

Abstract
Altered protease activity is considered important for tumour invasion and metastasis, processes in which the cysteine proteases cathepsin B and L are involved. Their natural inhibitor cystatin C is a secreted protein, suggesting that it functions to control extracellular protease activity. Because cystatins added to cell cultures can inhibit polio, herpes simplex and coronavirus replication, which are intracellular processes, the internalization and intracellular regulation of cysteine proteases by cystatin C should be considered. The extension, mechanism and biological importance of this hypothetical process are unknown. We investigated whether internalization of cystatin C occurs in a set of human cell lines. Demonstrated by flow cytometry and confocal microscopy, A‐431, MCF‐7, MDA‐MB‐453, MDA‐MB‐468 and Capan‐1 cells internalized fluorophore‐conjugated cystatin C when exposed to physiological concentrations (1 μm). During cystatin C incubation, intracellular cystatin C increased after 5 min and accumulated for at least 6 h, reaching four to six times the baseline level. Western blotting showed that the internalized inhibitor was not degraded. It was functionally intact and extracts of cells exposed to cystatin C showed a higher capacity to inhibit papain and cathepsin B than control cells (decrease in enzyme activity of 34% and 37%, respectively). The uptake of labelled cystatin C was inhibited by unlabelled inhibitor, suggesting a specific pathway for the internalization. We conclude that the cysteine protease inhibitor cystatin C is internalized in significant quantities in various cancer cell lines. This is a potentially important physiological phenomenon not previously described for this group of inhibitors.
Keywords: cancer, cysteine proteases, internalization, protease inhibitors, uptake
Abbreviations
- CLSM
confocal laser scanning microscopy
- DOL
degree of protein labelling
- PCI
potato carboxypeptidase inhibitor
Altered protease activity is thought to be important in tumour cell invasion and metastasis, and to have a profound role in angiogenesis. Implicated proteases belong to the serine, metallo‐, aspartic and cysteine protease classes. The latter comprises more than 30 protein families [1], including family C1 with mammalian enzymes like cathepsins B and L involved in cancer growth and metastasis [2]. Since the involvement of cathepsin B in cancer metastasis was originally described by Sloane et al. [3], cathepsins, and especially cathepsin B, have been studied thoroughly. The activity of the C1 family of cysteine proteases is balanced by tight‐binding inhibitors, the cystatins [4]. The cystatin protein family comprises three major groups of inhibitors: type 1 cystatins, also called stefins, which are intracellular proteins present in most cells (cystatin A and B); type 2 cystatins, which are extracellular inhibitors found in most body fluids (cystatin C, D, E/M, F, G, H, S, SA and SN); and type 3, which are multidomain proteins, the kininogens. Among the cystatins, cystatin C is the quantitatively most important and the best inhibitor of cathepsin B.
Various approaches have been used in order to understand the interplay between proteases and their inhibitors in the neoplastic state [5, 6, 7], how this interplay is regulated and its relevance. Extracellular activation of cathepsin B has been suggested in cancer [8] and several authors have reported altered cystatin levels in tumour tissue. However, the results are conflicting, depending on the type of cystatin and the cancer cell system studied. Overexpression of cystatin C has been shown to alter the metastatic properties of B16F10 melanoma cells [9] and to inhibit the motility and in vitro invasiveness of B16F10 [10] and SCC‐VII squamous carcinoma cells [11]. In vitro cystatin E/M has been found to diminish human breast carcinoma cell proliferation, migration, Matrigel invasion and adhesion to endothelial cells [12]. Cystatin E/M has been proposed as a candidate tumour suppressor gene for breast cancer [13]. Furthermore, control of breast tumour cell growth has been achieved by using a targeted synthetic cysteine protease inhibitor [14].
The potential cellular internalization of cystatins might be considered in various contexts [15], for example, to explain the results of experiments showing that coronavirus, herpes simplex virus and poliovirus replication were inhibited by different cystatins [16, 17, 18, 19]. A reasonable explanation for the inhibition of virus replication by the cysteine protease inhibitors is the inhibition of proteases involved in processing proteins coded by the virus genome, which is an intracellular process. However, the extension of the capacity for cellular uptake of cystatins, the mechanism by which uptake takes place and the biological importance of this hypothetical process are unknown. Because of the proposed role of cysteine proteases in the growth and spread of cancer cells, it is crucial that the interplay between cysteine proteases and their inhibitors in neoplasias is clarified. The aim of this study was: (a) to elucidate whether internalization of cystatin C occurs in a range of cancer cell lines; and (b) if uptake could be proven, to describe the general nature of this potentially important physiological phenomenon.
