

Abstract
Aim
Leukodystrophies are a group of inherited white matter disorders with clinical, genetic, and imaging heterogeneity, which usually pose a diagnostic challenge for physicians. We aimed to identify new clinical characteristics and novel pathogenic variants of hereditary leukodystrophies in this study.
Methods
Whole exome sequencing (WES) was performed in 28 unrelated patients clinically suspected with leukodystrophies. Leukocytes enzyme activity test, electroencephalogram (EEG), electromyography (EMG), and brain MRI were conducted. Functional analysis was performed, and the pathogenicity of variants was classified according to the American College of Medical Genetics and Genomics (ACMG) standards and guidelines.
Results
We made definite diagnosis in 8 probands with 12 pathogenic variants and reported new clinical characteristics and imaging features of these patients. Three novel pathogenic variants were identified, including a microdeletion variant c.2654_2654+3del within CSF1R, a nonsense variant c.1321C>T, and a missense variant c.166G>C within GALC.
Conclusion
Our results have deepened the understanding of clinical, genetic, and imaging heterogeneity of hereditary leukodystrophies, and expanded the spectrum of pathogenic variants and clinical features.
Keywords: clinical heterogeneity, genetic heterogeneity, hereditary leukodystrophy
1. INTRODUCTION
Leukodystrophies are a group of inherited white matter disorders with disparate genetic underpinnings that specifically affected the axon‐glia unit.1, 2 When considering a specific leukodystrophy case, genotype‐phenotype correlation often is not precise.1, 3 Patients with atypical clinical manifestations were usually misdiagnosed as spastic paraplegia, Charcot‐Marie‐Tooth disease (CMT), multiple sclerosis (MS), or cerebral autosomal dominant with subcortical infarcts and leukoencephalopathy (CADASIL). Therefore, early and accurate diagnosis of leukodystrophies usually poses a challenge to physicians.
Direct sequencing of certain suspected gene associated with leukodystrophies is neither effective nor economic, while whole exome sequencing (WES) can provide convenience to screen candidate genes in a short time.4 Discovery of causative genes leads to accurate prognosis and genetic counselling for the patients, and it also helps clinicians to get a better understanding of leukodystrophies.
Here, we performed WES in a cohort of Chinese patients with white matter abnormality of unknown etiology, dementia, or spastic paraparesis without pathogenic variants. We made definite diagnosis in 8 probands carrying 12 pathogenic variants and reported new clinical characteristics and imaging features of them.
2. MATERIAL AND METHODS
2.1. Patients
Patients who exhibit agnogenic white matter abnormality, unexplained spastic paraparesis, or early‐onset dementia were suggestive of leukodystrophies.1 Firstly we screened causative genes for CADASIL, adrenoleukodystrophy, hereditary spastic paraplegia (HSP), and familial Alzheimer's disease (FAD) on these patients in our previous studies,5, 6, 7, 8 and patients who carry pathogenic variants were excluded. Then, 28 unrelated patients (probands) were recruited consecutively between March 2015 and August 2018 from the Department of Neurology in Second Affiliated Hospital of Zhejiang University School of Medicine. Among them, 17 individuals had abnormal signals in brain white matter, and the other 11 individuals had been diagnosed with HSP. Leukocytes enzyme activity test, electroencephalogram (EEG), electromyography (EMG), and brain MRI were conducted. Written informed consents were obtained for participants or their legal guardians. The study was approved by the Ethics Committee of the Second Affiliated Hospital.
2.2. Whole exome sequencing
Genomic DNAs captured from peripheral blood were sequenced by WES. Details on library preparation, sequencing protocol, bioinformatics analysis, and filtering methods were conducted as described previously.9 All filtered variants were further validated by Sanger sequencing on an ABI 3500xL Dx Genetic Analyzer (Applied Biosystems) in the probands and the available family members.
