

Abstract
Background Angiotensin converting enzyme 2 (ACE2), a monocarboxylase that degrades angiotensin II to angiotensin 1–7, is also the functional receptor for severe acute respiratory syndrome (SARS) coronavirus (SARS‐CoV) and is highly expressed in the lungs and heart. Patients with SARS also suffered from cardiac disease including arrhythmias, sudden cardiac death, and systolic and diastolic dysfunction.
Materials and methods We studied mice infected with the human strain of the SARS‐CoV and encephalomyocarditis virus and examined ACE2 mRNA and protein expression. Autopsy heart samples from patients who succumbed to the SARS crisis in Toronto (Canada) were used to investigate the impact of SARS on myocardial structure, inflammation and ACE2 protein expression.
Results Pulmonary infection with the human SARS‐CoV in mice led to an ACE2‐dependent myocardial infection with a marked decrease in ACE2 expression confirming a critical role of ACE2 in mediating SARS‐CoV infection in the heart. The SARS‐CoV viral RNA was detected in 35% (7/20) of autopsied human heart samples obtained from patients who succumbed to the SARS crisis during the Toronto SARS outbreak. Macrophage‐specific staining showed a marked increase in macrophage infiltration with evidence of myocardial damage in patients who had SARS‐CoV in their hearts. The presence of SARS‐CoV in the heart was also associated with marked reductions in ACE2 protein expression.
Conclusions Our data show that SARS‐CoV can mediate myocardial inflammation and damage associated with down‐regulation of myocardial ACE2 system, which may be responsible for the myocardial dysfunction and adverse cardiac outcomes in patients with SARS.
Keywords: Angiotensin converting enzyme 2, heart, macrophage, SARS coronar virus, severe acute respiratory syndrome
Introduction
Human (and rodent) angiotensin converting enzyme 2 (ACE2) is an endothelium‐bound carboxymonopeptidase with single active‐site catalytic region whose expression is limited mainly to endothelial cells of the arteries, arterioles and venules in various organs including the heart, lungs and kidneys [1, 2]. Loss of ACE2 leads to an age‐dependent cardiomyopathy [1, 2, 3] and kidney disease [4] while also enhancing pulmonary [5], cardiac [6] and renal injuries [7]. In addition to its peptidase action, ACE2 also functions as the receptor for the ‘severe acute respiratory syndrome’ coronavirus (SARS‐CoV) [8, 9, 10] and ACE2 expression is necessary for the pulmonary infection by SARS‐CoV [10, 11, 12]. Severe acute respiratory syndrome (SARS) spread rapidly through the world leading to significant morbidity and acute, often lethal, lung failure with a mortality of approximately 10% despite modern‐day therapies [11, 12]. In Toronto, the SARS outbreak was associated with loss of lives, considerable burden on the healthcare system and a huge economic impact [13, 14, 15].
Fatal SARS is associated with a viraemic response suggesting that SARS‐CoV may affect other organs [16]. Interestingly, patients infected with the SARS‐CoV suffered from cardiac disease including systolic and diastolic dysfunction [17], arrhythmias and sudden death [18]. We hypothesized that the interaction between SARS‐CoV and ACE2 in the heart could contribute to SARS‐mediated myocardial inflammation and damage. We showed that pulmonary infection with the SARS‐CoV in mice leads to myocardial SARS‐CoV in an ACE2‐dependent manner coupled with down‐regulation of the myocardial Ace2 mRNA and loss of ACE2 protein. In patients who died from SARS, presence of SARS‐CoV in the heart was associated with greater macrophage infiltration and myocardial damage in association with decreased myocardial ACE2 protein expression. Our results demonstrate that ACE2 plays a key role in mediating SARS‐CoV infection in the heart, which in combination with down‐regulation of the myocardial ACE2 could be the underlying pathophysiological mechanism of SARS‐associated heart disease.
