

Abstract
Entelegyne spiders rarely show fusions yielding neo‐Y chromosomes, which M. J. D. White attributed to a constraint in spiders, namely their proximal chiasma localization acting to upset meiotic segregation in males with fusions. Of the 75 taxa of Habronattus and outgroups studied, 47 have X1X20 sex chromosomes in males, 10 have X1X2Y, 15 have X1X2X3Y, 2 have X0, and one has both X1X20 and X1X2X3Y. Chromosome numbers and behavior suggest neo‐Ys formed by an autosome‐X fusion to make X1X2Y, with a second fusion to an autosome to make X1X2X3Y. Phylogeny shows at least 8–15 gains (or possibly some losses) of neo‐Y (i.e., X‐autosome fusions), a remarkable number for such a small clade. In contrast to the many X‐autosome fusions, at most one autosome–autosome fusion is indicated. Origins of neo‐Y are correlated significantly with distal localization of chiasmata, supporting White's hypothesis that evolution of neo‐Y systems is facilitated by looser pairing (distal chiasmata) at meiosis. However, an alternative (or contributing) explanation for the correlation is that X‐autosome fusions were selected to permit isolation of male‐favored alleles to the neo‐Y chromosome, aided by distal chiasmata limiting recombination. This intralocus sexual conflict hypothesis could explain both the many X‐autosome fusions, and the stunning complexity of male Habronattus courtship displays.
Keywords: Chromosome evolution, intralocus sexual conflict, neo‐Y, phylogenetic correlation, segregation constraints, sexual antagonism
Chromosome numbers and forms might be expected to show relatively clear patterns among species with shifting evolutionary constraints and forces, as they are largely isolated from the complex variability of ecological pressures that affect many phenotypic characters. Although few have been examined by modern phylogenetic methods, many broad patterns have been described in the classical cytogenetic literature, with some clades notably conservative, and others labile (White 1973). For instance, a group of more than 10,000 acridoid grasshopper species is dominantly of a single karyotype with 23 one‐armed (acrocentric) chromosomes (White 1973). A clade of about 500 salamander species in six families has mostly biarmed (metacentric or submetacentric) chromosomes and stable numbers (Sessions 2008). In the carabid beetle genus Bembidion with more than 1000 species, almost all autosomes are biarmed, and 190 of the 205 species studied have 11 pairs of autosomes (D. Maddison 1985). In contrast, other clades show many evolutionary changes, such as the morabine grasshoppers with 11 or more origins of neo‐Y chromosomes (White 1969).
For some of these patterns, specific evolutionary constraints and forces have been suggested. White (1973, p. 671) proposed a constraint to explain the observation that most spiders, and almost all spiders of the large and familiar entelegyne clade, have acrocentric chromosomes and an X1X20 male/X1X1X2X2 female or related sex chromosome system (Araujo et al. 2005, 2012; Král et al. 2006). Rare in this clade are species with biarmed chromosomes and unknown to White were species with neo‐Y chromosomes from X‐autosome fusions. White noted that in spiders the crossing over points between the chromosomes of a pair tend to be placed close to the centromere (proximal chiasma localization). He argued that this constrains against the evolution of chromosome fusions that might generate metacentrics or neo‐Y chromosomes.
White's proposed constraint is illustrated in Figure 1, which shows how proximal versus distal chiasmata would appear in a complex fused system. The example used is the X1X2X3Y system of four sex chromosomes found by Maddison (1982) in Habronattus, the subject of this article. The X2 and X3 are paired with the Y whereas the X1 is to the side (and not relevant to the constraint). As with most spiders, there is a single chiasma in each pairing chromosome arm (White 1973, p. 671). To generate gametes properly, the Y must go to one pole at meiosis, the three Xs to the opposite pole. White (1973, p. 295) notes that for the highest probability of proper disjunction, the centromeres of such a three‐chromosome system (focusing on the linked X2, X3, and Y) need to arrange themselves into an isosceles triangle. In Figure 1A, the paired chromosomes are connected via chiasmata far from the centromere (i.e., distal), giving the chromosomes considerable space and allowing for them to easily achieve the regular arrangement. In Figure 1B, the chiasmata are close to the centromere, making a tightly paired configuration whose mechanical crowding could prevent the regular arrangement. White argues that chromosomes could often go to the incorrect pole, yielding aneuploid gametes and reduced fertility, and thus hindering the establishment of such complex fused systems in spiders, which generally have chiasmata like that of Figure 1B. In this way, White (1973) explained the lack of neo‐Y systems observed in spiders.
Figure 1.
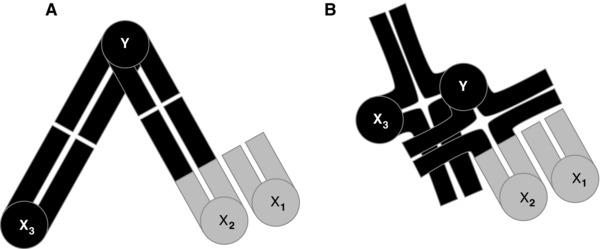
White's constraint hypothesis illustrated with a meiotic configuration of four sex chromosomes (X1X2X3Y, see also Fig. 3) as seen in some male Habronattus. Each chromosome's centromere is shown as a circle, and its chromatids as bars. The Y has two arms, each of which pairs with an X. (A) Chiasmata distal (at tips of chromatids), “loose pairing.” (B) Chiasmata proximal (near the centromere), “tight pairing.” Loose pairing gives enough room for proper segregation (Y to one pole, three Xs to the other); tight pairing is predicted to lead too often to improper disjunction.
White's hypothesis that proximal chiasmata impose a constraint against fusions yields a comparative prediction: species with fusions should tend to have chiasmata that are primarily distal. White (1973) notes that of 240 spider species then studied, about nine have metacentrics and “in most of these cases the chiasmata in the male are distal or interstitial rather than proximal.” However, such data have not yet been put into an appropriate phylogenetic framework to assess the strength of any correlation.
The discovery of neo‐Y sex chromosome systems in some entelegyne spiders (Maddison 1982; Rowell 1985; Král 2007) opens the group for an examination of the evolutionary interplay between chiasma localization and chromosome fusions. In Habronattus, Maddison (1982) found four species with X1X2X3Y sex chromosomes in three different species groups, hinting at the possibility of multiple origins via X‐autosome fusion. Using phylogenetic information on Habronattus (Maddison and Hedin 2003), we can therefore examine sex chromosome evolution and chiasma localization in the group to test White's constraint hypothesis by asking: are there multiple origins of neo‐Y systems in Habronattus, and do these origins appear to be associated with a lack of the localization constraint? That is, are origins of neo‐Ys primarily or only in lineages with distal, rather than proximal, chiasma localization?
Interspecific correlation of distal localization with neo‐Y chromosomes would be consistent with White's constraint hypothesis, but his hypothesis would not be a complete, nor would it be the only, explanation for such a correlation. If most of the fusions observed in Habronattus were between an X and an autosome, White's constraint lifting would not explain why there should be so many fusions of the same kind. Lifting the constraint would, in principle, allow a diversity of chromosome fusions, including autosome–autosome fusions. However, a mechanism such as intralocus sexual conflict could explain X‐autosome fusions in particular, as autosomes bearing loci whose alleles are favored differently in males versus females could be selected to fuse with sex chromosomes (Charlesworth and Charlesworth 1980). This hypothesis would at the same time explain a correlation of distal localization with neo‐Y chromosomes, as distal localization would limit recombination to permit the male‐favored allele to remain isolated to the Y.
Our goals are therefore to assess the number and types of chromosomal changes, and whether distal chiasmata are correlated with the origins of fusions.
