

Abstract
Polymorphisms in the MHC class II (MHCII) genes are strongly associated with rheumatoid arthritis, supporting the importance of autoreactive T helper (Th) cells for the development of this disease. Here, we used pristane‐induced arthritis (PIA), induced by the non‐antigenic hydrocarbon pristane, to study the impact of different MHCII alleles on T‐cell activation and differentiation. In MHCII‐congenic rats with disease‐promoting MHCII alleles, pristane primarily induced activation of Th1 cells, whereas activated T cells were Th17 biased in rats with protective MHCII alleles. Neutralization of IFN‐γ during T‐cell activation abrogated the development of disease, suggesting that Th1 immunity is important for disease induction. Neutralization of IL‐17, by contrast, suppressed arthritis only when performed in rats with established disease. Adoptive T‐cell transfers showed that T cells acquired arthritogenic capacity earlier in strains with a prevailing Th1 response. Moreover, upon pristane injection, these strains exhibited more Ag‐primed OX40+ and proliferating T cells of polyclonal origin. These data show that T cells are polarized upon the first encounter with peptide‐MHCII complexes in an allele‐dependent fashion. In PIA, the polyclonal expansion of autoreactive Th1 cells was necessary for the onset of arthritis, while IL‐17 mediated immunity contributed to the progression to chronic disease.
Keywords: Animal models, Autoimmunity, Immune responses, MHC, Rheumatoid arthritis, T helper (Th) cells
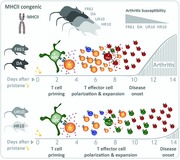
Using MHCII‐congenic rats injected with the hydrocarbon pristane, we show that Th‐differentiation is determined by the MHCII‐allele rather than by adjuvant‐induced cytokines. Strains with MHCII‐alleles that bias the immune response toward Th1 (FR61, DA) develop more severe arthritis with an earlier onset than strains with a Th17‐biased response (UR10, HR10).
Introduction
The MHC is located within a 3.6 Mb region on human chromosome 6 1, 2. The region encodes over 250 genes of which approximately 40% have immune related functions 1, 2, 3, 4. Extensive linkage disequilibrium 5, 6, 7, 8 and a high degree of sequence variation 9, 10 further contribute to the complexity of the MHC. The most polymorphic regions are found inside the MHCI and MHCII loci, which contain genes encoding for the MHC class I and II Ag‐presenting molecules. The MHCII locus also contains a number of mostly non‐polymorphic genes encoding proteins involved in the processing, editing and transportation of Ags for presentation on MHC molecules 11, 12, 13, 14.
Besides pathogen‐derived Ags, MHCII molecules present self‐Ags to CD4+ T cells, which is important for maintaining the size of the peripheral T‐cell pool 15, 16, 17. Tolerance to these self‐Ags is established through elimination of autoreactive T cells in the thymus, generation of T regulatory cells (Treg cells), as well as by several peripheral mechanisms of which anergy and deletion of autoreactive cells are the best characterized 18, 19. Nevertheless, in ∼3–5% of humans 20, 21, these control mechanisms are not sufficient and autoimmunity ensues.
The genetic risk associated with autoimmunity is largely governed by genes in the MHC 4, 22, in particular HLA‐DRB1, which accounts for approximately 36% of the heritability in rheumatoid arthritis (RA) 23, 24. To study the mechanisms underlying autoimmunity, animal models have been established in which self‐tolerance is broken by an active immunization with an Ag. In mice and rats, immunization with tissue‐specific Ags or even ubiquitously expressed proteins induces organ‐specific autoimmunity 25, 26, 27, 28. Such induced type of autoimmunity is often dependent on T cells recognizing a specific peptide‐MHCII complex; for example, in collagen‐induced arthritis (CIA), a peptide derived from type II collagen presented by H2‐Aq is crucial for the development of disease 29, 30, 31, 32. However, the ensuing autoimmunity in such models is not necessarily T‐cell‐driven 29, 33, 34, 35 or induced by an autologous T‐cell epitope: mice immunized with rat, bovine or chicken CII, for example, respond strongly to stimulation with the heterologous CII but relatively weakly to autologous mouse CII 36, 37.
A T‐cell‐driven arthritis model that is not dependent on the administration of exogenous Ags is pristane‐induced arthritis (PIA) in the rat 15, 18, 38. Pristane is a saturated, naturally occurring, hydrocarbon that is used for ascites production and the induction of lupus in mice 39, 40, 41. In DA rats, a single intradermal injection of pristane induces a chronic relapsing polyarthritis that can remain active for at least 200 days 18. The disease onset is early, highly synchronized between animals, and accompanied by elevated levels of IL‐6, rheumatoid factor, and a strong acute phase response 15, 18, 42, 43. The induction of arthritis does not appear to be dependent on B cells or the production of antibodies, since adoptive transfer of CD4 T cells from rats injected with pristane induces a disease in naïve rats that closely mimics the manifestations in PIA 14, 18, 23, 44.
Similar to RA, PIA is associated with multiple discrete loci in the MHC 15, 44 of which two have been mapped at high resolution; a 33‐kb locus in the MHCIII‐region (Ltab‐Ncr3), and a 0.2 Mb interval, denoted T cell selection QTL 2 (Tcs2) 12, in the MHCII‐region. Analysis of Tcs2‐congenic strains has demonstrated that certain MHCII alleles promote PIA whereas other alleles confer a protective effect 15. Functional studies and analysis of coding variants between different Tcs2‐congenic strains have suggested that RT1‐B, the rat orthologue to human HLA‐DQ, is responsible for the Tcs2‐association 15.
Here, we used Tcs2‐congenic strains to investigate how different MHCII alleles influence T‐cell differentiation in rats injected with pristane. We show that pristane administration induces a Th1 biased immune response in rats with disease‐promoting MHCII alleles and that IFN‐γ, a hallmark Th1 cytokine, is necessary for the development of PIA. Moreover, upon pristane administration, T cells in strains with promoting MHCII alleles acquired an arthritogenic capacity earlier, were more proliferative and contained a larger fraction of Ag‐experienced cells compared to strains with protecting MHCII alleles.
Results
Arthritis‐promoting MHCII alleles trigger an early expansion of pathogenic T cells
The 206 kb region that constitutes Tcs2 contains 12 genes of which RT1‐DMb (HLA‐DMB) and Btnl2 are located within the QTL borders (Fig. 1A) 12. We have previously shown that Tcs2‐congenic strains, that is, DA.1HR61, DA.1UR10, and DA.1FR9, differ in their susceptibility to PIA due to coding polymorphisms in the RT1‐B locus 15. DA.1HR61, and to a lesser extent, DA.1UR10 show a later disease onset compared to non‐congenic DA rats and are protected from severe PIA in the acute phase of the disease (i.e. the first ∼40 days after pristane injection). DA.1FR9, by contrast, develops a more severe form of arthritis with an early onset. In the present study, we also used two new strains, DA.1HR10 and DA.1FR61, which refine the congenic intervals in DA.1HR61 and DA.1FR9, respectively (for a physical map over all strains, see Tuncel at al. 12). The development of PIA in DA.1HR10 and DA.1FR61 was comparable to the arthritis development in the respective parental strains (Fig. 1B and C; see also Supporting Information Fig. 1 for a comparison of PIA in DA.1HR10 versus DA.1HR61 and DA.1FR61 versus DA.1FR9).
Figure 1.
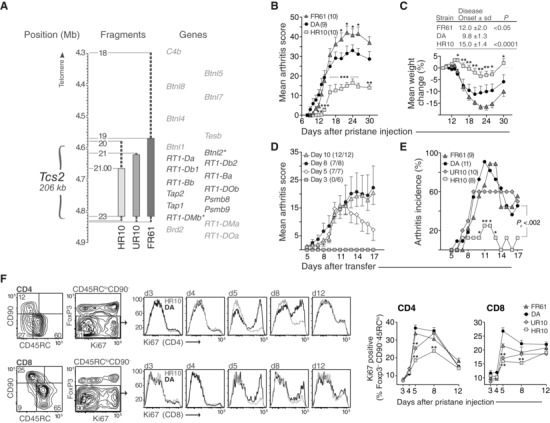
Tcs2 controls pathogenicity and expansion of T cells after pristane administration. (A) Physical map of the rat MHC‐II region. Asterisked genes are located within the flanking borders of T cell selection QTL 2 (Tcs2) while shaded genes are excluded. Congenic strains are depicted as vertical bars with dashed lines representing intervals of unknown genotype. Numbers next to fragments represent genetic markers and have been described previously 12. (B‐C) Clinical arthritis scores (B) and weight change (C) in DA and Tcs2‐congenic strains after pristane administration (max score = 60). Inset table shows day of disease onset (± standard deviation, S.D.) and p‐value versus DA. (D) Individual transfer T cells were adoptively transferred (one donor to one recipient) on day 3, 5, 8, or 10 after administration of pristane. Numbers in brackets depict recipients with arthritis out of total. (E) Individual transfer between Tcs2‐congenic strains. T cells were transferred on day 5 after pristane administration. Pc = p value for cumulative incidence. (F) Expression of Ki67 in CD4+ (upper row) and CD8+ (lower row) activated/memory T cells (CD90‐ CD45RClo Foxp3‐) in pristane dLNs at indicated time‐points after disease induction. Counter plots show gating strategy and adjacent histograms representative examples of Ki67 expression in DA (black) and DA.1HR10 (shaded); n = 5 per group. A–F: *p<0.05, **p<0.01 (by Mann–Whitney). (B–E) Data shown are representative of at least three independent experiments. (F) Data shown are pooled from five independent experiments. Error bars in B, C (graph), D, E, and F (graphs) represent standard error of the mean (SEM).
