

Abstract
Cryptic pockets, which are not apparent in crystallographic structures, provide promising alternatives to traditional binding sites for drug development. However, identifying cryptic pockets is extremely challenging and the therapeutic potential of cryptic pockets remains unclear. Here, we reported the discovery of novel inhibitors for striatal-enriched protein tyrosine phosphatase (STEP), a potential drug target for multiple neuropsychiatric disorders, based on cryptic pocket detection. By combining the use of molecular dynamics simulations and fragment-centric topographical mapping, we identified transiently open cryptic pockets and identified 12 new STEP inhibition scaffolds through structure-based virtual screening. Site-directed mutagenesis verified the binding of ST3 with the predicted cryptic pockets. Moreover, the most potent and selective inhibitors could modulate the phosphorylation of both ERK1/2 and Pyk2 in PC12 cells.
Keywords: Striatal-enriched protein tyrosine phosphatase, Cryptic pocket, Inhibitor, Virtual screening
Graphical Abstract
1. Introduction
Structure-based virtual screening (SBVS) has been widely employed for the identification of bioactive compounds[1, 2], However, SBVS has often been restricted by the static nature of X-ray crystal structure[1, 3, 4], It should be mentioned that many validated targets have cryptic pockets[5], which may not be obvious from structural information on the apo protein and become clear when a ligand is bound; thus, these cryptic pockets offer attractive opportunities for drug discovery[6, 7], Currently, the molecular mechanism by which cryptic pockets are formed is not clear, due to the rather static picture of protein-ligand complexes presented by crystal structures. Although ligands seem to be necessary in the opening of cryptic pockets, whether they act by a conformational selection, an induced-fit or a “mixed” mechanism is still debated. Because of these uncertainties and their hidden nature, cryptic pockets are difficult to identify by experimental methods. Recently, simulations and modeling play a significant role in the systematic search for cryptic pockets[8–12]. Tan et al. employed an explicit solvent ligand-mapping molecular dynamics (LMMD) approach to identify cryptic pocket in polo-like kinase 1 (Plkl), which lead to the design of a high affinity inhibitor for Plkl [13], More recently, Tan et al. further describe the use of LMMD to identify new binding sites for the design of portent peptide-based MDM2-p53 inhibitor[8]. Furthermore, classical all-atom molecular dynamics (MD) simulations of the p53 core domain also revealed a transiently open pocket in p53 core domain, which could be used for reactivation of mutant p53[14]. On the other hand, Bowman et al. identified multiple cryptic allosteric pockets by combining atomistic MD simulations and Markov State Models (MSM)[15, 16],
Reversible protein tyrosine phosphorylation, a major posttranslational modification mechanism, regulates numerous cellular processes[17–21]. Appropriate levels of protein tyrosine phosphorylation are dynamically maintained by protein tyrosine kinases (PTKs) and protein tyrosine phosphatases (PTPs)[22–24], PTPs play important roles in many human diseases, such as inflammation, cancer, metabolic and immunological disorders[18, 25–31], Encoded by the PTPN5 gene, striatal-enriched protein tyrosine phosphatase (STEP) is a brain-enriched PTP that is expressed specifically in the central nervous system[32–34], STEP activity can lead to the dephosphorylation and regulation of several neuronal signaling molecules [3 5, 36], Progeny null for STEP increased receptor levels on synaptic membranes and exhibited significant cognitive improvements [3 7, 38], Importantly, an inhibitor that exhibited specificity for STEP also reversed cognitive deficits in Alzheimer’s disease (AD) mouse model[36, 37], Because of the strong linkage of abnormal STEP activity to neuropsychiatrie disorders, there is increasing interest in developing small-molecule STEP inhibitors. However, a significant caveat regarding the development of STEP inhibitors is the widespread recognition that PTPs are extremely challenging targets due to the highly positively charged nature and high degree of sequence and structural homology of PTP active sites[29, 39, 40], Heretofore, only a few chemotypes for STEP inhibition have been reported, such as irreversible inhibitors and substrate-based inhibitors (Figure 1). For example, Xu et al. identified a reversible covalent STEP inhibitor TC-2153 with an IC50 of 24.6 nM[43], Ellman’s group discovered substrate-based STEP inhibitor 12t with nonhydrolyzable phosphate mimetic difluoromethylphosphonic acid (DFMP)[44], Further structural optimization lead to the identification of more potent STEP inhibitor with a Ki of 110 nM[45], In general, these known STEP inhibitors lack of enough druggabilities for further development. Thus, there is a need for developing more drug-like small molecular STEP inhibitors and evaluate their therapeutic potential.
Figure 1.

Structures and activities of reported STEP inhibitors.
Here, we describe a computational analysis of STEP structural models that suggests a transiently open cryptic pocket as a possible targeting direction for STEP inhibition. The cryptic pocket was identified in exosite (functionally important regions outside the active site) through all-atom molecular dynamics (MD) simulations, Markov State Models (MSMs)[46–50] and fragment-centric pocket mapping[51, 52], Structure-based virtual screening (SBVS) to target both catalytic and cryptic exosite pockets led to the identification of 12 novel low-molecular-weight STEP inhibitors with new scaffold. Combine use of interaction energy calculation as well as site-directed mutagenesis verified the binding of ST3 with the newly predicted cryptic pocket. Structure-activity relationships were explored, and the most potent compounds ST2–5 and ST3–5 exhibit competitive and reversible inhibition on STEP and possess selectivity for STEP against a panel of protein phosphatases, the ability to modulate the phosphorylation of STEP substrates (ERK1/2 and Pyk2), no toxicity in PC 12 cell cultures and an increased level of differentiation and migration functions.
2. Results and Discussion
2.1. Apo crystal structure-based virtual screening
At the start of this project, only the apo structure of STEP was available in the RCSB PDB (PDB code: 2BV5[53], 2BIJ[53] and 2CJZ[54]). Thus, our initial virtual screening was performed using the apo crystal structure of STEP (PDB: 2BV5). The commercial Specs database was screened using the HTVs, SP and XP modes of Glide[55] and rescored using AutoDock Vina[56] (Supporting Information, Figure S1.). After the evaluation of druggability and structural diversity, a total of 32 hit compounds were selected (Supporting Information Table S1), and their inhibitory activities were assessed through the inhibition of the STEP-catalyzed hydrolysis of p-nitrophenyl phosphate (pNPP) at pH 6.0 and 30 °C. Initial screening was conducted at a concentration of 100 μM. and compounds that exhibited more than 50% inhibition were further tested for concentration-dependent inhibition. Unfortunately, only one compound (ST1) showed weak inhibitory activity against STEP (IC50=70.4 μM) (Table 1). The low success rate (3.1%) of virtual screening using the apo crystal structure may be due to the lack of knowledge about the bound state structure of STEP. Notably, previous studies have suggested the existence of cryptic pockets in proteins for ligand binding, introducing more chemical space for drug design[9, 14, 57], Thus, investigation of the bound state structure of STEP as well as the detection of cryptic binding pockets would be helpful for the virtual screening of STEP inhibitors.
Table 1.
STEP inhibitors identified by apo-based (green background) and ensemble virtual screening (yellow background).
