

Abstract
As an interface with the environment, the skin is a complex ecosystem colonized by many microorganisms that coexist in an established balance. The cutaneous microbiome inhibits colonization with pathogens, such as Staphylococcus aureus, and is a crucial component for function of the epidermal barrier. Moreover, crosstalk between commensals and the immune system is now recognized because microorganisms can modulate both innate and adaptive immune responses. Host-commensal interactions also have an effect on the developing immune system in infants and, subsequently, the occurrence of diseases, such as asthma and atopic dermatitis (AD). Later in life, the cutaneous microbiome contributes to the development and course of skin disease. Accordingly, in patients with AD, a decrease in microbiome diversity correlates with disease severity and increased colonization with pathogenic bacteria, such as S aureus. Early clinical studies suggest that topical application of commensal organisms (eg, Staphylococcus hominis or Roseomonas mucosa) reduces AD severity, which supports an important role for commensals in decreasing S aureus colonization in patients with AD. Advancing knowledge of the cutaneous microbiome and its function in modulating the course of skin disorders, such as AD, might result in novel therapeutic strategies.
Keywords: Atopic dermatitis, biotherapy, commensal, host-microbiome interaction, immune regulation, microbiome, Staphylococcus aureus
A multitude of microbiota inhabit our human epithelial surfaces. Although there is increasing evidence that these microbiota, which live in and on our bodies, are important to human health and disease, the many functions and consequences of these resident microbiota remain poorly understood. Given the challenges in being able to adequately culture all microbes present in a given sample, technological advances in genome sequencing have increased the ability to interrogate human epithelial microbiomes (the full collection of microbiota). Several technical advances in the study of the composition and function of the microbiome have collectively enlightened our understanding of the role of the microbiome in both pathogenesis of atopic dermatitis (AD) and in disease modification (Fig 1).1
FIG 1.
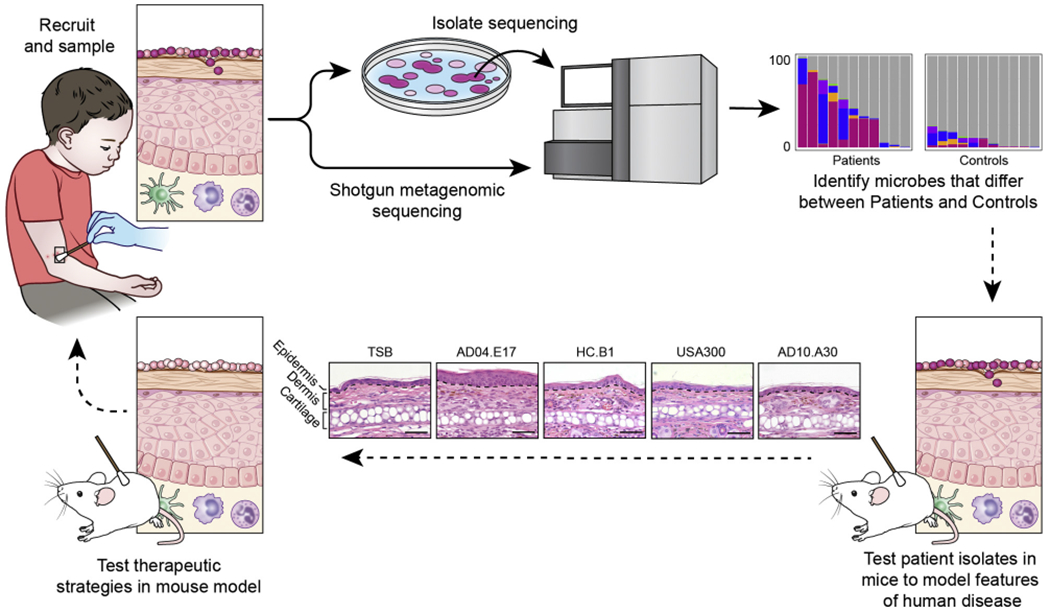
An example of a pipeline for skin microbiome studies in patients with AD. The pipeline can begin by posing a scientific question, with subsequent recruitment of patients and control subjects, phenotyping of patients with AD, and collection of clinical samples (top left). Microbiota from clinical samples can either be directly sequenced to study the complex communities of microbiota (the microbiome) or first cultivated to investigate individual clinical isolates through whole-genome sequencing (and/or with model systems). Shotgun metagenomic sequencing refers to the sequencing of all genetic material in a complex sample and can provide information on bacteria, fungi, and viruses within the sample; the functional potential of the mixed microbial communities; and the different microbial strains. Sequencing both cultivated isolates (whole-genome sequencing) and the complex communities of microbes (shotgun metagenomics sequencing) can provide complementary information. The bioinformatics analyses of microbial sequencing data can identify the microbiome differences between patients with AD and healthy control subjects, which is graphically represented by bar charts that indicate the relative abundances of different staphylococci shown as different colors. The y-axis of bar charts represents 0% to 100% relative abundance. Differences in the microbiota found on patients versus control subjects can be studied in mouse models using clinically relevant isolates to examine microbial strain-level differences. For example, differences in host responses can be observed in the histology (lower panels) from mice who undergo application of different patient-associated staphylococcal strains (AD10.A30, USA300, HC.B1, AD04.E17)1 versus tryptic soy broth (TSB) control; comparisons can be made in epidermal thickening and immune cell infiltrates elicited by different strains. Further studies might provide insight into the role of the skin microbiome in disease pathogenesis, which could lead to development of microbiome-targeted therapeutics for patients.
Although animal models cannot fully recapitulate the human microbiome and disease states, the use of model organisms to deeply investigate host-microbial relationships has elucidated intriguing biological mechanisms. The continued integration of knowledge gleaned from patient-derived microbiota, animal models, and host-microbial interactions will be critical for developing and understanding therapeutic approaches. Prior publications have extensively reviewed the differences in the human skin microbiome based on various factors, including anatomic skin sites, sexual maturity, and skin physiology; this review provides a broad overview of the different aspects of microbiome, immunology, microbiology, and barrier research as related to AD, in particular early host-microbiome events in patients with AD. Here we review the role of the cutaneous microbiome in healthy and AD skin.
THE MICROBIOME OF NORMAL SURFACE EPITHELIA
The complexities of human microbial communities are reflected in the distinct microbiomes observed in the human skin, gut, and respiratory tract, among other body sites. Furthermore, the microbiota in distinct niches undergo changes over the human lifespan. The continual advances in our understanding of the human microbiome and its potential roles in human disease might subsequently lead to preventative and/or therapeutic strategies.
Skin microbiome research has highlighted the diversity and skin site specificity of microbial communities on human subjects, such that the different regional skin surfaces have different compositions of microbial communities.2–5 The skin hosts the most diverse commensal communities in the body, with more than 1000 different bacterial species from 19 different phyla.3,6 Although there are unique features of specific skin sites, some shared features of skin microbial communities reflect shared skin physiology: sebaceous skin sites often have Cutibacterium acnes (formerly known as Propionibacterium acnes). Small studies in healthy adult volunteers have shown that skin microbiomes are relatively stable for months to years and that each person might possess a personalized skin microbiome.7
Studies have also demonstrated differences in the skin microbiomes of subjects at different life stages. For example, children who are less sexually mature have lower relative abundances of Corynebacterium and Cutibacterium species8 and greater diversity of skin fungi9 compared with more sexually mature subjects. The infant skin microbiome is a particularly active area of investigation because it might provide insights into early-life exposures.10–13 Children as young as 2 days old have site-specific differences in their skin microbiomes13 that might influence future development of disease.14,15
ESTABLISHMENT OF THE SKIN MICROBIOME IN EARLY LIFE
Early life is also characterized by rapid immunologic maturation. As such, it represents an active period during which host-commensal interactions can formatively affect how our immune system responds to our microbial brethren.16,17 Future success of microbially directed interventions to prevent or treat inflammatory skin disease will require a deeper understanding of the mechanisms responsible for development of a healthy symbiosis during this critical window.