Materials and methods
Experimental animals, in vivo SARS‐CoV infection and EMC myocarditis protocol
Mutant mice have been previously described [1] and only male littermate ACE2 mutant (Ace2−/y), wild‐type (Ace2+/y) controls were used in this study. All experiments were performed in accordance to institutional guidelines. The SARS‐CoV (Beijing strain, PUMC01 isolate) was used to infect mice via the intranasal route [10] and at day 2, mice were killed and the hearts were removed for further analyses. Wild‐type C57BL/6 mice (age 8–10 weeks) were maintained in a sterile, pathogen‐free environment and were injected peritoneally with 10 plaque‐forming units of encephalomyocarditis (EMC) virus or with normal saline as previously described [19]. Mice were killed at 3 days postinfection and hearts were aseptically removed and frozen in liquid nitrogen. All animal experiments were performed in accordance with the Institutional Guidelines and the Canadian Council on Animal Care.
Autopsies
In the greater Toronto area (Ontario, Canada), 44 patients died from SARS and 21 patients underwent autopsies. Dr J. Butany, a staff cardiovascular pathologist at the University Health Network, was the official pathologist of the SARS victims. The control group consisted of autopsies from age‐ and gender‐matched patients who were intubated and ventilated, and died from pneumonia and sepsis over the same time period. Our study was carried out in accordance with institutional research ethics board review.
Histology and immunohistochemistry
Trichrome and haematoxylin–eosin staining and visualization were carried out as previously described to assess for architectural alterations, inflammation and fibrosis [3, 20]. Macrophage and T‐lymphocyte specific staining using anti‐CD68 and anti‐CD3 antibodies, respectively, were carried out as previously described[20, 21]. Paraffin‐embedded sections were pretreated using the pepsin digestion method followed by treatment with mouse monoclonal anti‐CD68 antibody (Dako Labs, clone PGM1, 1 : 100 dilution, Mississauga, ON, Canada) or rabbit polyclonal anti‐CD3 antibody (Dako Labs, 1 : 100 dilution). Sections were then incubated with biotinylated multilink secondary antibody (ID Labs Biotechnology Inc., London, ON, Canada) and treated with horse peroxidase‐conjugated strepavidin labelling reagent (ID Labs Biotechnology Inc.). Macrophage counts, degree of inflammation, fibrosis and cardiomyocyte cross‐sectional area were obtained from three sections from each heart with five random views from each section as previously described [3]. Apoptosis was assessed by the terminal deoxynucleotidyl transferase‐mediated dNTP end‐labelling (TUNEL) using the ApopTag Plus kit (Intergen, Purchase, New York, NY, USA) [20]. Immunohistochemistry for ACE2 was carried out using anti‐ACE2 antibody as previously described [10] and quantified as previously described [3].
Real‐time PCR and Western blot analysis
Ethics approval and real‐time PCR analysis of human heart samples for the SARS‐CoV was performed as previously described [22, 23]. Reverse transcriptase polymerase‐chain reaction (RT‐PCR) analysis was in agreement with the diagnostic criteria for SARS as established by various agencies including the World Health Organization (WHO) and Centre for Disease Control (CDC). The homogenized heart samples were purified using Q1A shredder columns prior to RNA isolation by using the RNeasy Mini Kit (Qiagen Inc., Mississauga, ON, Canada). The RT‐PCR was carried out by using the Real Art HPA‐Coronavirus virus LightCycler RT Reagents Assay (Artus GmbH, Hamburg, Germany) with 80 LightCycler real‐time platform (Roche Diagnostics, Laval, Canada). The 80‐bp region of the SARS‐CoV RNA polymerase gene was amplified using previously published methods [22]. Western blot analysis for myocardial ACE2 protein was carried out using a primary mouse ACE2‐specific polyclonal antibody generated in our laboratory at a 1 : 1000 dilution and a secondary goat antimouse antibody (1 : 5000; BD Biosciences, Mississauga, ON, Canada) as previously described [1, 7]. Analysis of myocardial Ace2 mRNA expression was carried out using Taqman real‐time PCR using the forward primer: 5′‐GGATACCTACCCTTCCTACATCAGC‐3′, reverse primer: 5′‐CTACCCCACATATCACCAAGCA‐3′ and probe: 5′‐FAM‐CCACTGGATGCCTCCCTGCCC‐TAMRA‐3′ as previously described [7, 10].