We first asked whether the variation in disease onset between Tcs2‐congenic strains and DA was due to the extent of priming the T cells required to become arthritogenic. To test the arthritogenicity of T cells after pristane administration, we performed adoptive cell transfers where T cells from draining lymph nodes (dLNs) were transferred from one donor into one MHCII syngeneic recipient (here referred to as “individual transfer”; see Methods). In DA rats, donor T cells required a minimum of 5 days of priming in vivo to transfer arthritis and 8 days to become fully arthritogenic (Fig. 1D). Similarly, in DA.1FR61, T cells from eight out of nine donors (89%) transferred arthritis after 5 days of priming in vivo. In contrast, this 5‐day interval was only sufficient to prime T cells in 2/8 DA.1HR10 donors (25%, p<0.002 versus DA) and 6/10 DA.1UR10 (60%, p<0.2 versus DA) donors to transfer arthritis (Fig. 1E).
To identify whether the arthritogenic capacity of donor T cells was due to qualitative or quantitative differences between the Tcs2‐congenic strains and DA, we first assessed the frequency of proliferating (Ki67+) memory/activated T conventional (Tconv) cells (CD45RClo CD90– Foxp3– CD4+ 45) in pristane‐injected rats. The frequency of proliferating cells in this subset in DA and DA.1FR61 increased by ∼4‐fold from day 4 to day 5 and remained high until at least day 8 (Fig. 1F). DA.1UR10 showed fewer proliferating memory/activated Tconv cells than DA and DA.1FR61 at day 5 but not at day 8, while the proliferation of these cells in DA.1HR10 was significantly lower at both time‐points. This variation in T‐cell proliferation was not specific for memory/activated Tconv cells but was also seen among CD8 T cells (Fig. 1F) and in other CD4 subsets such as Treg cells and CD90+ recent thymic emigrants (RTEs) in DA.1HR10 (Supporting Information Fig. 2).
Taken together, the early onset in DA and DA.1FR61 compared to DA.1UR10 and DA.1HR10 correlated with a more rapid expansion of arthritogenic effector T cells in the pristane‐dLNs.
Pristane induces a polyclonal expansion of Tconv cells
The rate of T‐cell proliferation was increased in strains with an early arthritis onset. To assess the clonality of proliferating Tconv cells, we analyzed T cells in the dLN from pristane‐injected rats using available anti‐Vβ antibodies by flow cytometry. The frequency of Vβ10, Vβ16.1, Vβ8.5, and Vβ8.2/8.4 subsets, which together constituted ∼25% of the total Vβ repertoire, varied significantly between the strains (Fig. 2). Vβ10+ Tconv cells, for example, which have been shown to adoptively transfer PIA 23, were more abundant in DA and DA.1FR9 rats, while the frequency of Vβ16.1+ Tconv cells was highest in DA.1HR61 and DA.1UR10. However, except for Vβ8.5, which was more frequent in DA and DA.1FR9 after pristane administration, the relative frequency of each Vβ subset among total Tconv cells in dLNs was not affected by pristane (Fig. 2). Thus, this indicates that T cells of a broad clonal origin, of which some are likely to be autoreactive, expand in pristane‐dLNs.
Figure 2.
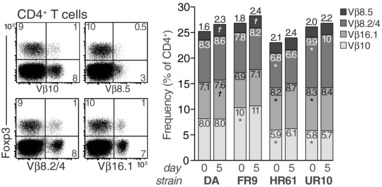
The TCR Vβ repertoire on CD4 T cells is largely unaffected by pristane administration. CD4+ T cells were analyzed for the relative frequency of Vβ10, Vβ16.1, Vβ8.5, and Vβ8.2/8.4+ subsets in dLNs before (day 0) and 5 days after pristane injection. Stacked bars show Vβ frequencies among total CD4+ T cells (no significant differences were observed between Foxp3+ and Foxp3‐ CD4 T cells). Numbers within bars represent mean frequencies. Data are representative of at least three independent experiments. n = 6 rats/ group; *p<0.01 compared to DA day 0, † p<0.01 compared to same strain day 0 (Mann–Whitney U).
Antigen‐experienced T cells are enriched in the dLNs of rats with arthritis‐promoting MHCII alleles
Flow cytometric analyses of Treg cells in naïve rats did not reveal any differences in frequencies or numbers between susceptible and protected strains (Supporting Information Fig. 3). After pristane injection, DA.1HR10 rats showed a modest increase in the frequency of Treg cells whereas there was a reduction of Treg cells in DA.1UR10 compared to DA and DA.1FR61 (Fig. 3A), indicating that the protective MHCII alleles in DA.1HR10 and DA.1UR10 did not simply favor a selection or expansion of Treg cells over Tconv cells. However, the proportion of Treg cells that were negative for the tumor necrosis factor receptor OX40 was almost 2‐fold higher in DA.1HR10 compared to all other strains (Fig. 3B, 3C). This may suggest that a significantly larger proportion of the Treg cells in DA.1HR10 are functionally suppressive since ligation of OX40 on Treg cells has been shown to turn off their suppressive capacity 29, 34.
Figure 3.
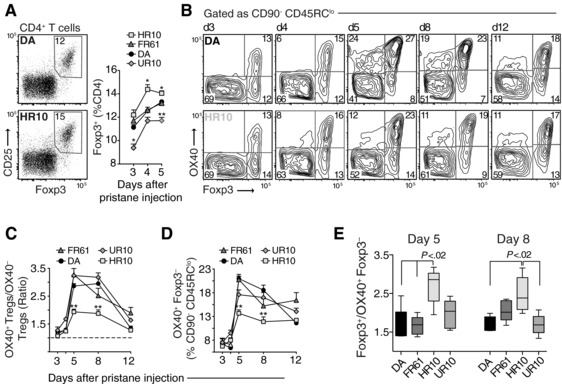
Variation in OX40+ Treg cells and T effector cells among DA and Tcs2‐congenic strains. (A) Frequency of Treg cells (CD25+ Foxp3+) among total CD4+ T cells in pristane dLNs. Dot plots show representative examples of Treg cells from dLN on day 4 after pristane administration in DA and DA.1HR10. (B) Expression of OX40 on Foxp3+ and Foxp3‐ T effector/memory cells (CD45RClo CD90‐ CD4+; see Fig. 1F for gating strategy). Counter plots show representative examples of OX40 expression at different time‐points after pristane administration in DA (top) and DA.1HR10 (bottom). Plots are representative of —three to five independent experiments. (C) Ratio of OX40+ Treg cells to OX40‐ Treg cells (gated as shown in (B)). (D) Frequency of Ag‐primed (OX40+) Foxp3‐ effector/memory T cells. (E) Ratio of total Treg cells to OX40+ Foxp3‐ effector/memory T cells at day 5 and 8 after pristane injection. n = 5 rats/group; *p<0.05, **p<0.01 (by Mann–Whitney); data shown are pooled from three to five independent experiments (A–E). Error bars represent standard error of the mean (SEM) (A, C, and D). In (E) each box represents the 25th to 75th percentiles; lines inside the boxes represent the median; lines outside the boxes represent the 10th and the 90th percentiles.
On Tconv cells, OX40 is expressed on effector cells that are Ag‐primed or have undergone TCR ligation 25, 27, 46. CD4 T cells in dLNs from DA and DA.1FR61 showed significantly higher proportions of OX40+ cells on day 5 and day 8 after pristane administration compared to DA.1HR10 (Fig. 3B and 3D). This early expansion of Ag‐primed T cells in DA and DA.1FR61 likely explains why T cells in these strains were more arthritogenic on day 5 than T cells in DA.1HR10 and DA.1UR10 (Fig. 1E). Moreover, when the variation in Treg cell frequency was considered (Fig. 3A), the Treg/T effector cell ratio was approximately twofold higher in DA.1HR10 compared to DA and DA.1FR61 on day 5 and 8 after pristane administration (Fig. 3E). Together, these data suggest that promoting MHCII alleles accelerate the onset of PIA by expanding Ag‐specific T cell clones in the pristane dLNs.
Activated T cells are predominantly of Th1 type in rats with disease‐promoting MHCII alleles
To assess differences in T‐cell responses between the strains, we determined mRNA levels of cytokines and Th‐lineage‐defining transcription factors in purified CD4 T cells from pristane‐dLNs. This showed that IFN‐γ was upregulated in strains with promoting MHCII alleles and, surprisingly, a distinct upregulation of Th17 signature genes (IL‐17A, IL‐22, and RORγt) in T cells from DA.1HR10 (Fig. 4A). A similar shift in Th1/Th17 associated cytokines was observed when stimulating T cells from pristane‐immunized DA and DA.1HR10 rats with anti‐CD3/CD28 in vitro (Fig. 4B).
Figure 4.