Materials and Methods
TAXON SAMPLING
Seventy one species of Habronattus and two additional subspecies were sampled for chromosomes (Table S1, Fig. 4). Informal names for undescribed species match those used by Maddison and Hedin (2003). The specimens are not the same, and the localities are not necessarily the same, as those used for molecular phylogenetic work by Maddison and Hedin (2003). Because geographic variability in some species hints to the possibility that some populations may represent distinct species, morphologically or karyotypically distinctive populations were kept separate for the analyses. This required a linking of the chromosome‐sampled populations onto the molecularly sampled populations so that the chromosomes could be interpreted on the molecular phylogeny. Chromosome data were linked to the geographically nearest and morphologically most similar population in the molecular phylogeny, indicated in Table S1 by the “Link” column. Chromosome data of the outgroup Pellenes peninsularis were linked to the molecular voucher from the very closely related (if not conspecific) Pellenes cf. apacheus.
Figure 4.
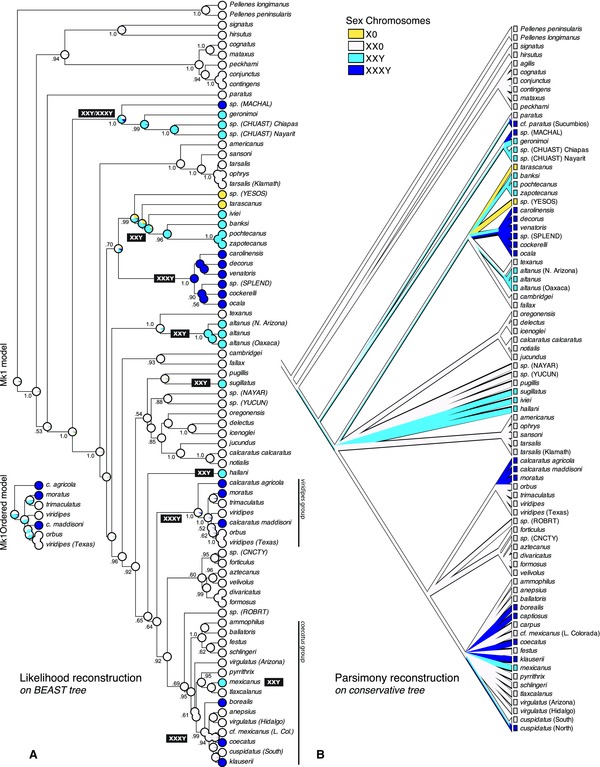
Evolution of sex chromosome systems. (A) Ancestral states reconstructed by likelihood (Mk1 model; estimated rate 1.5616) on BEAST maximum clade credibility tree, with inset showing reconstruction from Mk1Ordered model. Estimated posterior probabilities for clades shown. (B) Ancestral states reconstructed by parsimony on highly conservative tree from Maddison and Hedin (2003), treating the polytomies as soft and the character states as ordered X0 ‐ X1X20 ‐ X1X2Y ‐ X1X2X 3Y.
Data from Maddison (1982) for one species of Pellenes (Pellenes peninsularis) and 11 species of Habronattus (formerly placed in Pellenes) are included in this analysis. Maddison (1982) used now‐antiquated names for some of the species. His “Pellenes cf. agilis” is H. cognatus; “Pellenes cf. brunneus” is Habronattus cuspidatus; “Pellenes cf. calcaratus” is Habronattus calcaratus maddisoni.
CHROMOSOME METHODS
Meiotic chromosomes were observed in the testes of adult and subadult males using Feulgen staining, following the methods of Maddison (1982), except that no colchicine was used. We obtained good squashes from some specimens stored for more than 15 years in 3:1 ethanol:acetic acid in a −20°C (or colder) freezer. Nuclei were photographed with a Pentax 6‐megapixel camera with flash to minimize vibration blurring, under a 100× oil‐immersion objective, usually using phase contrast.
Chromosome number and form were determined primarily by examining diakinesis or metaphase I of male meiosis. A single nucleus with easily interpretable chromosomes was considered sufficient to determine chromosome count and sex chromosome system for a species, although in almost all cases multiple nuclei were available. For some specimens, the sex chromosome system was easily interpretable even if the autosome complement was not determined because of overlap. Sex chromosomes could be determined by their differential condensation (heteropycnosis) or behavior in pairing and movement.
Chiasma position was measured from the digital photographs using the straight line measurement tool of ImageJ 1.43u (Rasband 2012). Only nuclei with a clear orientation of the metaphase plate were used. Except where noted, chromosomes participating in the sex chromosome system are not included in the chiasma localization data reported.
By metaphase, typically each bivalent has opened up to form two rods parallel to the spindle axis if the chiasma is distal, a single‐rod perpendicular to the spindle axis and with medial bump if the chiasma is proximal, and cross‐shaped if the chiasma is in an interstitial position (e.g., see Fig. 2). Therefore, except where the chiasma is fully distal, there is a point, along the length from the centromere to the tips, at which the chromosome bends. This is taken as the point of the chiasma. The lengths of the chromosome segments between centromere and bend, and between bend and tip, were measured. Chiasma position was calculated as (length centromere to bend)/(total length). Measurements were taken either along the interior or along the exterior of the chromosome arms, depending on the picture's clarity. The ratios of the measurements for each of the four chromatids were averaged to determine the chiasma position for the whole chromosome bivalent. If no bend could be identified because the chiasma position was extreme, the position was determined to be proximal (0.0) or distal (1.0), depending on the bivalent's orientation relative to the spindle. For a species to be included in the analysis, we required at least 40 scored chromosomes.
Figure 2.
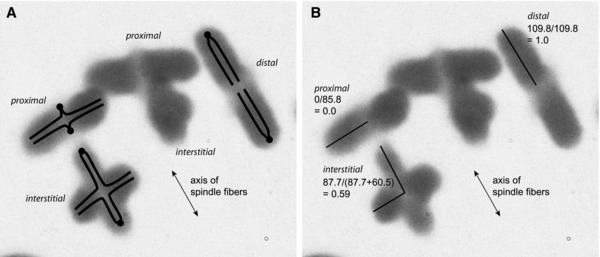
Interpretation and measurement of chiasma position in metaphase of male meosis. (A) Interpretation of bivalents using spots for centromeres and lines for chromatids, showing proximal, interstitial, and distal chiasmata. (B) Measurement of chiasma position. Spindle axis is shown for orientation, to distinguish proximal from distal chiasmata.
To avoid bias during chiasma position measurements, digital files of photographs of individual nuclei were renamed by random numbers, sorted, and scored in this anonymous sequence. Unusual sex chromosome systems could still be seen in the photographs, but at least for X1X20 species there was no way to bias localization measurements according to particular species groups.
In many nuclei, there were some bivalents that could not be confidently scored because of orientation or overlap. These were excluded, but we were concerned that this could introduce a bias if proximal chiasmata more often yield confusing forms of bivalents than distal chiasmata. To reduce this effect and yet still retain enough nuclei and species for a comprehensive analysis, we included a nucleus only if at least 60% of its autosomes were scorable.
The chiasma localization of a species was determined by pooling measured positions of all chromosomes in all nuclei in all specimens. Both mean position and median position were calculated. In general, we use the median, as it draws sharper distinctions among species.
PHYLOGENETIC TREE RECONSTRUCTION
Data from Maddison and Hedin (2003) were reanalyzed to provide a phylogenetic framework for this study. Their data include two gene regions, the mitochondrial 16sND1 and the nuclear Ef1‐alpha. The noncoding portion of 16sND1 was realigned with the Opal and Opalescent packages (Wheeler and Kececioglu 2007) in Mesquite (Maddison and Maddison 2011), using default alignment costs (costs 260 open, 100 terminal open, 69 extension, 66 terminal extension). The 16sND1 and EF1‐alpha alignments were concatenated into a single matrix and analyzed by Bayesian phylogenetic methods.