2.3. Reverse transcription PCR
RNAs were extracted from leukocyte of proband 2 which carries the CSF1R splicing variant and normal controls by RNAiso Plus (Takara). Mutant CSF1R mRNA of proband 2, wild type CSF1R mRNA (NM_005211) of normal controls, and wild type (WT) EIF2B3 (NM_020365) mRNA were reversely transcribed into cDNA by PrimeScript™ II 1st Strand cDNA Synthesis Kit (Takara) and amplified by high‐fidelity DNA polymerase KOD‐Plus‐Neo (TOYOBO). EIF2B3 is one of the subunits of eIF2B.10 Reduced eIF2B activity enhances the translation of ATF4 (NM_182810) mRNA due to the presence of upstream open reading frames (uORFs) in its 5′ untranslated regions (UTRs).11 In order to evaluate the effects of mutant EIF2B3 on eIF2B, wild type 5′ UTRs of ATF4 were obtained from a normal control with aforementioned means. Following primers were used: CSF1R (5′‐CTCTGAGCAAGACCTGGACAAG‐3′, 5′‐TACTCCCTGTCGTCAACTCC‐3′), EIF2B3 (5′‐ATGGAATTTCAAGCAGTAGTGATGG‐3′, 5′‐TCAGATCTCCATGAGCT GGTC‐3′) and ATF4 5′UTRs (5′‐TTTCTACTTTGCCCGCCCAC‐3′, 5′‐GTTGCGGT GCTTTGCTGGAATC‐3′).
2.4. Plasmid constructs
We cloned the WT EIF2B3 cDNA into p3 × Flag‐cmv‐10 vector (p3 × flag‐EIF2B3) by ClonExpress® II One Step Cloning Kit (Vazyme Biotech). Mutant constructs of EIF2B3, c.22A>T, and c.1037T>C were, respectively, introduced into the WT plasmid by site‐directed mutagenesis using KOD‐Plus‐Neo (Toyobo). To verify whether the translation led by ATF4 mRNA 5′UTRs was affected, we inserted 5′UTR before enhanced green fluorescent protein (EGFP) into the vector pEGFP‐N1 (pEGFP‐ATF4‐5′UTR) like literature described.11 All plasmids were fully sequenced after construction or mutagenesis.
2.5. Cell culture and transfection
For analysis of eIF2B3 expression, HEK293T cells were cultured at 37°C in Dulbecco's modified Eagle's medium (DMEM) (HyClone) supplemented with 10% fetal bovine serum (GIBCO) and cotransfected with WT or mutant EIF2B3 plasmids with pEGFP‐N1 as exogenous control using Lipofectamine 3000 (Invitrogen) according to the manufacturer's instructions. Simultaneously, since the activity of eIF2B holocomplex could be inhibited by phosphorylated α subunit of eIF2 at Ser51 (eIF2αP),12, 13 this experiment was also used to detect eIF2α phosphorylation level to evaluate the effects of mutant EIF2B3 on eIF2B indirectly. For study on the ATF4‐5′UTR‐linked report, HEK293T cells were cotransfected with pEGFP‐ATF4‐5′UTR and p3 × flag‐EIF2B3.
2.6. Western blot
HEK293T cells were harvested for 48 hours after transfection, then collected and lysed. Protein samples were separated by 10% SDS‐PAGE and transferred to a PVDF membrane (Biorad). The membrane was incubated with mouse anti‐Flag (Abmart), mouse anti‐GFP (Santa Cruz biotechnology), rabbit anti‐eIF2α (Cell signaling technology), rabbit anti‐eIF2αP (Cell signaling technology), and mouse anti‐actin (Santa Cruz biotechnology) followed by horseradish peroxidase (HRP)‐conjugated secondary antibody (Merck Millipore). Then, the protein was visualized by enhanced chemiluminescent substrates (Thermo Scientific).
3. RESULTS
3.1. Genetic findings and pathogenicity classification of variants
After variant screening via WES and verification by Sanger sequencing, we found 12 distinct variants (Table 1) in eight unrelated patients (Figure 1A). Among these 12 variants, three variants (Figure 1B,C) are novel and absent in dbSNP, gnomAD, and ExAC. All of 12 variants are absent in our WES database that contain 500 Chinese controls. As shown in Table 1, c.2654_2654+3del within CSF1R is a microdeletion, c.1321C>T within GALC is a nonsense variant, and c.166G>C in GALC is a missense variant predicted to be deleterious by SIFT, Polyphen‐2, and CADD. According to ACMG, the variant c.166G>C within GALC is a variant of uncertain significance. However, the GALC enzyme activity of leukocytes in proband 8 carrying this variant decreased to 2.77 nmol/17 h mg (normal range 12.89‐100.93 nmol/17 h mg protein), and thus, we inferred that this variant is pathogenic. The microdeletion variant c.2654_2654+3del located in the splicer donor site of exon 20 in CSF1R, which was confirmed to affect mRNA processing. The RT‐PCR fragment spanning exons 18‐22 of CSF1R across the microdeletion yielded two products including the WT fragment (349 bp) and the smaller one (near 250 bp). The smaller one was found to contain a 100 bp in‐frame deletion corresponding to the entire length of exon 20, presenting as the total skipping of exon 20. The junction between nucleotides c.2554 (exon 19) and c.2655 (exon 21) gave rise to an acid substitution (glycine to aspartic) at residue 852 followed by a frameshift that was predicted to lead to the premature termination of translation (p.G852Dfs*67) (Figure 1D,E).