Results
Flow cytometry
Based on indications that the cell internalization of cystatins could be a physiological pathway and thus might be important in processes such as the inhibition of virus replication and tumour growth, we addressed the question of whether there is cystatin C uptake in human cells. We initially chose cell lines with different characteristics such as a human epidermoid carcinoma cell line (A‐431) and a human mammary tumour cell line (MCF‐7). In order to allow us to delineate any potential uptake mechanism we also selected two mammary cancer cell lines (MDA‐MB‐453 and MDA‐MB‐468) which, according to the American Type Culture Collection (ATCC), express different cell surface receptors. Finally, we added another type of cell from a human pancreas adenocarcinoma (Capan‐1).
Initial experiments were carried out by the addition to cell cultures of various concentrations of fluorophore‐labelled cystatin C (data not shown). In this study 1 μm cystatin C was used, as this is within the physiological concentration range in different human body fluids (0.1–4 μm) [20]. Following incubation with cystatin C, cells were detached from the bottom of the wells by trypsin, which also meant that labelled protein attached to the cell surface was cleaved and could be washed away. Flow cytometry using the fluorophore Alexa‐488 as the protein label was used in these experiments. The resulting scattergram showed a dominating, easily defined group of cells that could be gated (Fig. 1A). Typically < 15% of the events were excluded. The reproducibility of the experiments was high and internalization of the labelled protein could be demonstrated easily (Fig. 1B). All five human cancer cell lines internalized Alexa‐488‐labelled cystatin C (Fig. 2A–C). Incubation for 10 s was used to ensure that the washing conditions were sufficient, and demonstrated that cystatin bound at the cell surface was cleaved by trypsin and washed away. After 5 min an increase in some of the cell lines could be detected and after 30 min the cell fluorescence had increased in all five strains. The pattern of internalization was more or less equal in the five strains. Two unrelated proteins acting as protein inhibitors, potato carboxypeptidase inhibitor (PCI; 4.3 kDa) and equistatin (22.3 kDa), were studied for comparison. Compared with cystatin C, a very low level of uptake of these molecules was detected in the different cell lines. To investigate whether the internalization of cystatin C was an active process, a similar experiment was carried out at 4 °C. None of the three cell lines tested showed any uptake of labelled cystatin C under these conditions (Fig. 2C–E).
Figure 1.
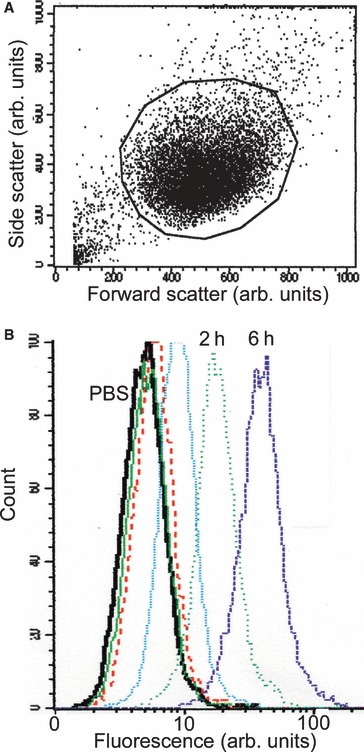
Flow cytometry to measure internalized fluorophore‐conjugated cystatin C. (A) Scattergram from a FACS Calibur flow cytometer. Subconfluent A‐431 cells were incubated for 6 h in medium containing NaCl/Pi (control). Cells were then trypsinized and analysed. At least 3000 events were measured. This experiment shows the typical distribution of cells in all internalization experiments in which this methodology was used. The y‐axis depicts side scattering and the x‐axis depicts forward scattering. (B) Distribution of cells in relation to cell fluorescence. Subconfluent A‐431 cells were incubated in medium containing fluorescence‐labelled cystatin C and analysed after being trypsinized. The y‐axis depicts cell count and the x‐axis depicts the amount of cell fluorescence (488 nm). The curves represent the result of the analysis of each cell population incubated with NaCl/Pi (black line), cystatin C for 10 s (green line), 5 min (red dotted line), 30 min (light blue dotted line), 2 h (green dotted line) and 6 h (dark blue dotted line), respectively.