Because there are signals of hybridization in the data (Maddison and Hedin 2003) and because incomplete lineage sorting may have occurred in this shallow phylogeny, any phylogeny reconstructed with just these two gene regions will have considerable uncertainty. Concatenating the genes may not be ideal, but more genes would be needed to use a gene tree approach effectively. Even a phylogeny from extensive genomic data might fail to explain karyotype evolution if, for example, sex chromosomes occasionally introgressed. As some of these chromosome data have already waited 30 years for publication, we have chosen to explore alternative trees cautiously rather than not at all.
The clear examples of hybridization found by Maddison and Hedin (2003) involved taxa not sampled for chromosomes, and so may have little effect on our analysis. However, their unexplained placement of Habronattus notialis, Habronattus jucundus, and H. calcaratus calcaratus far from the other members of the morphologically distinctive viridipes group, which may be because of distant introgression, could be problematical for our analyses. The complex morphological and behavioral synapomorphies uniting these with the other viridipes group members are compelling (Griswold 1987; Maddison and Hedin 2003; W. Maddison, unpubl. data).
Some taxa sampled here were not represented in Maddison and Hedin's (2003) molecular phylogeny study (Habronattus agilis, Habronattus carpus, Habronattus captiosus, Habronattus cf. paratus (Sucumbios), and the northern population of H. cuspidatus). For this reason they cannot be used in the analyses that rely on the branch lengths in the molecular phylogeny. However, for parsimony ancestral state reconstructions, they are included where relationships are clear enough to include them.
BEAST version 1.62 (Drummond and Rambaut 2007) was used on Maddison and Hedin's data to estimate a phylogeny with branch lengths proportional to time. We chose this for two reasons: first, the characters we studied may not follow the rate variation of the genes used for phylogeny, and a tree proportional to time is arguably more relevant. Second, preliminary runs with GARLI (likelihood; Zwickl 2006) and MrBayes (Bayesian; Ronquist and Huelsenbeck 2003) resulted in trees with some very short branches, including in parts of the tree variable in sex chromosomes. We were concerned these very short branches could act as outliers distorting analyses if change was implied along them; we found they were stretched to more reasonable lengths in the time‐proportional trees.
The data were divided into two partitions (mitochondrial vs. nuclear). For each partition, MrModelTest version 2.3 (Nylander 2004) was used with PAUP* 4.0b10 (Swofford 2002) to choose appropriate models using AIC with default options. BEAST was run for 400 million generations with Habronattus constrained monophyletic (to place the Pellenes species as outgroups), echoing state each 10,000 generations, Yule process prior for the tree, uncorrelated lognormal relaxed clock, and with default priors except yule.birthRate = uniform[0, 10000] initial = 1; 16sND1.ucld.mean = uniform[0,1] initial = 1; EF1alpha.ucld.mean = uniform[0,1] initial = 1. TreeAnnotator version 1.62 (Drummond and Rambaut 2007) was used to summarize the resulting trees by constructing a maximum clade credibility tree with median heights, discarding the first 4000 of the 40,000 trees as burn‐in.
After analysis of Maddison and Hedin's full dataset, those taxa not represented with chromosome data were trimmed from the tree.
ANCESTRAL STATE RECONSTRUCTION
Ancestral states of sex chromosome systems were reconstructed by Mesquite (Maddison and Maddison 2011) using both likelihood and parsimony. For parsimony, the sex chromosome character was treated as ordered according to the hypothesis of chromosome fusions (Fig. 3E). For likelihood, two models were used: the Mk1 model, which assumes the same rate of transitions between any two states; and what we call the Mk1Ordered model, which assumes the character is ordered as for parsimony. In the Mk1Ordered model, transition rates are constrained to be the same between adjacent states (0–1, 1–2, etc.) but zero between nonadjacent states (0–2, etc.).
Figure 3.
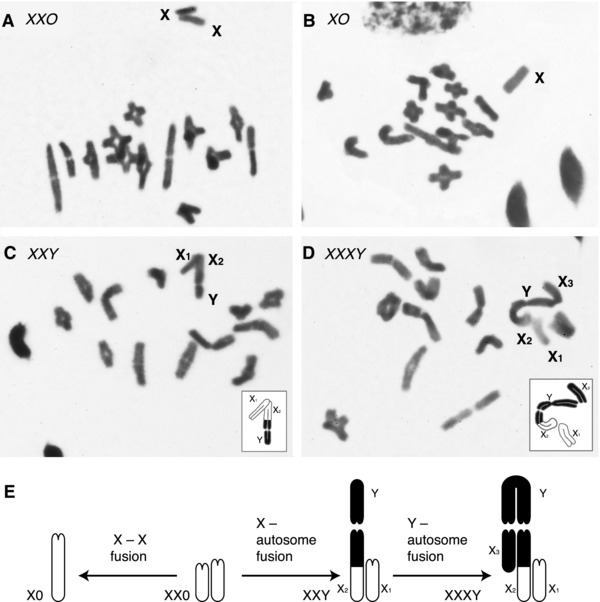
Examples of meiotic metaphase and diakinesis in males with different sex chromosomes, and interpretation of evolutionary transformations. Autosomal bivalents are all acrocentric. (A) A total of 13 pairs of autosomes + X1X20 (Habronattus pugillis). (B) A total of 13 pairs of autosomes + X0 (Habronattus tarascanus). (C) A total of 12 pairs of autosomes + X1X2Y (Habronattus altanus). (D) A total of 11 pairs of autosomes + X1X2X3Y (Habronattus borealis). Insets show interpretation of arms of sex chromosomes: white, ancestral X material; black, ancestral autosome material. (E) Evolutionary interpretation, with X1X20 as ancestral, showing fusions hypothesized to generate X0, X1X2Y, and X1X2X3Y systems.
CHARACTER CORRELATION
To test for an association among species between chiasma localization and the sex chromosome system, two analyses were done on a reduced molecular phylogeny of 44 taxa for which both types of data were available.
MCMCglmm
To accommodate mixed continuous (chiasma localization) and categorical (sex chromosomes) data, we used a Bayesian approach to generalized linear mixed models (GLMM) that accounts for phylogeny as a covariance structure (Hadfield and Nakagawa 2010), via the R package MCMCglmm (Hadfield 2010, version 2.16). The tree used was the BEAST tree, trimmed to include only those taxa for which both chiasma localization and sex chromosome data are available (Fig. 6). Tests were done in both directions, with chiasma localization predicting neo‐Y presence, and with neo‐Y presence predicting chiasma localization. First, median or mean chiasma localization was treated as the predictor, and the response variable was the sex chromosome system, recorded as a binary variable (neo‐Y chromosome present vs. absent). For this test, we used a binomial GLMM with a logit link function, run as follows:
PriorC = list(R=list(V=1,fix=1),G=list(G1=list(V=1,nu=0.002))) Ainv = inverseA(tree)$Ainv mC = MCMCglmm(ychrom∼medianchiloc, random=∼species, ginverse=list(species=Ainv), prior=PriorC, data=data, family='categorical', pl=T, nitt = 2000000000, thin = 400, burnin = 1000000)
Figure 6.