Table 1.
Twelve pathogenic variants identified in 8 probands with hereditary leukodystrophies
| Proband No. | Gene | Nucleotide change | Amino acid change | Genotype | gnomAD | ExAC | SIFT | Polyphen‐2 | CADD | ACMG |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | AARS2 | c.595C>T | p.R199C | Hom | 8.9e‐05 | 0.0001 | D | Pro_D | D | P(PS1+PM1+PM2+PM3+PP1+PP3+PP5) |
| 2 | CSF1R | c.2654_2654+3del | p.G852Dfs*67 | Het | 0 | 0 | NA | NA | NA | P(PVS1+PM1+PM2+PM4+PP3) |
| 3 | CSF1R | c.2381T>C | p.I794T | Het | 4.1e‐06 | 8.2e‐06 | D | Pro_D | D | P(PS3+PM1+PM2+PP3+PP4+PP5) |
| 4 | EIF2B3 | c.22A>T | p.M8L | Het | 0.0001 | 9.8e‐05 | T | B | D | US(PM2+PM3) |
| 4 | EIF2B3 | c.1037T>C | p.I346T | Het | 0 | 0 | T | B | D | LP(PM1+PM2+PP3+PP5) |
| 5 | GALC | c.599C>A | p.S200X | Het | 0 | 0 | NA | NA | D | P(PVS1+PM2+PP1+PP3+PP5) |
| 5 | GALC | c.1586C>T | p.T529M | Het | 9.8e‐05 | 6.6e‐05 | T | Pro_D | D | LP(PM1+PM2+PM3+PP1+PP3+PP5) |
| 6 | GALC | c.1321C>T | p.Q441X | Het | 0 | 0 | NA | NA | D | LP (PM1+PM2+PM3+PM4+PP3) |
| 6&7 | GALC | c.1901T>C | p.L634S | Het | 0.0006 | 0.0007 | D | Pro_D | D | LP(PM2+PM3+PP1+PP3+PP5) |
| 7 | GALC | c.2041G>A | p.V681M | Het | 0.0002 | 0.0002 | D | Pro_D | D | LP(PM2+PM3+PP1+PP3+PP5) |
| 8 | GALC | c.166G>C | p.D56H | Het | 0 | 0 | D | D | D | P (PS3+PM1+PM2+PM3+PP3) |
| 8 | GALC | c.461C>A | p.P154H | Het | 1.219e‐05 | 8.3e‐06 | D | Pro_D | D | LP(PM1+PM2+PM3+PP1+PP3) |
Novel variants are in bold.
Abbreviations: ACMG, American College of Medical Genetics and Genomics; B, benign; D, damaging, deleterious, or disease‐causing; ExAC, Exome Aggregation Consortium; gnomAD, The Genome Aggregation Database; LP, likely pathogenic; NA, not applicable; P, pathogenic; Polyphen‐2, Polymorphism Phenotyping v2; Pro_D, probably damaging; SIFT, Sorting Tolerant From Intolerant; T, tolerated; US, uncertain significance.
Figure 1.