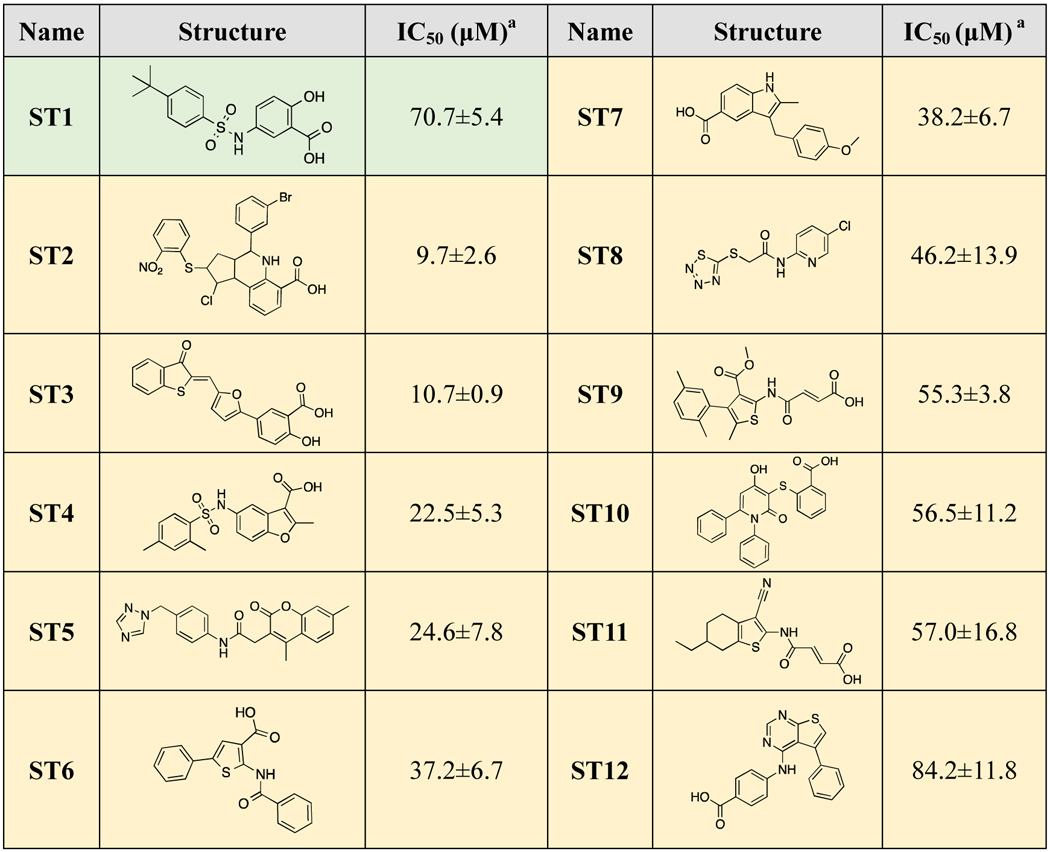 |
All measurements were performed using pNPP as a substrate at pH 7.0, 30°C, and I=0.15M.
2.2. Computational detection of cryptic pockets for inhibitor binding
Most recently, Michael et al. reported crystal structures of STEP complexed with substrate-based inhibitors (PDB code: 50VR, 50YX and 50W1)[45], However, the holo crystal structure displayed high structural similarity to the apo crystal structure, according to the calculated per-residue RMSD values (Supporting Information, Figure S2). We also compared the ligand binding pockets between the apo and holo crystal structures using AlphaSpace and observed similar pocket distributions (Supporting Information Figure S2). The high similarity between the apo and holo crystal structures of STEP may be due to the nature of the substrate-based inhibitor used in crystallization. The substrate-based inhibitor occupied only 241.4 Å3 of space (occupancy=25%) in the active site of STEP, and the phosphate group in this inhibitor may be the greatest contributor to its inhibitory activity (Ki=7.8 μ). Considering that the experimental holo crystal structure did not provide any new pockets for inhibitor binding, we sought to explore the existence of cryptic pockets in STEP, which depend mainly on protein flexibility and may open only transiently.
To detect potential chemical space for STEP inhibitor binding, we utilized multiple computational methodologies to detect cryptic pockets. First, we performed two 1-μs MD simulations using STEP with or without inhibitor (ST1) bound to fully explore the structural flexibility of STEP (Supporting Information Figure S3). Calculations of per-residue RMSF values indicated that the most flexible region in the MD simulations (both STEP-apo and STEP-ST1 system) is located in the WPD-loop (Supporting Information Figure S4). As expected, the binding of ST1 stabilized the helical region adjacent to the catalytic site as well as two distal loop regions (Supporting Information Figure S4). Importantly, we observed salt-bridge interactions between K439 and D272 (or E519), and the transformation of K439-D272 or K439-E519 salt-bridge interactions (Supporting Information Figure S5) governed the protein dynamics of WPD-loop, which play important role in the recognition of substrate/ligand. For MD simulation of STEP-apo system, K439 swing between D272 and E519 (Supporting Information Figure S5). On the other hand, the K439-E519 salt-bridge became the main interaction in STEP-ST1 system after 0.2 μs MD simulations (Supporting Information Figure S5). Fragment-centric topographical mapping of selected MD snapshots, which represent either K439-D272 or K439-E519 salt-bridge interaction, indicated the occurrence of cryptic pockets in exosite of STEP-apo system (Figure 2). Different from the exosite pockets in crystal structure and STEP-ST1 system, which are separated from catalytic pockets, the cryptic exosite pockets observed from MD simulations of STEP-apo system are well contiguous with catalytic pockets (Figure 2). These results revealed the existence of cryptic pockets in the exosite site of STEP and provide more valuable chemical space for the binding of small molecular inhibitor (Supporting Information Video S1).
Figure 2.

Fragment-centric topographical mapping of binding pockets in (A) apo crystal structure, (B) MD snapshot of STEP-ST1 system with K439-E519 salt-bridge interaction, (C) MD snapshots of STEP-apo system with K439-E519 salt-bridge interaction and (D) K439-D272 salt-bridge interaction. The pockets in catalytic site are shown as green spheres. The exosite pockets are shown as light blue spheres in crystal structure, red spheres in MD snapshot of STEP-ST1, and orange spheres in MD snapshots of STEP-apo. Residues D272, K439 and E519 are presented in stick model (yellow).
To better understand the occurrence of cryptic pockets in the STEP catalytic domain, we further constructed MSMs of the conformational space on the basis of the distances between D272, K439 and E519. Time-lagged independent component analysis (TICA)-based structure clustering gave three metastable states (macrostates) for apo STEP (apo-MSM-1, apo-MSM-2 and apo-MSM3) and two macrostates for inhibitor-bound STEP (holo-MSM-1 and holo-MSM-2) (Supporting Information Figure S6). We observed lower energy barriers between different macrostates in STEP-ST1 system (~2.5 kT) than STEP-apo system (~7.0 kT). Furthermore, we analyzed potential ligand binding pockets using representative snapshots of five macrostates obtained from MSMs (Supporting Information Figure S7). Interestingly, we observed the complete “open-to-closed” transition of the exosite pockets during MD simulations of apo STEP (apo-MSM-1, apo-MSM-2 and apo-MSM3), implying the existence of conformation selection mechanism. Specially, the exosite pockets are contiguous with catalytic pockets in the most dominant apo-MSM-3 (90%), providing good starting point for virtual screening of more potent STEP inhibitors. On the other hand, only the half-open exosite pocket (holo-MSM-1) and closed exosite pocket (holo-MSM-2) were observed during MD simulations of inhibitor-bound STEP. The most dominant holo-MSM-2 (85%) represents the “closed” state of exosite pockets which may due to the induced-fit conformational changes upon inhibitor binding.
2.3. Ensemble structure-based virtual screening
Since our unbiased MD simulations and pocket analysis suggested novel cryptic exosite pockets, which provide new opportunities to discover STEP inhibitors with heightened potency, we performed second-round virtual screening to search for ligands that target both catalytic pockets and exosite pockets. An ensemble of the structures from apo-MSM-3, which possess the most accessible exosite pockets (Supporting Information Figure S6–S7), was selected based on the calculation of pocket space and pocket scores (Supporting Information Figure S7C) and subsequently employed in second-round virtual screening (Figure 3). By taking the docking score, druggability and chemical diversity into consideration, a total of 32 compounds were purchased for biological tests (Supporting Information Figure S1 and Table S2). As expected, a total of 11 novel STEP inhibitors were identified (Table 1) with better inhibitory activities (IC50 values ranging from 9.7 to 47.7 μM) than that of the hit compound from the initial virtual screening using the apo structure (ST1, IC50=70.7 μM). Obviously, the success rate of virtual screening using the ensemble structures (34.4%) was significantly improved over that of the initial virtual screening using the apo structure (3.1%).