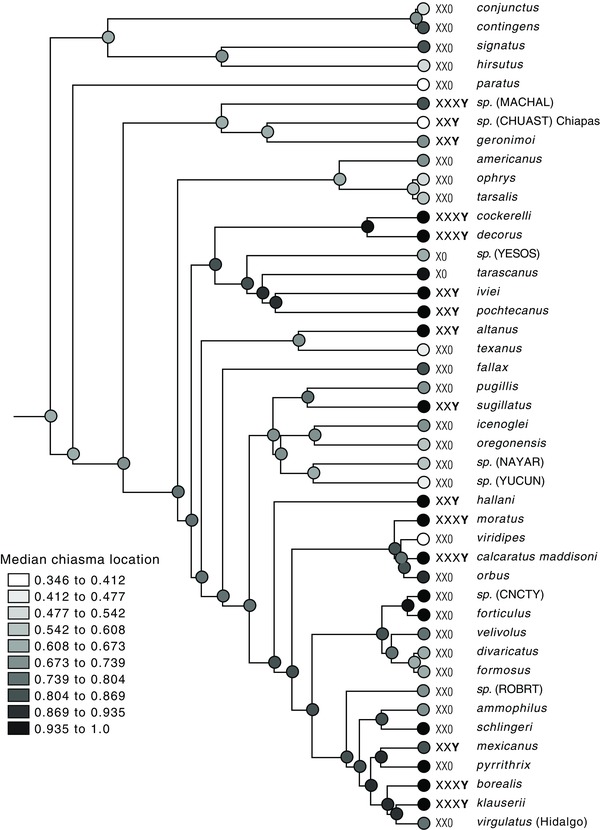
Evolution of chiasma localization. Ancestral states reconstructed using squared change parsimony weighted by branch lengths. Sex chromosomes indicated.
Otherwise, default settings of the package were used.
A secondary analysis was also performed with three taxa deleted because of concern that possibly‐false resolution in the viridipes and coecatus groups may be inflating independent origins of neo‐Y. Omitted were Habronattus calcaratus maddisoni, Habronattus borealis, and Habronattus klauserii, neo‐Y species representing possibly‐false independent origins and having the highest values for distal localization, and thus which would have been sample points most strongly in support of the tested correlation.
Second, the sex chromosome system was treated as a binary predictor variable, and median chiasma localization as the response, run as follows:
PriorA = list(R=list(V=1,nu=0.002), G=list(G1=list(V=1,nu=0.002))) Ainv = inverseA(tree)$Ainv mA = MCMCglmm(medianchiloc∼ychrom, random=∼species, ginverse=list(species=Ainv), prior=PriorA, data=data, nitt = 1000000000, burnin = 10000)
As a check of the validity of this second test, a generalized least squares model was fit by REML using the R function gls, with the command gls(medianchiloc∼ychrom, data, correlation = corPagel(1,tree).
Concentrated changes test
The concentrated changes test (Maddison 1990) was originally inspired by preliminary data on chiasma localization in Habronattus (Maddison 1990, p. 541). It asks whether evolutionary origins of Y chromosomes are more concentrated than expected by chance in regions of the phylogeny with distal localization. As this test requires two binary variables, chiasma localization was discretized by coding as 1 ( = distally localized) those taxa with median chiasma position 0.8 or over, those below as 0. This threshold matches a gap in the distribution of chiasma localization among species (Fig. S1). The test was applied using MacClade 4.08 (Maddison and Maddison 2005) with the BEAST tree, and with discretized chiasma localization as the independent variable and the presence of neo‐Y as the dependent. The exact calculations overflowed, and hence 10,000 simulations were done.
Results and Discussion
KARYOTYPES
Table S1 and Figure 4 show the scored karyotypes for specimens of 73 species and two additional subspecies. Five basic male karyotypes were observed (Fig. 3):
26 + X1X20, the karyotype typical for salticids, 26 acrocentric autosomes plus two acrocentric Xs (Fig. 3A);
26 + XO (Fig. 3B).
24 + X1X2Y (Fig. 3C);
22 + X1X2Y; and
22 + X1X2X3Y (Fig. 3D).
The appearance and behavior of the species with X1X2O and X1X2X3Y are similar to those for the few species reported by Maddison (1982). Karyotypes (2), (3), and (4) are newly reported for Habronattus.
All autosomes observed were acrocentrics. In X1X20 species, the two Xs are acrocentrics (one‐armed). In X1X2X3Y species, the Xs are all acrocentrics, and following Maddison (1982) these are named X1 (pairs achiasmately with X2), X2 (pairs achiastmately with X1 and chiasmately with one arm of Y) and X3 (pairs chiasmately with other arm of Y). We do not assume that X1 from one evolutionary origin of a neo‐Y is homologous to the X1 from a different evolutionary origin and likewise for X2s and X3s. The Y is a biarmed. In X1X2Y species, the Y is an acrocentric. All bivalents, or nearly all, showed a single chiasma.
NEO‐Y ORIGIN BY FUSION
Several lines of evidence suggest that the X1X2Y and X1X2X3Y systems arose from X1X20 systems by fusions between X chromosomes and autosomes, a mechanism that has commonly generated neo‐Y chromosomes (White 1973). First, the X1 and the proximal half of the X2 in X1X2Y and X1X2X3Y systems show the metaphase behavior of ancestral X material, because they lie side by side without chiasmata, and because they appear less condensed (heteropycnotic) than the autosomes (e.g., Fig. 3D)—as do the X1 and X2 in X1X20 species. In contrast, the distal half of the X2, all of the X3, and the Y of X1X2Y and X1X2X3Y systems behave like the autosomes in pairing and in condensation patterns. Second, with the exception of Habronattus banksi and Habronattus pochetecanus, X1X2Y species have 12 pairs of acrocentric autosomes and X1X2X3Y have 11 pairs.
A simple model (Fig. 3E) explaining this is that the modified X2 (in X1X2Y and X1X2X3Y species) represents a tandem fusion between an ancestral X and an autosome, with each portion retaining its ancestral behavior. The autosome's homologous partner therefore became the Y, and the number of uninvolved autosomes was reduced from 13 to 12 pairs. In X1X2X3Y species the Y would, in addition, have had a centric fusion with a second autosome to become biarmed, and this second autosome's homologous partner became the X3, further reducing the number of uninvolved autosomes from 12 to 11 pairs. This evolutionary model implies an ordered sequence: one step from X1X20 to X1X2Y and one step from X1X2Y to X1X2X3Y (Fig. 3E).
We do not have an explanation as to why two of the X1X2Y species (H. banksi and H. pochetecanus) have only 22 autosomes instead of the expected 24. Although this reduction may have occurred by an autosome–autosome fusion, these species have typical acrocentric autosomes with no observed sign of fusion. Except for these two species, there are no hints of autosome–autosome fusions or other karyotypic changes apart from those involved in sex chromosome systems. The two X0 species retain 26 autosomes, and thus their sex chromosomes may have evolved through simple X1–X2 fusions.
PHYLOGENY
The estimated substitution models for the two partitions were GTR + I+ GAMMA for 16sND1, and HKY + I + GAMMA for EF1‐alpha. The maximum clade credibility tree from BEAST is shown in Figure 4A, with estimated posterior probabilities. This tree agrees substantially with that reconstructed by Maddison and Hedin (2003), showing a few differences in middle depths as well as in some of the shallower clades.
INDEPENDENT ORIGINS OF X‐AUTOSOME FUSIONS
Multiple origins of Y chromosomes are implied by the distribution of sex chromosome types on the BEAST‐reconstructed phylogeny (Fig. 4A) and on a more conservative low‐resolution tree (Fig. 4B). The ancestral state in Habronattus is an X1X20 sex chromosome system by both likelihood and parsimony. This is consistent with the widespread occurrence of X1X20 system in salticids and other spiders (Araujo et al. 2005, 2012; Král et al. 2006). The Mk1 likelihood reconstruction on the BEAST tree (Fig. 4A) shows X1X20 on most internal nodes, with X1X2Y or X1X2X3Y systems evolving independently 13 times, six of which are in the viridipes and coecatus groups. The Mk1Ordered reconstruction is nearly the same, except that ambiguity in the viridipes group suggests as an alternative a single origin of Y (via X1X2Y) within the group (Fig. 4A, inset). If H. cf. paratus (Sucumbios), not included because of lack of molecular data, were included on this tree in its presumed location as sister to H. paratus, the number of origins of neo‐Y systems would rise to 14. Similarly, inclusion of the northern X1X2X3Y population of H. cuspidatus would likely raise the number of origins to 15. The apparent phylogenetic misplacement of H. notialis, H. jucundus and H. calcaratus calcaratus has little effect on our results, as this introduced no false origins of neo‐Y (all have X1X20) and none of these species were measured for chiasma localization.