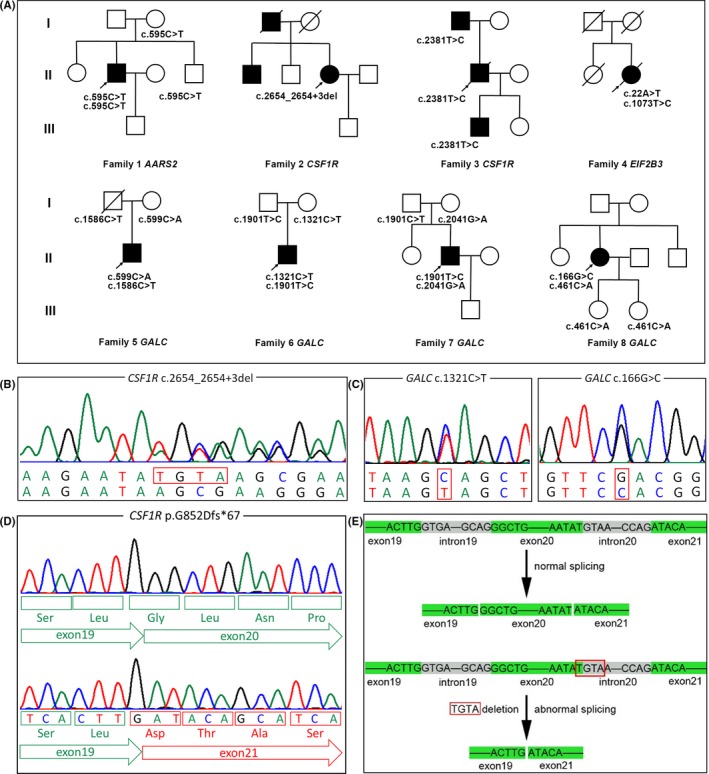
Pedigrees and novel mutations. A, Pedigree charts of the eight families. Squares indicate males, circles indicate females, black symbols indicate affected individuals, and arrows indicate the probands. B and C, Sequencing chromatograms of three novel variants. D and E, The sequencing chromatogram of c.2654_2653+3del within CSF1R and schematic diagram of splicing results
In the remaining nine known variants, eight variants (Figure 2A right, Figure S1) were previously reported as pathogenic variants, except EIF2B3 c.22A>T (Figure 2A, left). Given that EIF2B3 c.1037T>C (Figure 2A, right) was a known pathogenic variant, we investigated whether the functional influence of c.22A>T was similar to that of c.1037T>C. From our study, neither c.22A>T nor c.1037T>C was identified to appreciably destabilize the protein (Figure 2B). Both c.22A>T and c.1037T>C enhanced eIF2α phosphorylation without increase of total eIF2α (Figure 2C) and increased the level of expression of EGFP regulated by the uORFs (Figure 2D). In addition, this patient had the onset of cognitive deterioration and ataxia at 57, with a bilateral, diffuse and symmetric involvement of the cerebral white matter (Figure 2E). She was bedridden with two fractures because of her unsteady walk. Her disease lasted 4 years, and she died at 61. Combined the results of functional experiment with diagnostic clinical manifestations, we deduced that EIF2B3 c.22A>T was also a disease‐causing variant.
Figure 2.
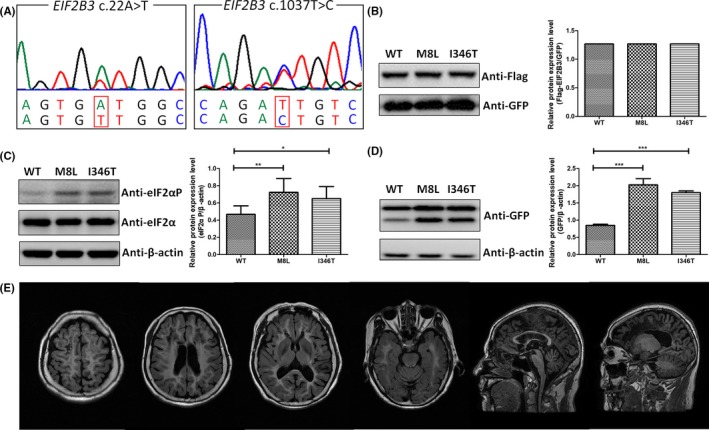
Functional analysis of variants c.22A>T and c.1037T>C within EIF2B3 and brain MRI of the patient. A, Sequencing chromatograms of variants c.22A>T (p.M8L) and c.1037T>C (p.I346T) within EIF2B3. B, HEK293T cells were cotransfected with p3 × flag‐EIF2B3 (WT or mutant) with pEGFP‐N1 as exogenous control. Neither p.M8L nor p.I346T destabilized the protein appreciably. C, Both p.M8L and p.I346T enhanced eIF2α phosphorylation without increase of total eIF2α. *P < .05 and **P < .01. D, HEK293T cells were cotransfected with p3 × flag‐EIF2B3 (WT, or mutant) with (pEGFP‐ATF4‐5′UTR). After incubated with anti‐GFP antibody, the upper bands reflected the fusion protein containing the product translated from the AUG within ATF4 5′UTR and EGFP, and the lower bands reflected only EGFP, which were regulated by the uORFs in ATF4 5′UTR. Both p.M8L and p.I346T increased the level of expression of EGFP than WT. ***P < .001. E, Brain MRI of proband 4 at her 60 showed extensive white matter hyperintense on FLAIR and markedly hypointense on T1‐weighted images
3.2. Novel clinical and imaging features
Ultimately, 8 probands were diagnosed as hereditary leukodystrophies, including AARS2‐related adult‐onset leukoencephalopathy with axonal spheroids and pigmented glia (ALSP) (proband 1), CSF1R‐related ALSP (probands 2 and 3), leukoencephalopathy with vanishing white matter (VWM) (proband 4), and Globoid cell leukodystrophy (GLD) (probands 5‐8). Their primary clinical manifestations were summarized in Table 2. Novel clinical and imaging features are described as follows.
