‐integrin mAb (Fig. 3A–C). Clinical outcomes (weight loss, diabetes insipidus, and death) were not significantly different between recipient mice treated with the anti‐VCAM‐1 mAbs or with the control IgG.
Figure 3.
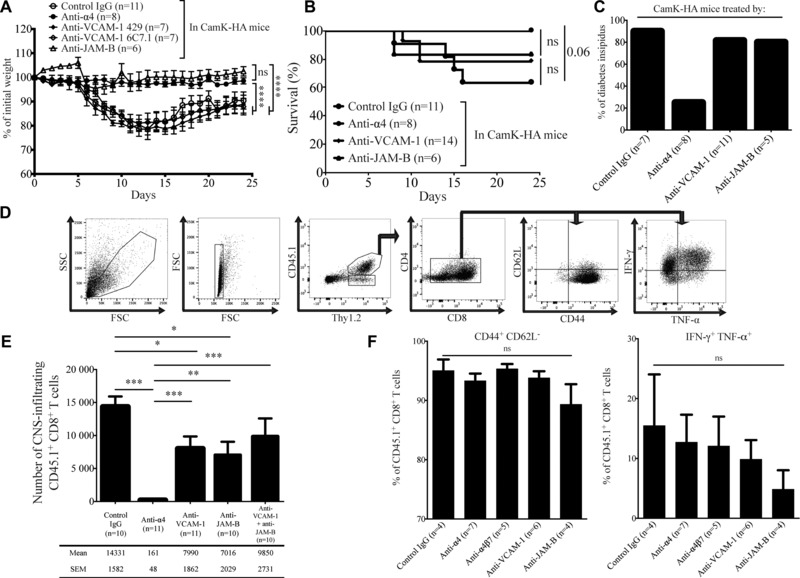
VCAM‐1 blockade retains the clinical phenotype and only partly decreases CD8 T cell CNS infiltration. (A–C) Adoptive transfer of 5 × 106 cytotoxic HA‐specific CD8 T cells in CamK‐HA mice treated at day 0, 4, and 8 by an anti‐α4‐integrin, a control IgG, anti‐VCAM‐1 (clone 429 or 6C7.1), or an anti‐JAM‐B mAb. Impact on the weight (A), survival (B), and on development of diabetes insipidus (C). Pool of two independent experiments, each involving three to six mice per group. (A) One‐way ANOVA ***p < 0.0001. (B) Log‐rank (Mantel–Cox) test, ns. Data represent mean ± SEM. (D–F) FACS analysis of CNS‐infiltrating cells at day 8 after adoptive transfer of 5 × 106 CD45.1+ cytotoxic HA‐specific CD8 T cells in CD45.2+ CamK‐HA mice treated at day 0 and 4 by a control IgG, an anti‐α4‐integrin, an anti‐VCAM‐1 (clone 6C7.1), an anti‐JAM‐B, or both an anti‐VCAM‐1 and anti‐JAM‐B mAbs. (D) Illustration of the general gating strategy. (E) Absolute number of CD45.1+ CD8+ T cells infiltrating the CNS. Pool of two to three independent experiments using three to five mice per group. Mann–Whitney test, *p < 0.05, **p < 0.001, ***p < 0.0001. Data represent mean ± SEM. (F) Percentage of CD45.1+ CD8+ T cells that are CD44+ CD62L− and that produce IFN‐γ and TNF‐α. Pool of two independent experiments using two to three mice per group. One‐way ANOVA, ns. Data represent mean ± SEM.
To assess the impact of VCAM‐1 inhibition on CD8 T‐cell infiltration of the CNS, and to unambiguously identify and enumerate the transferred cells, CD45.1+ HA‐specific CD8 T cells were injected in CD45.2+ CamK‐HA mice (Fig. 3D). Flow cytometry analysis of CNS‐infiltrating cells from CamK‐HA recipients 8 days after the adoptive transfer revealed a significant reduction in the number of transferred CD45.1+ CD8 T cells in mice treated with anti‐VCAM‐1 mAbs compared to mice receiving the control IgG (7990 ± 1862 versus 14 331 ± 1582 cells, Mann–Whitney test, p = 0.01; Fig. 3E). However, VCAM‐1 blockade reduced the number of CNS‐infiltrating transferred CD8 T cells to a much lesser extent than the use of anti‐α4‐integrin mAb (161 ± 48 cells, Mann–Whitney test, p < 0.0001). Collectively, these data indicate that blockade of VCAM‐1 partially inhibits the α4β1‐dependent migration of CD8 T cells across the BBB, but does not protect mice from developing CNS tissue damage, suggesting that additional vascular ligands of α4β1‐integrin might be implicated in CD8 T cell trafficking into the CNS.
JAM‐B is a ligand for α4β1‐integrin implicated in CD8 T‐cell migration to the CNS
Because junctional adhesion molecule‐B (JAM‐B) had been reported to be a ligand for α4β1‐integrin on endothelial cells 21, we assessed the contribution of JAM‐B as another putative BBB endothelial ligand for α4β1‐integrin in CD8 T‐cell migration into the CNS. Indeed, CNS microvascular endothelial cells express JAM‐B in addition to PECAM‐1 (Fig. 4). We investigated whether JAM‐B could be potential ligand for α4β1‐integrin expressed on CD8 T cells. We treated CamK‐HA recipients with an anti‐JAM‐B mAb at day 0, 4, and 8 after the adoptive transfer of 5 × 106 cytotoxic HA‐specific CD8 T cells. While CamK‐HA recipients treated by the anti‐JAM‐B mAb displayed significantly less weight loss compared to recipients treated by control IgG or anti‐VCAM‐1 mAbs, they displayed the same incidence of diabetes insipidus and death (Fig. 3A–C). Flow cytometry analysis of the CNS of CamK‐HA recipients treated by anti‐JAM‐B mAb showed a significant decrease in the number of infiltrating CD45.1+ CD8 transferred cells compared to mice receiving the control IgG (7016 ± 2029 versus 14 331 ± 1582 cells/CNS, Mann–Whitney test, p = 0.017; Fig. 3E). This decrease was similar to that achieved when blocking VCAM‐1 (Mann–Whitney test, ns), but clearly inferior to the inhibition achieved by the anti‐α4‐integrin mAb (Mann–Whitney test, p = 0.0017; Fig. 3E). Increased dose of anti‐JAM‐B mAb did not improve the reduction of CNS‐infiltrating CD45.1+ CD8+ transferred cells, suggesting that saturating concentrations of the antibody were obtained (Supporting Information Fig. 3). Therefore, JAM‐B expressed by BBB endothelial cells might represent a ligand for α4β1‐integrin‐mediated migration of CD8 T cells into the CNS. In order to test the hypothesis that VCAM‐1 and JAM‐B are complementary ligands for α4β1‐integrin, we treated recipients with both anti‐JAM‐B and anti‐VCAM‐1 mAbs. Coadministration of both mAbs did not demonstrate a synergistic reduction in the number of CNS‐infiltrated CD45.1+ CD8 transferred T cells (Fig. 3E). We finally investigated the phenotype of CD8 T cells that entered the CNS of CamK‐HA recipients after blockade of α4‐integrin, α4β7‐integrin, VCAM‐1, or JAM‐B. Whereas their absolute numbers differed importantly between groups, the proportion of transferred CD45.1+ CD8 T cells that are CD44+ CD62L−, and that produce both IFN‐γ and TNF‐α did not significantly differ between groups (Fig. 3F).