Fifteen independent origins of neo‐Y chromosomes may very well be an overestimate, an artifact of arbitrary resolution in some clades. Phylogeny within the viridipes and coecatus groups was viewed by Maddison and Hedin (2003) as uncertain. In these clades, the BEAST tree is resolved, but with short branches having low posterior probabilities. Such short branches are highly problematic for subsequent analyses such as ancestral state reconstructions: we do not trust the branches, and yet if the clade is variable, they may imply homoplasy and force the interpretation of high rates of change. The implication from Figure 4A of three independent origins of X1X2X3Y in each of the coecatus and viridipes groups should be considered doubtful, or at least, unsupported.
We therefore examined sex chromosome evolution on Maddison and Hedin's (2003) highly conservative summary tree, which collapses the coecatus and viridipes groups to polytomies, and collapses many of the other deeper nodes in the tree. This tree has added to it those taxa for which we have karyotype data but not molecular sequence data, including two (H. cf. paratus [Sucumbios], H. cuspidatus [North]) that add extra independent origins of the X1X2X3Y system. A parsimony reconstruction on this tree (Fig. 4B) shows multiple origins of X1X2Y or X1X2X3Y, but considerably fewer than on the BEAST trees. This low‐resolution tree implies at least 15 evolutionary steps: one to X0, and the remainder to X1X2Y and X1X2X3Y. Most of the reduction from 29 steps in the fully resolved tree comes from deresolving the viridipes and coecatus groups, which saves 13 steps total.
The low‐resolution tree with parsimony reconstruction permits X1X2Y as equally parsimonious to X1X20 on many deeper nodes of Habronattus. X1X20 on these nodes would imply the neo‐Y was independently evolved many times; X1X2Y on these nodes would imply a gain of a Y followed by subsequent losses back to X1X20. Because the transition from X1X20 to X1X2Y involves a gain of a Y as well as a reduction in autosome number, reversion to X1X20 would require the X2 be dissociated at the old X‐autosome boundary and a centromere reconstituted, so as to recover the extra autosome pair. The complexity and required precision of this reversion suggests that X1X20 may be a more reasonable ancestral state, given that X1X20 and X1X2Y are otherwise equally parsimonious as ancestral.
To focus on the specific issue of origins of Y chromosome, we used additive binary recoding to generate a character with state 1 = neo Y present and 0 = absent. Mapped on the low‐resolution tree (Fig. 4B), this character shows by parsimony at least eight evolutionary gains or losses of a neo‐Y in Habronattus. Two considerations make us conclude that most of these changes were likely gains (origins of neo‐Y). First, reversions to X1X20 appear unlikely, as argued above. Second, the low‐resolution Maddison and Hedin (2003) tree lacks some clades highly supported in Figure 4A (e.g., Habronattus tarascanus to Habronattus zapotecanus) that would have shifted the balance to a reconstruction of all gains.
In a clade with 100 species of which 75 were sampled, 8–15 gains of neo‐Y would be a remarkable rate of X‐autosome fusions. It is difficult to assess how unusual this is, given that most of the extensive classical cytogenetics literature (e.g., White 1973) has yet to be placed into an explicit phylogenetic perspective. White (1969) does not present his phylogenetic reasoning, but concludes that morabine grasshoppers have had 11 independent X‐autosome fusions and five independent Y‐autosome fusions among the 170 sampled species. Explicitly phylogenetic studies include Flores et al. (2008) and Henning et al. (2011), who both found two independent origins of sex chromosome–autosome fusions in their respective clades. Leaché and Sites (2010) reconstructed at least three Y‐autosome fusions among the 53 sampled species of Sceloporus lizards. Colombo et al. (2005) report a density of X‐autosome fusions comparable to that of Habronattus, at least five independent fusions in a clade of acridid grasshoppers with 27 studied species.
Although theory predicts that Y chromosomes should eventually degrade (Charlesworth et al. 2005), the neo‐Y chromosomes of Habronattus show no notable differences in size or behavior from the portions of Xs with which they pair (and with which they were autosomal homologues before fusion). This and the phylogenetic distribution of neo‐Ys in many isolated species (Fig. 4) are consistent with recent origins. Three clades, however, have multiple species that share an apparently homologous Y chromosome: the H. dorotheae group (2 species, Habronattus geronimoi and H. sp. [CHUAST]), the H. banksi group (four species), and the Habronattus decorus group (six species). This suggests that the origin of the Y is old enough to have preceded the diversification of each of these groups.
CHIASMA LOCALIZATION
Median and mean chiasma locations measured in 46 taxa are shown in Table S2, and compiled in Figures 5 and S1. Although a few species show chiasmata primarily near the centromere, most species have more than half of the chiasmata in the distal half of the arms (Fig. 5), including 15 species that have the median chiasma location fully distal (Fig. S1). A typical X1X20 nucleus shows a diversity of chiasma positions among its chromosomes, whereas a typical X1X2X3Y nucleus shows more uniformity, with most chiasmata terminal. Thus, the between‐nuclei SD of the nucleus's mean chiasma position is higher for X1X20 individuals than X1X2X3Y individuals (0.165 vs. 0.122, nuclei from all species combined).
Figure 5.
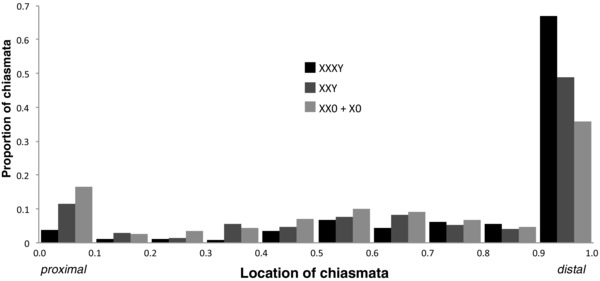
Proportion of chromosomes with various chiasma positions, summed across all nuclei and species of a sex chromosome type (X1X20, X1X2Y, or X1X2X3Y).
CORRELATION BETWEEN DISTAL LOCALIZATION AND X‐AUTOSOME FUSIONS
Figure 6 suggests that individuals with X1X2X3Y tend to have chiasmata especially distally localized, those with X1X20 less so, and those with X1X2Y intermediate. However, Figure 5 pools chromosomes from all individuals of a type without regard to phylogenetic placement, and we must consider whether the apparent pattern is a result of phylogenetic pseudoreplication.
Mapped on the BEAST phylogeny (Fig. 6), there is a hint that species with X1X2Y or X1X2X3Y tend to have chiasmata especially distally localized, with the exception of H. sp. (CHUAST) Chiapas. An informal analysis suggests that the pattern is phylogenetically replicated. We divided the BEAST tree into six independent phylogenetic regions, namely (1) a paraphyletic group of H. paratus + the dorotheae group (H. sp. [MACHAL] to H. geronimoi in Figure 6); (2) a clade of the decorus group and banksi group (Habronattus cockerelli through H. pochetecanus in Figure 6); (3) Habronattus altanus plus Habronattus texanus; (4) a clade of Habronattus oregonensis and others (Habronattus pugillis through H. sp. (YUCUN) in Figure 6); (5) the Habronattus viridipes group (Habronattus moratus through Habronattus orbus in Figure 6); and (6) the Habronattus coecatus group (Habronattus ammophilus through Habronattus virgulatus in Figure 6). In each of these six groups, the average of the median chiasma location is higher in species with a Y than species without (Fig. S2). The sample sizes of species with or without Y are very small within each group, but nonetheless there is a phylogenetically replicated and consistent pattern of distal localization being greater with a Y.