Table 2.
Clinical and imaging features of 8 probands identified with hereditary leukodystrophies
| Proband No. | Sex | Age at onset | Age at present | Gene | Variants | Predominant symptoms | MRI appearances | Diagnosis |
|---|---|---|---|---|---|---|---|---|
| 1 | M | NA | 43 | AARS2 | c.595C>T; c.595C>T | Learning difficulty, declining in memorizing and calculating, ataxia, nystagmus, and knee aching | Parietal lobes atrophy, WM abnormality in posterior horns of the LV, posterior CC, and splenium | AARS2‐related ALSP |
| 2 | F | 43 | 50 | CSF1R | c.2654_2654+3del | Memory and personality change, inattention and acalculia, gait difficulties, dysarthria, and dysphagia | Frontal prominence WM change, thin CC, cortical atrophy | CSF1R‐related ALSP |
| 3 | M | 37 | 38 | CSF1R | c.2381T>C | Memory and cognitive regression, gait difficulties, personality change | Widespread T2 hyperintensity in WM and cortical atrophy | CSF1R‐related ALSP |
| 4 | F | 57 | 61 dead | EIF2B3 | c.22A>T; c.1037T>C | Dizziness, dementia, gait abnormality, ataxia, and positive Babinski signs | Extensive WM T2 hyperintensity and T1 hypointensity, thin CC, and cerebellar atrophy | VWM |
| 5 | M | 1 | 23 | GALC | c.599C>A; c.1586C>T | Febrile seizures, unstable walking, cognitive regression, and visual field defect | Partly confluent T2 hyperintensity in WM near LV horns and posterior limbs of internal capsule | GLD |
| 6 | M | 12 | 28 | GALC | c.1321C>T; c.1901T>C | Shuffling gait, spastic paraplegia, and ataxia | No visible abnormality | GLD |
| 7 | M | 26 | 31 | GALC | c.1901T>C; c.2041G>A | Spastic paraplegia | No visible abnormality | GLD |
| 8 | F | 35 | 46 | GALC | c.166G>C; c.461C>A | Shuffling gait and asymmetric weakness in lower limbs | Symmetric corticospinal tract T2 hyperintensity | GLD |
Novel variants are in bold.
Abbreviations: ALSP, adult‐onset leukoencephalopathy with axonal spheroids and pigmented glia; CC, corpus callosum; GLD, globoid cell leukodystrophy; LL, lateral ventricles; VWM, leukoencephalopathy with vanishing white matter; WM, white matter.
The AARS2‐related ALSP patient (proband 1) was a 43‐year‐old farmer with pain in the left knee for 5 years and numbness in the right lower limb for 1 year, but still able to do his daily farm work. There was no discernible change of cognition from his wife's point of view though he had a history of learning difficulty in childhood. He was diagnosed with left anterior cruciate ligament injury, medial collateral ligament injury, and lumbar disk herniation. Brain MRI examination at age 43 indicated cortical atrophy, most pronounced in bilateral parietal lobes (Figure 3A,B). Fluid‐attenuated inversion recovery (FLAIR) hyperintensity and T1‐wighted images hyperintensity predominantly occur near the posteior horns of lateral ventricle (Figure 3A‐D).
Figure 3.