Statistical analysis
All data are shown as mean ± SEM. All statistical analyses were performed using spss software (Chicago, IL, USA; Version 10·1). We used anova followed by the Student’s Neuman–Keuls test for multiple comparison testing while the Student’s t‐test was used for comparisons between any two groups.
Results
ACE2 mediates myocardial SARS‐CoV infection and leads to its down‐regulation
Angiotensin converting enzyme 2 is the functional receptor for the SARS‐CoV and mediates SARS‐CoV pulmonary infection [8, 10]. We examined the myocardial response to pulmonary infection with the SARS‐CoV. Within 2 days following SARS‐CoV infection, there was a marked presence of the SARS‐CoV in the heart of wild‐type (Ace2+/y) mice, while SARS‐CoV levels were reduced in the hearts of Ace2−/y mice providing the first evidence for dependence of myocardial SARS‐CoV infection on ACE2 expression (Fig. 1a). Interestingly, this process lead to a partial down‐regulation of Ace2 mRNA expression in wild‐type mice (Fig. 1b) in association with a complete loss of myocardial ACE2 protein levels (Fig. 1c,d). These results show that in a well‐validated pulmonary murine model of SARS [10], SARS‐CoV can clearly infect the heart and modulate ACE2 expression. To decipher whether the effects on ACE2 expression is mediated directly by SARS‐CoV vs. a secondary myocarditis process, we studied the encephalomyocarditis (EMC) virus‐induced murine model of viral myocarditis. Consistent with the impact of SARS‐CoV on myocardial Ace2 mRNA expression, EMC viral myocarditis was associated with a significant decrease in Ace2 mRNA (Fig. 1e). In contrast, myocardial ACE2 protein levels showed a significant increase (Fig. 1f,g) suggesting an increased transcriptional efficiency of Ace2 mRNA and/or reduced proteolytic processing of ACE2. These results demonstrate that pulmonary infection with SARS‐CoV in our murine model leads to myocardial SARS‐CoV in an ACE2‐dependent manner in association with decreased myocardial Ace2 mRNA expression and a specific reduction in myocardial ACE2 protein levels.
Figure 1.
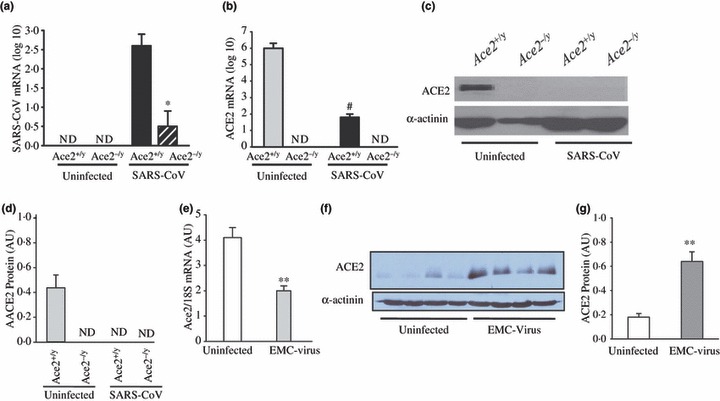
Pulmonary SARS‐CoV infection leads to myocardial SARS‐CoV infection and down‐regulation of myocardial ACE2 expression. (a–d) Human SARS‐CoV mRNA in the hearts of infected mice showing a clear dependence on ACE2 for myocardial SARS‐CoV infection (a) with down‐regulation of myocardial Ace2 mRNA expression based on real‐time PCR (b) and myocardial ACE2 protein levels shown by Western blot analysis (c) and quantification (d) in response to pulmonary SARS‐CoV infection. *P < 0·01 compared with infected Ace2+/y group; #P < 0·01 compared with uninfected group, n = 5, ND, not detectable. (e–g) Discordant changes in myocardial Ace2 mRNA and myocardial ACE2 protein levels in encephalomyocarditis (EMC) virus‐induced myocarditis with real‐time PCR showing reduced myocardial Ace2 mRNA (e) with increased myocardial ACE2 protein levels based on Western blot analysis (f) and quantification (g). **P < 0·01 compared with placebo group, n = 5.