MCMCglmm analysis
All tests showed a significant association between chiasma localization and the presence of a neo‐Y, regardless of the direction of the prediction. The test with median chiasma location as a predictor variable required a long MCMC run of 2 × 109 to achieve an adequate effective sample size, because the values of the effect of median chiasma location were fairly unstable. However, they were convincingly above 0 (posterior mean = 1048; 95% confidence interval [CI] = 97–2098, pMCMC = 5 × 10−5; effective sample size = 1271), demonstrating a significant effect of chiasma location on the presence of a Y. In the secondary analysis with three species deleted to reduce possibly‐false convergences, the effect of chiasma location remains significant (posterior mean = 1043; 95% CI = 16–2369; pMCMC = 0.0008; effective sample size = 939).
The MCMC run for the test with neo‐Y as the predictor variable achieved large effective sample sizes in 106 generations. The effect of neo‐Y was significant (posterior mean = 0.237; 95% CI = 0.120–0.358; pMCMC = 0.0003; effective sample size = 93103). The generalized least squares model fit with a significant effect of Y chromosome (correlation = −0.261; coefficient value = 0.239; with standard error = 0.06; P = 0.0002). That MCMCglmm achieved reasonable results is supported by the fact that lambda estimated in the Pagel model (0.432) is close to that implied by the intraclass correlation coefficient from MCMCglmm (0.422 = species mean/[species mean + units mean]).
Concentrated changes test
On the tree of Figure 6, the discretized chiasma localization character (median location greater than 0.8 [ = distal] vs. not [ = proximal]) shows Habronattus as ancestrally proximal, with four origins of distal, in H. sp. (MACHAL), Habronattus signatus, Habronattus contingens, and in the ancestor of a large clade (Habronattus cockerelli through H. klauserii in Fig. 6). In that large clade, distal is homologous throughout, with a reversion to proximal nine times. This reconstruction was used as the independent variable to ask the question whether origins of a Y are more concentrated on the branches reconstructed as distal than expected by chance.
There are two equally parsimonious reconstructions for Y absence versus presence on the reduced tree of Figure 6, one showing 11 independent gains, of which 10 occur on branches reconstructed as distal, one on proximal. The second reconstruction shows nine gains and two losses, with eight of the losses on branches reconstructed as distal. The first reconstruction, equivalent to the MINSTATE reconstruction (Maddison and Maddison 2005), was used in a concentrated changes test to ask whether MINSTATE reconstructed changes are expected to be so concentrated on branches reconstructed as distally localized. Only eight of the 10,000 simulated cases resulted in 10 or more gains on distal‐reconstructed branches, estimating P = 0.0008. The second reconstruction, equivalent to MAXSTATE, was analyzed analogously. Only 43 of the 10,000 simulated cases resulted in eight or more gains on distal‐reconstructed branches, estimating P = 0.0043. By the concentrated changes test, there is therefore a significant association between localization and the evolution of a Y.
DO SEGREGATION PROBLEMS EXPLAIN THE CORRELATION?
The correlation tests show that lineages with a Y chromosome tend to have more distal chiasma localization than those without a Y, but the tests do not isolate a single mechanism underlying the correlation. Because chiasma position was measured on chromosomes other than Xs and Ys, we can rule out the direct mechanical hypothesis, namely that mechanical forces during meiosis directly and immediately cause the chiasmata in the sex chromosomes to be pushed distally without the need for an evolutionary change (e.g., Bidau 1990, 1993). The localization across all autosomes suggests there was an evolutionary change that affected all chromosomes, as found also by Colombo (1989). In this regard, our data differ from those of Bidau et al. (2001), who found that distal localization was confined to those chromosomes involved in the fusions. The one species for which a direct mechanical response remains a possibility is H. sp. (CHUAST) Chiapas. Its autosomal chiasmata are proximally localized, but the chiasmata between the X and neo‐Y are fully distal in the several nuclei available.
White's constraint‐lifting hypothesis invokes segregation problems when chiasmata are proximal in fused chromosome systems. It holds that chiasmata shifted to distal first, freeing a constraint and permitting fusions to evolve. An alternative, however, is that fusions occurred first, after which the chiasma localization evolved to relieve a burden of low fertility (the accommodation hypothesis; Colombo 1987, 1989). Our tests using generalized linear mixed models did not distinguish between these: we detected a correlation in both directions, whether we treated chiasmata location or neo‐Y as the predictor variable.
If the phylogeny had suggested that chiasma localization shifted first, followed by the evolution of a Y, then we might have preferred the constraint‐lifting hypothesis. However, there is no obvious indication from the phylogeny that distal localization came first (Fig. 6), and the generalized linear mixed models approach is not designed to detect such a pattern. The concentrated changes test (Maddison 1990) was designed precisely to detect a pattern of constraint lifting followed by enhanced rate of change, but it is too crude to be relied upon—it fails to incorporate a good stochastic model and requires ancestral states reconstructed in advance.
Other spiders are said to have strong proximal localization (White 1973), which is not true of most Habronattus. This may suggest that Habronattus as a whole had the constraint partly lifted before any fusions arose. However, any such pattern is not yet well documented, because we lack comprehensive and quantitative chiasma localization data from other salticid spiders.
One weak hint that repeated chiasma localization shifts may have preceded evolution of neo‐Ys can be seen in a modified correlation analysis in which all species with Y chromosomes are removed, but their sister species (which actually have X1X20 or X0) are recoded as having a Y. In our data, this requires deletion of 15 neo‐Y taxa and the fictitious assignment of a Y to H. tarascanus, H. texanus, H. pugillis, H. orbus, H. virgulatus, and Habronattus pyrrithrix. By applying the MCMCglmm analysis with chiasma location as a predictor as done for the full dataset, we are therefore asking whether distal chiasma localization in a species predicts its having a close relative with a Y. The accommodation hypothesis could not explain such a result, but the constraint‐lifting hypothesis could. The results obtained with our data for this modified test are suggestive, but not significant (effect of median chiasma localization, median = 1531; 95% CI = −442 to 5073; pMCMC = 0.068; effective sample size = 753).
We grouped X1X2Y and X1X2X3Y species together as “neo‐Y,” and did not analyze formally the differences in chiasma localization between them. However, distal localization appears to be slightly stronger in X1X2X3Y species (Fig. 5). White (1973) claims that chiasma localization explains the lack of Ys in spiders in general, and it is on this basis that we sought the correlation between chiasmata and neo‐Ys of either form (X1X2Y or X1X2X3Y). However, his mechanistic explanation of segregation problems is inapplicable to X1X2Y systems. He argues that proper segregation is best served by a regular triangular arrangement of centromeres, but X1X2Y can have no such triangular arrangement, because the two Xs pair and go to the same pole. Although this may be merely a failure of explanation, it could also raise questions as to whether we should have expected that chiasma localization constrains against X1X2Y.
COULD INTRALOCUS SEXUAL CONFLICT EXPLAIN THE CORRELATION?
Although White's hypothesis of segregation problems can explain the correlation between distal localization and neo‐Y, it does not well explain two key aspects of our results: why there are so many changes, and why they invariably (or almost invariably) involve X chromosomes. The lifting of White's segregation constraint should permit fusions in general, including autosome–autosome fusions. We reconstructed at least 8–15 origins or losses of neo‐Y, probably mostly origins, involving X‐autosome fusions. In contrast there was at most one hint of a simple autosome–autosome fusion (in H. banksi and H. pochetecanus), and one hint of an X–X fusion (in H. tarascanus and H. sp. [YESOS]). With 13 pairs of autosomes and only two X chromosomes, the probability that random fusions would all or mostly involve X chromosomes is small. There are several independent cases of fusion of autosomes to ancestral autosomal material—the second autosome‐Y fusion to go from X1X2Y to X1X2X3Y (Fig. 3E)—but in each of these cases the autosome fused to autosomal material that was already bound to the sex chromosomes. Thus, there is a significant bias to involvement of sex chromosomes in the reconstructed fusions.