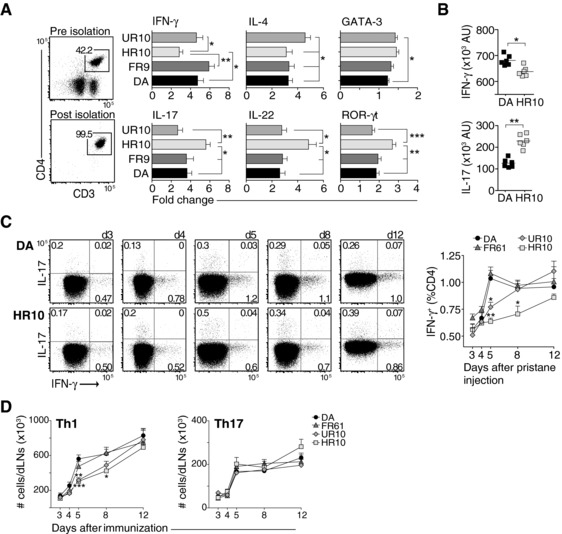
Administration of pristane drives Th1‐differentiation in rats with disease promoting MHCII alleles. (A) Messenger RNA expression (as determined by qPCR) of cytokine and Th‐lineage‐defining transcription factors from isolated CD4 T cells harvested on day 8 after pristane injection. Horizontal bars show mean values ±SEM. Variation is expressed as fold‐change differences between individuals, n = 9–10 per group. (B) Cytokine levels after in‐vitro stimulation of dLN cells from DA and DA.1HR10 with anti‐CD3/CD28. Cells were harvested 8 days after pristane administration. (C) Frequencies of IFN‐γ+ (Th1) and IL‐17+ (Th17) cells among non‐RTE (CD90‐) CD4+ T cells. Dot plots show DA (upper row) and DA.1HR10 (lower row). Data are summarized in adjacent line chart. n = 5 rats/group. (D) Total number of CD90‐ Th1 and Th17 cells in dLNs. AU = arbitrary units. (A–D) *p < 0.05; **p < 0.01; ***p < 0.001 (by Mann–Whitney). Results shown are representative of —two to three independent experiments (A and B). Data shown in (C and D) are pooled from five independent experiments. Error bars represent SEM (A, C, and D); vertical line represents mean (B).
Flow cytometric analyses of CD4 T cells showed that IFN‐γ producing Th1 cells increased by approximately twofold between day 3 and day 5 in DA and DA.1FR61 rats (Fig. 4C). By contrast, the frequency of Th1 cells in DA.1HR10 and DA.1UR10 was only marginally increased during the first 5 days of PIA. Further, the total number of Th1 cells was significantly increased in dLNs from DA and DA.1FR61 compared to DA.1HR10 and DA.1UR10 on day 5 and 8 after immunization. (Fig. 4D). Unlike unstimulated cells (Fig. 4A) and cells stimulated with anti‐CD3/CD28 (Fig. 4B), PMA/ionomycin stimulation did not reveal any significant differences in IL‐17 expression between the strains (data shown for total numbers of Th17 cells in Fig. 4D).
Taken together, these data show that after pristane administration T cells differentiate predominantly into Th1 lineage in rats with arthritis susceptible MHCII alleles.
Pristane‐induced arthritis is dependent on IFN‐γ for T‐cell priming and IL‐17 for disease perpetuation
A significant expansion of Th1 cells was observed in strains with an early‐onset PIA. To probe whether IFN‐γ is critical for the induction of arthritis, we treated rats with anti‐IFN‐γ and anti‐IL‐17 neutralizing antibodies at day 2, 4, and 6 after pristane administration.
All rats, regardless of MHCII haplotype, that were treated with anti‐IFN‐γ Abs developed significantly milder disease compared to non‐treated control rats (Fig. 5A), whereas no effect was observed when treating with anti‐IL‐17 Abs (Fig. 5B). In addition, the onset of PIA was delayed by up to 4 days in rats treated with anti‐IFN‐γ Abs (Fig. 5C), suggesting that T‐cell priming in PIA is IFN‐γ‐dependent also in strains associated with a Th17 biased immune response, such as DA.1HR10.
Figure 5.
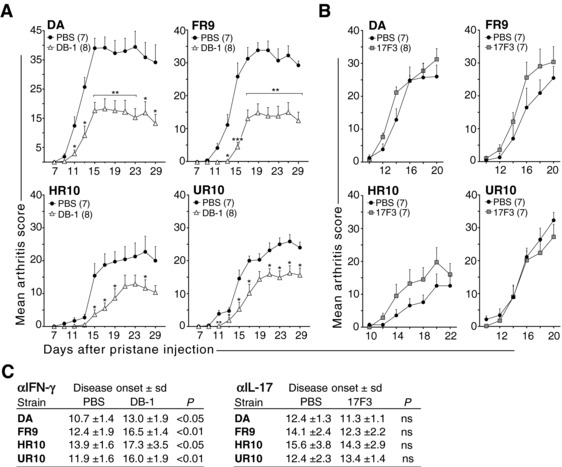
Pristane‐induced arthritis is dependent on IFN‐γ for T‐cell priming. (A‐B) Antibodies to INF‐γ (DB‐1) (A) and IL‐17 (17F3) (B) were administrated s.c. on day 2, 4, and 6 after pristane administration (control rats were injected with PBS; since, DB‐1 and 17F3 are both of mouse IgG1 isotype). (C) Day of disease onset for rats treated with DB‐1 (left panel) and 17F3 (right panel). n = 7–8 rats/group as indicated in figure; *p < 0.05; **p < 0.01; ***p < 0.001 (Mann–Whitney). Data are shown as mean + SEM and are pooled from two independent experiments with similar results.
We next assessed whether IFN‐γ and/or IL‐17 was required for the progression of established arthritis by injecting neutralizing antibodies at day 8, 10, 12, and 14 after pristane administration (Fig. 6). The effect of IFN‐γ neutralization on arthritis progression was negligible or promoting (Fig. 6A–6C) in all strains except in DA.1UR10 where it slowed disease development (Fig. 6D). Neutralization of IL‐17, by contrast, was highly efficient in suppressing the progression of disease in all strains (Fig. 6A–D). This effect of IL‐17 neutralization was followed by a twofold reduction in circulating neutrophils (Fig. 6E), suggesting that IL‐17 operates on granulopoiesis and the recruitment of neutrophils to the joints in PIA.
Figure 6.
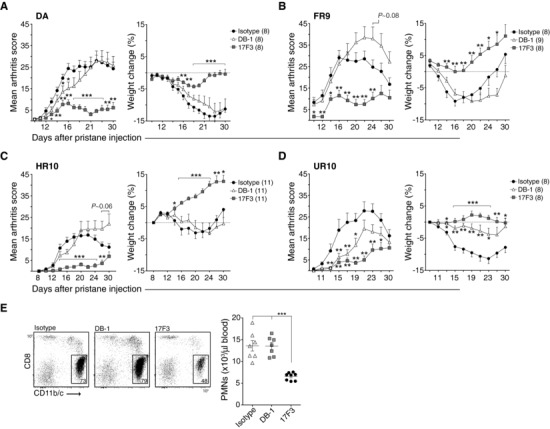
IL‐17 more than IFN‐γ is required for the progression of arthritis in PIA. (A–D) Neutralizing antibodies to IFN‐γ (DB‐1) and IL‐17 (17F3) were injected i.v. into rats at day 8, 10, 12, and 14 after pristane administration using MOPC‐21 as an isotype control. Disease development (left) and weight change (% versus day of onset, right) are shown for DA (A), DA.1FR9 (B), DA.1HR10 (C) and DA.1UR10 (D); n = 8–11 per group as indicated in figures, *p < 0.05; **p < 0.01; ***p < 0.001 (compared to isotype); p‐values for trend in (B) and (D) represent differences between DB‐1 and MOPC‐21 (All data in A–D was analyzed by Mann‐Whitney). (E) Frequencies (gates in dot plots) and numbers (scatter plot) of CD11 b/c+ CD8a‐ polymorphonuclear leukocytes (PMNs) in blood were determined in rats shown in (A) at day 16 post pristane injection; ***p < 0.001 (compared to isotype and DB‐1, Mann–Whitney). Data shown in A–D are pooled from two independent experiments with similar results. Error bars in (A–D) represent SEM; vertical line and error bars in (E) represent mean and standard deviation, respectively.
Discussion
There is compelling evidence that IL‐17 contributes to the pathology in a number of autoimmune diseases 47, 48. IL‐17 (or IL‐23) deficient mice, for example, are protected from arthritis and depletion of IL‐17 has been shown to suppress both priming of T‐cell responses as well as the progression of ongoing disease 49, 50, 51, 52. In addition, IL‐17 is enriched in the blood and synovial fluid of patients with RA, suggesting that IL‐17 might be important also in human RA 53, 54, 55. In comparison, the role of IFN‐γ in autoimmune arthritis has been more controversial, probably as its modus operandi can be both pro‐inflammatory and anti‐inflammatory depending on context 56. Here, we show that activation of autoreactive T cells through administration with a non‐antigenic hydrocarbon skews the immune response toward Th1 and that neutralization of IFN‐γ protects rats from developing severe arthritis. Importantly, we found that an early Th1 signature dominated in rats expressing arthritis‐promoting MHCII alleles, providing a link between effector T‐cell differentiation, disease phenotype and MHCII genotype.