Presence of SARS‐CoV in autopsied heart samples in association with myocardial damage
Interestingly, severe cardiac dysfunction has also been described in patients with SARS [17, 18]. Given the high expression of ACE2 in the heart [1, 24] coupled with the observation that SARS‐CoV is detectable in the plasma of SARS patients [12, 25], we examined whether pulmonary SARS‐CoV infection leads to myocardial SARS‐CoV infection in patients who died from SARS. We examined the archived postmortem autopsy heart tissues from patients who had succumbed to the SARS crisis for the evidence of myocardial SARS‐CoV infection. Twenty patients, all of whom had confirmed diagnoses of SARS with SARS‐CoV being detected in their lungs, were studied. Reverse transcriptase‐polymerase chain reaction analysis showed that 35% of the patients (7 of 20) had positive SARS‐CoV genome detected in the heart (Fig. 2a) with an average viral load of 4·07 × 106 copies of SARS‐CoV per gram of heart tissue (age: 70 ± 12 years; 3M/4F). Importantly, the duration of the illness was significantly shortened in patients with detectable SARSCoV in their hearts (n = 7; 3·9 ± 2·3 days) vs. patients without SARS‐CoV in the heart (n = 13; 43·2 ± 9·7 days) (P < 0·05) (Fig. 2a). These results indicate that in patients with SARS, pulmonary SARS‐CoV infection can clearly lead to SARS‐CoV infection in the heart resulting in a more aggressive illness and earlier death. Trichrome‐staining showed increased myocardial inflammation and interstitial fibrosis in patients who had SARS‐CoV detected in their hearts (Fig. 2a–e). The presence of myocardial inflammation and reduced ACE2 expression in response to myocardial SARS‐CoV infection was associated with pathological hypertrophy as shown by increased cardiomyocyte cross‐sectional area (Fig. 2f). Assessment of apoptosis using TUNEL staining revealed no increased apoptosis in the autopsied heart samples (Fig. 2g,h).
Figure 2.

Detection of SARS‐CoV genome in postmortem human heart samples with evidence of myocardial inflammation and damage. (a) Presence of SARS‐CoV genome in the heart of 35% of the patients who died from SARS (+SARS‐CoV, open bar, n = 7) and its negative impact on illness duration compared with patients who died from SARS without SARS‐CoV in the heart (‐SARS‐CoV, closed bar, n = 13); #P < 0·05 compared with +SARS‐CoV group. (b–h) Representative trichrome‐stained myocardial section obtained from a patient who died from non‐SARS related sepsis (bacterial pneumonia) (b), SARS with evidence of SARS‐CoV in the heart (c), SARS without evidence of SARS‐CoV in the heart (d) showing increased interstitial fibrosis and inflammation (e) and cardiomyocyte hypertrophy based on myocyte cross‐sectional area (MCSA) (f) without evidence of apoptosis in patients who died from SARS with (g) and without (h) evidence of SARS‐CoV in the heart. Scale bar represents 50 μM. *P < 0·01 compared with all other groups. VL‐ve and VH‐ve = patients who died from a non‐SARS related sepsis (open bar), VL+ve and VH+ve = patients who died from SARS with SARS‐CoV in the heart (grey bar) and VL+ve and VH‐ve = patients who died from SARS without SARS‐CoV in the heart (closed bar), n = 7 per group.
Myocardial macrophage infiltration in patients with myocardial SARS‐CoV
The presence of SARS‐CoV in the heart suggests that this could lead to myocardial inflammation. We used a positive control group that consisted of 13 patients who died from SARS without evidence of SARS‐CoV in the heart (age 67 ± 12 years; 6M/7F) and negative control group consisting of patients who died from bacterial pneumonia and did not have SARS (n = 7; age 68 ± 9 years; 3M/4F). Immunohistochemical staining of postmortem myocardial tissue using a macrophage‐specific (CD‐68) cell‐surface marker revealed a significant amount of macrophage infiltration (Fig. 3a–d), which was clearly increased in patients who had SARS‐CoV in their hearts with only a minor elevation in patients without SARS‐CoV in the heart (Fig. 3e). In contrast, immunohistochemical staining of T‐cell specific (CD‐3) cell‐surface marker showed that myocardial lymphocytic infiltration was minimal (Fig. 3f–h) with no significant difference in lymphocyte count between groups (Fig. 3i).