Among the forces proposed to select for X‐autosome fusions (Yoshida and Kitano 2012), one possibility is particularly intriguing for Habronattus: intralocus sexual conflict (White 1957; Charlesworth and Charlesworth 1980). Habronattus males are known for their elaborate courtship ornaments and dances, in some species rivaling in complexity those of birds of paradise (Elias et al. 2012; Fig. S3). The possibility of an antagonistic co‐evolutionary arms race between males and females in Habronattus has already been proposed based on this complexity (Elias et al. 2012) and observations of xenophilia (Hebets and Maddison 2005) and hybridization (Maddison and Hedin 2003). If there is strong intralocus sexual conflict, with some alleles favored in males and others in females, then X‐autosome fusions could be selected for (White 1957; Charlesworth and Charlesworth 1980; Van Doorn and Kirkpatrick 2007; Kitano et al. 2009; Kitano and Peichel 2012).
It may not be coincidental that the species groups with the most complex courtship behavior, the coecatus, viridipes, and clypeatus species groups (e.g., Elias et al. 2012) may have a large number of origins of X1X2X3Y, a system that gives two pairs of autosomes the opportunity to be sex‐linked (Fig. 4A). The second fusion of an autosome, to the neo‐Y generated by the first fusion, would be particularly prone to selection under intralocus sexual conflict (Charlesworth and Charlesworth 1980). Intersexual conflict could be particularly strong in these species groups, provoking both maximal courtship complexity and maximal sex linkage.
We note that there is no obvious correlation between courtship traits in a species and the presence of a neo‐Y. Within the coecatus species group, for example, all members except H. borealis have extremely complex behaviors and ornaments (Elias et al. 2012), whether or not they have neo‐Y chromosomes. Within H. cuspidatus, the northern X1X2X3Y population is not notably different in courtship ornaments from the southern X1X20 population. If X‐autosome fusions are promoted by new sex‐linked traits, it is possible that these traits are hidden among the many others that are not linked to sex chromosomes, or it is possible that the sex‐linked traits concern the less‐studied female responses.
Loci involved in intersexual conflict would provide a second selective advantage for distal chiasmata in neo‐Y systems, thus potentially explaining the correlation between neo‐Y and distal chiasmata. Distal chiasmata in the neo‐Y chromosome would suppress recombination through much of the chromosome and thus permit the male‐favored versus female‐favored alleles to remain associated with the appropriate respective sex chromosome. This advantage of distal chiasmata would apply for both X1X2Y and X1X2X3Y systems.
Conclusions
Of the four hypotheses to explain the correlation between distal chiasma localization and the presence of neo‐Y, only the direct mechanical hypothesis can be ruled out by our data. A direct and immediate mechanical response to shift chiasmata after a fusion occurs cannot explain why chiasmata are shifted on autosomes uninvolved in the fusion. White's constraint‐lifting hypothesis and the accommodation hypothesis are relatively minor variants on one another, as they differ in evolutionary sequence but share the basic mechanism of segregation problems constraining against proximal chiasma localization in neo‐Y species. The fourth hypothesis, involving intralocus sexual conflict, suggests that chiasmata shift distally to limit recombination in neo‐Ys with sexually antagonistic loci.
Intralocus sexual conflict can explain the high number of fusions biased strongly to X‐autosome fusions, which White's segregation constraints hypothesis cannot. Intersexual conflict may also explain the coincidence of these fusions in Habronattus, a group with remarkably diverse and complex courtship behavior, but this is only a single data point phylogenetically. However, White's segregation constraint hypothesis may explain why spiders apart from Habronattus show so few neo‐Y chromosomes, which after all might be expected to happen occasionally even without strong intralocus sexual conflict. It is therefore possible that both sexual antagonism and segregation constraints were involved. Successful establishment of X‐autosome fusions, selected by intralocus sexual conflict, may have been more likely in species that had already evolved some distal localization, as this would avoid segregation problems and facilitate the isolation of alleles on the Y. Alternatively or in addition, distal localization could have been strengthened after a selected fusion to improve both segregation and isolation of alleles.
Even though distal chiasma localization is correlated with the presence of neo‐Y, the observed distal localization occurs across the entire chromosome complement. This suggests that if distal localization were the result of selection on chiasmata on the Y, the mechanism by which it was achieved was not localized to the Y. One implication of this is that the attendant reduction in recombination occurs in the autosomes as well. Thus, sex chromosome evolution may broadly restructure linkage in male Habronattus.
Supporting information
Figure S1. Numbers of taxa with various median chiasma locations (from: Habronattus spp., Maddison and Leduc‐Robert 2013).
Figure S2. Chiasma localization by species group and sex chromosome type.
Figure S3. Adult males of various Habronattus species, indicating chromosome complement (number of autosomes and sex chromosomes).
Table S1. Karyotypes of Habronattus and Pellenes specimens examined.
Table S2. Chiasma localization of Habronattus species.
ACKNOWLEDGMENTS
This work began in 1980 in the laboratory of K. Rothfels, whose dedication to classical cytogenetics research and its still‐untapped treasures were inspiring. We are indebted to M. Hedin for his important partnership on field expeditions and in conceiving the broad outline of the Habronattus project, and for his contributions to Habronattus phylogeny. D. Maddison assisted with collecting specimens and did some of the chromosome preparations. Tila M. Pérez helped to obtain Mexican permits and specimens, which were collected with the assistance of M. Hedin, J. L. Castelo, F. Alvarez, and R. Ayala. R. Maia helped considerably in helping us navigate MCMCglmm. We received helpful comments on the manuscript from G. Blackburn, J. Zhang, E. Piascik, and two anonymous reviewers. This work was supported by an NSERC Discovery Grant and an National Science Foundation grant (#DEB‐9707368) to WPM.