The MHCII locus confers the highest genetic risk in RA 57 and the major impact of permissive MHCII alleles is likely to be on the early disease development. Although poorly characterized in humans, the preclinical stage of arthritis is likely to involve a series of linked events that eventually cause a breech in tolerance, activation of T and B cell responses, and an attack on peripheral joints. However, these events do not necessary need to unfold in the joints. In fact, telomeric length studies have shown that T cells are recruited to the joints in a pre‐activated state 1 suggesting that the joint is not the primary organ for a cognate T‐cell response. It has also been suggested that joint‐infiltrating T cells are of polyclonal origin; however, the Ag‐specificities of these T cells remain largely unknown even if post‐translationally modified peptides 3, and peptides with weak affinity to MHCII 5, 7 could serve as potential target Ags.
A similar series of events, albeit in a much‐compressed time frame, may lead to the development of PIA in rats. First, pristane, being a hydrocarbon, cannot bind to MHCII molecules and it is unlikely that pristane operates directly on T cells or is presented by CD1 9. Instead, functional and genetic data have suggested that stimulation of innate signals and pattern‐recognition receptors (PRRs), such as Toll‐like receptors and C‐type lectin receptors, are important for the triggering of an adaptive response in PIA 11, 13, 14. Although it has never formally been excluded that pristane cannot be presented by CD1, blocking of MHCII‐TCR interactions during T‐cell priming (2–4 days post pristane) is sufficient to ameliorate PIA 15. Second, activation of skin‐resident dendritic cells appears to be important for the priming of autoreactive T cells in PIA 18. Third, injections of 14C‐labelled hexadecane have shown that mineral oils accumulate within the dLNs with no detectable oil being found in the joints themselves 20, suggesting that the primary site of T‐cell activation is within the lymphoid organs. Our data suggest that the primary sites of T‐cell activation are LNs draining the injection site as T cells at these locations could transfer arthritis as early as 5 days after pristane administration. We show that expanded T cells in dLNs are of broad clonality, which is consistent with previous studies showing that arthritogenic T cells in PIA use a diverse repertoire of TCR Vβ chains 23.
The co‐stimulatory molecule OX40 is specifically upregulated on Ag‐primed T cells 25, 27 and has been shown to promote clonal expansion and survival of T effector and memory cells 29, 31. We assessed early OX40 expression in pristane dLNs and found that OX40 positive T effector cells (CD45RClo CD90– Foxp3–) were enriched in the dLNs of rats carrying arthritis‐promoting MHCII alleles. OX40 expression peaked around day 5 after pristane injection, which coincides with the emergence of arthritogenic T cells in DA and DA.1FR61 rats and the establishment of a Th1 signature in these strains. Together, these findings provided a possible explanation for the early arthritis onset associated with certain MHCII alleles; namely, the rapid expansion and differentiation of Ag‐primed T cells to arthritogenic effector cells.
In contrast to its role on Tconv cells, the role of OX40 on Treg cells remains controversial. Several studies have shown that ligation of OX40 on Treg cells dampens their regulatory activity 29, 34, and, in the presence of IFN‐γ and IL‐4, favors expansion of T effector cells over Treg cells 36. This could explain the expansion of T effector cells we observed in DA and DA.1FR61 after pristane administration, where the local cytokine milieu, being high in IFN‐γ, would favor T effector over Treg cell proliferation. Moreover, the high expression of OX40 on Treg cells in DA and DA.1FR61 could be associated with a poor immune regulatory function in these strains, which could amplify an ongoing expansion of autoreactive T cells even further. Consistent with this notion, also CD8 T cells, which are not restricted by MHCII or essential for the development of PIA 15, showed a greater proliferation in strains with arthritis promoting MHCII alleles. Such “by‐stander” proliferation could be associated with OX40 expression on Treg cells, since IL‐2 and other growth factors could become more available in the absence of sufficient Treg suppression. Further, this could mean that Treg cells in DA.1HR10, which consisted of a larger proportion of OX40 negative cells, are better suppressor cells than Treg cells in DA and DA.1FR61. However, OX40 deficiency in Treg cells has also been associated with a lack of immune suppression, as shown for T‐cell‐induced colitis 39. Hence, what implications the differential expression of OX40 may have on Treg cells in PIA and what relevance this may have for the protection observed in DA.1HR10 remains to be determined.
Effector T cells in rats carrying disease promoting MHCII alleles differentiated to Th1 cells upon pristane administration. This Th1 signature is likely to be responsible for the early onset and severe progression of arthritis in DA and DA.1FR61 since neutralization of IFN‐γ in pre‐arthritic rats delayed the onset and reduced the severity of PIA. Hence, depleting IFN‐γ during T‐cell priming in DA and DA.1FR61 rats seems to phenocopy the characteristic “DA.1HR10 disease”. Unlike DA and DA.1FR61, DA.1HR10 rats did not demonstrate a propensity for developing Th1 immunity. Instead, CD4 T cells from DA.1HR10 expressed higher levels of IL‐17, IL‐22 and ROR‐γt compared to DA and DA.1FR61, suggesting that pristane drives the expansion of Th17 cells in DA.1HR10. Importantly, this expansion of IL‐17 producing T cells did not seem to be pathogenic since depletion of IL‐17 in pre‐arthritic rats did not influence the onset or progression of PIA in DA.1HR10 or in any of the other strains. Interestingly, the frequency and total number of IFN‐γ producing Th1 cells increased over time in both DA.1HR10 and DA.1UR10, and reached the levels of DA and DA.1FR61 by the time of arthritis onset. This suggests that it is the expansion of autoreactive Th1 cells that ultimately determines the onset of PIA.
Th1 type immune responses have previously been demonstrated to be important for the induction of G6PI‐induced arthritis 58 and neutralization of IFN‐γ inhibits arthritis in proteoglycan‐induced arthritis (PGIA) 59, 60. Recently, the formation of neutrophil extracellular traps (NETs) was shown to induce expansion of Th1 cells and accelerate the onset of CIA in mice 61. Interestingly, pristane has been shown to be a potent inducer of NETs 62, suggesting that NETs could serve as an inducer of Th1 immunity in PIA. Th1 mediated immunity may also be relevant in early RA; studies have demonstrated that the preclinical phase of RA is dominated by Th1‐associated cytokines and chemokines (IFN‐γ, IL‐12, and CXCL10) 63, 64. Moreover, lymph node biopsies taken from individuals with an increased RA‐risk (that is, IgM‐RF and/or ACPA positive individuals with arthralgia but without any evidence of arthritis) and from patients with early RA showed elevated levels of T cells with a Th1 profile (CXCR3+ CCR6− CCR4−) as compared to healthy controls, whereas no such differences were found for T cells with a Th17 or Th2 profile 65. In established RA, however, the presence of Th1 cells seems to be associated with disease protection rather than exacerbation. Likewise, in the PGIA model, IFN‐γ deficient mice eventually succumb to severe arthritis, probably as a result of increasing IL‐17 levels 59. In PIA, DA.1HR10 rats develop the same severe chronic arthritis as non‐congenic DA rats 15, which is consistent with the finding here that the onset of disease marks the transition from an IFN‐γ to an IL‐17 dependent disease. Whether Th17 cells are the primary source of IL‐17 in established PIA remains to be determined. Unpublished findings from our lab show that both IL‐17 and T cells are important for the perpetuation of late‐stage chronic PIA, suggesting that Th17 cells may play a specific role in the transition to chronic disease (J.T., S.H., and R.H., unpublished results). This would be consistent with the notion that synovial infiltration of Th17 cells may play a specific role in the transition from acute synovitis to chronic inflammation in RA 49, 66.
We have shown that a Th1 type immune response is essential for the development of PIA and that Th1 skewing is solely dependent on the MHCII allele. Tcs2‐congenic rats carrying MHCII alleles that skewed the immune response toward Th1 had more Ag‐primed T cells in the dLNs, developed a more severe arthritis and showed an earlier disease onset than rats carrying MHCII alleles that skewed the immune response toward Th17. While a Th1‐type immune response was essential for the onset of PIA, IL‐17 mediated immunity was necessary for the establishment of chronic arthritis.
Materials and methods
Animals
DA/OlaHsd (Harlan Europe, The Netherlands) were maintained in a barrier facility by sister‐brother mating and were specific pathogen free (SPF) according to the current FELASA guidelines 67, which includes pathogens such as Parvovirus, Sendai virus, Hantaan virus, Coronavirus, Reovirus, Cytomegalovirus, Pasteurellaceae and Mycoplasma pulmonis. Animals were kept in a climate‐controlled environment with 14 h light/ 10 h dark cycles, housed in individually ventilated microisolator‐cages (IVC) containing wood shavings and fed standard rodent chow and filtered water ad libitum. The derivation of congenic strains has been described previously 12. All rat strains are available as an open resource at the Rat Resource and Research Center, Columbia, MO.