Figure 3.

Increased macrophage infiltration in the absence of increased lymphocytic infiltration in the left ventricle of patients who died from SARS. (a–e) Representative anti‐CD68‐stained immunohistochemistry section from a patient who died from non‐SARS related sepsis (bacterial pneumonia) (a), SARS with evidence of SARS‐CoV in the heart (b), SARS without evidence of SARS‐CoV in the heart (c) and a positive control section obtained from human spleen (d) with quantification of myocardial macrophage count (e). (f–i) Representative anti‐CD3 immunohistochemistry illustrating a representative section from a patient who died from SARS with evidence of SARS‐CoV in the heart (f), SARS without evidence of SARS‐CoV in the heart (g) and a positive control section obtained from human spleen (h) with quantification of myocardial lymphocyte count (i). Scale bar represents 50 μM. #P < 0·05 compared with all other groups. VL‐ve and VH‐ve = patients who died from a non‐SARS related sepsis (open bar), VL+ve and VH+ve = patients who died from SARS with SARS‐CoV in the heart (grey bar) and VL+ve and VH‐ve = patients who died from SARS without SARS‐CoV in the heart (closed bar), n = 7 per group.
Reduced ACE2 protein expression in patients with myocardial SARS‐CoV
The ability of SARS‐CoV to utilize ACE2 as a receptor in vivo, and the presence of SARS‐CoV in the heart suggests that SARS‐CoV can interact with the myocardial ACE2 system. We hypothesized that the presence of SAR‐CoV in the hearts could be associated with decreased ACE2 protein expression. Consistent with our observations in our murine model, immunohistochemistry for myocardial ACE2 protein expression showed that the presence SARS‐CoV in the heart was associated with marked down‐regulation of ACE2 protein expression (Fig. 4a–c), which was quantified and shown in Fig. 4f. The specificity of our anti‐ACE2 antibody was shown by pre‐incubation with human recombinant ACE2 (1 mg mL−1, kindly provided by Apeiron Biologics, Austria), which blocked most of the immunostaining for the native ACE2 protein (Fig. 4e).
Figure 4.

Reduced ACE2 protein expression in patients who died from SARS and had SARS‐CoV detected in their hearts. (a–e) Representative sections showing staining for ACE2 in the heart from a patient who died from non‐SARS related sepsis (bacterial pneumonia) (a), SARS with evidence of SARS‐CoV in the heart (b), SARS without evidence of SARS‐CoV in the heart (c), a negative control section (d) while pre‐incubation with recombinant human ACE2 (1 mg mL−1) prevented ACE2 staining (e). Scale bar represents 50 μM. (f) Quantification of ACE2 immunohistochemical staining showing reduced ACE2 protein expression in patients who died from SARS with SARS‐CoV in their hearts. *P < 0·05 compared with all other groups; VL‐ve and VHve = patients who died from a non‐SARS related sepsis (open bar), VL+ve and VH+ve = patients who died from SARS with SARS‐CoV in the heart (grey bar) and VL+ve and VH‐ve = patients who died from SARS without SARS‐CoV in the heart (closed bar), n = 7 per group.
Discussion
Patients who were infected with the SARS virus suffered from cardiac disease ranging from systolic and diastolic dysfunction [17], arrhythmias and sudden death [18]. In this study, we have provided evidence that ACE2 functions as the myocardial SARS‐CoV receptor in vivo. Respiratory SARS‐CoV infection in our murine model leads to an ACE2‐dependent SARS‐CoV infection in the heart and decreased myocardial Ace2 mRNA expression. Cytokine‐mediated transcriptional down‐regulation of Ace2 mRNA could have lead to decreased Ace2 mRNA level in SARS‐CoV and EMC‐mediated viral myocarditis[26]. Virus‐mediated receptor down‐modulation has been described for multiple viruses including HIV, measles and pulmonary SARS‐CoV [10]. The complete loss of ACE2 protein in hearts infected with SARS‐CoV could be secondary to the activation of ADAM‐17/TACE by the SARS spike protein, which is known to cleave and release ACE2 [27] and/or due to SARS‐CoV binding to ACE2 in the endothelial cells leading to endocytosis of the ligand/receptor complex [12, 28] and subsequent intracellular degradation of ACE2.