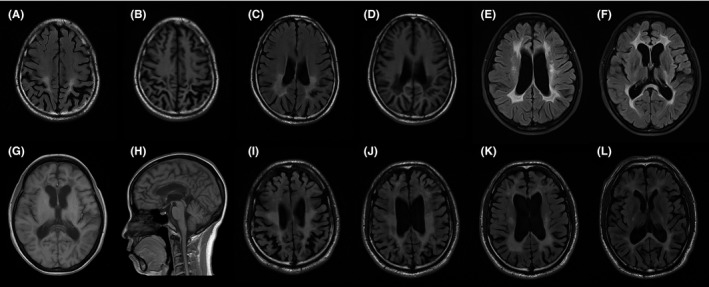
Brain MRI of patients with ALSP. A, B, C and D, MRI of proband 1 at his 43 showed the broadening and deepening of sulus with occipital prominence, and occipital periventricular white matter hyperintense on FLAIR and hypointense on T1‐weighted images. E, F, G and H, MRI of proband 2 at her 46 showed the white matter adjacent to the lateral ventricle hyperintense on FLAIR and hypointense on T1‐weighted images, and thinning of the corpus callosum. I, J, K and L, MRI of proband 3 at his 38 showed widespread white matter hyperintense and obvious cortical atrophy on FLAIR
The brother of a CSF1R‐related ALSP patient (proband 2) had similar, but much milder, symptoms. The other CSF1R‐related ALSP patient (proband 3) received allogeneic stem cell transplantation and died of graft‐versus‐host disease and uncontrolled infection 15 days after the transplantation. His father carried the same pathogenic variant but was not unresponsive and sluggish until he was 60 years old, 23 years later than proband 3. The MRI of the proband 2 at her 46 showed confluent demyelination changes of the white matter adjacent to the lateral ventricles symmetric cortical atrophy (Figure 3E‐G) and thinning of the corpus callosum (Figure 3H), which was less severe than the MRI of proband 3 (Figure 3I‐L).
Juvenile (proband 6) and adult‐onset (probands 7 and 8) GLD patients were all diagnosed with spastic paraplegia before. EMG results of proband 6 and 7 did not show obvious peripheral nerve injury while proband 8 showed dysmyelination of peripheral nervous system, which led to his diagnosis of CMT. From probands 5 to 8, the GALC enzyme activities in leukocytes were 3.41 nmol/17 h mg, 2.07 nmol/17 h mg, 5.9 nmol/17 h mg, and 2.77 nmol/17h mg (normal range 12.89‐100.93 nmol/17 h mg), respectively. T2‐weighted MRI of probands 5‐8 on the level of centrum semiovale, corona radiate, and internal capsule were presented in Figure 4. There was no visible abnormality in the brain MRI images of proband 6 (Figure 4D‐F) and 7 (Figure 4G‐I) when they were 28 years old. Two consecutive brain MRI of proband 8 were performed at the ages of 41 and 46, respectively, showing no significant progression. The image at the age of 46 showed only involvements of symmetric corticospinal tract (Figure 4J‐L).
Figure 4.
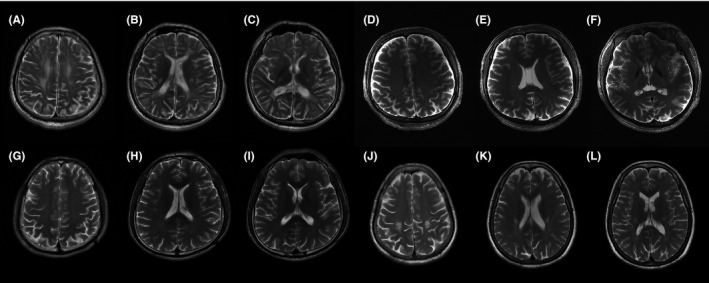
T2‐weighted MRI on the same three levels (centrum semiovale, corona radiate, and internal capsule) of patients with GLD. A, B and C, MRI of proband 5 at his 21 showed partly confluent hyperintense areas in white matter near the lateral ventricle and posterior limbs of internal capsule. D, E and F, MRI of proband 6 at his 28 showed no visible abnormality. G, H and I, MRI of proband 7 at his 28 showed no visible abnormality. J, K and L, MRI of proband 8 at her 46 showed symmetric hyperintense in bilateral corticospinal tracts
4. DISCUSSION
It is well established that hereditary leukodystrophies are highly heterogeneous disorders. In this study, we made definitive genetic diagnosis for 8 probands with rare hereditary leukodystrophies by WES and identified 12 pathogenic variants including three novel ones. The patients we described here did show a significant variability of phenotypes.