PIA induction
The induction and evaluation of PIA in DA rats have been described recently 18. In brief, PIA was induced by a single intradermal injection of 100 μL pristane (2,6,10,14‐tetramethylpentadecane, 95%, Acros Organics, Morris Plains, NJ, USA) at the dorsal side of the tail base. All experiments were performed in 8–11 week‐old, sex and age‐matched (±5 days) rats. Disease protected and non‐protected strains (or different treatment groups) were housed together in cages from the time‐point of weaning or for at least 2 weeks before the start of the experiment. Arthritis development was monitored daily or every second day using a macroscopic scoring system of 1–60 points per rat as described previously 18. In brief, 1 point was given for each inflamed knuckle or toe and up to 5 points were given for an affected ankle (in total 15 points per paw). All scoring was performed blinded and by different investigators. Weight loss of pristane‐injected rats was included as an objective measure of the disease severity. Weight changes in figures are indicated as % of body weight at disease onset. All experiments were approved and performed in accordance with the guidelines from the Swedish National Board for Laboratory Animals and the European Community Council Directive (86/609/EEC).
Individual transfer
Adoptive PIA transfer has been described previously 23 but was modified here as follows: A single cell suspension from a pair of inguinal LNs was prepared at RT in 5 ml transfer medium containing 3 μg/mL Concanavalin A (Con A, Sigma Aldrich, C5275), 5% FCS (Gibco), 2.4 mg/mL HEPES, 3.9 μg/mL 2‐ME, 104 IU/mL penicillin, and 10 mg/mL streptomycin (both Invitrogen Life Technologies) in Dulbecco's MEM (Gibco), using a 40 μm cell strainer (BD Falcon). The cell strainer was rinsed with 3 mL of transfer medium, and 1.5 × 108 cells were added to a 75 cm2 culture flask (2 × 106/cm2). Transfer medium was added to a final volume of 50 mL and the cells were thereafter incubated for 70 h in a humidified 5% CO2 atmosphere at 37°C. Con A‐stimulated cells were transferred to centrifugation tubes (BD Falcon), pelleted at 300 x g for 12 min at RT, and washed once in sterile PBS. The pelleted cells were gently resuspended (without adding new PBS) and then transferred to a 40 μm cell strainer placed over a clean 50 mL centrifugation tube. After washing the original tube with 200 μL of PBS, the tube was centrifuged (with the cell strainer attached) for 10 s at 200 x g to collect cells. The cell concentration was adjusted with PBS to 108 cells/mL. Naive (non‐irradiated) MHC‐II syngenic recipients were injected intravenously (i.v.) with 4 × 107 cells.
Flow cytometry
Cells were washed in cold EDTA‐FACS buffer containing 1% bovine serum albumin [BSA, Sigma‐Aldrich], 2 mM Na2EDTA [Merck, Darmstadt, Germany] and 0,02% NaN3 in PBS‐D and resuspended in the same buffer. For staining with MAbs, 106 cells/well were added in duplicates to 96‐well v‐bottom polypropylene plates (BD Falcon). For cytokine staining, cells were resuspended in transfer medium (see above) without Con A and seeded at 106 cells/well (100 μL) in duplicates in tissue‐culture treated 96‐well u‐bottom plates. The cells were mixed with an equal volume of the same medium containing 10 ng/mL Phorbol 12‐Myristate 13‐Acetate (PMA), 0.6 μg/mL ionomycin and 10 μg/mL Brefeldin A (all from Sigma‐Aldrich) and incubated for 4 h at 37°C in a 5% CO2 atmosphere. PMA‐stimulated cells were transferred to v‐bottom plates, washed once in cold EDTA‐FACS buffer, stained extracellularly with MAbs, fixed and permeabilized in BD Cytofix/Cytoperm for 20 min at RT followed by intracellular staining. Cells cultured in the absence of PMA and ionomycin served as negative controls. For the intracellular detection of Ki67 and Foxp3, cells were incubated for 45 min at RT in Fixation/Permeabilization buffer (eBioscience) and thereafter washed twice in a saponin‐containing buffer (BD Perm/Wash) before staining with anti‐Foxp3 and anti‐Ki67. Fluorescence‐minus‐one (FMO) controls were used in all experiments. LIVE/Dead Violet (Invitrogen, Carlsbad, CA) was used to gate live cells during analyses. Duplicate samples were pooled prior to acquisition on a BD LSR‐II Flow Cytometer. All data were analyzed with FlowJo (Tree Star Inc., Ashland, OR).
Antibodies for flow cytometry
The following FITC, Alexa Fluor 488, PE, Pe‐Cy5, PerCP‐Cy5.5, APC, Alexa Fluor 648, APC‐Cy7, Qdot‐655 and biotin conjugated antibodies were used for flow cytometry: CD4 (OX‐35), CD8a (OX‐8), CD25 (OX‐39), CD45RC (OX‐22), and CD3 (1F4) were purchased from BD Pharmingen (San Diego, CA); IFN‐γ (DB‐1), CD45 (OX‐1), CD90/Thy1 (OX7), TCR Vβ8.2/8.4 (R78) and αβTCR (R73) were purchased from BioLegend (San Diego, CA); TCR Vβ16.1 (His42) and TCR Vβ10 (G101) were purchased unlabeled from LSBio (Seattle, WA, USA) and used with a secondary polyclonal APC‐Cy7‐labelled goat anti‐mouse antibody (BioLegend); Foxp3 (FJK‐16s), Ki67 (SolA15) and CD11b/c (OX42) were purchased from eBioscience (San Diego, CA); Polyclonal goat‐anti mouse IL‐17 was purchased from R&D Systems (IC421P).
Treatment with anti‐IFN‐γ and anti‐IL‐17
DB‐1 (anti‐INF‐γ, mouse IgG1) was produced from B cell‐hybridoma (kindly provided by Dr. Tomas Olsson) as described previously 15. Anti‐mouse IL‐17A antibody (clone 17F3; mouse IgG1) and isotype control antibody (clone MOPC‐21) was obtained from Bio X Cell (West Lebanon, NH, USA). Cross‐reactivity to rat IL‐17 was evaluated by ELISA using supernatant from Con A‐activated cell cultures. Antibodies were administrated s.c. in 0.1 mL sterile PBS (1 mg/mL) at each flank of the tail base (0.2 mL/ 0.2 mg in total per rat and time point) as described previously 18 to target the pristane dLNs during the priming phase of PIA (day 2, 4, 6 after pristane injection). Antibodies for treatment of early‐established disease (day 8, 10, 12 and 14 after pristane injection) were administrated i.v. using the same volumes and concentrations as used for s.c. injections.
In vitro stimulation and cytokine ELISA
For the stimulation with plate‐bound anti‐CD3 (1F4, 2 μg/mL, coated overnight at 4°C in PBS), iLN cells were collected from rats at day 8 post pristane‐injection, seeded at 5.0 × 105 cells/well and cultured for 60 h at 37°C. Soluble anti‐CD28 (JJ319) was added to the cultures from start at a finale concentration of 5 μg/mL. IFN‐γ and IL‐17 in culture supernatants were quantified by ELISA using 2 μg/mL anti‐rat IFN‐γ (DB‐1, BioLegend) or 2 μg/mL anti‐mouse IL‐17 (PN 840525, R&D Systems) as capture antibodies together with biotinylated 0.5 μg/mL anti‐rat IFN‐γ (Poly5109, BioLegend) or 0.4 μg/mL anti‐mouse IL‐17 (PN 840526, R&D Systems) for detection. Eu3+‐conjugated streptavidin (DELFIA, PerkinElmer) was used as a secondary reagent. Fluorescence was detected on a Synergy 2 Multi‐Mode Microplate Reader. All values are represented as arbitrary units (AU).
Isolation of CD4 T cells
Single cell suspensions were prepared from iLNs in PBS‐D (–Ca2+ and –Mg2+), washed once and resuspended in 10 mL cold MACS buffer (0.5% BSA, 2 mM EDTA in PBS‐D) at 1 × 107 cells/mL. Ten million cells (1.0 mL) were incubated with antibodies to NK1.1 (0.1 μg, 10/78), γ/δ TCR (0.3 μg, V65), CD45RA (1 μg, OX33) and CD8a (2.7 μg, OX8) for 10 min at 4°C (all antibodies from BD). Cells were washed once in MACS buffer and resuspended in 1.0 mL of the same buffer. Dynabeads Pan Mouse IgG (DYNAL, Oslo, Norway) was added at 50 μL/sample and cells were incubated rotating for 30 minutes at 4°C and then washed once in MACS buffer. NK cells, NK T cells, B cells, γ/δ T cells, and CD8+ cells were removed by magnetic depletion. Non‐depleted cells were washed once and resuspended in 80 μL MACS buffer and incubated for 15 minutes at 8°C with 20 μL CD6‐coated microbeads (Miltenyi Biotec, GTF, Goteborg, Sweden). Labelled cells were positively selected on MS‐columns (Miltenyi Biotec). Isolated CD4 T cells were stored at −80°C until used. The purity of CD4 T cells was assessed by flow cytometry after staining with anti‐CD4 (OX35) and anti‐CD3 (1F4) and was typically ∼ 99%.