In patients who succumbed to SARS, SARS‐CoV was detected in the heart of 35% of the subjects suggesting that SARS‐CoV is capable of infecting the myocardium in susceptible individuals. We showed that patients who had SARS‐CoV in their hearts died considerably earlier suggesting that myocardial SARS‐CoV infection was associated with a more aggressive course of illness. SARS‐CoV interaction with ACE2 led to SARS‐associated cardiomyopathy and likely contributed significantly to the morbidity and mortality in patients with SARS [17, 18]. Consistent with the cardiac disease in patients who succumbed to SARS, we previously reported that bilateral pulmonary oedema in association with bilateral pleural effusions was frequently observed at the time of autopsy [22]. Increased myocardial interstitial fibrosis in patients with SARS‐CoV in the heart is consistent with recent findings showing that the SARS‐CoV nucleocapsid (N) protein potentiates transforming growth factor‐beta‐mediated fibrosis [29]. Consistent with observations in lung tissue from patients affected by SARS [23], CD 68 positive macrophages were detected in SARS‐positive hearts with an absence of lymphocytic invasion. In contrast to classic viral myocarditis as seen with the enteroviruses and herpesvirus type 6, which are associated with a prominent lymphocytic infiltration [30], SARS‐CoV‐induced myocardial inflammation is mediated predominantly by macrophages and the resultant production of chemokines [23, 31].
The deleterious effects of myocardial SARS‐CoV infection could be perpetuated by the prompt and severe down‐regulation of ACE2 resulting in increased Ang II action and/or loss of the cardioprotective effects from Ang 1–7 [1, 3, 5] and/or activation of ADAM‐17/TACE by the SARS spike protein leading to an increased release of TNF‐α [20, 27]. Myocardial dysfunction could also be secondary to the strong interferon‐mediated immunopathological events associated with the immune response in patients with SARS [31, 32]. The cardiovascular involvement could explain the marked age dependence of mortality seen in patients with SARS [11, 12] because of decreased cardiovascular reserve with ageing. Consistent with ACE2 being a key SARS‐CoV receptor, patients with diabetes mellitus in whom ACE2 expression is increased [7] showed greater susceptibility to SARS [13, 14]. Our results imply that interactions between infectious agents with key receptor/signalling pathways in the cardiovascular system may confer unique sensitivity to SARS‐CoV and potentially other viral infections. Indeed, interaction and subsequent activation of the PI3K signalling by influenza A virus are an important determinant of viral replication and may lead to adverse cardiovascular effects in the setting of an avian influenza epidemic [33].
Address
Division of Cardiology, Department of Medicine (G. Y. Oudit); Mazankowski Alberta Heart Institute (G. Y. Oudit, Z. Kassiri); Department of Physiology, University of Alberta, Edmonton, Canada (Z. Kassiri); National Key Laboratory of Medical Molecular Biology, Peking Union Medical College, Beijing, China (C. Jiang); Division of Cardiology, University Health Network, University of Toronto, Toronto, Canada (P. P. Liu); Division of Infectious Disease, Mount Sinai, Hospital, Toronto, Canada (S. M. Poutanen); IMBA, Institute for Molecular Biotechnology of the Austrian Academy of Sciences, Vienna, Austria (J. M. Penninger); Division of Cardiovascular Pathology, Department of Pathology, University Health Network, Toronto, Canada (J. Butany).
Acknowledgements
We acknowledge the financial support from the Canadian Institute for Health Research (G.Y.O. Grant 86602), Alberta Heritage Foundation for Medical Research (G.Y.O.), the EuGeneHeart (EU 6th Framework programs), the Austrian National Bank and IMBA (J.M.P.) and the Division of Pathology, University Health Network, Toronto (J.B.).