Figure 2.
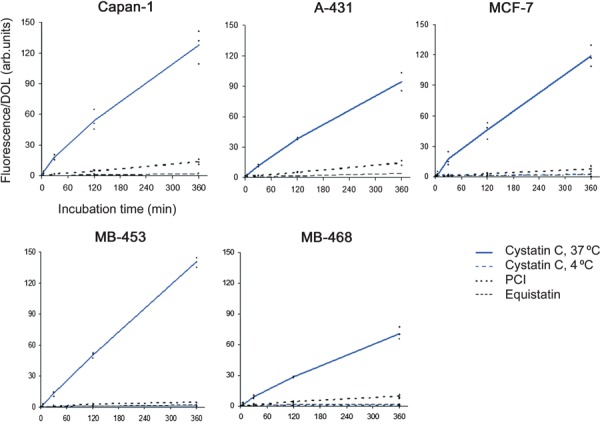
Internalization of cystatin C in cancer cell lines measured by flow cytometry. Five human cancer cell lines were used: MCF‐7, MDA‐MB‐453, MDA‐MB‐468, A‐431 and Capan‐1. Subconfluent cells were incubated in medium containing fluorescence‐labelled protein, either cystatin C, PCI or equistatin. NaCl/Pi was used as control. Cells were incubated for 10 s, 5 min, 30 min, 2 h or 6 h, respectively. Cell fluorescence was measured and median cell fluorescence was calculated, corrected for the control value and then related to the degree of labelling (DOL) of the protein used. The MCF‐7, MDA‐MB‐453 and MDA‐MB‐468 cell lines were in addition incubated with labelled cystatin C at 4 °C as described above. Each of the diagrams shows the results of three independent experiments (A‐431 experiments were carried out twice). The lines are drawn through the average value of the three results at each time point.
Microscope analyses
Microscope analyses were used to qualitatively visualize and thereby confirm cystatin C internalization in situ. A‐431 cells were selected initially because they showed negligible auto‐fluorescence when incubated for 6 h with 5 μm Alexa‐488‐labelled cystatin C. In principle, all cells possessed detectable cystatin C labelling. Confocal laser scanning microscopy (CLSM) analyses further demonstrated that cystatin C was present within the cells, in both cell bodies and processes, but was not detected in the plasma membrane. A large number of A‐431 and MCF‐7 cells (Figs 3A and S1) showed widespread low‐signal cystatin C fluorescence in the cytoplasm, although fewer also contained larger fluorescence accumulations (in ∼ 10% of the cells). Similar intracellular localization of cystatin C was detected when cells were exposed to unlabelled cystatin C followed by immunolabelling of the cystatin C (endogenous and internalized), using a primary antibody against cystatin C and a secondary antibody conjugated with Alexa‐568 fluorophore (Fig. S2). There was no fluorescence labelling in control cells or in cells used in antibody specificity tests (see Experimental procedures for a description of the control experiments). Live imaging experiments clearly showed uptake within 5 min and the Alexa‐488‐labelled cystatin C co‐localized with lysosome‐like structures stained by LysoTracker (Fig. 3B–D).
Figure 3.
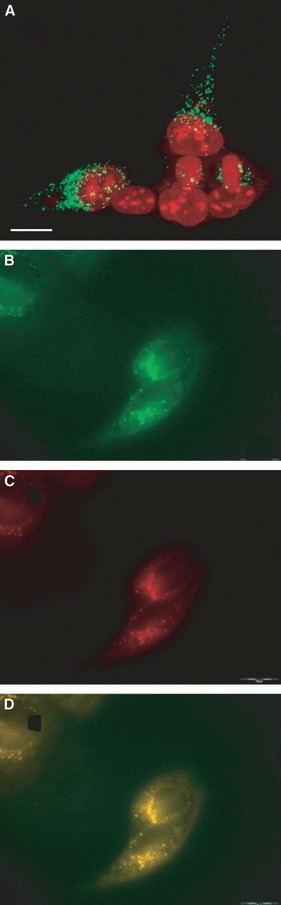
Microscopic examination of A‐431 cells incubated with labelled cystatin C. (A) The image shows confocal laser scanning microscopy of A‐431 cells, incubated for 6 h with Alexa‐488‐conjugated cystatin C (green) and with nuclei stained by propidium iodide (red). In the cells, Alexa‐488 labelling comprised high quantities of relatively large accumulations of fluorescence, distributed in different parts of the cell. Scale bar = 10 μm. (B–D) Live imaging of A‐431 cells incubated 15 min with cystatin C‐Alexa‐488 followed by LysoTracker incubation. (B) Visualization of the Alexa‐488 label. (C) Visualization of acidic compartments by LysoTracker. (D) Overlay of B and C, indicating co‐localization of cystatin C and LysoTracker.
Quantification of cystatin C
Cystatin C was quantified under normal cell culture conditions to obtain reference levels for its production and distribution in the cells studied, when grown under the conditions used in the internalization experiments. Capan‐1 cells were incubated for 6 or 24 h and secreted cystatin C in the medium and in cell extracts representing intracellular cystatin C were quantified by ELISA. The intracellular cystatin C level did not change from 6 to 24 h (Fig. 4) implying a steady‐state level, of ∼ 20 ng cystatin C·mg cell protein−1, within the cells. By contrast, the cystatin C concentration in the medium increased from ∼ 15 to ∼ 45 ng cystatin C·mg cell protein−1 (Fig. 4), as expected for a protein secreted as a result of the cellular production of cystatin C.
Figure 4.
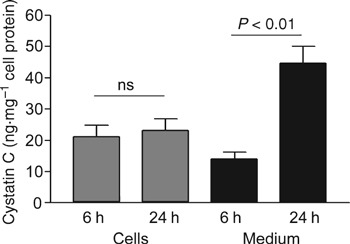
Cellular and secreted cystatin C in Capan‐1 cells. The presence of secreted endogenous cystatin C in the medium as well as the content of endogenous cystatin C in the cell extract was quantified by ELISA. The cystatin C level of the cell lysate and medium were correlated to the protein concentration of the corresponding cell lysate. Results are expressed as mean ± SD (all groups n = 6). Statistical analysis was carried out using Mann–Whitney U‐test.
Cellular levels of cystatin C after incubation of the cells in medium containing 1 μm cystatin C in a time‐scale manner, for up to 6 h, were then measured. The concentration chosen is within the physiological range, between that in cerebrospinal fluid (0.5 μm) and seminal plasma (3.7 μm) [20]. These experiments clearly showed that the cystatin C content of the cells increased rapidly during the first 5 min and then continued to accumulate for at least 6 h, which was the final time‐point of these experiments (Fig. 5A). Repeated experiments showed that after 6 h the cystatin C level had increased to four to six times baseline steady‐state levels, as shown by ELISA. A plateau noted for the increase in cystatin C at longer incubation times is likely due to increased competition with cystatin C‐binding proteins (i.e. target cysteine proteases) when the extract concentrations of cystatin C approach the equilibrium constants for enzyme binding [4], affecting cystatin C‐directed antibodies used in the assay and leading to an underestimation of the real intracellular cystatin C concentration.
Figure 5.
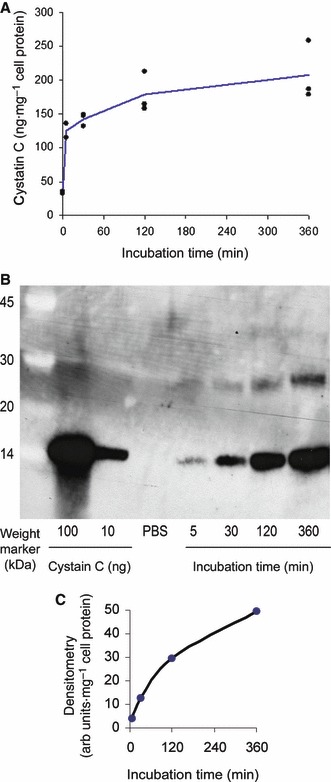
ELISA and western blotting of internalized cystatin C. (A) Capan‐1 cells were incubated with 1 μm recombinant human cystatin C for up to 6 h. The cystatin C content of the cell extract (representing intracellular cystatin C) was quantified by ELISA and the cystatin C level was correlated to the protein content of the cell lysate. The lines are drawn through the average value of the three wells at each time point (at 5 min only two wells were measured). The result presented is representative for two identical experiments. (B) Capan‐1 cells incubated for 5, 30 min, 2 or 6 h with 1 μm recombinant human cystatin C. NaCl/Pi was used as a control. Cystatin C was concentrated by immunoprecipitation, separated in a 4–12% SDS/PAGE gel and finally blotted to a membrane. The blotted proteins were immunodetected using a polyclonal rabbit‐anti‐(human cystatin C) serum. As a secondary antibody a horseradish‐peroxidase conjugated goat anti‐(rabbit IgG) fraction was used. Blotted proteins were visualized by chemoluminiscence. Lanes: molecular mass marker, 100 and 10 ng cystatin C, cells incubated with NaCl/Pi (equivalent to endogenous cystatin C), cells incubated with cystatin C 5, 30, 120 and 360 min, respectively. The ∼ 28 kDa immunorective band seen in addition to the main 14 kDa cystatin C band represents dimeric cystatin C, which may form in the intracellular milieu or as a result of slight denaturation when samples are prepared for SDS/PAGE [15, 22]. (C) Result from densitometric scanning of the western blot bands.
Western blotting
Western blotting was used to ensure that the fluorescence seen in the confocal experiments, and that the cystatin C molecules measured by ELISA after various incubation times, represented intact cystatin C molecules. The results clearly showed an increase in cellular cystatin C content and that the molecules were intact, as judged by maintenance of the same molecular mass (Fig. 5B). No degradation products were noted. In order to estimate cystatin C uptake, the western blot was scanned and bands representing cystatin C were semi‐quantified by densitometry (Fig. 5C). The results were in good agreement with those from ELISA experiments at shorter incubation times, but were clearly higher at longer incubation times, supporting the conclusion that the quantitative ELISA results are underestimates and that the increase in cystatin C continues throughout the 6 h incubation. This is also in good agreement with flow cytometry results (Fig. 2).
Assessment of papain and cathepsin B inhibition capacity
Evaluation of the balance between the cysteine proteases and their inhibitors was carried out by measuring the cysteine protease inhibitory capacity in extracts from cells that had been incubated in cystatin C‐containing medium (1 μm). In order to determine the concentration of active papain used in the assay, a fixed amount of enzyme was incubated with various concentrations of E‐64. A titration curve was drawn and the amount of active papain calculated. The baseline cysteine protease inhibitory capacity of the Capan‐1 cell lysate was then approximated by analysing various volumes of an extract from cells incubated for 24 h with NaCl/Pi, i.e. cells that had not been exposed to cystatin C. Before the experiment, the extract was boiled to denature all proteases and abolish their activity, a procedure that does not affect cystatins, which can withstand high temperatures without losing their inhibitory activity. It showed a concentration of ∼ 200 pmol cysteine protease inhibitor·mg protein−1. To elucidate whether the total cysteine protease inhibitory capacity changed after exposing cells to cystatin C, cells were incubated for 24 h with 1 μm cystatin C. The cysteine protease inhibitor concentration in these cell extracts was estimated to be 250 pmol·mg protein−1 by papain titration, indicating a substantial increase caused by the uptake of cystatin C, affecting the total cysteine protease inhibitory capacity (which should be mainly due to cytoplasmic cystatin B) within the cells [4].
To quantify and statistically test the increased intracellular cysteine protease inhibitory activity due to cystatin C internalization, the experiment was repeated at an optimal lysate volume (according to the titration curve above). The inhibitory capacity of cystatin C‐exposed cells and non‐exposed cells was compared. The results showed significantly higher inhibitory activity in cells exposed to cystatin C as compared with control cells when both papain and cathepsin B were analysed (enzyme activity decreased by 34% and 37%, respectively) (Fig. 6). In this context, papain was used because of its resemblance to cathepsin L, a protease involved in the propagation of cancer [2].
Figure 6.
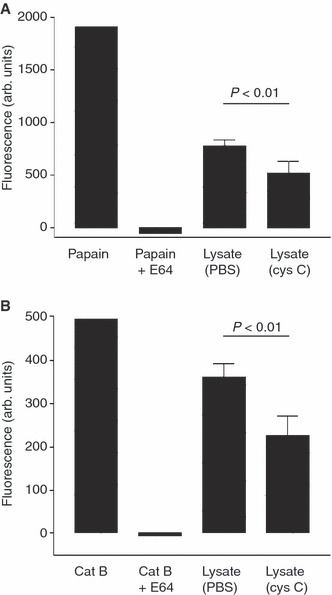
Papain and cathepsin B inhibition assay. Capan‐1 cells were cultured and incubated for 24 h with or without the addition of 1 μm cystatin C to the medium. After lysate preparation, centrifugation and heat denaturation of the endogenous cysteine proteinases, the cysteine protease inhibitory capacity of the cell lysate was determined by measuring the inhibition of (A) papain and (B) cathepsin B. As positive and negative controls, instead of cell lysate, Brij or E‐64 was used (n = 1). Z‐Phe‐Arg‐AMC was used as the substrate and the fluorescence was measured after 30 min incubation. Each experiment consisted of three wells for each condition. An average of the result from two samples from each well was calculated. The same experiment was then repeated another day. Results are expressed as mean ± SD (n = 6). Statistical analysis was carried out using Mann–Whitney U‐test.
Influences of preincubation by non‐labelled inhibitor
In order to address the uptake mechanism, Capan‐1 cells were incubated with labelled cystatin C after preincubation with various concentrations of unlabelled inhibitor. Uptake of 1 μm Alexa‐488‐labelled cystatin C decreased substantially as the concentration of the unlabelled inhibitor increased (Fig. 7A), indicating an active and specific pathway for cystatin C internalization.
Figure 7.
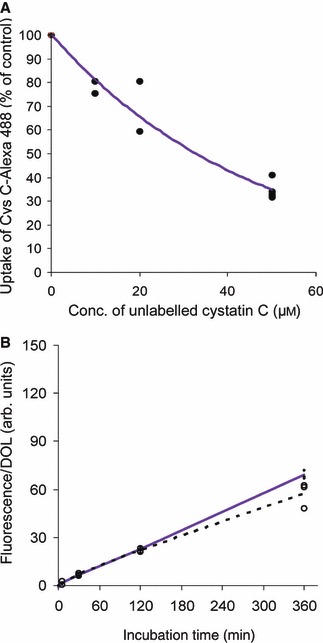
Uptake competition of labelled cystatin C in Capan‐1 cells. (A) Unlabelled cystatin C (10 μm, n = 2; 20 μm, n = 2; 50 μm, n = 5) was added to sub‐confluent Capan‐1 (pancreas adenocarcinoma) cells just before the addition of 1 μm labelled cystatin C. Cells were then incubated for 4 h at 37 °C and the fluorescence measured by flow cytometry. Data points are shown for each individual result. A line is drawn through the average value of the wells from each specified cystatin C concentration. (B) Internalization of cystatin C variants. Capan‐1 cells were incubated for 10 s, 5 min, 30 min, 2 h or 6 h at 37 °C in medium containing fluorescence‐labelled (R8G,L9G,V10G,W106G)–cystatin C (solid blue line) or N39K–cystatin C (dashed black line). Cell fluorescence was measured by flow cytometry and the median of the fluorescence of the cell population was calculated, corrected for the control value and then related to the degree of labelling of the protein used. Dashes and rings represent every single result. Lines are drawn through the average value of results at each time point (n = 3).
Internalization of cystatin C variants
To learn more about the structural requirements for the uptake studied, we used two cystatin C variants produced using site‐directed mutagenesis. One of the variants, (R8G,L9G,V10G,W106G)–cystatin C, essentially lacks the ability to inhibit C1 family cysteine proteases like papain and cathepsin B, because of the removal of side chains involved in the interaction with these enzymes [21]. The other, N39K–cystatin C, lacks inhibitory activity against the C13 family cysteine protease, legumain, because of removal of the key amino acid in the legumain‐binding site of cystatin C [22]. The amino acid substitutions reside in opposing parts of the cystatin C molecule, which makes these protein variants interesting. Experiments were carried out in cells from a human pancreas adenocarcinoma (Capan‐1) using Alexa‐488 labelling of the cystatin C variants. Both variants of the inhibitor were internalized (Fig. 7B). Thus, the experiment provided evidence that possible surface‐located target proteases are not involved in the uptake process and indicated that the protease‐reactive sites do not overlap with the site promoting internalization of cystatin C.
Discussion
Experiments demonstrating the inhibition of virus replication after adding cysteine protease inhibitors to the cell medium [16, 17, 18, 19] led to our proposal that some of these inhibitors have an intracellular fate. The issue merits further interest because changes in protease activity might be of importance in cancer cell growth, metastasis and angiogenesis. It is not clear, however, to what extent the implicated proteases function intra‐ or extracellularly. Recently, it has also been proposed that cystatin C has the capacity to promote astrogenesis and suppress oligodendrogenesis and these functions seemed to be independent of its cysteine protease inhibitor activity [23]. Thus, it is clear that cystatin C has various effects on the cell. Some of these seem to be independent of its cysteine protease inhibitory activity and some might be associated with its intracellular activity. Nevertheless, a specific cystatin pathway implicating cellular uptake by active internalization of extracellularly located cystatins has not been delineated.
We demonstrated that cystatin C is internalized in five different cancer cell lines by using conceptually different techniques such as CLSM, flow cytometry, western blotting, quantification of internalized inhibitor by immunological methods and measurement of the cysteine inhibitory capacity of cells. The human cancer cell lines chosen included one epidermoid carcinoma cell line, three mammary tumour cell lines and one human pancreas adenocarcinoma. All five cell lines exhibited cystatin C internalization when exposed to extracellular cystatin C. In most experiments uptake could be detected after 30 min, but in some cases it could be seen after 5 min. The uptake curves were very similar in the cell strains studied and showed that all cells exhibited fluorescence after 6 h exposure to 1 μm fluorescence‐conjugated cystatin C. In addition, flow cytometry recorded that the uptake of a specific labelled protein in one specific cell strain was highly reproducible within, as well as between, runs. However, the fluorometric method used to measure the degree of protein labelling is not precise, although it provides a good estimate, and detailed comparisons of the quantity of uptake can therefore not be concluded. The lack of uptake in experiments carried out at 4 °C, as well as the competition experiments supported the idea that internalization is an active process and not a passive flow of molecules into the cells [24], thus suggesting receptor‐mediated uptake.
The flow cytometry and ELISA results clearly show relatively rapid cystatin C uptake (2, 5) during the first minutes, but the increase appears to be much slower when it is measured by ELISA than when monitored by flow cytometry. Therefore, we used western blotting to verify the internalization of cystatin C. Western blots developed with cystatin C‐specific antibodies were scanned and each band representing cystatin C was semi‐quantified by densitometry. These results agreed well with the flow cytometry data and suggest a linear uptake rate over a relatively long period (Fig. 5C).
In microscope analysis, the tested cell lines showed different patterns of internalization and intracellular distribution of cystatin C. In the MCF‐7 and A‐431 cell lines, CLSM demonstrated that internalized cystatin C was distributed through all parts of the cytoplasm, visualized as both smaller and larger accumulations. In addition, CLSM analyses indicated that ∼ 10% of the cells contained relatively high levels of cystatin C, which appeared to be localized in discrete cellular compartments, co‐localized with LysoTracker, thus indicating localization in acidic compartments such as lysosomes. Differences between cell types were also observed in the amount of cystatin C uptake and/or its intracellular localization. The heterogeneous cell morphology shown by microscopy further supports that subpopulations of cells possess different abilities in cystatin C uptake.