RNA extraction and qRT‐PCR
Cell pellets were allowed to thaw slightly before being resuspended in 300 μL RLT buffer (QIAGEN Nordic, Ballerup, Denmark) containing 10 μL/mL 2‐ME. Automated RNA isolation was performed on a QIACube robot using RNeasy extraction reagents (Qiagen) with on‐column DNase I digestion (Qiagen). RNA quantity and purity was assessed on a NanoDrop ND‐1000 (NanoDrop Technologies, Wilmington, DE). RNA samples were diluted to 10 ng/μL in DEPC treated water (Ambion). Complementary DNA (cDNA) was synthesized using High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA) in 50 μL reactions. Primers were designed in Primer‐BLAST (http://ncbi.nlm.nih.gov/tools/primer‐blast/index.cgi) or obtained from RTPrimerDB (medgen.ugent.be/rtprimerdb) (Table S1). SYBR‐Green PCR master mix (Applied Biosystems, Foster City, CA) was used for all PCR reactions according to the manufacture's recommendation. Quantitative RT‐PCR was performed on an ABI Prism 7900 HT Sequence Detection System (Applied Biosystems). Specificity and efficiency of all primers were validated prior to use by absolute quantification. The expression fold‐change of each target gene was determined by the relative quantification method (ΔΔCt) after normalization to the geometric mean of the reference genes Arbp, β‐Actin and GusB.
Conflict of interest
The authors have no conflict of interest to declare.
Abbreviations
- CIA
Collagen‐induced arthritis
- dLN
Draining lymph nodes
- MHCII
MHC class II
- PIA
Pristane‐induced arthritis
- RA
Rheumatoid arthritis
- Tconv cells
T conventional
- Tcs2
T cell selection QTL 2
- Th cells
T helper
- Treg cells
T regulatory cells
Supporting information
FIGURE S1. Evaluation of PIA in new MHCII congenic strains, DA.1FR61 and DA.1HR10. (A) The development of PIA in DA.1FR61 was compared to its parental congenic strain, DA.1FR9, and to DA. Arthritis clinical scores (left) and weight change (in % versus day of disease onset, right) is shown; #, significant (p<0.05) DA versus DA.1FR61; *, significant (p<0.05) DA versus DA.1FR9. (B) Arthritis clinical scores and weight change in pristane immunized DA.1HR10, DA.1HR61 and DA; *P < 0.05. All statistic comparisons in (A) and (B) were determined by Mann‐Whitney; n = 5‐11 per group as indicated in figure.
FIGURE S2. Proliferation of Foxp3+ Tregs and recent thymic emigrants (RTEs) in dLNs after pristane administration. (A) Histograms show representative expression of Ki67 in CD4+ Foxp3+ Tregs from DA (blue) and DA.1HR10 at day 4 and 5 after pristane administration. Adjacent line chart shows summarized data from all strains. (B) Corresponding data (as shown in A) for RTEs (CD90+ CD4+ T cells) in pristane dLNs. (A‐B) n = 5 per group; **p<0.01 (by Mann‐Whitney).
FIGURE S3. Variation in Foxp3+ Tregs in naive MHCII‐congenic rats does not correlate with disease susceptibility. (A) Representative counter plots of CD4 T cells in spleen of naive rats. Summarized data in (B) show frequencies of Tregs among CD4 T cells, and in (C) total number of Tregs per spleen. P‐values depicted in graphs were determined by Mann‐Whitney (n=4 per strain).
Table S1. Primers for quantitative PCR.
Supporting Information
Acknowledgements
We thank Carlos Palestro, Kristina Palestro, and Evelina Wernersson for animal care. This work was supported by grants from the Crafoord Foundation, the Kock and Österlund Foundation, the Knut and Alice Wallenberg Foundation, the Swedish Association against Rheumatism, the Swedish Medical Research Council, and the Swedish Foundation for Strategic Research. The research leading to these results has received funding from the European Community's Seventh Framework Program (FP7/2007‐2013) under the grant agreement N° HEALTH‐F4‐2010‐241504 (EURATRANS), as well as the EU Innovative Medicine initiative BeTheCure grant (IMI 115142).
Contributor Information
Jonatan Tuncel, Email: jonatan.tuncel@ki.se.
Rikard Holmdahl, Email: rikard.holmdahl@ki.se.
References
- 1. Wagner, U. G. , Koetz, K. , Weyand, C. M. and Goronzy, J. J. , Perturbation of the T cell repertoire in rheumatoid arthritis. Proc. Natl. Acad. Sci. U.S.A. 1998. 95: 14447–14452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Complete sequence and gene map of a human major histocompatibility complex . The MHC sequencing consortium. Nature 1999. 401: 921–923. doi: 10.1038/44853. [DOI] [PubMed] [Google Scholar]
- 3. Mahdi, H. , Fisher, B. A. , Källberg, H. , Plant, D. , Malmström, V. , Rönnelid, J. , Charles, P. et al., Specific interaction between genotype, smoking and autoimmunity to citrullinated alpha‐enolase in the etiology of rheumatoid arthritis. Nat. Genet. 2009. 41: 1319–1324. doi: 10.1038/ng.480. [DOI] [PubMed] [Google Scholar]
- 4. Trowsdale, J. and Knight, J. C. , Major histocompatibility complex genomics and human disease. Annu. Rev. Genomics Hum. Genet. 2013. 14: 301–323. doi: 10.1146/annurev-genom-091212-153455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Stadinski, B. D. , Zhang, L. , Crawford, F. , Marrack, P. , Eisenbarth, G. S. and Kappler, J. W. , Diabetogenic T cells recognize insulin bound to IAg7 in an unexpected, weakly binding register. Proc. Natl. Acad. Sci. U.S.A. 2010. 107: 10978–10983. doi: 10.1073/pnas.1006545107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jeffreys, A. J. , Kauppi, L. and Neumann, R. , Intensely punctate meiotic recombination in the class II region of the major histocompatibility complex. Nat. Genet. 2001. 29: 217–222. doi: 10.1038/ng1001-217. [DOI] [PubMed] [Google Scholar]
- 7. Yang, J. , Chow, I.‐T. , Sosinowski, T. , Torres‐Chinn, N. , Greenbaum, C. J. , James, E. A. , Kappler, J. W. et al., Autoreactive T cells specific for insulin B:11‐23 recognize a low‐affinity peptide register in human subjects with autoimmune diabetes. Proc. Natl. Acad. Sci. U.S.A. 2014. 111: 14840–14845. doi: 10.1073/pnas.1416864111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gabriel, S. B. , Schaffner, S. F. , Nguyen, H. , Moore, J. M. , Roy, J. , Blumenstiel, B. , Higgins, J. et al., The structure of haplotype blocks in the human genome. Science 2002. 296: 2225–2229. doi: 10.1126/science.1069424. [DOI] [PubMed] [Google Scholar]
- 9. De Libero, G. and Mori, L. , Recognition of lipid antigens by T cells. Nat. Rev. Immunol. 2005. 5: 485–496. doi: 10.1038/nri1631. [DOI] [PubMed] [Google Scholar]
- 10. Traherne, J. A. , Horton, R. , Roberts, A. N. , Miretti, M. M. , Hurles, M. E. , Stewart, C. A. , Ashurst, J. L. et al., Genetic analysis of completely sequenced disease‐associated MHC haplotypes identifies shuffling of segments in recent human history. PLoS Genet. 2006. 2: e9. doi: 10.1371/journal.pgen.0020009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Brenner, M. , Meng, H.‐C. , Yarlett, N. C. , Joe, B. , Griffiths, M. M. , Remmers, E. F. , Wilder, R. L. et al., The non‐MHC quantitative trait locus Cia5 contains three major arthritis genes that differentially regulate disease severity, pannus formation, and joint damage in collagen‐ and pristane‐induced arthritis. J. Immunol. 2005. 174: 7894–7903. [DOI] [PubMed] [Google Scholar]
- 12. Tuncel, J. , Haag, S. , Yau, A. C. Y. , Norin, U. , Baud, A. , Lönnblom, E. , Maratou, K. et al., Natural polymorphisms in Tap2 influence negative selection and CD4∶CD8 lineage commitment in the rat. PLoS Genet. 2014. 10: e1004151. doi: 10.1371/journal.pgen.1004151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rintisch, C. , Kelkka, T. , Norin, U. , Lorentzen, J. C. , Olofsson, P. and Holmdahl, R. , Finemapping of the arthritis QTL Pia7 reveals co‐localization with Oia2 and the APLEC locus. Genes Immun. 2010. 11: 239–245. doi: 10.1038/gene.2010.2. [DOI] [PubMed] [Google Scholar]
- 14. Hoffmann, M. H. , Skriner, K. , Herman, S. , Baumann, C. , Steiner, C.‐W. , Ospelt, C. , Meyer, B. et al., Nucleic acid‐stimulated antigen‐presenting cells trigger T cells to induce disease in a rat transfer model of inflammatory arthritis. J. Autoimmun. 2011. 36: 288–300. doi: 10.1016/j.jaut.2011.02.007. [DOI] [PubMed] [Google Scholar]