AARS2 and CSF1R are two uncovered genes responsible for ALSP. Most patients diagnosed with ALSP shared cognitive decline, neuropsychiatric disturbance, and pyramidal and extrapyramidal signs. AARS2‐related ALSP is an autosomal recessive neurodegenerative disease while CSF1R‐related ALSP is inherited in a dominant manner. Clinical features of ALSP are very heterogeneous, showing a significant intra‐ and inter‐kindred variability of phenotype and disease duration.14 We reported one AARS2‐related ALSP and two CSF1R‐related ALSP cases. The AARS2‐related ALSP patient shared the same AARS2 pathogenic variant with a Turkey middle‐aged patient who presented a rapid progression and died in 1 year after symptoms onset.15 But there were no evident dystonia, dysarthria, cognitive deterioration, or neuropsychiatric symptoms in our patient as the Turkey patient presented.15 The two CSF1R‐related ALSP patients showed great clinical heterogeneity, especially in the patients with CSF1R c.2381T>C. The proband (son) had disease onset 23 years earlier than his father. These phenomena reminded us that even patients carrying same pathogenic variants would present totally different phenotypes. The remarkable distinction of clinical manifestation among these genotype‐identical ALSP patients may be due to environmental or other genetic factors that play an important role and cannot be replaced.
Leukoencephalopathy with VWM is an autosomal recessive neurodegenerative disease due to pathogenic variants in the five genes (EIF2B1 to 5) encoding the five subunits of the eucaryotic initiation factor 2B (eIF2B).16 The variation in disease severity is extremely wide with stress‐provoked episodes of rapid deterioration.1 Survival increased with increasing age of onset and 80% of patients with an age of onset over 5 years were expected to be without severe disability at 14 years of disease evolution.16 In this study, we identified c.22A>T and c.1037T>C variants within EIF2B3 in the proband of family 4 and firstly confirmed the pathogenicity of c.22A>T, which was never reported to be associated with VWM. Both of c.22A>T and c.1037T>C were indirectly proved to inhibit eIF2B activity.11, 12, 13 However, the proband had the onset symptom at 57 and died at 61. Such a short duration was inconsistent with the literature. The two fractures could act as factors provoking the episodes of rapid neurological deterioration.
Globoid cell leukodystrophy comprises a spectrum with more severe infantile‐onset disease to milder juvenile or adult‐onset disease. Juvenile and adult‐onset GLD patients were easily misdiagnosed as spastic paraplegia. Globoid cell leukodystrophy is the most common leukodystrophy in our study, and all four GLD patients presented pyramidal signs. Proband 6 and 7 shared one same pathogenic variant, c.1901T>C in GALC, which was supposed to contribute to a mild phenotype.17 However, the phenotype of proband 6 was more severe than that of proband 7, and we supposed that the nonsense variant c.1321C>T was more deleterious than the missense variant c.2041G>A. It should be noted that there was no significant abnormality in brain MRI of proband 6 and 7 at the age of 28, while another 55‐year‐old asymptomatic individual carrying homozygous c.1901T>C showed selective pyramidal tract impairment in brain MRI.18 These MRI features suggested that the demyelinating degree in conventional MRI may not reflect the severity of clinical symptoms in GLD patients.
In summary, we made definite diagnosis in 8 probands with 12 pathogenic variants including three novel ones and reported new clinical characteristics and imaging features of these patients. Our results have deepened the understanding of clinical, genetic, and imaging heterogeneity of hereditary leukodystrophies and expanded the spectrum of pathogenic variants and clinical profiles.
CONFLICT OF INTEREST
All authors reported no potential conflicts of interest.
Supporting information
ACKNOWLEDGMENTS
We sincerely thank the participants for their cooperation and willingness to participate in this study. We thank Ms Wan‐Qing Xu for editing the manuscript.
Xie J‐J, Ni W, Wei Q, et al. New clinical characteristics and novel pathogenic variants of patients with hereditary leukodystrophies. CNS Neurosci Ther. 2019;26:567–575. 10.1111/cns.13284
Funding information
This study was supported by the Fundamental Research Funds for the Central Universities (2019XZZX001‐01‐04) and the Research foundation for distinguished scholar of Zhejiang University to Zhi‐Ying Wu (188020‐1938 10101/089).
Xie and Ni contributed equally to this work.