Figure 3.

Virtual screening of STEP inhibitors using apo crystal structure (left) and ensemble structures from MSMs (right). Catalytic pockets and cryptic exosite pockets are detected using AlphaSpace and illustrated by green and orange surfaces.
Moreover, we performed a time-dependent inhibition test against the two most potent inhibitors (ST3 and ST2) to examine whether they inhibit STEP reversibly. Reversible binding of ST3 and ST2 were examined by varying the pre-incubation time, in order to determine whether there is a time-dependent inhibition. Irreversibly binding could lead to the decrease of Kcat (inhibitor) as time, whereas the Kcat (inhibitor) remain constant when the inhibitor binding reversible. With our previously reported PTP covalent inhibitor A25[58] as a positive control, our results indicated that the kinetics of STEP inhibition by compounds ST3 and ST2 suggested non-time-dependent inhibition with no increase in the ratio of Kcat (control) over Kcat (inhibitor) (Supporting Information Figure S8).
2.4. Binding of ST3 with predicted cryptic pocket
Furthermore, 0.5-μs MD simulation was performed for the most active hit compound, ST3, to verify whether ST3 could target STEP through the binding of both catalytic pockets and exosite pockets. The top-scored docking pose of compound ST3, in which both catalytic pockets and exosite pockets are occupied, served as the starting complex for MD simulation. As shown in Supporting Information Figure S3 and Video S2, the predicted binding pose of compound ST3 is very stable during 0.5-μs MD simulations. Additionally, both the calculated protein-ligand interaction energies and AlphaSpace pocket analysis further indicated that compound ST3 inhibited STEP by targeting both catalytic pockets and exosite pockets.
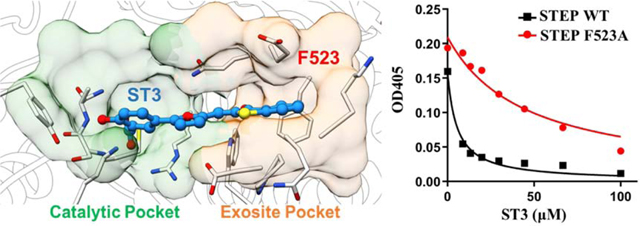
To further verify the interactions between ST3 and exosite pocket, we have constructed specific STEP mutant (F523A) and measured the inhibitory activity of ST3 against this mutant. As expected, the inhibitory activity of ST3 was decreased in STEP F523A mutant (IC50=99.7±10.9 μM) compared with wide type STEP (IC50= 10.7±0.9 μM) (Figure 4E). The results above help us understand why ST3 has better inhibitory activity than ST1, which targets only catalytic pockets (Figure 4). Additionally, the inhibitory activity of compound ST2 was also decreased in STEP F523A mutant (Supporting Information Table S4).Detailed protein-ligand interactions for (C) ST1 and (D) ST3. Hydrogen bond interactions are marked with dotted red line. (E) Graph shows dose-response inhibition of STEP wide type or STEP F523A mutant for compound ST3. (F) Space occupied by ST1 and ST3 in catalytic pockets and exosite pockets during MD simulations. (G) Calculated interaction energy between two inhibitors (ST1 and ST3) and residue F523 in exosite pocket.
Figure 4.

Comparison of predicted binding modes for ST1 and ST3. Fragment-centric mapping of binding pockets for (A) ST1 and (B) ST3 using representative snapshots from MD simulations.
2.5. Structure-activity relationship analysis of compounds ST2 and ST3
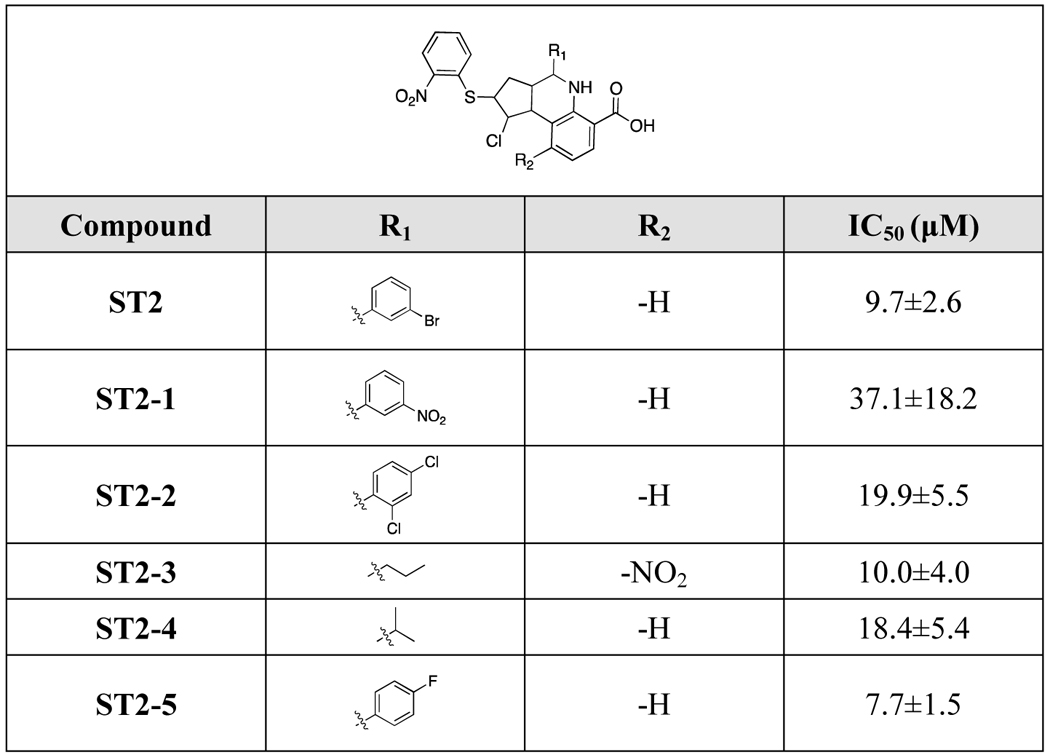
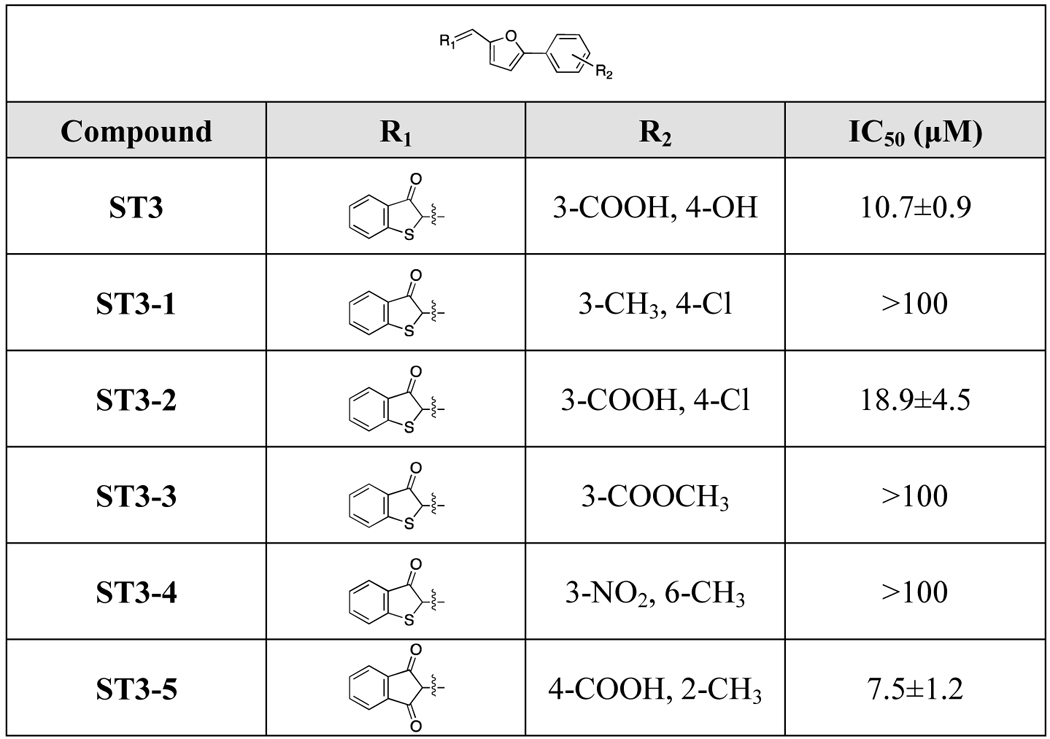
To gain further understanding of the structure-activity relationships and search for inhibitors with improved potency, hit-based substructure searches were performed using the two most active STEP inhibitors (ST2 and ST3). Taking structural diversity into consideration, five ST2 analogs and five ST3 analogs were purchased from the Specs database (Supporting Information Table S3). As shown in Table 2, removal of the carboxyl group and hydroxyl group (ST3–1, ST3–3) significantly decreased the inhibitory activity of ST3 analogs. Exchanging the hydroxyl group to a chlorine atom gave an inhibitor with decreased activity (ST3–2, IC50=18.9 μM). Interestingly, moving the meta-substituted carboxylic acid group to the para-position and introducing a methyl group in the ortho-position in the R1-substituent as well as exchanging the R2-substituent to a 1,3-indandione group gave the most active compound ST3–5 (IC50=7.5 μM). This SAR information on the ST3 analogs is consistent with our predicted binding model of STEP-ST3, where the carboxyl group forms multiple hydrogen bond interactions with the STEP catalytic site (Figure 4D). In terms of ST2 analogs, although we observed only slight changes upon modification of the R1- and R2-substituents, the R1-substituent seems to be more important. The most active ST2 analog (ST2–5) exhibited better potency than ST2. We further tested the inhibitory activities of ST2–5 and ST3–5 against STEP F523A mutant. Similar with results for ST2 and ST3, the inhibitory activities of ST2–5 and ST3–5 were also decreased against F523A mutant. (Supporting Information Table S4).
Table 2.
Structure-activity relationship of ST2 and its analogs.
 |
2.6. Selectivity against other protein phosphatases
The four most active STEP inhibitors (ST2, ST2–5, ST3 and ST3–5) were ultimately chosen for further biological evaluation. The Lineweaver-Burk plots of the most potent inhibitors, shown in Figure 5, indicate that these compounds are competitive inhibitors for STEP with low micromolar IQ values (3.7±0.5, 5.4±0.6, 2.2±0.8 and 2.3±0.8 μM). Because all classical PTPs nossess common nhosnhotvrosine binding sites, the identification of small inhibitors that selectively block a particular member of PTP family proteins is difficult. Currently, the relatively low selectivity of PTP inhibitors represents the major hurdle for their development and clinical usage[39, 59], Therefore, we measured the inhibition selectivity of ST2, ST3, ST2–5 and ST3–5 against a panel of protein phosphatases, including VHR, LYP, PTP IB, HePTP, YopH, PP1, GLEEP, PRL, SSH2, PPM1B and PPM1G. As shown in Table 4, ST2 showed at least 2.7-fold selectivity for other PTPs except for PTP1B. The more potent derivative ST2–5 exhibited the highest selective for STEP over PTP1B (8.8-fold) among these four inhibitors. Compound ST3 showed similar inhibitory activities against STEP, VHR and LYP. Moreover, the selectivity of ST3 against YopH and PP1 is significantly better than ST2 and ST2–5. Importantly, the most active STEP inhibitor ST3–5 displayed the highest selectivity for STEP over VHR (19.6-fold) and YopH (44.4-fold).
Figure 5.

Kinetic analysis of STEP inhibition by (A) ST2, (B) ST3, (C) ST2–5 and (D) ST3–5. The Lineweaver-Burk plot displays a characteristic pattern of intersecting lines that indicates competitive inhibition.
Table 4.
Selectivity studies of STEP inhibitors with a panel of protein phosphatases.
| PTPs | Inhibitory activity (μM) |
|||
|---|---|---|---|---|
| ST2 | ST2–5 | ST3 | ST3–5 | |
| STEP | 9.7±2.6 | 7.7±1.5 | 10.7±0.9 | 7.5±1.2 |
| VHR | 56.9±9.6 | 26.0±5.0 | 9.1±1.1 | 147.3±25.0 |
| LYP | 33.5±6.1 | 14.5±2.7 | 10.4±2.5 | 19.8±6.4 |
| PTP1B | 13.3±3.9 | 67.6±3.3 | 15.0±5.5 | 12.3±2.3 |
| HePTP | 48.2±11.3 | 20.1±4.0 | 28.7±3.0 | 39.8±4.7 |
| YopH | 26.2±4.4 | 15.3±2.3 | 108.5±25.0 | 333.0±33.6 |
| PP1 | 33.0±4.9 | 32.5±3.3 | 131.1±38.0 | 87.8±21.8 |
| PPM1B | 165.0±21.5 | 191.6±28.6 | 152.8±18.4 | 223.0±10.3 |
| PPM1G | 138.5±23.0 | 188.8±22.7 | 166.9±23.3 | 98.7±21.5 |
| Gleep | 18.3±2.9 | 41.9±6.4 | 213.9±2.0 | 32.3±2.0 |
| PRL | 195.6±21.5 | 288.3±12.3 | >500 | 282.5±9.3 |
The data presented are the average of at least three independent measurements. Data were fitted to the Michaelis-Menten equation using nonlinear least squares regression (GraphPad Prism).
2.7. Effects on the phosphorylation level of ERK1/2 and Pyk2
Given the good potency and selectivity of ST2, ST3, ST2–5 and ST3–5 toward STEP in vitro, we further evaluated their abilities to inhibit STEP-dependent signaling at the cellular level. Previous studies have already demonstrated that STEP decreases the nerve growth factor (NGF)-stimulated phosphorylation of ERK1/2 and Pyk2[36, 60, 61], Therefore, the inhibition of STEP would increase the tyrosine phosphorylation of ERK1/2 and Pyk2 (Figure 6A). The effects of four new STEP inhibitors on the phosphorylation of ERK1/2 and Pyk2 were tested in PC12 cells, which express high levels of NGF receptor. Briefly, PC12 cells were serum deprived for 12 h followed by incubation with vehicle or 20 μM inhibitor compounds for 2 h. The cells were then stimulated with NGF at 20 ng/mL for 5 min, the total cellular lysates were resolved by SDS-PAGE, and the levels of phospho-ERKl/2 and phosphor-Pyk2 were examined. As shown in Figure 6, all four compounds significantly increased the NGF-induced phosphorylation of ERK1/2 and Pyk2. Specifically, ST3–5 and ST2–5 showed more potent inhibition than ST3 and ST2, which is consistent with their enzyme inhibition activity in vitro (Figure 6).
Figure 6.

(A) Inhibition of STEP effects the phosphorylation level of ERK and Pyk2. (B) Effects of ST3, ST3–5, ST2 and ST2–5 on the NGF-induced phosphorylation of PYK2 pY412 and ERK pT202 and pY204 as detected by the phosphorylation-specific antibody. The GAPDH level was used as a control. PC12 cells were treated with DMSO or 20 μM ST3, ST3–5, ST2, or ST2–5 for 1 h and analyzed by Western blotting. (B) Statistical analysis of the phosphorylation of PYK2 pY412 in PC 12 cells treated with NGF and preincubated with different inhibitors. All experiments were repeated at least in triplicate. (C) Statistical analysis of the NGF-induced phosphorylation of ERK pT202 and pY204 in PC12 cells preincubated with different inhibitors. All experiments were repeated in triplicate. (**) p<0.01; (***) p<0.001; (****) p<0.0001 compared with control cells.
3. Conclusion
Because of the critical role of STEP in neuronal signaling, the development of STEP inhibitors for the treatment of multiple neural disorders has attracted considerable interest. Currently, only a limited number of small-molecule STEP inhibitors have been reported, which encouraged us to identify new chemotypes for STEP inhibition. However, due to the lack of bound-state structures, our initial virtual screening using the apo crystal structure of STEP identified only one inhibitor (ST1) with an IC50 of 70.7±5.4 μM. Witten et al. recently reported the crystal structure of STEP complexed with a sub strate-derived inhibitor [42]; however, this holo structure of STEP showed high structural similarity to the apo structure and did not reveal new pockets for inhibitor binding.
In the current study, we have combined the use of microsecond MD simulations, MSMs and fragment-centric pocket mapping to reveal the structural dynamics of STEP proteins and identify new cryptic pockets in exosite during MD simulation of apo STEP structure. We have also proposed a mixed binding mechanism for STEP inhibitors depending on the size of the small molecules, virtual screening using the ensemble structures efficiently improved the success rate compared with that of virtual screening using the apo structure and led to the identification of 11 new STEP inhibitors (ST2–ST12) with improved inhibitory activities (IC50=9.7–47.7 μM). Further study demonstrated the reversible binding and competitive inhibition of ST2 and ST3. Importantly, the molecualr dynamics simulation of STEP-ST3 complex as well as site-directed mutagenesis verified the binding of ST3 with the newly predicted cryptic pocket. Structure-activity relationship studies of ST2 analogs and ST3 analogs discovered compounds with improved potency (ST2–5 and ST3–5). Moreover, these new inhibitors also exhibit a certain degree of selectivity against a panel of protein phosphatases. Further biological studies in PC12 cells validated their effects on the phosphorylation of ERK1/2 and Pyk2 by the specific inhibition of STEP and simulated the cell migration and differentiation function.
In summary, our results demonstrated the value of integrated strategies combining multiple computational approaches for the identification of cryptic pockets and highlighted the feasibility of using MD simulations and MSMs to expose draggable protein regions that otherwise may not be evident. Moreover, these newly identified STEP inhibitors provide novel scaffold for selective STEP inhibition and serve as a good starting point for future drag development.
4. Materials and Methods
4.1. Materials.
pNPP was purchased from Sangon Biotech Co., Ltd. Ni-NTA agarose was obtained from Amersham Pharmacia Biotech. The selected virtual screening hit compounds were purchased from Specs with purity confirmed by the supplier (LC-MS or 1H-NMR). All other chemicals and reagents were purchased from Sigma. The histidine-tagged phosphatase constructs, His-STEP, have been described previously[58, 62, 63],
4.2. Molecular dynamics simulation.
Molecular dynamics (MD) simulations were performed using the Amber 14 package[64] with the AMBER14SB force field[65] and TIP3P water model. The apo crystal structure of STEP (PDB: 2BV5) served as the starting structure for the STEP-apo system. Two holo STEP systems (STEP-ST1 and STEP-ST3–5) were constructed using top-scored poses from the molecular docking results. The topology file for each ligand was generated using Antechamber with general AMBER force field (GAFF) [66], and partial charges were fit with HF/6–31 G(d) calculations using the Gaussian 09 package[67]. The RESP module in the Amber package was employed to fit the charges to each atomic center. Long-range electrostatic interactions were treated using the Particle Mesh Ewald (PME) method and non-bonded interactions were cut off at 12 Å. The SHAKE algorithm was applied to constrain all bonds involving hydrogen atoms. After a series of minimizations and equilibrations, MD simulations were performed on GPUs using the CUDA version of PMEMD[68] with periodic boundary conditions. The Berendsen thermostat method was used to control the system temperature at 300 K. We performed 1-μs MD simulations for the STEP-apo and STEP-ST1 systems to explore protein structural dynamics with or without an inhibitor bound. For the STEP-ST3 system, a 0.5-μs MD simulation was carried out to examine the binding stability of ST3. Other parameters were the default values.
4.3. Construction of Markov State Model.
The MD simulation data was used to construct Markov State Model (MSM) using the pyEMMA software (version 2.5.2)[69], A total of 2-μs of MD simulation data were used to construct the MSM for STEP-apo and STEP-ST1, respectively. Since we found two salt-bridge interactions (K439-D272 and K439-E519) governed the formation of exosite pockets, the minimum distances between D272, K439 and E519 were used as input features, yielding three dimensions. Time-lagged independent component analysis (TICA) were performed at a TICA lag time of 20 ns. The TICA coordinates were then clustered into 250 microstates using k-means algorithm, and the produced clusters were then lumped together into 3 metastable macrostates for STEP-apo and 2 metastable macrostates for STEP-ST1 using the PCCA algorithm [70], The implied timescale plot was used to identify a suitable MSM lag time (τ = 20 ns) for building the MSM.
4.4. AlphaSpace pocket analysis.
Fragment-centric pocket mapping was calculated using AlphaSpace[51, 52], which employed a geometric model based on Voronoi tessellation to identify and represent all concave interaction space across the protein surface. The concave spaces are represented as a set of alpha-atom/alpha-space pairs, and then clustered into discrete fragment-centric pockets. The occupation status of each individual alpha-space within each pocket is evaluated based on the distance between its associated alpha-atom and the nearest atom from the ligand, using a 1.6 Å cutoff. The total pocket occupation by ligand is calculated by taking the sum of all occupied alpha-space volumes associated with ligand atoms.
4.5. Virtual screening.
Structure-based virtual screenings were performed according to the workflow in Figure S1 (Supporting Information). Glide in Schrodinger suite as well as Autodock Vina were used for all molecular dockings [55, 56, 71], In general, the docking parameters for both Glide and Vina were kept to their default values. The size of the docking grid was 60 Å×60 Å×60 Å, which encompassed the active site of STEP structure. The top-ranked compounds were clustered based on the FCFP_6 fingerprints calculation and then selected manually. The drug-likeness of hit compounds were evaluated according to the metrics of Lipinski’s role of five were excluded from selection.
4.6. Protein expression and purification.
The catalytic domain of STEP with an N-terminal His tag was prepared and used for in vitro studies. The STEP mutant F523A was generated by PCR reaction with the QuickChange site-directed mutagenesis kit from Stratagene. The PAGE-purified oligonucleotide primers were obtained from Beijing Genomics Institute. BL21 (DE3) E. coli cells were transformed with the expression plasmids and cultured in LB medium with shaking at 37 °C. The culture temperature was adjusted to 18°C at OD600=0.6, and expression was induced for 12 h with 0.3 mM isopropyl b-D-l-thiogalactopyranoside (IPTG) once OD600 reached 0.8. The cells were harvested by centrifugation and resuspended in lysis buffer (50 mL, 20 mM Tris pH 8.0, 150 mM NaCl). After centrifugation, the supernatant was incubated with Ni2+-NTA resin with end-to-end mixing at 4 °C for 1 h. The beads were collected, washed once with buffer (20 mL, 20 mM Tris pH 8.0, 150 mM NaCl, and 5 mM imidazole) and eluted with an imidazole gradient (20 mM Tris pH 8.0, 150 mM NaCl, and 20–200 mM imidazole). After purification, the protein was further concentrated and stored at −80 °C. The expression and purification of the other His-tagged proteins were performed similarly.
4.7. IC50 measurements.
All 64 compounds were purchased from the Specs database, and the purity of these compounds was greater than 95% (confirmed by Specs; data are summarized in Supporting Information Table S1). Kinetic assays for STEP-catalyzed pNPP hydrolysis in the presence of small-molecule inhibitors were performed as previously described[58, 63, 72], The effect of each inhibitor on STEP-catalyzed pNPP hydrolysis was determined at 30 °C in reaction buffer (50 mM DMG buffer with anionic strength of 0.15 M adjusted by NaCl). The Km value of STEP for pNPP hydrolysis (0.7 mM for STEP) was used to determine the IC50. The reaction was detected by monitoring the absorbance of pNP at 405 nm. The IC50 values were obtained by fitting the data to Eq. 1 using GraphPad Prism 6 (GraphPad Software, Inc., La Jolla, CA, USA) as follows:
| (Eq. 1) |
4.8. Reversible binding.
Reversible binding of compounds ST2 and ST3 was conducted as previously described[58, 63], STEP was preincubated with compounds ST2 and ST3 at 80 μM for various times. The time-dependent ratio Kcat(control)/Kcat (inhibitor) was determined. The time-dependent inhibition of LYP by its irreversible inhibitor A25 was taken as a positive control.
4.9. Data analysis and software.
The data were analyzed using GraphPad Prism software. All experiments were performed at least in triplicate, and the data are expressed as the mean±SD. Statistical comparisons were performed with ANOVA tests using GraphPad Prism 5.
4.10. Ki measurements.
The STEP-catalyzed hydrolysis of pNPP in the presence of inhibitors was assayed at 30 °C in assay buffer (pH 6.0 with an ionic strength of 0.15 M). The reaction was initiated by the addition of pNPP (ranging from 0.2 to 5 times the Km) to a reaction mixture containing STEP and various fixed concentrations of the inhibitors and then quenched by the addition of 1 M NaOH. The inhibition constant, Ki, and the inhibition pattern were evaluated by fitting the data to the Michaelis-Menten equation for competitive inhibition (Eq. 2 and 3) using a Lineweaver-Burk plot.
| (Eq. 2) |
| (Eq. 3) |
4.11. Cell culture and immunoblotting assay.
PC12 cells were grown at 37 °C in RPMI 1640 medium supplemented with 10% FBS as described previously[73]. The PC12 cells were preincubated with a small-molecule inhibitor (20 μM) or DMSO for 1 h. The cells were then stimulated with 25 ng/ml NGF or medium for 5 min and lysed in lysis buffer (50 mM Tris, pH 7.5, 150 mM NaCl, 10 mM NaF, 2 mM EDTA, 10% glycerol, 1% NP-40, 0.25% sodium deoxycholate, 1 mM NaVO4 and a protease cocktail). The protein concentrations of the lysates were measured using the BCA Protein Quantitation Kit (Beyotime). Equal amounts of each cell lysate were denatured in 2× SDS loading buffer and boiled for 10 min. The protein samples were subjected to western blotting.
Supplementary Material
Table 3.
Structure-activity relationship of ST3 and its analogs.
 |
Highlights.
Integrated computational approaches were employed in the identification of cryptic pockets in exosite of STEP;
The application of cryptic pockets increased the success rate of structure-based virtual screening against STEP;
A total of 12 new STEP inhibitors were identified and preliminary structure-activity relationship was explored;
Site-directed mutagenesis verified the binding of ST3 with predicted cryptic pocket;
The most potent and selective inhibitors modulate the phosphorylation of ERK1/2 and Pyk2 in PC12 cells.
Acknowledgment
This work was supported by grants from National Natural Science Foundation of China (Grant 21672127 and 81874288 to H.F. and Grant 31470789 and 81773704 to J.P.S.), US National Institute of Health (R35-GM127040) to Y.Z., Key Research and Development Project of Shandong Province (Grant No. 2017CXGC1401), Fundamental Research Funds of Shandong University (Grant No. 2019GN045) and The Joint Research Funds for Shandong University and Karolinska Institute (SDU-KI-2019-06). The authors gratefully acknowledge NYU-ITS and NYUAD for providing computational resources.
Abbreviations
- AD
Alzheimer’s disease
- STEP
striatal-enriched protein tyrosine phosphatase
- PTP
protein tyrosine phosphatases
- SBDD
structure-based drug design
- SBVS
structure-based virtual screening
- MD
molecular dynamics
- MSM
Markov State Model
- TICA
Time-lagged independent component analysis
- pNPP
p-nitrophenyl phosphate
Footnotes
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Appendix A. Supplementary data
Supplementary data associated with this article can be found in the online version. Data include figures and tables for additional results, 1H-NMR spectral information for representative compounds (PDF); MD simulation of STEP-apo system (MPG); MD simulation of STEP-ligand system (MPG).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Sledz P, Caflisch A, Protein structure-based drug design: from docking to molecular dynamics, Curr Opin Struct Biol, 48 (2018) 93–102. [DOI] [PubMed] [Google Scholar]
- [2].Lionta E, Spyrou G, Vassilatis DK, Cournia Z, Structure-based virtual screening for drug discovery: principles, applications and recent advances, Curr Top Med Chem, 14 (2014) 1923–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Bermudez M, Mortier J, Rakers C, Sydow D, Wolber G, More than a look into a crystal ball: protein structure elucidation guided by molecular dynamics simulations, Drug Discov Today, 21 (2016) 1799–1805. [DOI] [PubMed] [Google Scholar]
- [4].Stank A, Kokh DB, Fuller JC, Wade RC, Protein Binding Pocket Dynamics, Accounts Chem Res, 49 (2016) 809–815. [DOI] [PubMed] [Google Scholar]
- [5].Beglov D, Hall DR, Wakefield AE, Luo L, Allen KN, Kozakov D, Whitty A, Vajda S, Exploring the structural origins of cryptic sites on proteins, Proc Natl Acad Sci USA, 115 (2018) E3416–E3425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Lama D, Brown CJ, Lane DP, Verma CS, Gating by Tryptophan 73 Exposes a Cryptic Pocket at the Protein-Binding Interface of the Oncogenic elF4E Protein, Biochemistry-Us, 54 (2015) 6535–6544. [DOI] [PubMed] [Google Scholar]
- [7].Comitani F, Gervasio FL, Exploring Cryptic Pockets Formation in Targets of Pharmaceutical Interest with SWISH, J Chem Theory Comput, 14 (2018) 3321–3331. [DOI] [PubMed] [Google Scholar]
- [8].Tan YS, Reeks J, Brown CJ, Thean D, Gago FJF, Yuen TY, Goh ETL, Lee XEC, Jennings CE, Joseph TL, Lakshminarayanan R, Lane DP, Noble MEM, Verma CS, Benzene Probes in Molecular Dynamics Simulations Reveal Novel Binding Sites for Ligand Design, J Phys Chem Lett, 7 (2016) 3452–3457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Oleinikovas V, Saladino G, Cossins BP, Gervasio FL, Understanding Cryptic Pocket Formation in Protein Targets by Enhanced Sampling Simulations, J Am Chem Soc, 138 (2016) 14257–14263. [DOI] [PubMed] [Google Scholar]
- [10].Ghanakota P, van Vlijmen H, Sherman W, Beuming T, Large-Scale Validation of Mixed-Solvent Simulations to Assess Hotspots at Protein-Protein Interaction Interfaces, Journal of Chemical Information and Modeling, 58 (2018) 784–793. [DOI] [PubMed] [Google Scholar]
- [11].Bowman GR, Bolin ER, Hart KM, Maguire BC, Marqusee S, Discovery of multiple hidden allosteric sites by combining Markov state models and experiments, Proc Natl Acad Sci USA, 112 (2015) 2734–2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Bowman GR, Geissler PL, Equilibrium fluctuations of a single folded protein reveal a multitude of potential cryptic allosteric sites, Proc Natl Acad Sci USA, 109 (2012) 11681–11686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Tan YS, Sledz P, Lang S, Stubbs CJ, Spring DR, Abell C, Best RB, Using Ligand-Mapping Simulations to Design a Ligand Selectively Targeting a Cryptic Surface Pocket of Polo-Like Kinase 1, Angew Chem Int Edit, 51 (2012) 10078–10081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Wassman CD, Baronio R, Demir O, Wallentine BD, Chen CK, Hall LV, Salehi F, Lin DW, Chung BP, Hatfield GW, Chamberlin AR, Luecke H, Lathrop RH, Kaiser P, Amaro RE, Computational identification of a transiently open L1/S3 pocket for reactivation of mutant p53, Nat Commun, 4 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Bowman GR, Geissler PL, Equilibrium fluctuations of a single folded protein reveal a multitude of potential cryptic allosteric sites, P Natl Acad Sci USA, 109 (2012) 11681–11686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Bowman GR, Bolin ER, Hart KM, Maguire BC, Marqusee S, Discovery of multiple hidden allosteric sites by combining Markov state models and experiments, P Natl Acad Sei USA, 112 (2015) 2734–2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Rhee I, Veillette A, Protein tyrosine phosphatases in lymphocyte activation and autoimmunity, Nat Immunol, 13 (2012) 439–447. [DOI] [PubMed] [Google Scholar]
- [18].Tonks NK, Protein tyrosine phosphatases--from housekeeping enzymes to master regulators of signal transduction, Febs J, 280 (2013) 346–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Lee H, Yi JS, Lawan A, Min K, Bennett AM, Mining the function of protein tyrosine phosphatases in health and disease, Semin Cell Dev Biol, 37 (2015) 66–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Hendriks WJ, Elson A, Harroch S, Pulido R, Stoker A, den Hertog J, Protein tyrosine phosphatases in health and disease, Febs J, 280 (2013) 708–730. [DOI] [PubMed] [Google Scholar]
- [21].Tsou RC, Bence KK, Central regulation of metabolism by protein tyrosine phosphatases, Front Neurosci-Switz, 6 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Alonso A, Sasin J, Bottini N, Friedberg I, Friedberg I, Osterman A, Godzik A, Hunter T, Dixon J, Mustelin T, Protein tyrosine phosphatases in the human genome, Cell, 117 (2004) 699–711. [DOI] [PubMed] [Google Scholar]
- [23].Hubbard SR, Till JH, Protein tyrosine kinase structure and function, Annu Rev Biochem, 69 (2000) 373–398. [DOI] [PubMed] [Google Scholar]
- [24].Chen MJ, Dixon JE, Manning G, Genomics and evolution of protein phosphatases, Sei Signal, 10 (2017). [DOI] [PubMed] [Google Scholar]
- [25].Barr AJ, Protein tyrosine phosphatases as drug targets: strategies and challenges of inhibitor development, Future Med Chem, 2 (2010) 1563–1576. [DOI] [PubMed] [Google Scholar]
- [26].He RJ, Yu ZH, Zhang RY, Zhang ZY, Protein tyrosine phosphatases as potential therapeutic targets, Acta Pharmacol Sin, 35 (2014) 1227–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Bohmer F, Szedlacsek S, Tabernero L, Ostman A, den Hertog J, Protein tyrosine phosphatase structure-function relationships in regulation and pathogenesis, Febs J, 280 (2013) 413–431. [DOI] [PubMed] [Google Scholar]
- [28].Li H, Yang D, Ning S, Xu Y, Yang F, Yin R, Feng T, Han S, Guo L, Zhang P, Qu W, Guo R, Song C, Xiao P, Zhou C, Xu Z, Sun JP, Yu X, Switching of the substrate specificity of protein tyrosine phosphatase N12 by cyclin-dependent kinase 2 phosphorylation orchestrating 2 oncogenic pathways, FasebJ, 32 (2018) 73–82. [DOI] [PubMed] [Google Scholar]
- [29].Wang HM, Xu YF, Ning SL, Yang DX, Li Y, Du YJ, Yang F, Zhang Y, Liang N, Yao W, Zhang LL, Gu LC, Gao CJ, Pang Q, Chen YX, Xiao KH, Ma R, Yu X, Sun JP, The catalytic region and PEST domain of PTPN18 distinctly regulate the HER2 phosphorylation and ubiquitination barcodes, Cell Res, 24 (2014) 1067–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Yao Z, Darowski K, St-Denis N, Wong V, Offensperger F, Villedieu A, Amin S, Malty R, Aoki H, Guo H, Xu Y, lorio C, Kotlyar M, Emili A, Jurisica I, Neel BG, Babu M, Gingras AC, Stagljar I, A Global Analysis of the Receptor Tyrosine Kinase-Protein Phosphatase Interactome, Mol Cell, 65 (2017) 347–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Tonks NK, Protein tyrosine phosphatases: from genes, to function, to disease, Nat Rev Mol Cell Biol, 7 (2006) 833–846. [DOI] [PubMed] [Google Scholar]
- [32].Boulanger LM, Lombroso PJ, Raghunathan A, During MJ, Wahle P, Naegele JR, Cellular and molecular characterization of a brain-enriched protein tyrosine phosphatase, J Neurosci, 15 (1995) 1532–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Braithwaite SP, Paul S, Nairn AC, Lombroso PJ, Synaptic plasticity: one STEP at a time, Trends Neurosci, 29 (2006) 452–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Lombroso PJ, Murdoch G, Lerner M, Molecular Characterization of a Protein-Tyrosine-Phosphatase Enriched in Striatum, P Natl Acad Sei USA, 88 (1991) 7242–7246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Li R, Xie DD, Dong JH, Li H, Li KS, Su J, Chen LZ, Xu YF, Wang HM, Gong Z, Cui GY, Yu X, Wang K, Yao W, Xin T, Li MY, Xiao KH, An XF, Huo Y, Xu ZG, Sun JP, Pang Q, Molecular mechanism of ERK dephosphorylation by striatal-enriched protein tyrosine phosphatase (vol 128, pg 315, 2014), J Neurochem, 134 (2015) 978–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Xu J, Chatterjee M, Baguley TD, Brouillette J, Kurup P, Ghosh D, Kanyo J, Zhang Y, Seyb K, Ononenyi C, Foscue E, Anderson GM, Gresack J, Cuny GD, Glicksman MA, Greengard P, Lam TT, Tautz L, Nairn AC, Ellman JA, Lombroso PJ, Inhibitor of the tyrosine phosphatase STEP reverses cognitive deficits in a mouse model of Alzheimer’s disease, Plos Biol, 12 (2014) el001923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Zhang Y, Kurup P, Xu J, Carty N, Fernandez SM, Nygaard HB, Pittenger C, Greengard P, Strittmatter SM, Nairn AC, Lombroso PJ, Genetic reduction of striatal-enriched tyrosine phosphatase (STEP) reverses cognitive and cellular deficits in an Alzheimer’s disease mouse model, Proc Natl Acad Sei USA, 107 (2010) 19014–19019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Zhang Y, Kurup P, Xu J, Anderson GM, Greengard P, Nairn AC, Lombroso PJ, Reduced levels of the tyrosine phosphatase STEP block beta amyloid-mediated GluAl/GluA2 receptor internalization, J Neurochem, 119 (2011) 664–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Zhang ZY, Drugging the Undruggable: Therapeutic Potential of Targeting Protein Tyrosine Phosphatases, Acc Chem Res, 50 (2017) 122–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Li H, Yang F, Liu C, Xiao P, Xu Y, Liang Z, Liu C, Wang H, Wang W, Zheng W, Zhang W, Ma X, He D, Song X, Cui F, Xu Z, Yi F, Sun JP, Yu X, Crystal Structure and Substrate Specificity of PTPN12, Cell Rep, 15 (2016) 1345–1358. [DOI] [PubMed] [Google Scholar]
- [41].Baguley TD, Xu HC, Chatterjee M, Nairn AC, Lombroso PJ, Ellman JA, Substrate-based fragment identification for the development of selective, nonpeptidic inhibitors of striatal-enriched protein tyrosine phosphatase, J Med Chem, 56 (2013) 7636–7650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Witten MR, Wissler L, Snow M, Geschwindner S, Read JA, Brandon NJ, Nairn AC, Lombroso PJ, Kack H, Ellman JA, X-ray Characterization and Structure-Based Optimization of Striatal-Enriched Protein Tyrosine Phosphatase Inhibitors, J Med Chem, 60 (2017) 9299–9319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Xu J, Chatterjee M, Baguley TD, Brouillette J, Kurup P, Ghosh D, Kanyo J, Zhang Y, Seyb K, Ononenyi C, Foscue E, Anderson GM, Gresack J, Cuny GD, Glicksman MA, Greengard P, Lam TT, Tautz L, Nairn AC, Ellman JA, Lombroso PJ, Inhibitor of the Tyrosine Phosphatase STEP Reverses Cognitive Deficits in a Mouse Model of Alzheimer’s Disease, Plos Biol, 12 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Baguley TD, Xu HC, Chatterjee M, Nairn AC, Lombroso PJ, Ellman JA, Substrate-Based Fragment Identification for the Development of Selective, Nonpeptidic Inhibitors of Striatal-Enriched Protein Tyrosine Phosphatase, J Med Chem, 56 (2013) 7636–7650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Witten MR, Wissler L, Snow M, Geschwindner S, Read JA, Brandon NJ, Nairn AC, Lombroso PJ, Kack H, Ellman JA, X-ray Characterization and Structure-Based Optimization of Striatal-Enriched Protein Tyrosine Phosphatase Inhibitors, J Med Chem, 60 (2017) 9299–9319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Shukla D, Hernandez CX, Weber JK, Pande VS, Markov State Models Provide Insights into Dynamic Modulation of Protein Function, Accounts Chem Res, 48 (2015) 414–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Unarta IC, Zhu LZ, Tse CKM, Cheung PPH, Yu J, Huang XH, Molecular mechanisms of RNA polymerase II transcription elongation elucidated by kinetic network models, Curr Opin Struc Biol, 49 (2018) 54–62. [DOI] [PubMed] [Google Scholar]
- [48].Zhu LZ, Sheong FK, Zeng XZ, Huang XH, Elucidation of the conformational dynamics of multi-body systems by construction of Markov state models, Phys Chem Chem Phys, 18 (2016) 30228–30235. [DOI] [PubMed] [Google Scholar]
- [49].Jiang HL, Sheong FK, Zhu LZ, Gao X, Bernauer J, Huang XH, Markov State Models Reveal a Two-Step Mechanism of miRNA Loading into the Human Argonaute Protein: Selective Binding followed by Structural Re-arrangement, Plos Computational Biology, 11 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Hart KM, Ho CM, Dutta S, Gross ML, Bowman GR, Modelling proteins’ hidden conformations to predict antibiotic resistance, Nat Commun, 7 (2016) 12965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Rooklin D, Wang C, Katigbak J, Arora PS, Zhang Y, AlphaSpace: Fragment-Centric Topographical Mapping To Target Protein-Protein Interaction Interfaces, J Chem Inf Model, 55 (2015) 1585–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Rooklin D, Modell AE, Li H, Berdan V, Arora PS, Zhang Y, Targeting Unoccupied Surfaces on Protein-Protein Interfaces, J Am Chem Soc, 139 (2017) 15560–15563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Eswaran J, Von Kries JP, Marsden B, Longman E, Debreczeni JE, Ugochukwu E, Turnbull A, Lee WH, Knapp S, Barr AJ, Crystal structures and inhibitor identification for PTPN5, PTPRR and PTPN7: a family of human MAPK-specific protein tyrosine phosphatases, Biochem J, 395 (2006) 483–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Barr AJ, Ugochukwu E, Lee WH, King ONF, Filippakopoulos P, Alfano I, Savitsky P, Burgess-Brown NA, Muller S, Knapp S, Large-Scale Structural Analysis of the Classical Human Protein Tyrosine Phosphatome, Cell, 136 (2009) 352–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE, Francis P, Shenkin PS, Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy, J Med Chem, 47 (2004) 1739–1749. [DOI] [PubMed] [Google Scholar]
- [56].Trott O, Olson AJ, Software News and Update AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading, J Comput Chem, 31 (2010) 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM, K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions, Nature, 503 (2013) 548–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Hou XB, Li R, Li KS, Yu X, Sun JP, Fang H, Fast Identification of Novel Lymphoid Tyrosine Phosphatase Inhibitors Using Target-Ligand Interaction-Based Virtual Screening, J Med Chem, 57 (2014) 9309–9322. [DOI] [PubMed] [Google Scholar]
- [59].Yu ZH, Zhang ZY, Regulatory Mechanisms and Novel Therapeutic Targeting Strategies for Protein Tyrosine Phosphatases, Chem Rev, 118 (2018) 1069–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Li R, Xie DD, Dong JH, Li H, Li KS, Su J, Chen LZ, Xu YF, Wang HM, Gong Z, Cui GY, Yu X, Wang K, Yao W, Xin T, Li MY, Xiao KH, An XF, Huo Y, Xu ZG, Sun JP, Pang Q, Molecular mechanism of ERK dephosphorylation by striatal-enriched protein tyrosine phosphatase, J Neurochem, 128 (2014) 315–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Xu J, Kurup P, Bartos JA, Patriarchi T, Hell JW, Lombroso PJ, Striatal-enriched protein-tyrosine phosphatase (STEP) regulates Pyk2 kinase activity, J Biol Chem, 287 (2012) 20942–20956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Li R, Xie DD, Dong JH, Li H, Li KS, Su J, Chen LZ, Xu YF, Wang HM, Gong Z, Cui GY, Yu X, Wang K, Yao W, Xin T, Li MY, Xiao KH, An XF, Huo YQ, Xu ZG, Sun JP, Pang Q, Molecular mechanism of ERK dephosphorylation by striatal-enriched protein tyrosine phosphatase, J Neurochem, 128 (2014) 315–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Ge L, Li KS, Li MM, Xiao P, Hou XB, Chen X, Liu HD, Lin A, Yu X, Ren GJ, Fang H, Sun JP, Identification of a benzo imidazole thiazole derivative as the specific irreversible inhibitor of protein tyrosine phosphatase, Bioorg Med Chem Lett, 26 (2016) 4795–4798. [DOI] [PubMed] [Google Scholar]
- [64].Case D, Darden T, Cheatham T, Simmerling C, Wang J, Duke R, AMBER 14. 2014, University of California, San Francisco. [Google Scholar]
- [65].Maier JA, Martinez C, Kasavajhala K, Wickstrom L, Hauser KE, Simmerling C, ffl4SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB, J Chem Theory Comput, 11 (2015) 3696–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA, Development and testing of a general amber force field, J Comput Chem, 25 (2004) 1157–1174. [DOI] [PubMed] [Google Scholar]
- [67].Frisch M, Trucks G, Schlegel H, Scuseria G, Robb M, Cheeseman J, Scalmani G, Barone V, Mennucci B, Petersson G, Gaussian 09; Gaussian, Inc: Wallingford, CT, 2009, There is no corresponding record for this reference. [Google Scholar]
- [68].Gotz AW, Williamson MJ, Xu D, Poole D, Le Grand S, Walker RC, Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 1. Generalized Born, J Chem Theory Comput, 8 (2012) 1542–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Scherer MK, Trendelkamp-Schroer B, Paul F, Perez-Hernandez G, Hoffmann M, Plattner N, Wehmeyer C, Prinz JH, Noe F, PyEMMA 2: A Software Package for Estimation, Validation, and Analysis of Markov Models, J Chem Theory Comput, 11 (2015) 5525–5542. [DOI] [PubMed] [Google Scholar]
- [70].Weber M, Kube S, Robust Perron Cluster Analysis for various applications in computational life science, Computational Life Sciences, Proceedings, 3695 (2005) 57–66. [Google Scholar]
- [71].Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT, Banks JL, Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening, J Med Chem, 47 (2004) 1750–1759. [DOI] [PubMed] [Google Scholar]
- [72].Yu X, Sun JP, He YT, Guo XL, Liu SJ, Zhou B, Hudmon A, Zhang ZY, Structure, inhibitor, and regulatory mechanism of Lyp, a lymphoid-specific tyrosine phosphatase implicated in autoimmune diseases, P Natl Acad Sci USA, 104 (2007) 19767–19772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Pan C, Liu HD, Gong Z, Yu X, Hou XB, Xie DD, Zhu XB, Li HW, Tang JY, Xu YF, Yu JQ, Zhang LY, Fang H, Xiao KH, Chen YG, Wang JY, Pang Q, Chen W, Sun JP, Cadmium is a potent inhibitor of PPM phosphatases and targets the Ml binding site, Sci Rep-Uk, 3 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.