- 15. Haag, S. , Tuncel, J. , Thordardottir, S. , Mason, D. E. , Yau, A. C. Y. , Dobritzsch, D. , Bäcklund, J. et al., Positional identification of RT1‐B (HLA‐DQ) as susceptibility locus for autoimmune arthritis. J. Immunol. 2015. 194: 2539–2550. doi: 10.4049/jimmunol.1402238. [DOI] [PubMed] [Google Scholar]
- 16. Kirberg, J. , Berns, A. and Boehmer von, H. , Peripheral T cell survival requires continual ligation of the T cell receptor to major histocompatibility complex‐encoded molecules. J. Exp. Med. 1997. 186: 1269–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bender, J. , Mitchell, T. , Kappler, J. and Marrack, P. , CD4+ T cell division in irradiated mice requires peptides distinct from those responsible for thymic selection. J. Exp. Med. 1999. 190: 367–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tuncel, J. , Haag, S. , Hoffmann, M. H. , Yau, A. C. Y. , Hultqvist, M. , Olofsson, P. , Bäcklund, J. et al., Animal models of rheumatoid arthritis (I): pristane‐induced arthritis in the rat. PLoS ONE. 2016. 11: e0155936. doi: 10.1371/journal.pone.0155936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Xing, Y. and Hogquist, K. A. , T‐cell tolerance: central and peripheral. Cold Spring Harb. Perspect. Biol. 2012. 4. doi: 10.1101/cshperspect.a006957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kleinau, S. , Dencker, L. and Klareskog, L. , Oil‐induced arthritis in DA rats: tissue distribution of arthritogenic 14C‐labelled hexadecane. Int. J. Immunopharmacol. 1995. 17: 393–401. [DOI] [PubMed] [Google Scholar]
- 21. Marrack, P. , Kappler, J. and Kotzin, B. L. , Autoimmune disease: why and where it occurs. Nat. Med. 2001. 7: 899–905. doi: 10.1038/90935. [DOI] [PubMed] [Google Scholar]
- 22. Vingsbo, C. , Sahlstrand, P. , Brun, J. G. , Jonsson, R. , Saxne, T. and Holmdahl, R. , Pristane‐induced arthritis in rats: a new model for rheumatoid arthritis with a chronic disease course influenced by both major histocompatibility complex and non‐major histocompatibility complex genes. Am. J. Pathol. 1996. 149: 1675–1683. [PMC free article] [PubMed] [Google Scholar]
- 23. Holmberg, J. , Tuncel, J. , Yamada, H. , Lu, S. , Olofsson, P. and Holmdahl, R. , Pristane, a non‐antigenic adjuvant, induces MHC class II‐restricted, arthritogenic T cells in the rat. J. Immunol. 2006. 176: 1172–1179. [DOI] [PubMed] [Google Scholar]
- 24. Eyre, S. , Bowes, J. , Diogo, D. , Lee, A. , Barton, A. , Martin, P. , Zhernakova, A. et al., High‐density genetic mapping identifies new susceptibility loci for rheumatoid arthritis. Nat. Genet. 2012. 44: 1336–1340. doi: 10.1038/ng.2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Endl, J. , Rosinger, S. , Schwarz, B. , Friedrich, S.‐O. , Rothe, G. , Karges, W. , Schlosser, M. et al., Coexpression of CD25 and OX40 (CD134) receptors delineates autoreactive T‐cells in type 1 diabetes. Diabetes 2006. 55: 50–60. [PubMed] [Google Scholar]
- 26. Matsumoto, I. , Staub, A. , Benoist, C. and Mathis, D. , Arthritis provoked by linked T and B cell recognition of a glycolytic enzyme. Science 1999. 286: 1732–1735. [DOI] [PubMed] [Google Scholar]
- 27. Klinger, M. , Kim, J. K. , Chmura, S. A. , Barczak, A. , Erle, D. J. and Killeen, N. , Thymic OX40 expression discriminates cells undergoing strong responses to selection ligands. J. Immunol. 2009. 182: 4581–4589. doi: 10.4049/jimmunol.0900010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tuncel, J. , Haag, S. , Carlsen, S. , Yau, A. C. Y. , Lu, S. , Burkhardt, H. and Holmdahl, R. , Class II major histocompatibility complex‐associated response to type XI collagen regulates the development of chronic arthritis in rats. Arthritis Rheum. 2012. 64: 2537–2547. doi: 10.1002/art.34461. [DOI] [PubMed] [Google Scholar]
- 29. Croft, M. , So, T. , Duan, W. and Soroosh, P. , The significance of OX40 and OX40L to T‐cell biology and immune disease. Immunol. Rev. 2009. 229: 173–191. doi: 10.1111/j.1600-065X.2009.00766.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wooley, P. H. , Luthra, H. S. , Stuart, J. M. and David, C. S. , Type II collagen‐induced arthritis in mice. I. Major histocompatibility complex (I region) linkage and antibody correlates. J. Exp. Med. 1981. 154: 688–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Webb, G. J. , Hirschfield, G. M. and Lane, P. J. L. , OX40, OX40L and autoimmunity: a comprehensive review. Clin. Rev. Allergy Immunol. 2015. doi: 10.1007/s12016-015-8498-3. [DOI] [PubMed] [Google Scholar]
- 32. Bäcklund, J. , Li, C. , Jansson, E. , Carlsen, S. , Merky, P. , Nandakumar, K. S. , Haag, S. et al., C57BL/6 mice need MHC class II Aq to develop collagen‐induced arthritis dependent on autoreactive T cells. Ann. Rheum. Dis. 2012. doi: 10.1136/annrheumdis-2012-202055. [DOI] [PubMed] [Google Scholar]
- 33. Yoshino, S. and Cleland, L. G. , Depletion of alpha/beta T cells by a monoclonal antibody against the alpha/beta T cell receptor suppresses established adjuvant arthritis, but not established collagen‐induced arthritis in rats. J. Exp. Med. 1992. 175: 907–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Vu, M. D. , Xiao, X. , Gao, W. , Degauque, N. , Chen, M. , Kroemer, A. , Killeen, N. et al., OX40 costimulation turns off Foxp3+ Tregs. Blood. 2007. 110: 2501–2510. doi: 10.1182/blood-2007-01-070748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Korganow, A. S. , Ji, H. , Mangialaio, S. , Duchatelle, V. , Pelanda, R. , Martin, T. , Degott, C. et al., From systemic T cell self‐reactivity to organ‐specific autoimmune disease via immunoglobulins. Immunity. 1999. 10: 451–461. [DOI] [PubMed] [Google Scholar]
- 36. Ruby, C. E. , Yates, M. A. , Hirschhorn‐Cymerman, D. , Chlebeck, P. , Wolchok, J. D. , Houghton, A. N. , Offner, H. et al., Cutting edge: OX40 agonists can drive regulatory T cell expansion if the cytokine milieu is right. J. Immunol. 2009. 183: 4853–4857. doi: 10.4049/jimmunol.0901112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bäcklund, J. , Treschow, A. , Bockermann, R. , Holm, B. , Holm, L. , Issazadeh‐Navikas, S. , Kihlberg, J. et al., Breaking T cell tolerance against self type II collagen in HLA‐DR4‐transgenic mice and development of autoimmune arthritis. Eur. J. Immunol. 2002. 32: 3776–3784. doi: . [DOI] [PubMed] [Google Scholar]
- 38. Yau, A. C. Y. and Holmdahl, R. , Rheumatoid arthritis: identifying and characterising polymorphisms using rat models. Dis. Model Mech. 2016. 9: 1111–1123. doi: 10.1242/dmm.026435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Griseri, T. , Asquith, M. , Thompson, C. and Powrie, F. , OX40 is required for regulatory T cell‐mediated control of colitis. J. Exp. Med. 2010. 207: 699–709. doi: 10.1084/jem.20091618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Overkamp, D. , Mohammed‐Ali, S. , Cartledge, C. and Landon, J. , Production of polyclonal antibodies in ascitic fluid of mice: technique and applications. J. Immunoassay. 1988. 9: 51–68. doi: 10.1080/01971528808053210. [DOI] [PubMed] [Google Scholar]
- 41. Satoh, M. and Reeves, W. H. , Induction of lupus‐associated autoantibodies in BALB/c mice by intraperitoneal injection of pristane. J. Exp. Med. 1994. 180: 2341–2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Olofsson, P. , Nordquist, N. , Vingsbo‐Lundberg, C. , Larsson, A. , Falkenberg, C. , Pettersson, U. , Akerström, B. et al., Genetic links between the acute‐phase response and arthritis development in rats. Arthritis Rheum. 2002. 46: 259–268. doi: . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rintisch, C. , Ameri, J. , Olofsson, P. , Luthman, H. and Holmdahl, R. , Positional cloning of the Igl genes controlling rheumatoid factor production and allergic bronchitis in rats. Proc. Natl. Acad. Sci. U.S.A. 2008. 105: 14005–14010. doi: 10.1073/pnas.0803956105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yau, A. C. Y. , Tuncel, J. , Haag, S. , Norin, U. , Houtman, M. , Padyukov, L. and Holmdahl, R. , Conserved 33‐kb haplotype in the MHC class III region regulates chronic arthritis. Proc. Natl. Acad. Sci. U.S.A. 2016. 113: E3716–24. doi: 10.1073/pnas.1600567113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bunce, C. and Bell, E. B. , CD45RC isoforms define two types of CD4 memory T cells, one of which depends on persisting antigen. J. Exp. Med. 1997. 185: 767–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Weinberg, A. D. , Bourdette, D. N. , Sullivan, T. J. , Lemon, M. , Wallin, J. J. , Maziarz, R. , Davey, M. et al., Selective depletion of myelin‐reactive T cells with the anti‐OX‐40 antibody ameliorates autoimmune encephalomyelitis. Nat. Med. 1996. 2: 183–189. [DOI] [PubMed] [Google Scholar]
- 47. Hirota, K. , Hashimoto, M. , Yoshitomi, H. , Tanaka, S. , Nomura, T. , Yamaguchi, T. , Iwakura, Y. et al., T cell self‐reactivity forms a cytokine milieu for spontaneous development of IL‐17+ Th cells that cause autoimmune arthritis. J. Exp. Med. 2007. 204: 41–47. doi: 10.1084/jem.20062259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Korn, T. , Bettelli, E. , Oukka, M. and Kuchroo, V. K. , IL‐17 and Th17 cells. Annu. Rev. Immunol. 2009. 27: 485–517. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- 49. Lubberts, E. , Koenders, M. I. , Oppers‐Walgreen, B. , van den Bersselaar, L. , Coenen‐de Roo, C. J. J. , Joosten, L. A. B. and van den Berg, W. B. , Treatment with a neutralizing anti‐murine interleukin‐17 antibody after the onset of collagen‐induced arthritis reduces joint inflammation, cartilage destruction, and bone erosion. Arthritis Rheum. 2004. 50: 650–659. doi: 10.1002/art.20001. [DOI] [PubMed] [Google Scholar]
- 50. Nakae, S. , Nambu, A. , Sudo, K. and Iwakura, Y. , Suppression of immune induction of collagen‐induced arthritis in IL‐17‐deficient mice. J. Immunol. 2003. 171: 6173–6177. [DOI] [PubMed] [Google Scholar]
- 51. Nakae, S. , Saijo, S. , Horai, R. , Sudo, K. , Mori, S. and Iwakura, Y. , IL‐17 production from activated T cells is required for the spontaneous development of destructive arthritis in mice deficient in IL‐1 receptor antagonist. Proc. Natl. Acad. Sci. U.S.A. 2003. 100: 5986–5990. doi: 10.1073/pnas.1035999100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lubberts, E. , The IL‐23‐IL‐17 axis in inflammatory arthritis. Nat. Rev. Rheumatol. 2015. 11: 415–429. doi: 10.1038/nrrheum.2015.53. [DOI] [PubMed] [Google Scholar]
- 53. Wilson, N. J. , Boniface, K. , Chan, J. R. , McKenzie, B. S. , Blumenschein, W. M. , Mattson, J. D. , Basham, B. et al., Development, cytokine profile and function of human interleukin 17‐producing helper T cells. Nat. Immunol. 2007. 8: 950–957. doi: 10.1038/ni1497. [DOI] [PubMed] [Google Scholar]
- 54. Chabaud, M. , Durand, J. M. , Buchs, N. , Fossiez, F. , Page, G. , Frappart, L. and Miossec, P. , Human interleukin‐17: a T cell‐derived proinflammatory cytokine produced by the rheumatoid synovium. Arthritis Rheum. 1999. 42: 963–970. doi: . [DOI] [PubMed] [Google Scholar]
- 55. van Hamburg, J. P. , Corneth, O. B. J. , Paulissen, S. M. J. , Davelaar, N. , Asmawidjaja, P. S. , Mus, A. M. C. and Lubberts, E. , IL‐17/Th17 mediated synovial inflammation is IL‐22 independent. Ann. Rheum. Dis. 2013. doi: 10.1136/annrheumdis-2012-202373. [DOI] [PubMed] [Google Scholar]
- 56. Dardalhon, V. , Korn, T. , Kuchroo, V. K. and Anderson, A. C. , Role of Th1 and Th17 cells in organ‐specific autoimmunity. J. Autoimmun. 2008. 31: 252–256. doi: 10.1016/j.jaut.2008.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Raychaudhuri, S. , Sandor, C. , Stahl, E. A. , Freudenberg, J. , Lee, H.‐S. , Jia, X. , Alfredsson, L. et al., Five amino acids in three HLA proteins explain most of the association between MHC and seropositive rheumatoid arthritis. Nat. Genet. 2012. 44: 291–296. doi: 10.1038/ng.1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Frey, O. , Mitera, T. , Kelchtermans, H. , Schurgers, E. , Kamradt, T. and Matthys, P. , Ameliorated course of glucose‐6‐phosphate isomerase (G6PI)‐induced arthritis in IFN‐γ receptor knockout mice exposes an arthritis‐promoting role of IFN‐γ;. J. Autoimmun. 2011. 36: 161–169. doi: 10.1016/j.jaut.2010.12.006. [DOI] [PubMed] [Google Scholar]
- 59. Doodes, P. D. , Cao, Y. , Hamel, K. M. , Wang, Y. , Rodeghero, R. L. , Mikecz, K. , Glant, T. T. et al., IFN‐gamma regulates the requirement for IL‐17 in proteoglycan‐induced arthritis. J. Immunol. 2010. 184: 1552–1559. doi: 10.4049/jimmunol.0902907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Finnegan, A. , Grusby, M. J. , Kaplan, C. D. , O'Neill, S. K. , Eibel, H. , Koreny, T. , Czipri, M. et al., IL‐4 and IL‐12 regulate proteoglycan‐induced arthritis through Stat‐dependent mechanisms. J. Immunol. 2002. 169: 3345–3352. [DOI] [PubMed] [Google Scholar]
- 61. Papadaki, G. , Kambas, K. , Choulaki, C. , Vlachou, K. , Drakos, E. , Bertsias, G. , Ritis, K. et al., Neutrophil extracellular traps exacerbate Th1‐mediated autoimmune responses in rheumatoid arthritis by promoting DC maturation. Eur. J. Immunol. 2016. 46: 2542–2554. doi: 10.1002/eji.201646542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Herman, S. , Kny, A. , Schorn, C. , Pfatschbacher, J. , Niederreiter, B. , Herrmann, M. , Holmdahl, R. et al., Cell death and cytokine production induced by autoimmunogenic hydrocarbon oils. Autoimmunity. 2012. 45: 602–611. doi: 10.3109/08916934.2012.719948. [DOI] [PubMed] [Google Scholar]
- 63. Kokkonen, H. , Söderström, I. , Rocklöv, J. , Hallmans, G. , Lejon, K. and Rantapää‐Dahlqvist, S. , Up‐regulation of cytokines and chemokines predates the onset of rheumatoid arthritis. Arthritis Rheum. 2010. 62: 383–391. doi: 10.1002/art.27186. [DOI] [PubMed] [Google Scholar]
- 64. Deane, K. D. , O'Donnell, C. I. , Hueber, W. , Majka, D. S. , Lazar, A. A. , Derber, L. A. , Gilliland, W. R. et al., The number of elevated cytokines and chemokines in preclinical seropositive rheumatoid arthritis predicts time to diagnosis in an age‐dependent manner. Arthritis Rheum. 2010. 62: 3161–3172. doi: 10.1002/art.27638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ramwadhdoebe, T. H. , Hähnlein, J. , Maijer, K. I. , van Boven, L. J. , Gerlag, D. M. , Tak, P. P. and van Baarsen, L. G. M. , Lymph node biopsy analysis reveals an altered immunoregulatory balance already during the at‐risk phase of autoantibody positive rheumatoid arthritis. Eur. J. Immunol. 2016. doi: 10.1002/eji.201646393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Lubberts, E. , Schwarzenberger, P. , Huang, W. , Schurr, J. R. , Peschon, J. J. , van den Berg, W. B. and Kolls, J. K. , Requirement of IL‐17 receptor signaling in radiation‐resistant cells in the joint for full progression of destructive synovitis. J. Immunol. 2005. 175: 3360–3368. [DOI] [PubMed] [Google Scholar]
- 67. Nicklas, W. , Deeny, A. , Diercks, P. , Gobbi, A. , Illgen‐Wilcke, B. and Seidelin, M. , FELASA guidelines for the accreditation of health monitoring programs and testing laboratories involved in health monitoring. Lab Anim (NY). 2010. 39: 43–48. doi: 10.1038/laban0210-43. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
FIGURE S1. Evaluation of PIA in new MHCII congenic strains, DA.1FR61 and DA.1HR10. (A) The development of PIA in DA.1FR61 was compared to its parental congenic strain, DA.1FR9, and to DA. Arthritis clinical scores (left) and weight change (in % versus day of disease onset, right) is shown; #, significant (p<0.05) DA versus DA.1FR61; *, significant (p<0.05) DA versus DA.1FR9. (B) Arthritis clinical scores and weight change in pristane immunized DA.1HR10, DA.1HR61 and DA; *P < 0.05. All statistic comparisons in (A) and (B) were determined by Mann‐Whitney; n = 5‐11 per group as indicated in figure.
FIGURE S2. Proliferation of Foxp3+ Tregs and recent thymic emigrants (RTEs) in dLNs after pristane administration. (A) Histograms show representative expression of Ki67 in CD4+ Foxp3+ Tregs from DA (blue) and DA.1HR10 at day 4 and 5 after pristane administration. Adjacent line chart shows summarized data from all strains. (B) Corresponding data (as shown in A) for RTEs (CD90+ CD4+ T cells) in pristane dLNs. (A‐B) n = 5 per group; **p<0.01 (by Mann‐Whitney).
FIGURE S3. Variation in Foxp3+ Tregs in naive MHCII‐congenic rats does not correlate with disease susceptibility. (A) Representative counter plots of CD4 T cells in spleen of naive rats. Summarized data in (B) show frequencies of Tregs among CD4 T cells, and in (C) total number of Tregs per spleen. P‐values depicted in graphs were determined by Mann‐Whitney (n=4 per strain).
Table S1. Primers for quantitative PCR.
Supporting Information