Neonatal immune responses demonstrate a reduced propensity for activation or inflammation compared with those in adults. We now appreciate that this is not only due to immaturity of the immune system but also due to the existence of regulatory mechanisms unique to this early window. In infants, compared with older children or adults, activation of Toll-like receptors (TLRs), key sensors of the innate immune system, results in greater production of IL-6 and IL-23 and less production of TNF-α and IL-1.16 Composition and function of the adaptive immune system evolves in parallel, with regulatory T (Treg) cells found in greater abundance during fetal life and infancy.17–19 Although these differences place neonates at greater risk for disseminated infection, they also promote immune tolerance to self-antigens and foreign antigens, thereby preventing inflammation disadvantageous to healthy tissue development.
Birth marks an abrupt transition, with increased exposure to microbial products and antigens. Composition of microbial communities in infants are distinct from those seen later in life and can be influenced, at least initially, by exogenous factors, such as birth delivery mode and maternal commensals.10,12 Notably, the identity and function of these microbes can shape host health trajectory. In animal models early-life immune responses to gut- and lung-resident microbes have been shown to influence adult susceptibility to colitis, asthma, and anaphylaxis.20 In human infants the presence or absence of certain gut bacteria has been associated with increased proinflammatory metabolites and heightened risk of asthma.21 Whether early disruption of the microbial community on skin directly affects future risk of inflammatory skin disease remains an open question. However, it is notable that recent longitudinal studies examining the skin microbiome in patients at risk for AD have found alterations in skin flora that predate disease onset.13,22
Until recently, little was known about the effect of early-life microbial exposures on skin immune function. Modeling this complex relationship in mice has taught us that these early-life interactions are likely to be of equal or greater significance in skin as in other tissues. When neonatal mice are colonized by the commensal bacterium (coagulase-negative Staphylococcus [CoNS]) Staphylococcus epidermidis, they develop a large percentage of Treg cells specific for S epidermidis and mount less inflammation to this microbe on rechallenge in later life. In contrast, delaying S epidermidis exposure until adulthood prevents this protective effect and promotes skin inflammation in responses to this otherwise “healthy” bacterium (Fig 2).22
FIG 2.
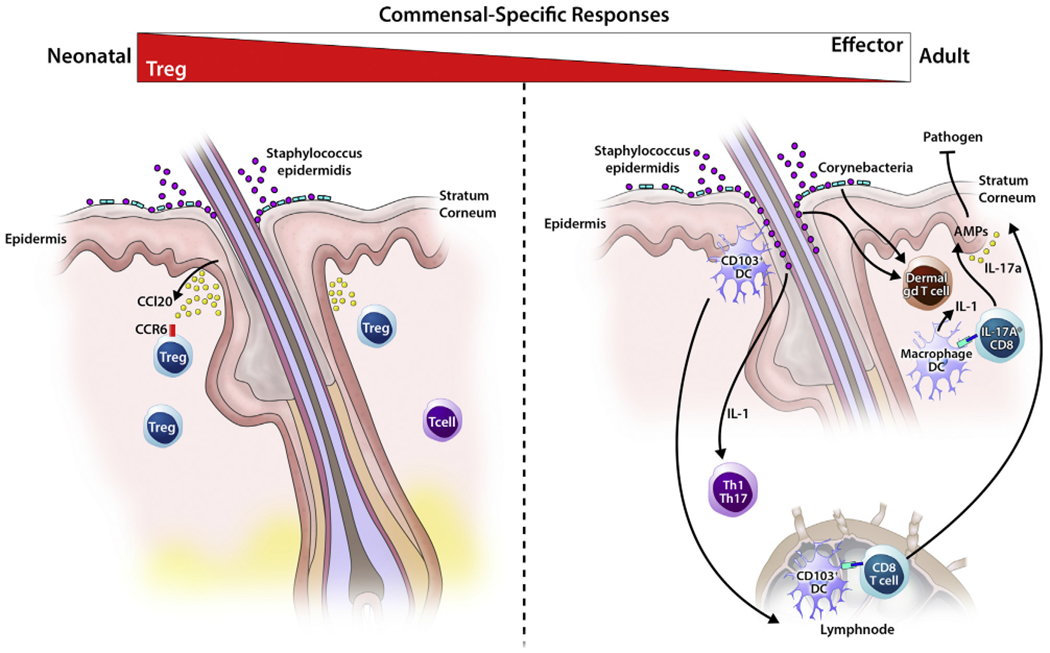
Age-dependent immune-commensal crosstalk in skin. Left panel, Some cutaneous immune cells, in particular CD4+ T cells generated in response to the commensal S epidermidis, are notably age dependent. Neonatal colonization by S epidermidis yields a population of antigen-specific CD4+ T cells dominated by Treg cells. In contrast, when exposure is delayed until adulthood, cytokine-producing effector CD4+ T cells predominate. Only early-life exposure is conducive to the establishment of antigen-specific immune tolerance and protection against skin inflammation on subsequent exposure. One factor accounting for this age-dependent response is the high density of Treg cells found in neonatal skin, which conditions the skin for tolerogenic interactions with the microbiota through yet undefined mechanisms. Neonatal skin Treg cells are preferentially localized around hairfollicles, a dense niche for commensal skin microbes. Right panel, Colonization of adult skin with specific microbiota results in an IL-1–meditated homeostatic effector immune response, including TH1 and TH17 cells, as well as dermal IL-17A+ γδ T cells. Certain strains of S epidermidis also induce commensal-specific populations of IL-17A+CD8+ T cells (TC17) through CD103+ dendritic cells.
At least 1 factor accounting for this age-dependent difference is the greater density of Treg cells found in neonatal compared with adult skin.23 Intriguingly, these Treg cells are markedly decreased in the skin of young mice raised under gnotobiotic (“germ-free”) conditions and in those lacking hair follicles, a major tissue niche for skin CoNS species. Indeed, colonization of hair follicles by commensal microbes appears to stimulate production by isthmus keratinocytes of a chemokine, CCL20, which then helps recruit these Treg cells into the skin (Fig 2, left panel).24
Thus animal models suggest that mechanisms promoting establishment of a healthy immunologic symbiosis with our skin microbiota are preferentially active early in life and can be facilitated by microbes themselves. Of course, there are notable differences between mice and human subjects with regard to timing of adaptive immune development, composition of skin bacterial communities, and skin structure. Thus how these findings translate to human biology and their implications for disrupting the composition of the neonatal skin microbiota will be a fruitful area of active investigation. Although detailed immunologic phenotyping of neonatal human skin has not yet been undertaken, Treg cells are enriched in pediatric compared with adult human skin.25
In considering potential translational applications of skin microbiome research, one can envision both corrective interventions to treat an established skin disease and preventative measures to reduce risk of disease onset or mitigate future severity. The latter might be especially relevant for conditions such as AD, in which variable penetrance based on genetic susceptibility and an early age of onset are defining features.26 Continued work to define the early-life influence of skin microbes on cutaneous immune function, both in the context of healthy and barrier-disrupted skin, will be critical to inform future development of prevention-oriented recommendations and microbe-based interventions.
IMMUNE-COMMENSAL CROSSTALK IN THE SKIN
The skin presents a physical barrier to harmful agents while establishing a unique innate immune system to regulate resident microbial communities. In contrast to other epithelial surfaces, such as the gut, which maintain physical separation from microbes through establishment of a mucous layer, the dense distribution of skin appendages creates a large surface area for close communication with microbes.27 The skin strictly regulates a sophisticated set of innate antimicrobial gene products that include antimicrobial peptides and proteins, lipids, a pH barrier, and free radical production to control the surface microbial community.28 A network of immune cells patrol the skin to reinforce the physical barrier because commensal and many potential pathogens can penetrate the epidermis after even a minor breach.29 The interplay between the epithelial barrier, immune defense, and the cutaneous microbiome has emerged as a key system for maintaining balance between health and disease.30,31 Mounting clinical and experimental evidence suggests that modulating the microbiome might be efficacious for the treatment of inflammatory skin conditions.32 However, the fundamental mechanisms underlying the immune-commensal crosstalk are only beginning to unfold. A nuanced understanding of the microbial factors that regulate skin immunity offers an opportunity to harness the power of the microbiome for therapeutic benefit.
Studies in germ-free mice have revealed that optimal immune cell function in healthy skin requires cues from indigenous microbes (Fig 2).33 For instance, the ability of effector T cells to make cytokines, such as IL-17A and IFN-γ, is dramatically abrogated in the absence of commensals. This defect is only restored with association of a skin commensal, S epidermidis, and not microbes residing in the intestine, highlighting the nonredundant role of skin-resident microbes in immune modulation.33,34 S epidermidis controls T-cell effector function by co-opting existing innate immune pathways, in this case IL-1α production from keratinocytes and dendritic cells. Although skin-derived innate signals are dispensable for the specification of T cells in the lymph node, these commensally induced molecules stimulate T cells on entry into the skin and sustain their effector functions. Importantly, this homeostatic tuning of skin T-cell function occurs in the absence of overt inflammation and in the context of an intact epidermal barrier.35
Several lines of evidence suggest that maintaining microbial diversity is advantageous to support the rich immune milieu of the skin. For instance, certain key microbes can elicit specific types of immune cells to the skin. Defined strains of S epidermidis induce IL-17A, producing CD8 (TC17) cells that reside in the epidermis.35 This cell population is actively generated on S epidermidis colonization through dendritic cell-dependent antigen presentation of bacterial N-formyl methionine peptides.36 In line with these experimental findings, human skin tropic T cells produce IL-17A and IFN-γ in response to stimulation with S epidermidis antigen.37 Moreover, TC17 cells are constitutively found in normal human skin33,38 and are enriched in squamous cell carcinomas39 and psoriatic plaques,40 suggesting that commensally induced cells can contribute to skin disease. Indeed, the demonstration of increased TH17-driven gene expression in the skin of healthy pediatric control subjects and the significantly greater Th17-related gene expression in the lesional and nonlesional skin of infants with recent-onset AD potentially reflect this early period of response to environmental commensals.41,42
Specificity of commensal interactions with the host immune system is not limited to cognate T-cell responses. Indeed, the first detailed molecular description of the ability of commensal skin bacteria to benefit skin immunity came with the identification of chemical moieties displayed on commensals that interact with innate immune receptors to drive certain responses. A TLR2 ligand, lipoteichoic acid, from a commensal strain of S epidermidis and not pathogenic bacteria is uniquely able to dampen skin inflammation.43 S epidermidis can also enhance innate immune defense by enhancing antimicrobial peptide expression.44 Several members of the commensal genus Corynebacterium have the cell envelope component mycolic acid, which can specifically induce IL-17A+ dermal γδ T cells (Fig 2).45 By contrast, CD4+ TH17 programs are broadly triggered by a wide array of microbes on skin colonization.35 Thus it is tempting to speculate that the cutaneous immune system has evolved to sense skin microbial complexity and to use this information as a rheostat to continuously calibrate its function.
The myriad of immune cells elicited by commensals have several contextual roles in reinforcing the epidermal barrier. Commensal-specific immune responses help provide heterologous protection against dermal pathogens. By augmenting epidermal antimicrobial function, commensal-specific T cells limit the ability of pathogens, such as Candida albicans, to establish infections.35 Additionally, effector T cells in the skin use commensal signals as natural adjuvants to amplify ongoing immune responses to pathogens.33,46 Skin T cells also play a crucial role in monitoring microbes in the steady state. Recombination-activating gene 1–deficient animals, which lack an adaptive immune system, have dramatic shifts in the composition of their skin microbial communities and bacterial translocation to skin lymph nodes.47 Patients with primary immunodeficiencies show similar alterations to commensal communities and a dramatic susceptibility to skin infections.48 Repair and regeneration are key features of inflammatory resolution. Commensal-specific TC17 cells aid in the healing process by promoting epidermal re-epithelialization.36 Thus one reason for maintaining such a rich milieu of microbes might be to maintain the complex repertoire of immune cell types and functions that aid not only in host defense but also in repair and regeneration of healthy skin.
The dynamic conversation between immune cells and commensals is disturbed when the skin barrier is compromised in patients with inflammatory diseases, such as AD, diabetic wounds, and skin infections.49,50 Such inflammatory reactions are often accompanied by a reduction in the complexity of microbial communities and outgrowth of pathogenic species, such as Staphylococcus aureus.51 If and how this reduction in microbial communities affects disease and whether pathogenic species expansion during inflammation causes disease or is merely a consequence is an area of active investigation. Untangling these observations and defining the microbial players that contribute to immune fitness will allow for the use of microbial interventions to manipulate immune responses and restore health.
S AUREUS AND AD
AD has a well-known association with altered skin microbiota, with a high prevalence of S aureus colonization and secondary infections that was recognized well before the application of DNA-sequencing approaches.52 Epidemiologic, metagenomic, and functional studies have since shown this complex host-pathogen relationship between S aureus and AD to be a sophisticated interaction between host and pathogen factors.53 Host factors include the hostile environment created by the physical, chemical, and antimicrobial properties of healthy skin, many of which are altered in AD skin. Pathogen-specific factors include highly evolved mechanisms facilitating adhesion, epidermal dissolution invasion, and proinflammatory mechanisms, which directly drive TH2, TH17, and innate immunity in the skin, promoting or exacerbating the inflammatory component of AD (Fig 3).53
FIG 3.
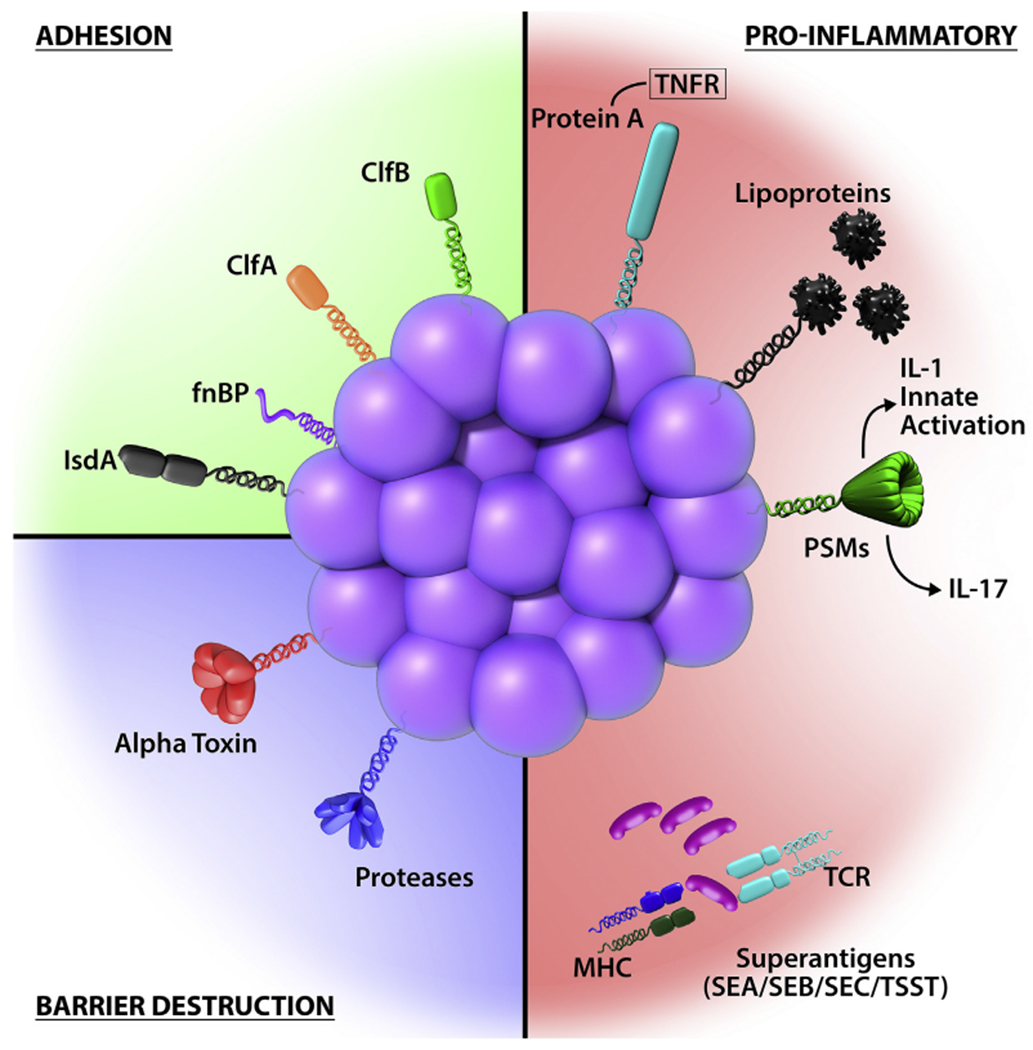
S aureus has highly evolved multiple cell-wall proteins and secreted factors that enable adhesion to human skin and barrier disturbance by using physical, chemical, and inflammatory mechanisms. Adhesion, S aureus has developed several surface molecules to adhere to the human stratum corneum, including clumping factors A and B (ClfA and ClfB), fibronectin-binding protein (fnBP), and iron-regulated surface determinant A (IsdA). Barrier destruction, S aureus α-toxin, a water-soluble cytotoxin, forms a heptameric β-barrel pore in host cell membranes. In the epidermis it directlyforms pores in keratinocytes, which erodes the integrity of the epidermal barrier. S aureus produces at least 10 proteases, a number of which facilitate dissolution of the stratum corneum. In addition to secreted proteases, S aureus can directly stimulate endogenous keratinocyte proteases, including KLK6, KLK13, and KLK14, highlighting an additional mechanism toward barrier destruction. Proinflammatory mechanisms, Cell-wall bound protein A, when solubilized, triggers inflammatory responses from keratinocytes through TNF receptor (TNFR). Staphylococcal superantigens, such as SEA, SEB, SEC, and toxic shock syndrome toxin-1 (TSST-1), trigger B-cell expansion and cytokine release. S aureus secretes PSMs, which are direct proinflammatory drivers with compartment-specific effects. In the epidermal compartment PSMs stimulate IL-36α-driven γδ T cell–mediated inflammation, whereasinthe dermal compartmentthey stimulate IL-1β-driven TH17 inflammation.
AD AND S AUREUS: EPIDEMIOLOGY
S aureus is commonly found on the skin of patients with AD,54–58 with reported rates of carriage varying from 30% to 100% depending on the type of patient, the sample size, and sampling and analysis methods, whereas in healthy control subjects the prevalence is about 20%. A recent meta-analysis of 95 observational studies of culture-based methods reported that the prevalence of S aureus carriage by patients with AD was 70% on lesional skin compared with 39% on nonlesional skin or healthy control skin within the same patient.58 The rate of S aureus colonization in this meta-analysis was related to disease severity.58 RT-PCR studies estimating the density of S aureus on both lesional and nonlesional skin have shown correlation with disease severity,57 confirming results from earlier culture-based studies.54,55
Analysis of the skin microbiota by using deep shotgun metagenomic sequencing and sequencing 16S rRNA genes has shown a reduction of microbial diversity during an AD flare (Fig 1). Microbiome diversity decreased in inflamed atopic skin, with reductions in the genera Streptococcus, Corynebacterium, and Cutibacterium and the phylum Proteobacteria toward the genus Staphylococcus in general and S aureus in particular.14,50,59–61 Shotgun metagenomic sequencing showed that patients with AD are usually colonized with a single strain of S aureus during a severe flare.59 Microbiome composition reverted to more normal diverse communities during treatment and recovery.50
S aureus strains isolated from patients with AD show differences to those isolated from unaffected carriers; clonal complex (CC1) strains are enriched among patients with AD, whereas the CC30 strains most frequently isolated from nasal carriers in the healthy population are less common in patients with AD.62–64 Although evidence that S aureus is directly causative of AD is lacking, there is abundant evidence that this bacterial species is highly influential in disease pathogenesis, is associated with severe disease flares, and significantly influences the disease phenotype. The mechanisms through which S aureus influences AD are beginning to be understood, as are the critical and dynamic relationships between S aureus and the microbiome, especially non–S aureus staphylococci. Here we briefly review mechanisms of adherence and colonization and mechanisms causing barrier destruction and S aureus–driven activation of inflammation.
MECHANISMS OF ADHERENCE AND COLONIZATION IN AD SKIN
S aureus has developed multiple mechanisms to gain purchase on the hostile and difficult environment presented by healthy and intact human skin (Fig 3). In contrast to healthy skin, AD skin is permissive for S aureus colonization. The antimicrobial peptides LL-37, β-defensins, and dermicidin are present at reduced levels in AD skin. One mechanism underlying this effect is the known inhibition of IL-4 and IL-13 on human β-defensin 2 and 3 gene expression.65 S aureus species grow poorly in acidic conditions, as seen in healthy stratum corneum, but grow much better in higher pH conditions, which are often seen in patients with AD.66 S aureus isolated from patients with AD binds more strongly to intact AD skin67 and in standard binding assays than S aureus isolated from unaffected carriers,62 an effect that is modulated by levels of filaggrin breakdown products (natural moisturizing factor) in human corneocytes.68 In patients with established AD, filaggrin deficiency, either genetic or acquired from TH2 skewing, leads to irregular or deformed corneocytes.69 S aureus isolates from patients with AD also bind more strongly to these corneocytes compared with isolates from unaffected control subjects in a clumping factor B-dependent fashion.62
DESTRUCTION OF THE EPIDERMAL BARRIER AND PROINFLAMMATORY S AUREUS MECHANISMS
In addition to having excellent adhesion and immune avoidance mechanisms, S aureus has well-developed resources to penetrate and disrupt the skin barrier (Fig 3). S aureus a-toxin is a cytotoxin secreted as a water-soluble monomer, which forms a heptameric β-barrel pore in host cell membranes.70,71 In the epidermis it directly forms pores in keratinocytes, which erodes the integrity of the epidermal barrier. α-Toxin is critical to formation of the S aureus biofilm,72 which makes elimination of S aureus much more difficult to achieve. S aureus produces at least 10 proteases, a number of which facilitate dissolution of and penetration through the stratum corneum.73 Importantly, the activity of these proteases is enhanced in the absence of filaggrin and in the presence of canonical TH2 cytokines.73 In addition to secreted proteases, S aureus can directly stimulate endogenous keratinocyte proteases, including kallikrein (KLK) 6, KLK13, and KLK14, highlighting an additional mechanism toward barrier destruction.30 S aureus α-toxin modulates the skin host to viral infection.74 These discoveries illustrate the complexity of interactions between host factors (TH2 immunity, barrier deficiency, and reduced antimicrobial peptides) and pathogen-driven mechanisms.
S aureus expresses several molecules that contribute to disease pathogenesis through proinflammatory mechanisms (Fig 3). These include α-toxin, a pore-forming toxin that directly causes cellular damage in keratinocytes with a resultant effect on skin barrier function and possible effects on susceptibility to viral infection.75 When solubilized, The cell wall–bound protein A triggers inflammatory responses from keratinocytes through the TNF receptor. Staphylococcal superantigens, such as staphylococcal enterotoxin (SE) A, SEB, SEC, and toxic shock syndrome toxin 1, trigger B-cell expansion and cytokine release.75
Proinflammatory staphylococcal lipoproteins induce thymic stromal lymphopoietin expression in primary human keratinocytes in a TLR2/TLR6-dependent manner, identifying another possible mechanism through which S aureus induces a Th2 response.76 Both mechanisms, barrier disruption and TH2 induction, make food allergy development more likely. S aureus incorporates short-chain unbranched fatty acids into its cytoplasmic membrane when growing in vivo.77 This increases membrane fluidity and could influence the expression of virulence factors78 and tolerance to host innate immunity, such as resistance to oxidative stress mediated by staphyloxanthin.78 Incorporation of skin fatty acids into bacterial lipoproteins increases their proinflammatory properties.79 S aureus secretes phenol-soluble modulins (PSMs), which are direct proinflammatory drivers with compartment-specific effects. In the epidermal compartment PSMa stimulates keratinocyte production of IL-36 and IL-36α-driven γδ T cell–mediated inflammation, whereas in the dermal compartment it stimulates IL-1β–driven80 and innate lymphoid cell (ILC)–driven81 TH17 inflammation. PSMγ (δ-toxin) also stimulates dermal mast cells and induces skin inflammation.82 S aureus mechanisms that contribute to AD pathogenesis have been comprehensively reviewed.53
CAN COMMENSAL ORGANISMS BE USED AS THERAPY FOR AD?
There are several lines of evidence suggesting that CoNS species could be therapeutically beneficial to AD. This approach is supported by birth cohort studies, which have shown that the presence of staphylococci, other than S aureus, at 2 months of age can protect infants against later development of AD.13,22
In addition, some CoNS species are able to fight against pathogens.83 Several previously unknown and potent anti–S aureus molecules have been discovered to be produced by skin CoNS species, such as S epidermidis, Staphylococcus hominis, and Staphylococcus lugdunensis.84–86 This anti–S aureus function of the commensal microbiome might be particularly useful in patients with AD because a high-throughput screen of CoNS species found that these antimicrobial strains are deficient on lesional and, to a lesser extent, nonlesional AD skin.85 Interestingly, these antimicrobials are only made by specific strains of CoNS species and were not initially predictable by using 16S sequencing. Also, because the antimicrobials produced by CoNS species synergize with the human antimicrobial peptide LL-37 and LL-37 is also relatively deficient in adult lesional AD skin,87 patients with chronic AD appear to have an exacerbated deficiency in innate antimicrobial defense against S aureus. Autoinducing peptide, which is produced by commensal organisms, can block S aureus accessory gene regulator–Quorum Sensing88 and therefore S aureus colonization and skin infection.89 S epidermidis has also been shown to suppress skin inflammation through TLR crosstalk.43 However, TLR ligands also exist in S aureus, so that the role of CoNS TLR ligands in reducing S aureus colonization is unclear.
In an attempt to address the deficiency of antimicrobial activity from the AD microbiome, a double-blind, placebo-controlled trial of CoNS species with antimicrobial activity was conducted. Results showed that topical application of antimicrobial CoNS bacteria was effective in mouse models of AD.85 CoNS species with anti–S aureus activity from patients with AD, which were far fewer than in healthy control subjects, were collected, expanded, applied to AD skin, and shown to reduce colonization by S aureus85; these studies are advancing to topical use of one lead strain of CoNS with anti–S aureus action (Fig 4). Furthermore, recent results from a similar 1-week trial have shown that this intervention can significantly improve local Eczema Area and Severity Index scores.90 Another recent open-label trial that evaluated Roseomonas mucosa found that topical application of this gram-negative organism decreased AD severity, pruritus, and the use of topical corticosteroids.91 This observation mirrors other reports of anti-inflammatory effects from environmental bacteria92 but lacks a known mechanism of action.
FIG 4.
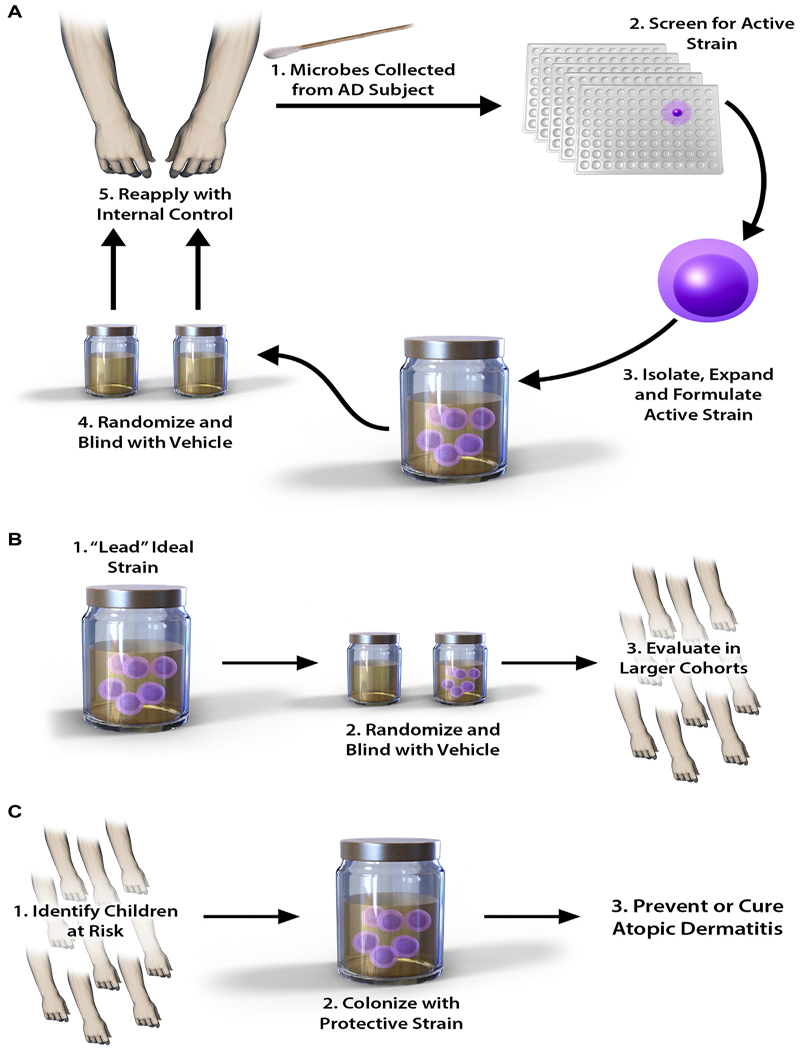
Potential role of biotherapy in patients with AD. Biotherapy for AD takes advantage of the natural antipathogen properties of human CoNS species. A, As a first step, microbes are grown out from swabs of an affected subject, with the most biologically active strains then selected for expansion. This generates the lead ideal strain, which is trialed back on the donor subject and compared with a randomized placebo vehicle. B, Ifsuccessful atthis stage,the lead strain istrialed in a largercohortof affected subjects. C, Finally, the strain is applied to high-risk children as a preventative measure.
Taken together, current preclinical and clinical trial results strongly support the use of commensal skin bacteria in the therapy of AD. However, these studies are still quite preliminary, and the effect on the cutaneous microbiome and safety, including long-term safety, of topical application of commensal organisms that target S aureus is unknown.
In summary, there is growing evidence of the key role of the microbiome in the pathogenesis of AD, from both the predominance of S aureus and the relative reduction of commensal organisms, which might play a role in controlling S aureus growth. However, many questions remain, including the following: (1) What is the effect of antiseptics or administration of systemic AD treatment on the skin microbiome? (2) How does short-term treatment with antibiotics affect the AD microbiome, and are certain antibiotics more deleterious? (3) How does therapy, including topical therapy, conventional nonspecific immunomodulators, and novel targeted therapy, such as dupilumab, affect the microbiome? (4) Are changes in the neonatal gut microbiome a risk factor for AD development? Successful treatment of AD in the future through microbiome manipulation with topically applied commensals has the potential not only to advance our understanding about AD pathogenesis but also to greatly expand the spectrum of topical treatment options for AD. Vaccines against S aureus also represent a possible innovative approach to manipulate the AD microbiome.93
Acknowledgments
The authors acknowledge the support of the International Eczema Council (IEC) in facilitating and sponsoring a symposium on the Microbiome in Atopic Dermatitis, San Diego, February 2018. This work reflects ideas and collaboration initiated in the IEC symposium.
Disclosure of potential conflict of interest: A. S. Palleris an investigator without personal compensation for AbbVie, Anaptysbio, Eli Lilly, Galderma, Incyte, Leo, Janssen, Novartis, and Sanofi-Regeneron and a consultant with honorarium for AbbVie, Amgen, Asana, Dermavant, Dermira, Galderma, Eli Lilly, Forte, Leo, Matrisys, Menlo, Morphosys/Galapagos, Novartis, Pfizer, and Sanofi-Regeneron. A. D. Irvine reports personal fees from AbbVie, Dermavant, Pfizer, Sanofi-Regeneron, AbbVie, and Eli Lilly and grants from Abbvie and Sanofi-Regeneron outside the submitted work. The rest of the authors declare that they have no relevant conflicts of interest.
Abbreviations used
- AD
Atopic dermatitis
- CoNS
Coagulase-negative Staphylococcus
- KLK
Kallikrein
- PSM
Phenol-soluble modulin
- SE
Staphylococcal enterotoxin
- TLR
Toll-like receptor
- Treg
Regulatory T
REFERENCES
- 1.Byrd AL, Belkaid Y, Segre JA. The human skin microbiome. Nat Rev Microbiol 2018;16:143–55. [DOI] [PubMed] [Google Scholar]
- 2.Findley K, Oh J, Yang J, Conlan S, Deming C, Meyer JA, et al. Topographic diversity of fungal and bacterial communities in human skin. Nature 2013;498: 367–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grice EA, Kong HH, Conlan S, Deming CB, Davis J, Young AC, et al. Topographical and temporal diversity of the human skin microbiome. Science 2009;324:1190–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Oh J, Byrd AL, Deming C, Conlan S, NISC program, Kong HH, et al. Biogeography and individuality shape function in the human skin metagenome. Nature 2014;514:59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Costello EK, Lauber CL, Hamady M, Fierer N, Gordon JI, Knight R. Bacterial community variation in human body habitats across space and time. Science 2009;326:1694–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kong HH, Segre JA. The molecular revolution in cutaneous biology: investigating the skin microbiome. J Invest Dermatol 2017;137:e119–22. [DOI] [PubMed] [Google Scholar]
- 7.Oh J, Byrd AL, Park M, NISC Comparative Sequencing Program, Kong HH, Segre JA. Temporal stability of the human skin microbiome. Cell 2016;165: 854–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Oh J, Conlan S, Polley EC, Segre JA, Kong HH. Shifts in human skin and nares microbiota of healthy children and adults. Genome Med 2012;4:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jo JH, Deming C, Kennedy EA, Conlan S, Polley EC, Ng WL, et al. Diverse human skin fungal communities in children converge in adulthood. J Invest Dermatol 2016;136:2356–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Capone KA, Dowd SE, Stamatas GN, Nikolovski J. Diversity of the human skin microbiome early in life. J Invest Dermatol 2011;131:2026–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Costello EK, Carlisle EM, Bik EM, Morowitz MJ, Relman DA. Microbiome assembly across multiple body sites in low-birthweight infants. MBio 2013;4, e00782–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dominguez-Bello MG, Costello EK, Contreras M, Magris M, Hidalgo G, Fierer N, et al. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc Natl Acad Sci U S A 2010;107:11971–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kennedy EA, Connolly J, Hourihane JO, Fallon PG, McLean WH, Murray D, et al. Skin microbiome before development of atopic dermatitis: early colonization with commensal staphylococci at 2 months is associated with a lower risk of atopic dermatitis at 1 year. J Allergy Clin Immunol 2017;139: 166–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shi B, Bangayan NJ, Curd E, Taylor PA, Gallo RL, Leung DYM, et al. The skin microbiome is different in pediatric versus adult atopic dermatitis. J Allergy Clin Immunol 2016;138:1233–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chu DM, Ma J, Prince AL, Antony KM, Seferovic MD, Aagaard KM. Maturation of the infant microbiome community structure and function across multiple body sites and in relation to mode of delivery. Nat Med 2017;23:314–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kollmann TR, Levy O, Montgomery RR, Goriely S. Innate immune function by Toll-like receptors: distinct responses in newborns and the elderly. Immunity 2012;37:771–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McGovern N, Shin A, Low G, Low D, Duan K, Yao LJ, et al. Human fetal dendritic cells promote prenatal T-cell immune suppression through arginase-2. Nature 2017;546:662–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang S, Fujikado N, Kolodin D, Benoist C, Mathis D. Immune tolerance. Regulatory T cells generated early in life play a distinct role in maintaining self-tolerance. Science 2015;348:589–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thome JJC, Bickham KL, Ohmura Y, Kubota M, Matsuoka N, Gordon C, et al. Early-life compartmentalization of human T cell differentiation and regulatory function in mucosal and lymphoid tissues. Nat Med 2015;22:72–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gensollen T, Iyer SS, Kasper DL, Blumberg RS. How colonization by microbiota in early life shapes the immune system. Science 2016;352:539–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fujimura KE, Sitarik AR, Havstad S, Lin DL, Levan S, Fadrosh D, et al. Neonatal gut microbiota associates with childhood multisensitized atopy and T cell differentiation. Nat Med 2016;22:1187–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meylan P, Lang C, Mermoud S, Johannsen A, Norrenberg S, Hohl D, et al. Skin colonization by Staphylococcus aureus precedes the clinical diagnosis of atopic dermatitis in infancy. J Invest Dermatol 2017;137:2497–504. [DOI] [PubMed] [Google Scholar]
- 23.Scharschmidt TC, Vasquez KS, Truong H- A, Gearty SV, Pauli ML, Nosbaum A, et al. A wave of regulatory t cells into neonatal skin mediates tolerance to commensal microbes. Immunity 2015;43:1011–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Scharschmidt TC, Vasquez KS, Pauli ML, Leitner EG, Chu K, Truong H- A, et al. Commensal microbes and hair follicle morphogenesis coordinately drive Treg migration into neonatal skin. Cell Host Microbe 2017;21:467–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cordoro KM, Hitraya-Low M, Taravati K, Sandoval PM, Kim E, Sugarman J, et al. Skin-infiltrating, interleukin-22-producing T cells differentiate pediatric psoriasis from adult psoriasis. J Am Acad Dermatol 2017;77:417–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weidinger S, Beck LA, Bieber T, Kabashima K, Irvine AD. Atopic dermatitis. Nat Rev Dis Primers 2018;4:1. [DOI] [PubMed] [Google Scholar]
- 27.Gallo RL. Human skin is the largest epithelial surface for interaction with microbes. J Invest Dermatol 2017;137:1213–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang LJ, Gallo RL. Antimicrobial peptides. Curr Biol 2016;26:R14–9. [DOI] [PubMed] [Google Scholar]
- 29.Nakatsuji T, Chiang HI, Jiang SB, Nagarajan H, Zengler K, Gallo RL. The microbiome extends to subepidermal compartments of normal skin. Nat Commun 2013;4:1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Williams MR, Nakatsuji T, Gallo RL. Staphylococcus aureus: master manipulator of the skin. Cell Host Microbe 2017;22:579–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Belkaid Y, Segre JA. Dialogue between skin microbiota and immunity. Science 2014;346:954–9. [DOI] [PubMed] [Google Scholar]
- 32.Grice EA. The skin microbiome: potential for novel diagnostic and therapeutic approaches to cutaneous disease. Semin Cutan Med Surg 2014;33:98–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Naik S, Bouladoux N, Wilhelm C, Molloy MJ, Salcedo R, Kastenmuller W, Deming C, et al. Compartmentalized control of skin immunity by resident commensals. Science 2012;337:1115–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Belkaid Y, Naik S. Compartmentalized and systemic control of tissue immunity by commensals. Nat Immunol 2013;14:646–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Naik S, Bouladoux N, Linehan JL1, Han SJ, Harrison OJ, Wilhelm C. Commensal-dendritic-cell interaction specifies a unique protective skin immune signature. Nature 2015;520:104–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Linehan JL, Harrison OJ, Han SJ, Byrd AL, Vujkovic-Cvijin I, Villarino AV, et al. Non-classical immunity controls microbiota impact on skin immunity and tissue repair. Cell 2018;172:784–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schlapbach C, Gehad A, Yang C, Watanabe R, Guenova E, Teague JE, et al. Human TH9 cells are skin-tropic and have autocrine and paracrine proinflammatory capacity. Sci Transl Med 2014;6:219ra8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Clark RA. Resident memory T cells in human health and disease. Sci Transl Med 2015;7:269rv1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roberts SJ, Ng BY, Filler RB, Lewis J, Glusac EJ, Hayday AC, et al. Characterizing tumor-promoting T cells in chemically induced cutaneous carcinogenesis. Proc Natl Acad Sci U S A 2007;104:6770–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cheuk S, Wikén M, Blomqvist L, Nylen S, Talme T, Stahle M, et al. Epidermal Th22 and Tc17 cells form a localized disease memory in clinically healed psoriasis. J Immunol 2014;192:3111–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brunner PM, Israel A, Zhang N, Leonard A, Wen HC, Huynh T, et al. Early-onset pediatric atopic dermatitis is characterized by TH2/TH17/TH22-centered inflammation and lipid alterations. J Allergy Clin Immunol 2018;141:2094–106. [DOI] [PubMed] [Google Scholar]
- 42.Esaki H, Brunner PM, Renert-Yuval Y, Czarnowicki T, Huynh T, Tran G, et al. Early-onset pediatric atopic dermatitis is TH2 but also TH17 polarized in skin. J Allergy Clin Immunol 2016;138:1639–51. [DOI] [PubMed] [Google Scholar]
- 43.Lai Y, Di Nardo A, Nakatsuji T, Leichtle A, Yang Y, Cogen AL, Wu ZR, et al. Commensal bacteria regulate Toll-like receptor 3-dependent inflammation after skin injury. Nat Med 2009;5:1377–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lai Y, Cogen AL, Radek KA, Park HJ, Macleod DT, Leichtle A, et al. Activation of TLR2 by a small molecule produced by Staphylococcus epidermidis increases antimicrobial defense against bacterial skin infections. J Invest Dermatol 2010; 130:2211–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ridaura VK, Bouladoux N, Claesen J, Chen YE, Byrd AL, Constantinides MG, et al. Contextual control of skin immunity and inflammation by Corynebacterium. J Exp Med 2018;215:785–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.de Oliveira MR, Tafuri WL, Nicoli JR, Vieira EC, Melo MN, Vieira LQ. Influence of microbiota in experimental cutaneous leishmaniasis in Swiss mice. Rev Inst Med Trop Sao Paulo 1999;41:87–94. [DOI] [PubMed] [Google Scholar]
- 47.Shen W, Li W, Hixon JA, Bouladoux N, Belkaid Y, Dzutzev A, et al. Adaptive immunity to murine skin commensals. Proc Natl Acad Sci U S A 2014;111: E2977–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Oh J, Freeman AF, NISC Comparative Sequencing Program, Park M, Sokolic R, Candotti F, et al. The altered landscape of the human skin microbiome in patients with primary immunodeficiencies. Genome Res 2013;23:2103–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gimblet C, Meisel JS, Loesche MA, Cole SD, Horwinski J, Novais FO, et al. Cutaneous Leishmaniasis induces a transmissible dysbiotic skin microbiota that promotes skin inflammation. Cell Host Microbe 2017;22:13–24.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kong HH, Oh J, Deming C, Conlan S, Grice EA, Beatson MA, et al. Temporal shifts in the skin microbiome associated with disease flares and treatment in children with atopic dermatitis. Genome Res 2012;22:850–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Grice EA, Snitkin ES, Yockey LJ, Bermudez DM. NISC Comparative Sequencing Program, Liechty KW, et al. Longitudinal shift in diabetic wound microbiota correlates with prolonged skin defense response. Proc Natl Acad Sci U S A 2010;107:14799–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Leyden JJ, Marples RR, Kligman AM. Staphylococcus aureus in the lesions of atopic dermatitis. Br J Dermatol 1974;90:525–30. [DOI] [PubMed] [Google Scholar]
- 53.Geoghegan JA, Irvine AD, Foster TJ. Staphylococcus aureus and atopic dermatitis: a complex and evolving relationship. Trends Microbiol 2018;26: 484–97. [DOI] [PubMed] [Google Scholar]
- 54.Higaki S, Morohashi M, Yamagishi T, Hasegawa Y. Comparative study of staphylococci from the skin of atopic dermatitis patients and from healthy subjects. Int J Dermatol 1999;38:265–9. [DOI] [PubMed] [Google Scholar]
- 55.Guzik TJ, Bzowska M, Kasprowicz A, Czerniawska-Mysik G, Wojcik K, Szmyd D, et al. Persistent skin colonization with Staphylococcus aureus in atopic dermatitis: relationship to clinical and immunological parameters. Clin Exp Allergy 2005;35:448–55. [DOI] [PubMed] [Google Scholar]
- 56.Park HY, Kim CR, Huh IS, Jung MY, Seo EY, Park JH, et al. Staphylococcus aureus colonization in acute and chronic skin lesions of patients with atopic dermatitis. Ann Dermatol 2013;25:410–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tauber M, Balica S, Hsu CY, Jean-Decoster C, Lauze C, Redoules D, et al. Staphylococcus aureus density on lesional and nonlesional skin is strongly associated with disease severity in atopic dermatitis. J Allergy Clin Immunol 2016;137:1272–4. [DOI] [PubMed] [Google Scholar]
- 58.Totte JE, van der Feltz WT, Hennekam M, van Belkum A, van Zuuren EJ, Pasmans SG. Prevalence and odds of Staphylococcus aureus carriage in atopic dermatitis: a systematic review and meta-analysis. Br J Dermatol 2016;175:687–95. [DOI] [PubMed] [Google Scholar]
- 59.Byrd AL, Deming C, Cassidy SKB, Harrison OJ, Ng WI, Conlan S, et al. Staphylococcus aureus and Staphylococcus epidermidis strain diversity underlying pediatric atopic dermatitis. Sci Transl Med 2017;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gonzalez ME, Schaffer JV, Orlow SJ, Gao Z, Li H, Alekseyenko AV, et al. Cutaneous microbiome effects of fluticasone propionate cream and adjunctive bleach baths in childhood atopic dermatitis. J Am Acad Dermatol 2016;75: 481–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Seite S, Flores GE, Henley JB, Martin R, Zelenkova H, Aguilar L, et al. Microbiome of affected and unaffected skin of patients with atopic dermatitis before and after emollient treatment. J Drugs Dermatol 2014;13:1365–72. [PubMed] [Google Scholar]
- 62.Fleury OM, McAleer MA, Feuillie C, Formosa-Dague C, Sansevere E, Bennett DE. Clumping factor B promotes adherence of Staphylococcus aureus to corneocytes in atopic dermatitis. Infect Immun 2017;85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Simpson EL, Villarreal M, Jepson B, Rafaels N, David G, Hanifin J, et al. Patients with atopic dermatitis colonized with Staphylococcus aureus have a distinct phenotype and endotype. J Invest Dermatol 2018;138:2224–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shi B, Leung DYM, Taylor PA, Li H. Methicillin-resistant Staphylococcus aureus colonization is associated with decreased skin commensal bacteria in atopic dermatitis. J Invest Dermatol 2018;138:1668–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hata TR, Kotol P, Boguniewicz M, Taylor P, Paik A, Jackson M, et al. History of eczema herpeticum is associated with the inability to induce human β-defensin (HBD)-2, HBD-3 and cathelicidin in the skin of patients with atopic dermatitis. Br J Dermatol 2010;163:659–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Miajlovic H, Fallon PG, Irvine AD, Foster TJ. Effect of filaggrin breakdown products on growth of and protein expression by Staphylococcus aureus. J Allergy Clin Immunol 2010;126:1184–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cho SH, Strickland I, Boguniewicz M, Leung DY. Fibronectin and fibrinogen contribute to the enhanced binding of Staphylococcus aureus to atopic skin. J Allergy Clin Immunol 2001;108:269–74. [DOI] [PubMed] [Google Scholar]
- 68.Feuillie C, Vitry P, McAleer MA, Kezic S, Irvine AD, Geoghegan JA, et al. Adhesion of Staphylococcus aureus to corneocytes from atopic dermatitis patients is controlled by natural moisturizing factor levels. mBio 2018;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Riethmuller C, McAleer MA, Koppes SA, Abdayem R, Franz J, Haftek M, et al. Filaggrin breakdown products determine corneocyte conformation in patients with atopic dermatitis. J Allergy Clin Immunol 2015;136:1573–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Song L, Hobaugh MR, Shustak C, Cheley S, Bayley H, Gouaux JE. Structure of staphylococcal alpha-hemolysin, a heptameric transmembrane pore. Science 1996;274:1859–66. [DOI] [PubMed] [Google Scholar]
- 71.Berube BJ, Bubeck Wardenburg J. Staphylococcus aureus alpha-toxin: nearly a century of intrigue. Toxins (Basel) 2013;5:1140–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Anderson MJ, Lin YC, Gillman AN, Parks PJ, Schlievert PM, Peterson ML. Alpha-toxin promotes Staphylococcus aureus mucosal biofilm formation. Front Cell Infect Microbiol 2012;2:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nakatsuji T, Chen TH, Two AM, Chun KA, Narala S, Geha RS, et al. Staphylococcus aureus exploits epidermal barrier defects in atopic dermatitis to trigger cytokine expression. J Invest Dermatol 2016;136:2192–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bin L, Kim BE, Brauweiler A, Goleva E, Streib J, Ji Y. Staphylococcus aureus a-toxin modulates skin host response to viral infection. J Allergy Clin Immunol 2012;130:683–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Spaulding AR, Salgado-Paboen W, Kohler PL, Horswill AR, Leung DY, Schlievert PM. Staphylococcal and streptococcal superantigen exotoxins. Clin Microbiol Rev 2013;26:422–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Vu AT, Baba T, Chen X, Le TA, Kinoshita H, Xie Y, et al. Staphylococcus aureus membrane and diacylated lipopeptide induce thymic stromal lymphopoietin in keratinocytes through the Toll-like receptor 2-Toll-like receptor 6 pathway. J Allergy Clin Immunol 2010;126:985–93. [DOI] [PubMed] [Google Scholar]
- 77.Sen S, Sirobhushanam S, Johnson SR, Song Y, Tefft R, Gatto C. Growth-environment dependent modulation of Staphylococcus aureus branched-chain to straight-chain fatty acid ratio and incorporation of unsaturated fatty acids. PLoS One 2016;11:e0165300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sun Y, Wilkinson BJ, Standiford TJ, Akinbi HT, O’Riordan MX. Fatty acids regulate stress resistance and virulence factor production for Listeria monocytogenes. J Bacteriol 2012;194:5274–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nguyen MT, Hanzelmann D, Hartner T, Peschel A, Gotz F. Skin-specific unsaturated fatty acids boost the Staphylococcus aureus innate immune response. Infect Immun 2015;84:205–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Liu H, Archer NK, Dillen CA, Wang Y, Ashbaugh AG, Ortines RV, et al. Staphylococcus aureus epicutaneous exposure drives skin inflammation via IL-36-mediated T cell responses. Cell Host Microbe 2017;22:653–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nakagawa S, Matsumoto M, Katayama Y, Oguma R, Wakabayashi S, Nygaard T, et al. Staphylococcus aureus virulent PSMa peptides induce keratinocyte alarmin release to orchestrate IL-17-dependent skin inflammation. Cell Host Microbe 2017;22:667–77.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nakamura Y, Oscherwitz J, Cease KB, Chan SM, Munoz-Planillo R, Hasegawa M, et al. Staphylococcus δ-toxin induces allergic skin disease by activating mast cells. Nature 2013;503:397–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cogen AL, Nizet V, Gallo RL. Skin microbiota: a source of disease or defence? Br J Dermatol 2008;158:442–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zipperer A, Konnerth MC, Laux C, Berscheid A, Janek D, Weidenmaier C, et al. Human commensals producing a novel antibiotic impair pathogen colonization. Nature 2016;535:511–6. [DOI] [PubMed] [Google Scholar]
- 85.Nakatsuji T, Chen TH, Narala S, Chun KA, Two AM, Yun T, et al. Antimicrobials from human skin commensal bacteria protect against Staphylococcus aureus and are deficient in atopic dermatitis. Sci Transl Med 2017;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Nakatsuji T, Chen TH, Butcher AM, Trzoss LL, Nam SJ, Shirakawa KT, et al. A commensal strain of Staphylococcus epidermidis protects against skin neoplasia. Sci Adv 2018;4:eaao4502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ong PY, Ohtake T, Brandt C, Strickland I, Boguniewicz M, Ganz T, et al. Endogenous antimicrobial peptides and skin infections in atopic dermatitis. N Engl J Med 2002;347:1151–60. [DOI] [PubMed] [Google Scholar]
- 88.Alexandra E, Paharik AE, Parlet CP, Chung N, Todd DA, Rodriguez EI, Van Dyke MJ, et al. Coagulase-negative staphylococcal strain prevents Staphylococcus aureus colonization and skin infection by blocking quorum sensing. Cell Host Microbe 2017;22:746–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ji G, Beavis R, Novick RP. Bacterial interference caused by autoinducing peptide variants. Science 1997;276:2027–30. [DOI] [PubMed] [Google Scholar]
- 90.Nakatsuji T, Tong Y, Butcher A, Hayashi A, Chun K, Shafiq F, et al. Clinical improvement in atopic dermatitis following autologous application of microbiome therapy targeting Staphylococcus aureus. J Invest Dermatol 2018; 138:S72. [Google Scholar]
- 91.Myles IA, Earland NJ, Anderson ED, Moore IN, Kieh MD, Williams KW, et al. First-in-human topical microbiome transplantation with Roseomonas mucosa for atopic dermatitis. JCI Insight 2018;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gueniche A, Philippe D, Bastien P, Reuteler G, Blum S, Castiel-Higounenc I, et al. Randomised double-blind placebo-controlled study of the effect of Lactobacillus paracasei NCC 2461 on skin reactivity. Benef Microbes 2014;5: 137–45. [DOI] [PubMed] [Google Scholar]
- 93.Clowry J, Irvine AD, McLoughlin R. Next generation anti-S. aureus vaccines: a potential new therapeutic option for atopic dermatitis? J Allergy Clin Immunol 2019;143:78–81. [DOI] [PubMed] [Google Scholar]