Figure 4.
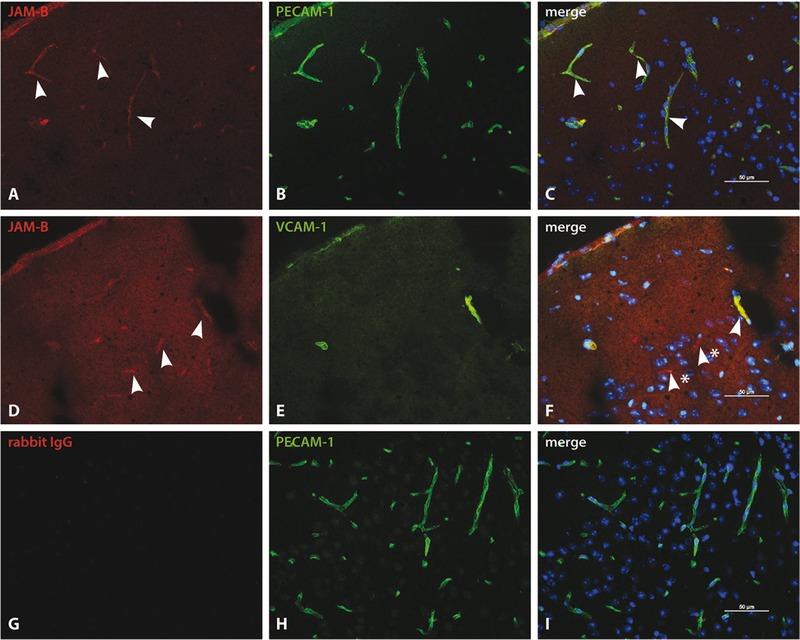
CNS microvascular endothelial cells express JAM‐B in addition to PECAM‐1. (A–C) Immunofluorescence staining for JAM‐B in the mouse CNS was evaluated on cryosections (6 μm) of brains of CamK‐HA mice day 8 after adoptive transfer of 5 × 106 cytotoxic HA‐specific CD8 T cells. Double immunofluorescence staining of JAM‐B (A) and PECAM‐1 (B) show colocalization of JAM‐B and PECAM‐1 staining (C) on all vessels in the mouse cortex. Arrowheads point to representable JAM‐B positive (A) and JAM‐B/PECAM‐1 double positive vessels (C). (D–F) Double immunofluorescence staining of JAM‐B (D, arrowheads) and VCAM‐1 (E) show a colocalization of JAM‐B and VCAM‐1 (F) on blood vessels (arrowhead), however JAM‐B immune staining can additionally be detected on endothelial cells staining negative for VCAM‐1. (F) Arrowheads with stars point to JAM‐B positive vessels that do not stain positive for VCAM‐1. (G–I) Isotype control staining with rabbit IgG control antibody (G) and PECAM‐1 (H) shows no staining for the rabbit IgG (G and I). The variability in background with the anti‐JAM‐B antibody observed in (A), (D), and (J) is related to the area in the CNS and the inflammatory status of the brain. All sections were counter‐stained with DAPI to show cell nuclei. Bars are 50 μm. Panel shown above constitutes one representative staining; in total three individual tissues were processed.
Discussion
Molecular cues used by CD8 T cells to migrate into the CNS are poorly defined. Using a murine model of CNS autoimmune neuroinflammation, we showed that the migration of cytotoxic CD8 T cells to the CNS relies on the α4β1‐integrin heterodimer. We also showed that VCAM‐1 and JAM‐B expressed by BBB endothelial cells are likely implicated in this process, but that their inhibition is insufficient to completely block CD8 T‐cell infiltration into the CNS or to mitigate the clinical encephalomyelitis signs.
Two studies using animal models of neuroinflammation and in vitro transmigration assays demonstrated that blocking the α4‐integrin decreases migration of CD8 T cells across CNS vascular structures 17, 18. However, because the α4‐integrin is common to α4β1‐ and α4β7‐integrin heterodimers 22, it is unknown which one is implicated. Our model showed that blockade of α4‐integrin, but not of the α4β7‐integrin heterodimer, prevents encephalomyelitis and tissue damage by interfering with cytotoxic CD8 T‐cell entry into the CNS. These results strongly suggest that the α4β1‐integrin is the essential heterodimer for the interaction of activated CD8 T cells with the BBB. Similarly, blocking α4‐integrin can abrogate the development of EAE mediated by encephalitogenic CD4 Th1 cells 7, 8, 9, 10. Those cells critically rely on β1‐integrin to accumulate in the CNS during EAE 6. Moreover, blocking the α4β7‐integrin heterodimer fails to inhibit EAE development in mice and Rhesus monkeys 7, 23, and ectopic expression of the α4β7‐integrin ligand MAdCAM‐1 in CNS endothelial cells fails to trigger CNS recruitment of α4β7‐expressing T cells 24. Thus, α4β1‐integrin rather than α4β7‐integrin mediates both CD4 and CD8 T‐cell interaction with CNS endothelium. However, as α4‐integrin blockade did not totally abrogate clinical signs and CNS infiltration of transferred CD8 T cells in our model, an α4β1‐independent route, although accessory, remains operative. Its molecular bases are still to be determined.