REFERENCES
- 1. Köhler W, Curiel J, Vanderver A. Adulthood leukodystrophies. Nat Rev Neurol. 2018;14(2):94‐105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. van der Knaap MS, Bugiani M. Leukodystrophies: a proposed classification system based on pathological changes and pathogenetic mechanisms. Acta Neuropathol. 2017;134(3):351‐382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ahmed RM, Murphy E, Davagnanam I, et al. A practical approach to diagnosing adult onset leukodystrophies. J Neurol Neurosurg Psychiatry. 2014;85(7):770‐781. [DOI] [PubMed] [Google Scholar]
- 4. Vanderver A, Simons C, Helman G, et al. Whole exome sequencing in patients with white matter abnormalities. Ann Neurol. 2016;79(6):1031‐1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chen S, Ni W, Yin XZ, et al. Clinical features and mutation spectrum in Chinese patients with CADASIL: a multicenter retrospective study. CNS Neurosci Ther. 2017;23(9):707‐716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Niu YF, Ni W, Wu ZY. ABCD1 mutations and phenotype distribution in Chinese patients with X‐linked adrenoleukodystrophy. Gene. 2013;522(1):117‐120. [DOI] [PubMed] [Google Scholar]
- 7. Lu C, Li LX, Dong HL, et al. Targeted next‐generation sequencing improves diagnosis of hereditary spastic paraplegia in Chinese patients. J Mol Med (Berl). 2018;96(7):701‐712. [DOI] [PubMed] [Google Scholar]
- 8. Jiang B, Zhou J, Li HL, et al. Mutation screening in Chinese patients with familial Alzheimer's disease by whole‐exome sequencing. Neurobiol Aging. 2019;76:215.e15‐215.e21. [DOI] [PubMed] [Google Scholar]
- 9. Li LX, Liu GL, Liu ZJ, Lu C, Wu ZY. Identification and functional characterization of two missense mutations in NDRG1 associated with Charcot‐Marie‐Tooth disease type 4D. Hum Mutat. 2017;38(11):1569‐1578. [DOI] [PubMed] [Google Scholar]
- 10. Kruger M, Beger C, Li QX, et al. Identification of eIF2Bgamma and eIF2gamma as cofactors of hepatitis C virus internal ribosome entry site‐mediated translation using a functional genomics approach. Proc Natl Acad Sci U S A. 2000;97(15):8566‐8571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Li W, Wang X, Van Der Knaap MS, Proud CG. Mutations linked to leukoencephalopathy with vanishing white matter impair the function of the eukaryotic initiation factor 2B complex in diverse ways. Mol Cell Biol. 2004;24(8):3295‐3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bogorad AM, Lin KY, Marintchev A. Novel mechanisms of eIF2B action and regulation by eIF2alpha phosphorylation. Nucleic Acids Res. 2017;45(20):11962‐11979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pavitt GD, Yang W, Hinnebusch AG. Homologous segments in three subunits of the guanine nucleotide exchange factor eIF2B mediate translational regulation by phosphorylation of eIF2. Mol Cell Biol. 1997;17(3):1298‐1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Stabile C, Taglia I, Battisti C, Bianchi S, Federico A. Hereditary diffuse leukoencephalopathy with axonal spheroids (HDLS): update on molecular genetics. Neurol Sci. 2016;37(9):1565‐1569. [DOI] [PubMed] [Google Scholar]
- 15. Lynch DS, Zhang WJ, Lakshmanan R, et al. Analysis of Mutations in AARS2 in a Series of CSF1R‐negative patients with adult‐onset leukoencephalopathy with axonal spheroids and pigmented Glia. JAMA Neurol. 2016;73(12):1433‐1439. [DOI] [PubMed] [Google Scholar]
- 16. Fogli A, Schiffmann R, Bertini E, et al. The effect of genotype on the natural history of eIF2B‐related leukodystrophies. Neurology. 2004;62(9):1509‐1517. [DOI] [PubMed] [Google Scholar]
- 17. Zhao S, Zhan X, Wang Y, et al. Large‐scale study of clinical and biochemical characteristics of Chinese patients diagnosed with Krabbe disease. Clin Genet. 2018;93(2):248‐254. [DOI] [PubMed] [Google Scholar]
- 18. Zhang T, Yan C, Ji K, et al. Adult‐onset Krabbe disease in two generations of a Chinese family. Ann Transl Med. 2018;6(10):174. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials