

Abstract
Peptide inhibitors against the SARS-CoV-2 coronavirus, currently causing a worldwide pandemic, are designed and simulated. The inhibitors are mostly formed by two sequential self-supporting α-helices (bundle) extracted from the protease domain (PD) of angiotensin-converting enzyme 2 (ACE2), which bind to the SARS-CoV-2 receptor binding domains. Molecular dynamics simulations revealed that the α-helical peptides maintain their secondary structure and provide a highly specific and stable binding (blocking) to SARS-CoV-2. To provide a multivalent binding to the SARS-CoV-2 receptors, many such peptides could be attached to the surfaces of nanoparticle carriers. The proposed peptide inhibitors could provide simple and efficient therapeutics against the COVID-19 disease.
Keywords: SARS-CoV-2, COVID-19, peptide inhibitors, molecular dynamics simulations, therapeutics
Graphical Abstract

Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), previously known as 2019 novel coronavirus (2019-nCoV),1 is causing a pandemic of coronavirus disease.2,3 SARS-CoV-2 shares about 80% of its genome identity with SARS-CoV, which emerged in 2002–2003.4 SARS-CoV-2 is highly contagious in humans, which has rapidly caused an unprecedented pandemic, with a large number of fatalities worldwide.
The SARS-CoV-2 virion, 50–200 nm in diameter, contains four structural proteins, known as the S (spike), E (envelope), M (membrane), and N (nucleocapsid) proteins.2 The S protein, imaged at the atomic level using cryo-electron microscopy,5 is responsible for the host attachment and fusion of the viral and host-cell membranes.6,7 This process is triggered when the S1 subunit of S protein binds to a host-cell receptor. To engage a host-cell receptor, the receptor-binding domain (RBD) of S1 undergoes transient hinge-like conformational motions (receptor-accessible or receptor-inaccessible states).8 The angiotensin-converting enzyme 2 (ACE2) is the host cellular receptor with a higher affinity to SARS-CoV-2 than to SARS-CoV.5 In the recognition of RBD, the protease domain (PD) of ACE2 mainly engages the α1-helix with a minor contribution from the α2-helix and the linker of the β3- and β4-sheets.8,9
In addition to a hectic search for vaccines against COVID-19, there is a very fast ongoing search for therapeutics acting on SARS-CoV-2. Depending on the activity, the therapies can be divided into several main categories: (1) preventing the viral RNA synthesis and replication, (2) blocking the virus from binding to human cell receptors, (3) restoring the host’s innate immunity, and (4) blocking the host’s specific receptors or enzymes.10 Despite many experimental and computational studies currently exploring all of these categories, to date, there is no confirmed effective treatment specifically available for COVID-19.
Computational approaches have been used to search potential therapeutics against SARS-CoV-2 protease (category 1).11 Analogous screening of potential drugs against the S protein of SARS-CoV-2 (category 2) provided small molecular compounds with a high binding affinity. Unfortunately, most of these compounds do not attach with the binding interface of the RBD–ACE2 complex. Hesperidin was predicted to lie on the surface of RBD, but it did not cover the whole interface.10 In the early attempts of SARS-CoV blocking, short peptide inhibitors were studied and amino acid mutations were implemented to the S protein of SARS-CoV.12,13 However, the proposed peptide was too short (8 residues) to maintain secondary structure, so it was unable to block the whole SARS-CoV binding surface.12 Broad-spectrum antiviral nanoparticles and cyclodextrins were designed, simulated, and implemented in blocking of other viruses.14–16 They are category 2 or 3 inhibitors, but their applicability to SARS-CoV-2 is unknown. Proteins or rigid peptides with specific (multivalent) binding domains and conformations matching RBD could be promising therapeutics for COVID-19. Overall, protein therapies show a high specificity, small interference with biological processes, good tolerance to human organisms, and faster FDA approval times.17
In this work, we design and simulate several peptide inhibitors against SARS-CoV-2, which included components from the virus-binding domains of ACE2; based on the recently released crystal structure (PDB code: 6M179). The inhibitors, which have relatively low molecular weights, are structurally stable, they conformationally match the S protein, and are highly specific to SARS-CoV-2. This study could provide a potential guidance in antigen recognition and structure-based designs of antibodies with high affinities. The proposed small peptides could be used as inhaled therapeutics for topical lung delivery, providing an efficient way to combat COVID-19.18
RESULTS AND DISCUSSION
Preparation of Inhibitors.
In the crystal structure of ACE2 and RBD of SARS-CoV-2 (PDB: 6M179), we first analyzed the interacting amino acids at the ACE2 and RBD interface. In total, 15 residues from ACE2 interact with RBD: residues 24(Q), 27(T), 30(D), 31(K), 34(H), 35(E), 37(E), 38(D), 41(Y), and 42(Q) are in α1, one residue (residue 82 M) comes from α2, residues 353(K), 354(G), 355(D), and 357(R) come from the linker between β3 and β4. Therefore, the 15 amino acids can be labeled as critical amino acids and α1, α2, β3, and β4 as critical binding components.
Because most of the interacting residues are from α1, we picked as inhibitor 1 the α1-helix alone. In particular, the 21–55 residues, shown in Figure 1a, were selected. Realizing that α1 (alone) might not even be stable, we next picked as inhibitor 2 both α1- and α2-helices (residues 21 to 88) and the residues 349 to 357 (residues between β3 and β4 shown in orange in Figure 1b). This selection included all 15 interacting residues from the crystal structure 6M17.9 As the two α-helices are closely joined on one side (Figure 1b), they stabilize each other. To connect the two helices (red) with the β-sheets with residues 349 to 357 (orange), as shown in Figure 1b, residues 45 (LEU) and 351 (LEU) were linked together by a side chain with a carbon–carbon bond, as shown in Figure 2b.
Figure 1.
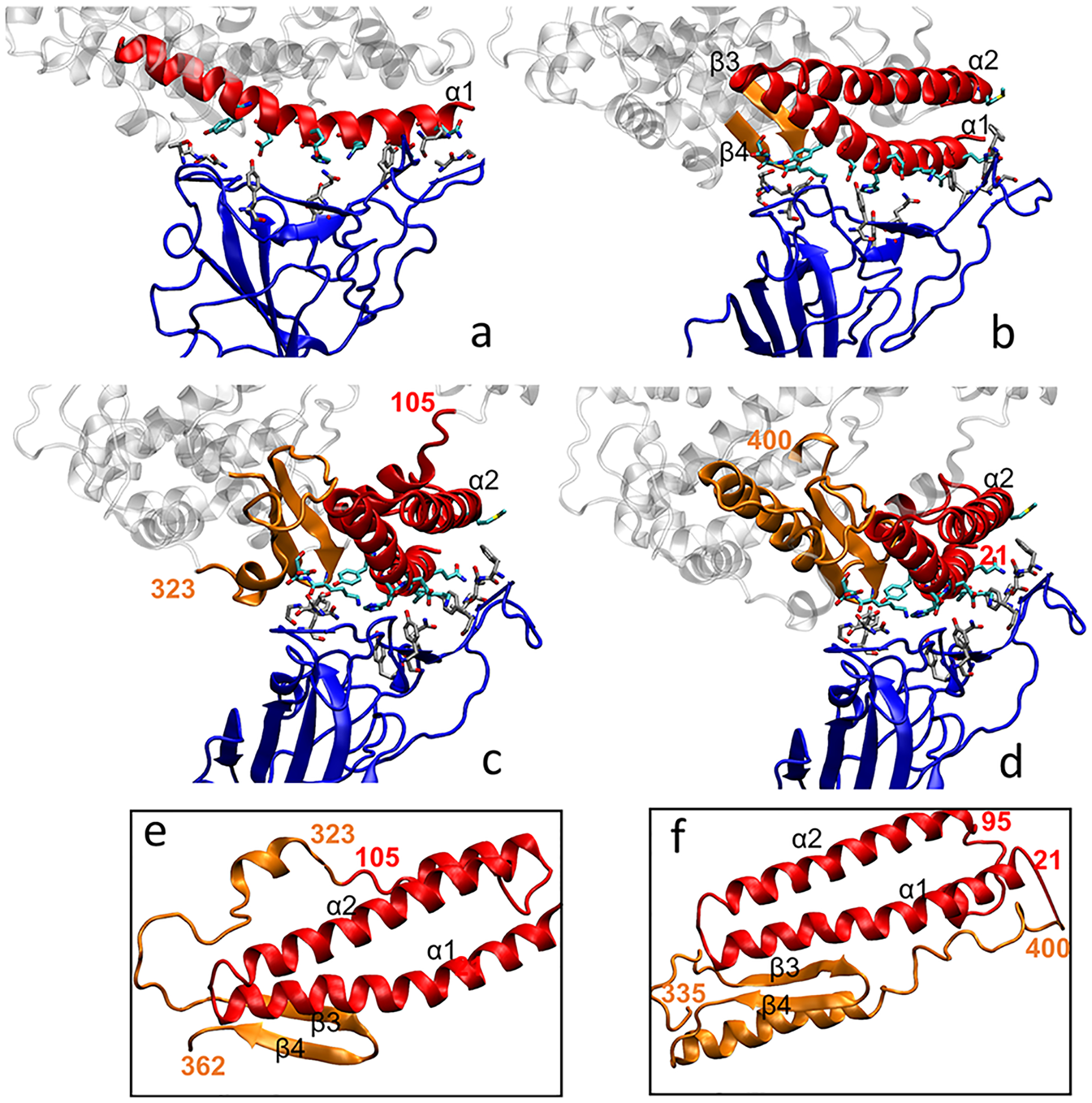
Structural components of the inhibitors designed: (a) inhibitor 1 is composed of α1 (residues 21 to 55); (b) inhibitor 2 is composed of α1, α2, and loose chain between β3 and β4 connected by a C–C bond between residues 45 and 351 (residues 21 to 88 and 349 to 357); (c) inhibitor 3 is composed of α1, α2, and β3, β4 (residues 21 to 105 and 323 to 362); (d) inhibitor 4 has the same composition as inhibitor 3 but different linkage (residues 21 to 95 and 335 to 400); (e) details of inhibitor 3 (c), reorganized with residue 323 connecting residue 105; (f) details of inhibitor 3 (d), reorganized with residue 21 connecting residue 400. (e-f) Conformation of the α-helices and β-sheets was maintained with the rest adapting to the connection. Coloring scheme: red, α-helices; orange, β-sheets or other linker components; blue, RBD of SARS-CoV-2; gray, other parts of ACE2; licorice, the initial contacting residues in the RBD–ACE2 interface.
Figure 2.
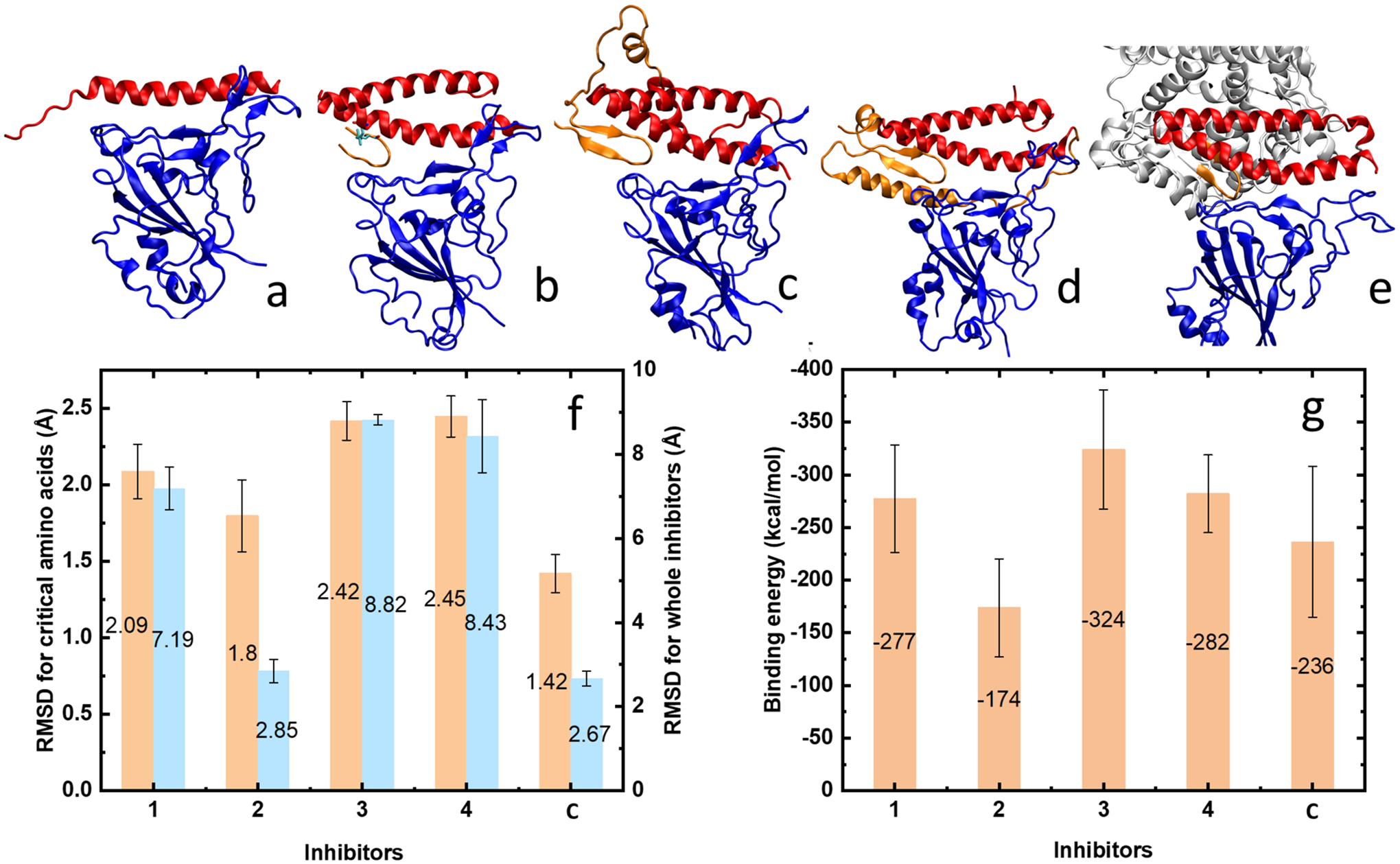
(a–e) Final conformations of inhibitors 1, 2, 3, 4, and control. (f) Averaged root-mean-square deviation for the critical amino acids in each inhibitor and for the whole inhibitors when binding with the RBD of SARS-CoV-2. Numbering scale: 1–4, inhibitors 1–4 with RBD; C, control system of PD from ACE2 and the RBD of SARS-CoV-2. (g) Average interaction energies between the contact residues of inhibitors 1–4 (or ACE2) and the RBD of SARS-CoV-2.
We have also designed other inhibitors that are closer to the ACE2 protein, whose parts are connected by peptide bonds, and which contain all 15 residues that initially bind to RBD in the 6M17 crystal structure.9 Figure 1c (detail in Figure 1e) shows inhibitor 3, where residues 323 to 362 (orange) include the two β-sheets and a random coil (residues 323 to 348), whereas residues 21 to 105 (red) include the two α-helices with another random coil (residues 89 to 105). The two sequences are joined together by a peptide bond between residues 105 and 323, and the two pieces of random coils were moved close to each other. Finally, Figure 1d (detail in Figure 1f) shows inhibitor 4, where two sequences including residues 21 to 95 (red) and residues 335 to 500 (orange) were selected. An extra peptide bond was made between residue 21 and residue 400 by adjusting the position of the corresponding sequences. The sequences of all inhibitors are shown in Table S1.
To examine how these potential inhibitors bind to RBD of SARS-CoV-2, we prepared these systems in the initial position known from the crystal structure (PDB: 6M17) and simulated them in physiological solution (Methods), as shown in Figure 2a–d. As a control, the PD of ACE2 (residues 19 to 615) and RBD of SARS-CoV-2 were also simulated (Figure 2e).
Binding Conformations.
In Figure 2a, 200 ns long simulations showed that the helical structure of inhibitor 1 deforms from the left side—loose end unfolding, although it still binds to the RBD of SARS-CoV-2. In Figure 2b–d, 120–300 ns long simulations revealed that inhibitors 2–4 bind in a stable way to the RBD of SARS-CoV-2, without α1 losing its structure. Due to different linkages among the critical binding components, the overall conformations of inhibitors 2–4 vary. Specifically, the α1-helix, which mostly contributes to the complementary sequence and conformational matching to RBD, is maintained in inhibitors 2–4 with different degrees of bending. The β-sheets in the structures of inhibitors 3 and 4 are also preserved. Overall, the critical binding components in inhibitors 2–4 bind to RBD in a manner very similar to that of the crystal structure. The simulated stable conformation of inhibitors 2, 3, and 4 correspond to their energy minima of folding, which would drive the folding process toward the stable direction.
Root-Mean-Square Deviation (RMSD) and Interaction Energies.
To further quantify the binding of these inhibitors to RBD, we calculated the RMSD for the 15 critical amino acids in each inhibitor and for the whole inhibitors. Figure 2f shows the average RMSD at the end of our simulations (see also Figure S1). Inhibitor 1 has larger RMSD for the critical amino acids compared to that of the control and the largest fluctuations for both the critical amino acids and the overall RMSD (Figure S1a,b). This can be attributed to unfolding of α1, shown in Figure 2a. A highly promising inhibitor 2 has a RMSD of the critical amino acids and the overall RMSD similar to those in the control (lowest). Inhibitor 3 has a RMSD of the critical amino acids and the overall RMSD higher than that of the control and inhibitors 1 and 2. However, Figure S1b shows that inhibitor 3 has a very smooth overall RMSD at later times. This may be due to a poor adaptation of their added connections at early times. Inhibitor 4 shows slightly bigger fluctuation for the overall RMSD but steady RMSD (Figure S1a) for the critical amino acids at later times, which indicates fluctuation shown in the overall structure comes from nonessential connection parts.
The interaction energies have van der Waals (vdW) and electrostatic components, calculated by the NAMD energy plugin. The total energies are shown in Figure 2 g and Figure S2 (detail). The residues which contribute to the interaction energies between inhibitors and SARS-CoV-2 are selected with a cutoff of 3 Å. The selections are updated in every frame. Inhibitors 1 and 4 show interaction energies similar to those of the control, with inhibitor 3 having slightly stronger binding than the control; however, inhibitor 2 shows an interaction energy slightly lower than that of the control. The larger interaction energy in inhibitor 1 might be due to nonspecific interactions caused by the deformed helix. The lower interaction energy in inhibitor 2 could be attributed to the total number of residues, which is less than those of inhibitor 3 and 4.
CONCLUSIONS
In summary, using classical molecular dynamics simulations, we have shown that peptide inhibitors extracted from ACE2 provide highly promising trails for SARS-CoV-2 blocking. The single α1-helix used in inhibitor 1 is less stable, whereas the α1,2-helices used in inhibitors 2–4 support each other and retain their bent shape, which provides a conformational matching to the RBD of SARS-CoV-2 and a full cover of the RBD surface. Precise conformational matching between the designed peptides and the virus provides room for improving the binding affinity, which should be considered in future inhibitor design protocols. Suitable inhibitors should have a selective binding with lower RMSD for critical amino acids and relatively high binding energies. The binding affinity could be further enhanced by a multivalent binding of multiple peptides attached to surfaces of nanoparticles, dendrimers, and clusters. In analogy to nanoparticle-based inhibitors,14 we could attach to the α1 helix a sulphonated ligand mimicking a heparane sulfate, which can attach to positively charged residues at the bottom of RBD. These inhibitors could be used as inhaled therapeutics, preventing the virus activation in lungs.
METHODS
The inhibitors and RBD of the virus were simulated by NAMD19 and the CHARMM36 protein force field.20 The particle mesh Ewald (PME) method was used for the evaluation of long-range Coulombic interactions.21 The time step was set to 2 fs. The simulations were performed in the NpT ensemble (p = 1 bar and T = 310 K), using the Langevin dynamics with a damping constant of 1 ps−1. After 2000 steps of minimization, ions and water molecules were equilibrated for 2 ns around proteins, which were restrained using harmonic forces with a spring constant of 2 kcal/(mol Å2). The last frames of restrained equilibration were used to start simulations of free inhibitors and partially constrained PD of ACE2 (two residues on the bottom). The simulations last for 120–300 ns due to different atom numbers in different systems and different computer power used.
Calculation of RMSD.
The time-dependent RMSD for the critical amino acids and the whole inhibitors (Figure S1) were calculated from
| (1) |
where Nα is the number of atoms whose positions are being compared, is the position of atom α at time tj, and is the initial coordinate. The selection of coordinates contains all of the atoms in the inhibitors or critical amino acids, excluding hydrogens.
The time-dependent RMSD was averaged over the last 50 ns of simulation time, which corresponds to the last 50 frames of each trajectory, as shown in Figure 2f. The standard deviations are shown by the error bars.
Calculation of Binding Energy.
The interacting residues from inhibitors and RBD of SARS-CoV-2 were first selected with a 3 Å cutoff distance. The electrostatic and vdW energy contributions between the interacting residues are calculated by the NAMD energy plugin. The electrostatic contribution is given by
| (2) |
where is the distance between the two charges, qi and qj; ε is the dielectric constant of the solvent which is set to 1. To increase the efficiency of the simulations, pairwise interaction calculations are not performed beyond a cutoff distance. Long-range electrostatic interactions are calculated by the PME method.21
The Lennard-Jones (LJ) 6–12 potential energies are used to describe the vdW interactions and close distance atomic repulsions:
| (3) |
where εij is the maximum stabilization energy for the ith and the jth atoms σij is the distance between ith and jth atoms at the minimum of the potential, and rij is the actual distance between the two atoms. The LJ parameters between different atom types are calculated using a mixing rule, such as and (Lorentz–Berthelot rules).
The time evolution of the interaction energy is shown in Figure S2, and the time-averaged interaction energy over the last 50 ns (50 frames) is shown in Figure 2g, with standard deviation shown by the error bar.
Supplementary Material
ACKNOWLEDGMENTS
We would like to thank Lela Vuković (UTEP) for useful discussions. Y.H. acknowledges the support from the Dean’s Scholar Fellowship (UIC).
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsnano.0c02857.
Sequences of inhibitors, RMSD for the critical amino acids in each inhibitor and for the whole inhibitors, and interaction energies between the contact residues of inhibitors (or ACE2) and SARSCoV-2 (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acsnano.0c02857
The authors declare no competing financial interest.
Contributor Information
Yanxiao Han, Department of Chemistry, University of Illinois at Chicago, Chicago, Illinois 60607, United States.
Petr Král, Department of Chemistry and Department of Physics, Biopharmaceutical Sciences, and Chemical Engineering, University of Illinois at Chicago, Chicago, Illinois 60607, United States.
REFERENCES
- (1).Gorbalenya AE; Baker SC; Baric RS; de Groot RJ; Drosten C; Gulyaeva AA The Species Severe Acute Respiratory Syndrome-Related Coronavirus: Classifying 2019-nCoV and Naming It SARS-CoV-2. Nat. Microbiol 2020, 5, 536–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Chen N; Zhou M; Dong X; Qu J; Gong F; Han Y; Qiu Y; Wang J; Liu Y; Wei Y; Xia J; Yu T; Zhang X; Zhang L Epidemiological and Clinical Characteristics of 99 Cases of 2019 Novel Coronavirus Pneumonia in Wuhan, China: A Descriptive Study. Lancet 2020, 395, 507–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Huang C; Wang Y; Li X; Ren L; Zhao J; Hu Y; Zhang L; Fan G; Xu J; Gu X; Cheng Z; Yu T; Xia J; Wei Y; Wu W; Xie X; Yin W; Li H; Liu M; Xiao Y; Gao H; Guo L; Xie J; Wang G; Jiang R; Gao Z; Jin Q; Wang J; Cao B Clinical Features of Patients Infected with 2019 Novel Coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Zhou P; Yang X-L; Wang X-G; Hu B; Zhang L; Zhang W; Si H-R; Zhu Y; Li B; Huang C-L; Chen H-D; Chen J; Luo Y; Guo H; Jiang R-D; Liu M-Q; Chen Y; Shen X-R; Wang X; Zheng X-S; Zhao K; Chen Q-J; Deng F; Liu L-L; Yan B; Zhan F-X; Wang Y-Y; Xiao G-F; Shi Z-L A Pneumonia Outbreak Associated with a New Coronavirus of Probable Bat Origin. Nature 2020, 579, 270–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Wrapp D; Wang N; Corbett KS; Goldsmith JA; Hsieh C-L; Abiona O; Graham BS; McLellan JS Cryo-EM Structure of the 2019-nCoV Spike in the Prefusion Conformation. Science 2020, 367, 1260–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Li F Structure, Function, and Evolution of Coronavirus Spike Proteins. Annu. Rev. Virol 2016, 3, 237–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Bosch BJ; van der Zee R; de Haan CAM; Rottier PJM The Coronavirus Spike Protein Is a Class I Virus Fusion Protein: Structural and Functional Characterization of the Fusion Core Complex. J. Virol 2003, 77, 8801–8811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Wan Y; Shang J; Graham R; Baric RS; Li F Receptor Recognition by the Novel Coronavirus from Wuhan: An Analysis Based on Decade-Long Structural Studies of SARS Coronavirus. J. Virol 2020, 94, e00127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Yan R; Zhang Y; Li Y; Xia L; Guo Y; Zhou Q Structural Basis for the Recognition of the SARS-CoV-2 by Full-Length Human ACE2. Science 2020, 367, 1444–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Wu C; Liu Y; Yang Y; Zhang P; Zhong W; Wang Y; Wang Q; Xu Y; Li M; Li X; Zheng M; Chen L; Li H Analysis of Therapeutic Targets for SARS-CoV-2 and Discovery of Potential Drugs by Computational Methods. Acta Pharm. Sin. B 2020, DOI: 10.1016/j.apsb.2020.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Wang J Fast Identification of Possible Drug Treatment of Coronavirus Disease-19 (COVID-19) Through Computational Drug Repurposing Study. ChemRxix, 10.26434/chemrxiv.11875446.v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Du Q; Wang S; Wei D; Sirois S; Chou K-C Molecular Modeling and Chemical Modification for Finding Peptide Inhibitor Against Severe Acute Respiratory Syndrome Coronavirus Main Proteinase. Anal. Biochem 2005, 337, 262–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Zhang Y; Zheng N; Nan P; Cao Y; Hasegawa M; Zhong Y Computational Simulation of Interactions between SARS Coronavirus Spike Mutants and Host Species-Specific Receptors. Comput. Biol. Chem 2007, 31, 134–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Cagno V; Andreozzi P; D’Alicarnasso M; Jacob Silva P; Mueller M; Galloux M; Le Goffic R; Jones ST; Vallino M; Hodek J; Weber J; Sen S; Janecek E-R; Bekdemir A; Sanavio B; Martinelli C; Donalisio M; Rameix Welti M-A; Eleouet J-F; Han Y; Kaiser L; Vukovic L; Tapparel C; Kral P; Krol S; Lembo D; Stellacci F Broad-Spectrum Non-Toxic Antiviral Nanoparticles with a Virucidal Inhibition Mechanism. Nat. Mater 2018, 17, 195–203. [DOI] [PubMed] [Google Scholar]
- (15).Jones ST; Cagno V; Janecek M; Ortiz D; Gasilova N; Piret J; Gasbarri M; Constant DA; Han Y; Vukovic L; Kral P; Kaiser L; Huang S; Constant S; Kirkegaard K; Boivin G; Stellacci F; Tapparel C Modified Cyclodextrins As Broad-Spectrum Antivirals. Sci. Adv 2020, 6, No. eaax9318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Sen S; Han Y; Rehak P; Vukovič L; Král P Computational Studies of Micellar and Nanoparticle Nanomedicines. Chem. Soc. Rev 2018, 47, 3849–3860. [DOI] [PubMed] [Google Scholar]
- (17).Leader B; Baca Q; Golan D Protein Therapeutics: A Summary and Pharmacological Classification. Nat. Rev. Drug Discovery 2008, 7, 21–39. [DOI] [PubMed] [Google Scholar]
- (18).Bodier-Montagutelli E; Mayor A; Vecellio L; Respaud R; Heuzé-Vourc’h N Designing Inhaled Protein Therapeutics for Topical Lung Delivery: What Are the Next Steps? Expert Opin. Drug Delivery 2018, 15, 729–736. [DOI] [PubMed] [Google Scholar]
- (19).Phillips JC; Braun R; Wang W; Gumbart J; Tajkhorshid E; Villa E; Chipot C; Skeel RD; Kalé L; Schulten K Scalable Molecular Dynamics with NAMD. J. Comput. Chem 2005, 26, 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).MacKerell AD; Bashford D; Bellott M; Dunbrack RL; Evanseck JD; Field MJ; Fischer S; Gao J; Guo H; Ha S; Joseph-McCarthy D; Kuchnir L; Kuczera K; Lau FTK; Mattos C; Michnick S; Ngo T; Nguyen DT; Prodhom B; Reiher WE; Roux B; Schlenkrich M; Smith JC; Stote R; Straub J; Watanabe M; Wiorkiewicz-Kuczera J; Yin D; Karplus M All-Atom Empirical Potential for Molecular Modeling and Dynamcis Studies of Proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [DOI] [PubMed] [Google Scholar]
- (21).Darden T; York D; Pedersen L Particle Mesh Ewald: An N.log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys 1993, 98, 10089–10092. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.