The most studied brain endothelial ligand for α4β1‐integrin is VCAM‐1 20. VCAM‐1 is constitutively expressed at low level on the BBB endothelium, and mediates both the initial capture and the subsequent G‐protein‐dependent arrest of CD4 Th1 cells upon interaction with the α4β1‐integrin 8. Expression of VCAM‐1 is upregulated in inflammatory conditions on endothelial cells of the BBB and of the blood‐leptomeningeal barrier, as well as on choroid plexus epithelium of the blood‐cerebrospinal fluid barrier, further increasing α4β1/VCAM‐1‐mediated T‐cell adhesion to inflamed CNS endothelium 25, 26. It was previously shown that VCAM‐1 blockade inhibits clinical or histopathological signs of EAE mediated by encephalitogenic CD4 Th1 cells 7. In a model of OVA‐expressing Listeria monocytogenes infection, access of OVA‐specific CD8 T cells to the CNS was inhibited by the antibody‐mediated blockade of VCAM‐1 to the same extent as did the α4‐integrin blockade 18. Unexpectedly, blockade of VCAM‐1 in our model only had a partial effect on CD8 T‐cell migration to the CNS, as the number of CNS‐infiltrated CD8 T cells was reduced by 46% compared to mice treated by the control IgG (Fig. 3D). This decrease was not clinically relevant as recipients treated with different clones of anti‐VCAM‐1 mAb experienced weight loss, death, and diabetes insipidus to the same extent as mice treated by the control IgG. Therefore, VCAM‐1 blockade was not sufficient in our model to inhibit α4β1‐expressing CD8 T‐cells migration into the CNS. Several differences between our study and that of Young et al. 18 might explain those discordant results, including the genetic background of the mice, BALB/c or (BALB/c × C57BL6) F1 versus C57BL6/J, the localization of the HA and OVA antigens in different cells, neurons and oligodendrocytes, respectively, and the state of differentiation of the encephalitogenic CD8 T cells (terminally differentiated highly cytotoxic versus shortly activated CD8 T cells). Moreover, the L. monocytogenes infection within the CNS in this model might induce additional triggers including increased expression of VCAM‐1 at the BBB, allowing for increased VCAM‐1‐dependence of CD8 T‐cell migration into the CNS. It was suggested that natalizumab, a humanized mAb against the human α4 subunit of α4β1‐integrin and α4β7, might also affect CD4 and CD8 T‐cell expression of α4‐integrin‐unrelated molecules such as CD62L, CXCR3, and αLβ2 in long‐term treated natalizumab patients 27, 28. Here, we have used a very short treatment. In addition, the high specificity of PS/2 for the murine α4‐integrin subunit makes this antibody unlikely to cross‐react with other integrin subunit or unrelated molecules such as selectins. Indeed, PS/2 recognizes the functional epitope B2 on the α4‐integrin subunit and was shown to specifically block α4β1‐mediated lymphocyte binding to VCAM‐1 and fibronectin as well as α4β7‐dependent binding to VCAM‐1 and MAdCAM‐1 in vitro 29, 30. Another explanation could be that, in our model, VCAM‐1 is not the sole, or even not the main, ligand for α4β1‐integrin.
In that respect, we investigated if other known α4β1‐integrin ligands might be implicated. At least two additional vascular ligands have been described, the CS1 domain of a spliced variant of fibronectin 31, 32, and JAM‐B 21, 33. Since no reliable blocking antibody against CS1 domain is currently available, the role of fibronectin in the migration process of CD8 T cells could not be explored yet. Because α4β1–JAM‐B interaction has only been investigated in vitro or in vivo in skin microvasculature 21, and because JAM‐B is expressed by the BBB in our model, we focused on this putative ligand for CD8 T‐cell migration to the CNS. Members of the classical JAM family are transmembrane type 1 proteins with two immunoglobulin domains highly expressed in cells that present well organized tight junctions, such as the endothelium of the BBB 34. JAM‐A, interacting with αLβ2‐integrin, is implicated in cell migration to the CNS. A blocking mAb directed to JAM‐A significantly inhibited leukocyte infiltration in the CSF and the brain parenchyma in a model of cytokine‐induced meningitis 35. However, those results were not confirmed in infectious meningitis 36. JAM‐B plays a central role in the regulation of paracellular permeability 34, but also impacts T‐cell rolling and firm adhesion 21. Although a lack of JAM‐B expression at steady state on brain endothelial cells was suggested 37, we show that JAM‐B is expressed on brain endothelial cells in our murine model of CNS autoimmunity. Because blockade of JAM‐B was associated to a partial protection against encephalomyelitis and a significant reduction in the number of infiltrated CD8 T cells, however to a lesser extent than α4β1‐integrin blockade, our data suggest that JAM‐B participates to the CD8 T‐cell migration process to the CNS.
The discovery of the role of α4β1–VCAM‐1 interaction in T‐cell migration to the CNS led to the development of natalizumab, which has proven to be highly beneficial in relapsing‐remitting MS 38. Unfortunately, natalizumab is associated with an increased risk of developing progressive multifocal leukoencephalopathy (PML), an opportunistic disease of the CNS caused by John Cunningham (JC) virus infection of oligodendrocytes 39]. Development of PML following natalizumab therapy likely relies on a multifactorial scenario including permissiveness for active JC virus replication and impaired immune surveillance 40, 41, 42, 43. Compared with untreated MS patients, natalizumab treatment induces a decrease in cerebrospinal fluid CD4 and CD8 T cells 44, 45. Because CD4 and CD8 T cells are crucial in controlling JC virus reactivation and dissemination 46, 47, our results suggest that development of PML is, at least in part, due to natalizumab‐mediated inhibition of CNS immunosurveillance by JC virus‐specific CD8 T cells. Furthermore, natalizumab‐associated PML in MS patients is often complicated after natalizumab withdrawal by IRIS, darkening again the prognosis 48. Although CD4 T cells might participate 49, a clear dominance of CD8 T cells in CNS‐infiltrates has been observed in natalizumab‐associated PML‐IRIS occurring in patients with MS 50, reminiscent of the PML‐IRIS pathology described in HIV‐infected patients 51. This indirectly supports the idea that the discontinuation of natalizumab allows migration of CD8 T cell to the CNS.
Migration of encephalitogenic CD8 T cells to the CNS is dependent on the α4β1‐integrin. α4β1‐Integrin blockade in MS is beneficial but exposes to opportunistic viral infections due to the disruption of CNS immune surveillance. Because CD8 T cells requirements for CNS migration are distinct from those of other immune cells, future work should help identify novel molecular targets to block specifically CNS recruitment of destructive immune cells while leaving the migration of protective immune cell subsets into the CNS unaffected.
Materials and methods
Mice
The CL4‐TCR transgenic mouse line expresses an H‐2Kd‐restricted TCR (Vα10; Vβ8.2) against the Influenza virus HA transmembrane peptide amino acids 512–520 (IYSTVASSL) 52. The CamK‐iCre 53 and the Rosa26tm(HA)1Lib 54 mice have been described previously. CamK‐HA mice were obtained by crossing the CamK‐iCre transgenic mice with the Rosa26tm(HA)1Lib mice 19. Cre‐mediated genomic DNA recombination and the resulting transcription of HA occurred only in CamK‐HA mice and were confined to the brain and spinal cord 19. Mice were backcrossed at least six times on the BALB/c background. In some experiments, to benefit from a congenic marker allowing the distinction between transferred HA‐specific CD8 T cells (CD45.1) and recipient T cells (CD45.2), donor and recipient mice on a (BALB/c × C57BL6) F1 background were used. Of note, the number of T cells infiltrating the CNS of CamK‐HA mice did not differ between the BALB/c and the (BALB/c × C57BL6) F1 backgrounds (Supporting Information Fig. 4). All mice were 6–9‐week‐old at the onset of the experiments. Mice were kept under specific pathogen‐free conditions. This study was carried out in strict accordance with EU regulations and with the recommendations of the French national chart for ethics of animal experiments (articles R 214‐87–90 of the “Code rural”). The protocol was approved by the committee on the ethics of animal experiments of the région Midi‐Pyrénées (permit numbers: 04‐U563‐DG‐06 and MP/18/26/04/04). All procedures were performed under deep anesthesia as described below and all efforts were made to minimize animal suffering.
In vitro differentiation and adoptive transfer of HA‐specific cytotoxic CD8 T cells
HA‐specific cytotoxic CD8 T cells were generated as previously described 55. Briefly, 5.105 purified naive CD8 T cells from CL4‐TCR mice were stimulated with 5.106 irradiated syngeneic splenocytes in DMEM supplemented with 10% FCS (Life Technologies, Paisley, Scotland) containing 1 mM HA peptide, 1 ng/mL IL‐2, and 20 ng/mL IL‐12. On day 5, cells were collected by Ficoll density separation and 5 × 106 living cytotoxic CD8 T cells were injected intravenously in recipient mice. Cells routinely contained >98% CD8+ CD3+ Vβ8.2+ T cells and >95% of them produce high levels of granzyme B and IFN‐γ.
Antibodies used for in vivo experiments
mAbs were injected intravenously. Entotoxin‐free rat mAb against mouse α4‐integrin (clone PS/2, IgG2b, 250 μg/injection), α4β7‐integrin (clone DATK32, IgG2a, 500 μg/injection), VCAM‐1 (clone 6C7.1, IgG1, 250 μg/injection), and antiendoglin (control IgG, clone MJ7/18, IgG2a, 250 μg/injection) were produced in B.E.'s lab. The rat anti‐CD4‐depleting mAb (clone GK1.5, IgG2b, 500 μg/injection) was produced in R.L.'s lab. The anti‐VCAM‐1 clone 429 (IgG2a, 100 μg/injection) and the anti‐JAM‐B clone 150005 (IgG2a, 100 μg/injection, or 200 μg/injection where indicated) were purchased from eBioscience and R&D Systems, respectively.
Purification of mononuclear cells
Mice were deeply anesthetized and perfused with 20 mL of PBS. For purification of mononuclear cells, brains were collected in PBS and dissociated using a glass Potter. Brain suspensions were enzymatically digested for 1 h at 37°C in RPMI 1640 medium (Invitrogen) containing collagenase D (1 mg/mL), and DNase I (10 mg/mL). The digested suspensions were filtered (70 mm cell strainer, Falcon), CNS‐infiltrating mononuclear cells were collected after Percoll density separation and directly used for FACS staining. For purification of mononuclear cells from spleen and cervical draining lymph nodes, organs were collected in PBS and dissociated using a glass Potter. After red blood cell lysis, mononuclear cells were used for FACS staining.
Ex vivo restimulation of HA‐specific CD8 T cells
Mononuclear cells from spleen and cervical lymph nodes (4 × 106) were plated in a 24‐well plate for 6 h and restimulated with 1 μM of HA peptide or of H‐2Kd‐binding noncognate antigen (Cw3 peptide, RYLKNGKETL). GolgiPlug™ (BD Pharmingen) was added for the last 2 h before mononuclear cells were used for FACS staining.
FACS analysis
In vitro differentiated cytotoxic HA‐specific CD8 T cells were stained using a viability dye and anti‐CD8α (53‐6.7, BD Pharmingen), anti‐α4‐integrin (hybridoma supernatant, clone PS/2), anti‐β7‐integrin (hybridoma supernatant, clone FIB504), or anti‐α4β7‐integrin heterodimer (hybridoma supernatant, clone DATK32). Mononuclear cells were stained using a viability dye and anti‐CD45 (30‐F11, BD Pharmingen), anti‐CD45.1 (A20, Biolegend), anti‐CD45.2 (104, BD Pharmingen), anti‐CD11b (M1/70, eBioscience), anti‐Thy1.2 (53‐2.1, eBioscience), anti‐CD4 (RM4‐5, BD Pharmingen), anti‐CD8 (53‐6.7, BD Pharmingen), anti‐MHC class II (M5/114.15.2, eBioscience), anti‐CD44 (IM7, Biolegend), anti‐CD62L (MEL‐14, BD Pharmingen), and anti‐CD25 (PC61, BD Pharmingen) mAbs. After fixation and permeabilization, cells were intracellularly stained for IFN‐γ (XMG1.2, BD Biosciences) and TNF‐α (MP6‐XT22, BD Pharmingen). Data were collected on an LSRII or FACSCalibur (Becton Dickinson) and analyzed with FlowJo software (Tree Star).
In vitro binding assay system
Wells of diagnostic microscope slides (ER‐202W‐CE24, Thermo Scientific) were coated with protein A (20 μg/mL in PBS pH 9) for 1 h at 37°C. After protein A incubation, wells were washed three times with PBS (pH 7.4) and blocked with 1.5% BSA in PBS for 30 min at 37°C. Subsequently, wells were washed once with PBS (pH 7.4) and recombinant proteins (100 nM) were incubated for 2 h at 37°C (recombinant DNER‐Fc chimera, R&D 2254‐DN; recombinant mouse ICAM‐1‐Fc chimera R&D 796‐IC; recombinant VCAM‐1‐Fc chimera, R&D 643‐VM). After recombinant protein‐coating, wells were washed twice with PBS (pH 7.4) and blocked with 1.5% BSA in PBS for 30 min at room temperature.
To perform the in vitro binding assay, HA‐specific cytotoxic CD8 T cells were collected at 5 × 106 cells/mL in assay medium (DMEM, 25 mM HEPES, 5% calf serum, 2% L‐glutamine) and incubated with either the blocking antibody (PS/2, 10 μg/mL) or the isotype control antibody (rat IgG2b, 10 μg/mL) for 20 min at room temperature. Antibodies were washed away by centrifugation for 3 min at 1400 rpm. Diagnostic microscope slides were placed on a rotating platform and 20 μL cell suspension (1 × 105 cells/well) was added and incubated on the recombinant proteins for 30 min. Slides were washed twice with PBS and fixed in 2.5% v/v glutaraldehyde in PBS.
The number of adherent cells was evaluated by counting bound cells per field of view under the microscope using a grid ocular.
Immunofluorescence staining
Preparation of mouse tissue and staining procedures were performed as published previously 56. Briefly, mice were perfused with PBS or 1% formaldehyde in PBS through the left ventricle of the heart. Dissected tissues were embedded in Tissue‐Tek (Sakura, the Netherlands), frozen, and stored at −80°C. For immunofluorescence staining of JAM‐B, 6 μm cryostat sections were fixed with ice‐cold methanol for 20 s, followed by blocking with blocking buffer (2% BSA, 1% FCS, 1% donkey serum in PBS). Subsequently, primary rabbit anti‐JAM‐B antibody (αJB829 1:250; kindly provided by Beat Imhof, Geneva, Switzerland) and either rat antimouse PECAM‐1 (Mec 13.3, 20 μg/mL) or rat antimouse VCAM‐1 (6C7.1, 10 μg/mL) were incubated with the sections for 1 h. In between and after antibody incubation, sections were washed with PBS. Sections were then incubated with secondary antibodies goat antirat Alexa 488 (1:200, Invitrogen MP A11006) and donkey antirabbit Cy3 (1:200, JIR 111‐165‐144) and DAPI (0.5 μg/mL, Invitrogen D3571) for 1 h and coverslipped with Mowiol (Sigma–Aldrich, USA). For immunofluorescence staining of CD8, cryostat sections were fixed in ethanol for 10 min at 4°C, followed by acetone for 1 min at room temperature. Subsequently, sections were dried for 30 min at room temperature. Blocking solution, skimmed milk (5% in TBS) was incubated for 20 min. Primary rat‐antimouse CD8 antibody (Lyt 2, eBiosciences) and secondary goat‐antirat IgG‐Cy3 antibody (eBiosciences) were diluted in blocking solution. Antibody incubation was performed for 1 h at room temperature. To visualize the cell nuclei, DAPI (0.5 μg/mL, Invitrogen D3571) was incubated for 5 min at room temperature. Sections were washed with TBS between incubation steps and finally mounted with Mowiol (Sigma–Aldrich) and analyzed using a Nikon Eclipse 600 fluorescence microscope.
Statistical analysis
Differences between two sets of data were evaluated by a two‐tailed Mann–Whitney U test. For analysis of scatter plots comparing ≥3 groups of mice, one‐way ANOVA with Bonferroni's posttest was used. Survival curves were plotted by the Kaplan–Meier method and compared with the log‐rank test. Data represent mean ± SEM. Probability values of p ≤ 0.05 were considered statistically significant. All tests were carried out using GraphPad Prism 5 (GraphPad Software, La Jolla, CA, USA).
Conflict of interest
The authors declare no financial or commercial conflict of interest.
Abbreviations
- BBB
Blood‐brain barrier
- IRIS
Immune reconstitution inflammatory syndrome
- JAM‐B
Junctional adhesion molecule‐B
- PML
Progressive multifocal leukoencephalopathy
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary Material
Acknowledgments
This work was supported by an international ARSEP grant (to B.E. and R.L.), the Toulouse University Hospital, the French National Institute for Medical Research (INSERM), the French National Research Agency (ANR‐13‐BSV3‐0003‐01), and European Union, FP7‐PEOPLE‐2012‐ITN NeuroKine to R.L., and the Swiss National Science Foundation (Grant No. 149420) and the Swiss Multiple Sclerosis Society to B.E. S.T. has been supported by a fellowship from the German Research Foundation. We thank Dr. Beat Imhof (Geneva, Switzerland) for kindly providing the anti‐JAM‐B antibody.
References
- 1. Galea, I. , Bechmann, I. and Perry, V. H. , What is immune privilege (not)? Trends Immunol. 2007. 28: 12–18. [DOI] [PubMed] [Google Scholar]
- 2. Liblau, R. S. , Gonzalez‐Dunia, D. , Wiendl, H. and Zipp, F. , Neurons as targets for T cells in the nervous system. Trends Neurosci. 2013. 36: 315–324. [DOI] [PubMed] [Google Scholar]
- 3. Wilson, E. H. , Weninger, W. and Hunter, C. A. , Trafficking of immune cells in the central nervous system. J. Clin. Invest. 2010. 120: 1368–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Engelhardt, B. and Ransohoff, R. M. , Capture, crawl, cross: the T cell code to breach the blood‐brain barriers. Trends Immunol. 2012. 33: 579–589. [DOI] [PubMed] [Google Scholar]
- 5. Bartholomaus, I. , Kawakami, N. , Odoardi, F. , Schlager, C. , Miljkovic, D. , Ellwart, J. W. , Klinkert, W. E. et al, Effector T cell interactions with meningeal vascular structures in nascent autoimmune CNS lesions. Nature 2009. 462: 94–98. [DOI] [PubMed] [Google Scholar]
- 6. Bauer, M. , Brakebusch, C. , Coisne, C. , Sixt, M. , Wekerle, H. , Engelhardt, B. and Fassler, R. , Beta1 integrins differentially control extravasation of inflammatory cell subsets into the CNS during autoimmunity. Proc. Natl. Acad. Sci. USA 2009. 106: 1920–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Engelhardt, B. , Laschinger, M. , Schulz, M. , Samulowitz, U. , Vestweber, D. and Hoch, G. , The development of experimental autoimmune encephalomyelitis in the mouse requires alpha4‐integrin but not alpha4beta7‐integrin. J. Clin. Invest. 1998. 102: 2096–2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Vajkoczy, P. , Laschinger, M. and Engelhardt, B. , Alpha4‐integrin‐VCAM‐1 binding mediates G protein‐independent capture of encephalitogenic T cell blasts to CNS white matter microvessels. J. Clin. Invest. 2001. 108: 557–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yednock, T. A. , Cannon, C. , Fritz, L. C. , Sanchez‐Madrid, F. , Steinman, L. and Karin, N. , Prevention of experimental autoimmune encephalomyelitis by antibodies against alpha 4 beta 1 integrin. Nature 1992. 356: 63–66. [DOI] [PubMed] [Google Scholar]
- 10. Rothhammer, V. , Muschaweckh, A. , Gasteiger, G. , Petermann, F. , Heink, S. , Busch, D. H. , Heikenwalder, M. et al, Alpha4‐integrins control viral meningoencephalitis through differential recruitment of T helper cell subsets. Acta Neuropathol. Commun. 2014. 2: 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rothhammer, V. , Heink, S. , Petermann, F. , Srivastava, R. , Claussen, M. C. , Hemmer, B. and Korn, T. , Th17 lymphocytes traffic to the central nervous system independently of alpha4 integrin expression during EAE. J. Exp. Med. 2011. 208: 2465–2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Martin‐Blondel, G. , Delobel, P. , Blancher, A. , Massip, P. , Marchou, B. , Liblau, R. S. and Mars, L. T. , Pathogenesis of the immune reconstitution inflammatory syndrome affecting the central nervous system in patients infected with HIV. Brain 2011. 134: 928–946. [DOI] [PubMed] [Google Scholar]
- 13. Saxena, A. , Martin‐Blondel, G. , Mars, L. T. and Liblau, R. S. , Role of CD8 T cell subsets in the pathogenesis of multiple sclerosis. FEBS Lett. 2011. 585: 3758–3763. [DOI] [PubMed] [Google Scholar]
- 14. Bien, C. G. , Vincent, A. , Barnett, M. H. , Becker, A. J. , Blumcke, I. , Graus, F. , Jellinger, K. A. et al, Immunopathology of autoantibody‐associated encephalitides: clues for pathogenesis. Brain 2012. 135: 1622–1638. [DOI] [PubMed] [Google Scholar]
- 15. Battistini, L. , Piccio, L. , Rossi, B. , Bach, S. , Galgani, S. , Gasperini, C. , Ottoboni, L. et al, CD8+ T cells from patients with acute multiple sclerosis display selective increase of adhesiveness in brain venules: a critical role for P‐selectin glycoprotein ligand‐1. Blood 2003. 101: 4775–4782. [DOI] [PubMed] [Google Scholar]
- 16. Cayrol, R. , Wosik, K. , Berard, J. L. , Dodelet‐Devillers, A. , Ifergan, I. , Kebir, H. , Haqqani, A. S. et al, Activated leukocyte cell adhesion molecule promotes leukocyte trafficking into the central nervous system. Nat. Immunol. 2008. 9: 137–145. [DOI] [PubMed] [Google Scholar]
- 17. Ifergan, I. , Kebir, H. , Alvarez, J. I. , Marceau, G. , Bernard, M. , Bourbonniere, L. , Poirier, J. et al, Central nervous system recruitment of effector memory CD8+ T lymphocytes during neuroinflammation is dependent on alpha4 integrin. Brain 2011. 134: 3560–3577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Young, K. G. , Maclean, S. , Dudani, R. , Krishnan, L. and Sad, S. , CD8+ T cells primed in the periphery provide time‐bound immune‐surveillance to the central nervous system. J. Immunol. 2011. 187: 1192–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Scheikl, T. , Pignolet, B. , Dalard, C. , Desbois, S. , Raison, D. , Yamazaki, M. , Saoudi, A. et al, Cutting edge: neuronal recognition by CD8 T cells elicits central diabetes insipidus. J. Immunol. 2012. 188: 4731–4735. [DOI] [PubMed] [Google Scholar]
- 20. Elices, M. J. , Osborn, L. , Takada, Y. , Crouse, C. , Luhowskyj, S. , Hemler, M. E. and Lobb, R. R. , VCAM‐1 on activated endothelium interacts with the leukocyte integrin VLA‐4 at a site distinct from the VLA‐4/fibronectin binding site. Cell 1990. 60: 577–584. [DOI] [PubMed] [Google Scholar]
- 21. Ludwig, R. J. , Hardt, K. , Hatting, M. , Bistrian, R. , Diehl, S. , Radeke, H. H. , Podda, M. et al, Junctional adhesion molecule (JAM)‐B supports lymphocyte rolling and adhesion through interaction with alpha4beta1 integrin. Immunology 2009. 128: 196–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Luo, B. H. , Carman, C. V. and Springer, T. A. , Structural basis of integrin regulation and signaling. Annu. Rev. Immunol. 2007. 25: 619–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Haanstra, K. G. , Hofman, S. O. , Lopes Estevao, D. M. , Blezer, E. L. , Bauer, J. , Yang, L. L. , Wyant, T. et al, Antagonizing the alpha4beta1 integrin, but not alpha4beta7, inhibits leukocytic infiltration of the central nervous system in rhesus monkey experimental autoimmune encephalomyelitis. J. Immunol. 2013. 190: 1961–1973. [DOI] [PubMed] [Google Scholar]
- 24. Doring, A. , Pfeiffer, F. , Meier, M. , Dehouck, B. , Tauber, S. , Deutsch, U. and Engelhardt, B. , TET inducible expression of the alpha4beta7‐integrin ligand MAdCAM‐1 on the blood‐brain barrier does not influence the immunopathogenesis of experimental autoimmune encephalomyelitis. Eur. J. Immunol. 2011. 41: 813–821. [DOI] [PubMed] [Google Scholar]
- 25. Laschinger, M. and Engelhardt, B. , Interaction of alpha4‐integrin with VCAM‐1 is involved in adhesion of encephalitogenic T cell blasts to brain endothelium but not in their transendothelial migration in vitro. J. Neuroimmunol. 2000. 102: 32–43. [DOI] [PubMed] [Google Scholar]
- 26. Steiner, O. , Coisne, C. , Cecchelli, R. , Boscacci, R. , Deutsch, U. , Engelhardt, B. and Lyck, R. , Differential roles for endothelial ICAM‐1, ICAM‐2, and VCAM‐1 in shear‐resistant T cell arrest, polarization, and directed crawling on blood‐brain barrier endothelium. J. Immunol. 2010. 185: 4846–4855. [DOI] [PubMed] [Google Scholar]
- 27. Jilek, S. , Mathias, A. , Canales, M. , Lysandropoulos, A. , Pantaleo, G. , Schluep, M. and Du Pasquier, R. A. , Natalizumab treatment alters the expression of T‐cell trafficking marker LFA‐1 alpha‐chain (CD11a) in MS patients. Mult. Scler. 2013. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 28. Schwab, N. , Schneider‐Hohendorf, T. , Posevitz, V. , Breuer, J. , Gobel, K. , Windhagen, S. , Brochet, B. et al, L‐selectin is a possible biomarker for individual PML risk in natalizumab‐treated MS patients. Neurology 2013. 81: 865–871. [DOI] [PubMed] [Google Scholar]
- 29. Berlin, C. , Berg, E. L. , Briskin, M. J. , Andrew, D. P. , Kilshaw, P. J. , Holzmann, B. , Weissman, I. L. et al, Alpha 4 beta 7 integrin mediates lymphocyte binding to the mucosal vascular addressin MAdCAM‐1. Cell 1993. 74: 185–195. [DOI] [PubMed] [Google Scholar]
- 30. Kamata, T. , Puzon, W. and Takada, Y. , Identification of putative ligand‐binding sites of the integrin alpha 4 beta 1 (VLA‐4, CD49d/CD29). Biochem. J. 1995. 305: 945–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Man, S. , Tucky, B. , Bagheri, N. , Li, X. , Kochar, R. and Ransohoff, R. M. , Alpha4 Integrin/FN‐CS1 mediated leukocyte adhesion to brain microvascular endothelial cells under flow conditions. J. Neuroimmunol. 2009. 210: 92–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. van der Laan, L. J. , van der Goes, A. , Wauben, M. H. , Ruuls, S. R. , Dopp, E. A. , De Groot, C. J. , Kuijpers, T. W. et al, Beneficial effect of modified peptide inhibitor of alpha4 integrins on experimental allergic encephalomyelitis in Lewis rats. J. Neurosci. Res. 2002. 67: 191–199. [DOI] [PubMed] [Google Scholar]
- 33. Cunningham, S. A. , Rodriguez, J. M. , Arrate, M. P. , Tran, T. M. and Brock, T. A. , JAM2 interacts with alpha4beta1. Facilitation by JAM3. J. Biol. Chem. 2002. 277: 27589–27592. [DOI] [PubMed] [Google Scholar]
- 34. Martin‐Padura, I. , Lostaglio, S. , Schneemann, M. , Williams, L. , Romano, M. , Fruscella, P. , Panzeri, C. et al, Junctional adhesion molecule, a novel member of the immunoglobulin superfamily that distributes at intercellular junctions and modulates monocyte transmigration. J. Cell Biol. 1998. 142: 117–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Del Maschio, A. , De Luigi, A. , Martin‐Padura, I. , Brockhaus, M. , Bartfai, T. , Fruscella, P. , Adorini, L. et al, Leukocyte recruitment in the cerebrospinal fluid of mice with experimental meningitis is inhibited by an antibody to junctional adhesion molecule (JAM). J. Exp. Med. 1999. 190: 1351–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lechner, F. , Sahrbacher, U. , Suter, T. , Frei, K. , Brockhaus, M. , Koedel, U. and Fontana, A. , Antibodies to the junctional adhesion molecule cause disruption of endothelial cells and do not prevent leukocyte influx into the meninges after viral or bacterial infection. J. Infect. Dis. 2000. 182: 978–982. [DOI] [PubMed] [Google Scholar]
- 37. Aurrand‐Lions, M. , Johnson‐Leger, C. , Wong, C. , Du Pasquier, L. and Imhof, B. A. , Heterogeneity of endothelial junctions is reflected by differential expression and specific subcellular localization of the three JAM family members. Blood 2001. 98: 3699–3707. [DOI] [PubMed] [Google Scholar]
- 38. Polman, C. H. , O'Connor, P. W. , Havrdova, E. , Hutchinson, M. , Kappos, L. , Miller, D. H. , Phillips, J. T. et al, A randomized, placebo‐controlled trial of natalizumab for relapsing multiple sclerosis. N. Engl. J. Med. 2006. 354: 899–910. [DOI] [PubMed] [Google Scholar]
- 39. Bloomgren, G. , Richman, S. , Hotermans, C. , Subramanyam, M. , Goelz, S. , Natarajan, A. , Lee, S. et al, Risk of natalizumab‐associated progressive multifocal leukoencephalopathy. N. Engl. J. Med. 2012. 366: 1870–1880. [DOI] [PubMed] [Google Scholar]
- 40. Bonig, H. , Wundes, A. , Chang, K. H. , Lucas, S. and Papayannopoulou, T. , Increased numbers of circulating hematopoietic stem/progenitor cells are chronically maintained in patients treated with the CD49d blocking antibody natalizumab. Blood 2008. 111: 3439–3441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. del Pilar Martin, M. , Cravens, P. D. , Winger, R. , Frohman, E. M. , Racke, M. K. , Eagar, T. N. , Zamvil, S. S. et al, Decrease in the numbers of dendritic cells and CD4+ T cells in cerebral perivascular spaces due to natalizumab. Arch. Neurol. 2008. 65: 1596–1603. [DOI] [PubMed] [Google Scholar]
- 42. Perkins, M. R. , Ryschkewitsch, C. , Liebner, J. C. , Monaco, M. C. , Himelfarb, D. , Ireland, S. , Roque, A. et al, Changes in JC virus‐specific T cell responses during natalizumab treatment and in natalizumab‐associated progressive multifocal leukoencephalopathy. PLoS Pathog. 2012. 8: e1003014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Warnke, C. , Mausberg, A. K. , Stettner, M. , Dehmel, T. , Nekrich, L. , Meyer zu Horste, G. , Hartung, H. P. et al, Natalizumab affects the T‐cell receptor repertoire in patients with multiple sclerosis. Neurology 2013. 81: 1400–1408. [DOI] [PubMed] [Google Scholar]
- 44. Schneider‐Hohendorf, T. , Rossaint, J. , Mohan, H. , Boning, D. , Breuer, J. , Kuhlmann, T. , Gross, C. C. et al, VLA‐4 blockade promotes differential routes into human CNS involving PSGL‐1 rolling of T cells and MCAM‐adhesion of TH17 cells. J. Exp. Med. 2014. 211: 1833–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Stuve, O. , Marra, C. M. , Jerome, K. R. , Cook, L. , Cravens, P. D. , Cepok, S. , Frohman, E. M. et al, Immune surveillance in multiple sclerosis patients treated with natalizumab. Ann. Neurol. 2006. 59: 743–747. [DOI] [PubMed] [Google Scholar]
- 46. Gasnault, J. , Kahraman, M. , de Goer de Herve, M. G. , Durali, D. , Delfraissy, J. F. and Taoufik, Y. , Critical role of JC virus‐specific CD4 T‐cell responses in preventing progressive multifocal leukoencephalopathy. AIDS 2003. 17: 1443–1449. [DOI] [PubMed] [Google Scholar]
- 47. Du Pasquier, R. A. , Schmitz, J. E. , Jean‐Jacques, J. , Zheng, Y. , Gordon, J. , Khalili, K. , Letvin, N. L. et al, Detection of JC virus‐specific cytotoxic T lymphocytes in healthy individuals. J. Virol. 2004. 78: 10206–10210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tan, I. L. , McArthur, J. C. , Clifford, D. B. , Major, E. O. and Nath, A. , Immune reconstitution inflammatory syndrome in natalizumab‐associated PML. Neurology 2011. 77: 1061–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Aly, L. , Yousef, S. , Schippling, S. , Jelcic, I. , Breiden, P. , Matschke, J. , Schulz, R. et al, Central role of JC virus‐specific CD4+ lymphocytes in progressive multi‐focal leucoencephalopathy‐immune reconstitution inflammatory syndrome. Brain 2011. 134: 2687–2702. [DOI] [PubMed] [Google Scholar]
- 50. Metz, I. , Radue, E. W. , Oterino, A. , Kumpfel, T. , Wiendl, H. , Schippling, S. , Kuhle, J. et al, Pathology of immune reconstitution inflammatory syndrome in multiple sclerosis with natalizumab‐associated progressive multifocal leukoencephalopathy. Acta Neuropathol. 2012. 123: 235–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Martin‐Blondel, G. , Bauer, J. , Cuvinciuc, V. , Uro‐Coste, E. , Debard, A. , Massip, P. , Delisle, M. B. et al, In situ evidence of JC virus control by CD8+ T cells in PML‐IRIS during HIV infection. Neurology 2013. 81: 964–970. [DOI] [PubMed] [Google Scholar]
- 52. Morgan, D. J. , Liblau, R. , Scott, B. , Fleck, S. , McDevitt, H. O. , Sarvetnick, N. , Lo, D. et al, CD8(+) T cell‐mediated spontaneous diabetes in neonatal mice. J. Immunol. 1996. 157: 978–983. [PubMed] [Google Scholar]
- 53. Casanova, E. , Fehsenfeld, S. , Mantamadiotis, T. , Lemberger, T. , Greiner, E. , Stewart, A. F. and Schutz, G. , A CamKIIalpha iCre BAC allows brain‐specific gene inactivation. Genesis 2001. 31: 37–42. [DOI] [PubMed] [Google Scholar]
- 54. Saxena, A. , Bauer, J. , Scheikl, T. , Zappulla, J. , Audebert, M. , Desbois, S. , Waisman, A. et al, Cutting edge: multiple sclerosis‐like lesions induced by effector CD8 T cells recognizing a sequestered antigen on oligodendrocytes. J. Immunol. 2008. 181: 1617–1621. [DOI] [PubMed] [Google Scholar]
- 55. Vizler, C. , Bercovici, N. , Heurtier, A. , Pardigon, N. , Goude, K. , Bailly, K. , Combadiere, C. et al, Relative diabetogenic properties of islet‐specific Tc1 and Tc2 cells in immunocompetent hosts. J. Immunol. 2000. 165: 6314–6321. [DOI] [PubMed] [Google Scholar]
- 56. Wyss, L. , Schafer, J. , Liebner, S. , Mittelbronn, M. , Deutsch, U. , Enzmann, G. , Adams, R. H. et al, Junctional adhesion molecule (JAM)‐C deficient C57BL/6 mice develop a severe hydrocephalus. PLoS One 2012. 7: e45619. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary Material