References
- 1. Crackower MA, Sarao R, Oudit GY, Yagil C, Kozieradzki I, Scanga SE et al. Angiotensinconverting enzyme 2 is an essential regulator of heart function. Nature 2002;417:822–8. [DOI] [PubMed] [Google Scholar]
- 2. Oudit GY, Crackower MA, Backx PH, Penninger JM. The role of ACE2 in cardiovascular physiology. Trends Cardiovasc Med 2003;13:93–101. [DOI] [PubMed] [Google Scholar]
- 3. Oudit GY, Kassiri Z, Patel MP, Chappell M, Butany J, Backx PH et al. Angiotensin II‐mediated oxidative stress and inflammation mediate the age‐dependent cardiomyopathy in ACE2 null mice. Cardiovasc Res 2007;75:29–39. [DOI] [PubMed] [Google Scholar]
- 4. Oudit GY, Herzenberg AM, Kassiri Z, Wong D, Reich H, Khokha R et al. Loss of angiotensinconverting enzyme‐2 leads to the late development of angiotensin II‐dependent glomerulosclerosis. Am J Pathol 2006;168:1808–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Imai Y, Kuba K, Rao S, Huan Y, Guo F, Guan B et al. Angiotensin‐converting enzyme 2 protects from severe acute lung failure. Nature 2005;436:112–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yamamoto K, Ohishi M, Katsuya T, Ito N, Ikushima M, Kaibe M et al. Deletion of angiotensinconverting enzyme 2 accelerates pressure overload‐induced cardiac dysfunction by increasing local angiotensin II. Hypertension 2006;47:718–26. [DOI] [PubMed] [Google Scholar]
- 7. Wong DW, Oudit GY, Reich H, Kassiri Z, Zhou J, Liu QC et al. Loss of angiotensin‐converting enzyme‐2 (Ace2) accelerates diabetic kidney injury. Am J Pathol 2007;171:438–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Li W, Moore MJ, Vasilieva N, Sui J, Wong SK, Berne MA et al. Angiotensin‐converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003;426:450–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Turner AJ, Hiscox JA, Hooper NM. ACE2: from vasopeptidase to SARS virus receptor. Trends Pharmacol Sci 2004;25:291–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kuba K, Imai Y, Rao S, Gao H, Guo F, Guan B et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus‐induced lung injury. Nat Med 2005;11:875–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Peiris JS, Yuen KY, Osterhaus AD, Stohr K. The severe acute respiratory syndrome. N Engl J Med 2003;349:2431–41. [DOI] [PubMed] [Google Scholar]
- 12. Peiris JS, Guan Y, Yuen KY. Severe acute respiratory syndrome. Nat Med 2004;10:S88–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Booth CM, Matukas LM, Tomlinson GA, Rachlis AR, Rose DB, Dwosh HA et al. Clinical features and short‐term outcomes of 144 patients with SARS in the greater Toronto area. JAMA 2003;289:2801–9. [DOI] [PubMed] [Google Scholar]
- 14. Fowler RA, Lapinsky SE, Hallett D, Detsky AS, Sibbald WJ, Slutsky AS et al. Critically ill patients with severe acute respiratory syndrome. JAMA 2003;290:367–73. [DOI] [PubMed] [Google Scholar]
- 15. Poutanen SM, Low DE, Henry B, Finkelstein S, Rose D, Green K et al. Identification of severe acute respiratory syndrome in Canada. N Engl J Med 2003;348:1995–2005. [DOI] [PubMed] [Google Scholar]
- 16. Farcas GA, Poutanen SM, Mazzulli T, Willey BM, Butany J, Asa SL et al. Fatal severe acute respiratory syndrome is associated with multiorgan involvement by coronavirus. J Infect Dis 2005;191:193–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li SS, Cheng CW, Fu CL, Chan YH, Lee MP, Chan JW et al. Left ventricular performance in patients with severe acute respiratory syndrome: a 30‐day echocardiographic follow‐up study. Circulation 2003;108:1798–803. [DOI] [PubMed] [Google Scholar]
- 18. Yu CM, Wong RS, Wu EB, Kong SL, Wong J, Yip GW et al. Cardiovascular complications of severe acute respiratory syndrome. Postgrad Med J 2006;82:140–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dong R, Liu P, Wee L, Butany J, Sole MJ. Verapamil ameliorates the clinical and pathological course of murine myocarditis. J Clin Invest 1992;90:2022–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kassiri Z, Oudit GY, Sanchez O, Dawood F, Mohammed FF, Nuttall RK et al. Combination of tumor necrosis factor‐alpha ablation and matrix metalloproteinase inhibition prevents heart failure after pressure overload in tissue inhibitor of metalloproteinase‐3 knock‐out mice. Circ Res 2005;97:380–90. [DOI] [PubMed] [Google Scholar]
- 21. Abbate A, Bonanno E, Mauriello A, Bussani R, Biondi‐Zoccai GG, Liuzzo G et al. Widespread myocardial inflammation and infarct‐related artery patency. Circulation 2004;110:46–50. [DOI] [PubMed] [Google Scholar]
- 22. Mazzulli T, Farcas GA, Poutanen SM, Willey BM, Low DE, Butany J et al. Severe acute respiratory syndrome‐associated coronavirus in lung tissue. Emerg Infect Dis 2004;10:20–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hwang DM, Chamberlain DW, Poutanen SM, Low DE, Asa SL, Butany J. Pulmonary pathology of severe acute respiratory syndrome in Toronto. Mod Pathol 2005;18:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Donoghue M, Hsieh F, Baronas E, Godbout K, Gosselin M, Stagliano N et al. A novel angiotensinconverting enzyme‐related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1‐9. Circ Res 2000;87:E1–9. [DOI] [PubMed] [Google Scholar]
- 25. Grant PR, Garson JA, Tedder RS, Chan PK, Tam JS, Sung JJ. Detection of SARS coronavirus in plasma by real‐time RT‐PCR. N Engl J Med 2003;349:2468–9. [DOI] [PubMed] [Google Scholar]
- 26. De Lang A, Osterhaus AD, Haagmans BL. Interferon‐gamma and interleukin‐4 downregulate expression of the SARS coronavirus receptor ACE2 in Vero E6 cells. Virol 2006;353:474–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Haga S, Yamamoto N, Nakai‐Murakami C, Osawa Y, Tokunaga K, Sata T et al. Modulation of TNF‐alpha‐converting enzyme by the spike protein of SARS‐CoV and ACE2 induces TNF‐alpha production and facilitates viral entry. Proc Natl Acad Sci USA 2008;105:7809–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang S, Guo F, Liu K, Wang H, Rao S, Yang P et al. Endocytosis of the receptor‐binding domain of SARS‐CoV spike protein together with virus receptor ACE2. Virus Res 2008;136:8–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhao X, Nicholls JM, Chen YG. Severe acute respiratory syndrome‐associated coronavirus nucleocapsid protein interacts with Smad3 and modulates transforming growth factor‐beta signaling. J Biol Chem 2008;283:3272–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Magnani JW, Dec GW. Myocarditis: current trends in diagnosis and treatment. Circulation 2006;113:876–90. [DOI] [PubMed] [Google Scholar]
- 31. Cameron MJ, Ran L, Xu L, Danesh A, Bermejo‐Martin JF, Cameron CM et al. Interferon‐mediated immunopathological events are associated with atypical innate and adaptive immune responses in patients with severe acute respiratory syndrome. J Virol 2007;81:8692–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cameron MJ, Bermejo‐Martin JF, Danesh A, Muller MP, Kelvin DJ. Human immunopathogenesis of severe acute respiratory syndrome (SARS). Virus Res 2008;133:13–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hale BG, Jackson D, Chen YH, Lamb RA, Randall RE. Influenza A virus NS1 protein binds p85beta and activates phosphatidylinositol‐3‐kinase signaling. Proc Natl Acad Sci USA 2006;103:14194–9. [DOI] [PMC free article] [PubMed] [Google Scholar]