LITERATURE CITED
Associate Editor: J. Mank
- Araújo, D. , Cella D. M., and Brescovit A. D.. 2005. Cytogenetic analysis of the neotropical spider Nephilengys cruentata (Araneomorphae, Tetragnathidae): standard staining, NORs, C‐bands and base‐ specific fluorochromes. Braz. J. Biol. 65:193–202. [DOI] [PubMed] [Google Scholar]
- Araújo, D. , Schneider M. C., Paula‐Neto E., and Cella D. M.. 2012. Sex chromosomes and meiosis in spiders: a review Pp. 87–108 in Swan A., ed. Meiosis—molecular mechanisms and cytogenetic diversity. Intech, Rijeka, Croatia. [Google Scholar]
- Bidau, C. J. 1990. The complex Robertsonian system of Dichroplus pratensis (Melanoplinae, Acrididae). II. Effects of the fusions polymorphisms on chiasma frequency and distribution. Heredity 64:145–159. [Google Scholar]
- Bidau, C. J. . 1993. Causes of chiasma repatterning due to centric fusions. Rev. Brasil. Genet. 16: 293–296. [Google Scholar]
- Bidau, C. J. , Gimenez M. D., Palmerà C. L., and Searle J. B.. 2001. The effects of Robertsonian fusions on chiasma frequency and distribution in the house mouse (Mus musculus domesticus) from a hybrid zone in northern Scotland. Heredity 87:305–313. [DOI] [PubMed] [Google Scholar]
- Charlesworth, D. , and Charlesworth B.. 1980. Sex differences in fitness and selection for centric fusions between sex‐chromosomes and autosomes. Genet. Res. 35:205–214. [DOI] [PubMed] [Google Scholar]
- Charlesworth, D. , Charlesworth B., and Marais G.. 2005. Steps in the evolution of heteromorphic sex chromosomes. Heredity 95:118–128. [DOI] [PubMed] [Google Scholar]
- Colombo, P. C. 1987. Effects of centric fusions on chiasma frequency and position in Leptysma argentina (Acrididae: Orthoptera) I. Spontaneous and stable polymorphic centric fusions. Genetica 72:171–179. [Google Scholar]
- Colombo, P. C. ‐. 1989. Chromosome polymorphisms affecting recombination and exophenotypic traits in Leptysma argentina (Orthoptera): a populational survey. Heredity 62:289–299. [Google Scholar]
- Colombo P., Cigliano M. M., Sequeira A. S., Lange C. E., Vilardi J. C., and Confalonieri V. A.. 2005. Phylogenetic relationships in Dichroplus Stål (Orthoptera: Acrididae: Melanoplinae) inferred from molecular and morphological data: testing karyotype diversification. Cladistics 21:375–389. [DOI] [PubMed] [Google Scholar]
- Drummond, A. J. , and Rambaut A.. 2007. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 7:214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias, D. O. , Maddison W. P., Peckmezian C., Cirard M. B., and Mason A. C.. 2012. Orchestrating the score: complex multimodal courtship in the H. coecatus group of Habronattus jumping spiders (Araneae: Salticidae). Biol. J. Linnaean Soc. 105:522–547. [Google Scholar]
- Flores, S. V. , Evans A. L., and McAllister B. F.. 2008. Independent origins of new sex‐linked chromosomes in the melanica and robusta species groups of Drosophila . BMC Evol. Biol. 8:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griswold, C. E. 1987. A revision of the jumping spider genus Habronattus F.O.P. Cambridge (Araneae: Salticidae) with phenetic and cladistic analyses. Univ. Calif. Publ. Entomol. 107:1–344. [Google Scholar]
- Hadfield, J. D. 2010. MCMC methods for multi‐response generalized linear mixed models: the MCMCglmm R package. J. Stat. Software 33:1–22. [Google Scholar]
- Hadfield, J. D. , and Nakagawa S.. 2010. General quantitative genetic methods for comparative biology: phylogenies, taxonomies and multi‐trait models for continuous and categorical characters. J. Evol. Biol. 23:494–508. [DOI] [PubMed] [Google Scholar]
- Hebets, E. A. , and Maddison W. P.. 2005. Xenophilic mating preferences among populations of the jumping spider Habronattus pugillis Griswold. Behav. Ecol. 16:981–988. [Google Scholar]
- Henning, F. , Moysés C. B., Calcagnotto D., Meyer A., and de Almeida‐Toledo L. F.. 2011. Independent fusions and recent origins of sex chromosomes in the evolution and diversification of glass knife fishes (Eigenmannia). Heredity 106:391–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitano, J. , and Peichel C. L.. 2012. Turnover of sex chromosomes and speciation in fishes. Environ. Biol. Fish. 94:549–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitano, J. , Ross J. A., Mori S., Kume M., Jones F. C., Chan Y. F., Absher D. M., Grimwood J., Schmutz J., Myers R. M., Kingsley D. M., and Peichel C. L.. 2009. A role for a neo‐sex chromosome in stickleback speciation. Nature 461:1079–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Král, J. 2007. Evolution of multiple sex chromosomes in the spider genus Malthonica (Araneae: Agelenidae) indicates unique structure of the spider sex chromosome systems. Chromosome Res. 15:863–879. [DOI] [PubMed] [Google Scholar]
- Král, J , Musilová J., Št’áhlavský F., Řezáč M., Akan Z., Edwards R. L., Coyle F. A. and Almerje C. R.. 2006. Evolution of the karyotype and sex chromosome systems in basal clades of araneomorph spiders (Araneae: Araneomorphae). Chromosome Res. 14:859–880. [DOI] [PubMed] [Google Scholar]
- Leaché, A. D. , and J. W. Sites, Jr . 2010. Chromosome evolution and diversification in North American Spiny Lizards (Genus Sceloporus). Cytogenet. Genome Res. 127:166–181. [DOI] [PubMed] [Google Scholar]
- Maddison, D. R. 1985. Chromosomal diversity and evolution in the ground beetle genus Bembidion and related taxa (Coleoptera: Carabidae: Trechitae). Genetica 66:93–114. [Google Scholar]
- Maddison, D. R. , and Maddison W. P.. 2005. MacClade version 4.08: analysis of phylogeny and character evolution. Sinauer Associates, Sunderland, Massachusetts. [Google Scholar]
- Maddison, W. P. 1982. XXXY sex chromosomes in males of the jumping spider genus Pellenes (Araneae: Salticidae). Chromosoma (Berl.) 85:23–37. [Google Scholar]
- Colombo, P. C. . 1990. A method for testing the correlated evolution of two binary characters: are gains or losses concentrated on certain branches of a phylogenetic tree? Evolution 44:539–557. [DOI] [PubMed] [Google Scholar]
- Maddison, W. P. , and Hedin M. C.. 2003. Phylogeny of Habronattus jumping spiders (Araneae: Salticidae), with consideration of genitalic and courtship evolution. Syst. Entomol. 28:1–21. [Google Scholar]
- Maddison, W. P. , and Maddison D. R.. 2011. Mesquite: a modular system for evolutionary analysis. Version 2.75. Available at http://mesquiteproject.org. Accessed September 30, 2011.
- Nylander, J. A. A. 2004. MrModeltest v2.3. Program distributed by the author. Evolutionary Biology Centre, Uppsala University. [Google Scholar]
- Rasband, W. S. 2012. ImageJ. U. S. National Institutes of Health, Bethesda, Maryland: Available at http://imagej.nih.gov/ij/. Accessed July 2, 2012. [Google Scholar]
- Ronquist, F. , and Huelsenbeck J. P.. 2003. MRBAYES 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19:1572–1574. [DOI] [PubMed] [Google Scholar]
- Rowell, D. M. 1985. Complex sex‐linked fusion heterozygosity in the Australian huntsman spider Delena cancerides (Araneae: Sparassidae). Chromosoma (Berl.) 93:169–176. [Google Scholar]
- Sessions, S. K. 2008. Evolutionary cytogenetics in salamanders. Chromosome Res. 16:183–201. [DOI] [PubMed] [Google Scholar]
- Swofford, D. L. 2002. PAUP.* Phylogenetic Analysis Using Parsimony (*and Other Methods). Version 4.0b10. Sinauer Associates, Sunderland, MA. [Google Scholar]
- Van Doorn, G. S. , and Kirkpatrick M.. 2007. Turnover of sex chromosomes induced by sexual conflict. Nature 449;7164:909–912. [DOI] [PubMed] [Google Scholar]
- Wheeler, T. J. , and Kececioglu J. D.. 2007. Multiple alignments by aligning alignments. Bioinformatics 23:559–568. [DOI] [PubMed] [Google Scholar]
- White, M. J. D. 1957. Some general problems of chromosomal evolution and speciation in animals. Surv. Biol. Progr. 3:109–147. [DOI] [PubMed] [Google Scholar]
- White, M. J. D. . 1969. Chromosomal rearrangements and speciation in animals. Annu. Rev. Genet; 3:75–98. [Google Scholar]
- White, M. J. D. . 1973. Animal cytology and evolution. 3rd ed. Cambridge Univ. Press, Cambridge. [Google Scholar]
- Yoshida, K. , and Kitano J.. 2012. The contribution of female meiotic drive to the evolution of neo‐sex chromosomes. Evolution 66:3198–3208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwickl, D. J. 2006. Genetic algorithm approaches for the phylogenetic analysis of large biological sequence datasets under the maximum likelihood criterion Ph.D. diss., The University of Texas at Austin, Texas. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Numbers of taxa with various median chiasma locations (from: Habronattus spp., Maddison and Leduc‐Robert 2013).
Figure S2. Chiasma localization by species group and sex chromosome type.
Figure S3. Adult males of various Habronattus species, indicating chromosome complement (number of autosomes and sex chromosomes).
Table S1. Karyotypes of Habronattus and Pellenes specimens examined.
Table S2. Chiasma localization of Habronattus species.