The inhibition capacity in lysates of cells incubated for 24 h without labelled cystatin C showed a level of ∼ 200 pmol cysteine protease inhibitor·mg protein−1 compared with ∼ 2 pmol cystatin C·mg cell protein−1 when measured using ELISA (4, 5). This suggests that cystatin C constitutes ∼ 1% of the total cysteine protease inhibitor capacity in cells grown in medium, which is reasonable because cystatin A and B are the dominating intracellular, cytoplasmatic, cysteine protease inhibitors [4]. After 24 h exposure to 1 μm cystatin C the total cysteine protease inhibitor capacity as well as the cystatin C concentration of the cells increased. As indicated by the experiment in which the uptake was measured by ELISA, the cystatin C level increased at least fivefold (Fig. 5). Thus, internalized cystatin C appears to influence the balance between proteases and their inhibitors in cancer metastasis and growth, particularly when considering that it is a more efficient cathepsin B inhibitor than are cystatins A and B. It is possible that the intracellular increase in inhibitory capacity has an even greater impact on this balance than is suspected at first, because the cystatin molecules responsible for the increase are probably localized to, and hence concentrated in, just some cell compartments (e.g. endosomes).
In additional experiments, Capan‐1 cells were incubated with two cystatin C variants (Fig. 7B), carrying inactivating substitutions of key amino acid side chains important for target enzyme binding, (R9G,L9G,V10G,W106G)–cystatin C and N39K–cystatin C. Both protein variants, which are essentially depleted of any inhibitory capacity against cathepsin B and other papain‐like enzymes, and legumain, respectively, were substantially internalized. This suggests that the uptake mechanism is not dependent on any of the residues central for the inhibitory capacity of cystatin C.
Our experiments were carried out on cell lines emanating from tumours with different origins, but which behaved identically regarding cellular cystatin C uptake. It seems possible that internalization will also be seen in normal cells, i.e. it is a general, physiological phenomenon, but this has to be investigated further. In conclusion, by using cell culture experiments and flow cytometry we were able to convincingly demonstrate that the cysteine protease inhibitor cystatin C is internalized in all five of the cancer cell lines investigated in this study. Using confocal microscopy, western blotting and quantification by ELISA, internalization of the cysteine inhibitor was verified and further delineated. Previously, target enzymes of this cysteine protease inhibitor have been shown to be involved in cell invasion and metastasis in cancer, and the cystatins have also been proposed to inhibit virus replication in cell cultures. Our findings open conceptually new lines of research in order to further elucidate the extension, the mechanism and the biological importance of this phenomenon.
Experimental procedures
Cells and reagents
Five different human cancer cell lines were used (from the German Collection of Micro‐organisms and Cell Cultures, Hamburg, Germany and the ATCC, Manassas, VA): MCF‐7, MDA‐MB‐453 and MDA‐MB‐468, (all three cell lines are human breast adenocarcinoma), A‐431 (epidermoid carcinoma) and Capan‐1 (human pancreas adenocarcinoma). Cell culture medium used was Dulbecco’s modified Eagle’s medium with 4500 mg·L−1 glucose, GlutaMAX‐I and pyruvate supplemented with 10% fetal calf serum, penicillin G, streptomycin and in some experiments amphotericin B (all from Invitrogen, Grand Island, NY, USA). Cells were lysed in 0.2% Triton‐X 100 in calcium‐ and magnesium‐free NaCl/Pi (lysis buffer). To all cell lysates and culture medium samples a preservation cocktail was added to a final concentration of 5 mm benzamidinium hydrochloride, 15 mm NaN3 and 10 mm EDTA.
Internalization measured by flow cytometry
Recombinant human cystatin C [25], cystatin C with the amino acid substitutions Arg8Gly, Leu9Gly, Val10Gly and Trp106Gly [here (R8G,L9G,V10G,W106G)–cystatin C] [21], cystatin C with the amino acid substitution Asn39Lys (N39K–cystatin C) [22] and PCI [26] were fluorescently labelled with an Alexa Fluor 488 Protein Labeling Kit (Molecular Probes, Eugene, OR, USA). Equistatin [27] was labelled with fluorescein. The degree of protein labelling (DOL) was then estimated by the formula recommended by the manufacturer of the labelling kit. The DOL values for cystatin C and PCI were between 0.20 and 0.32. The cystatin C variants (R8G,L9G,V10G,W106G)–cystatin C and N39K–cystatin C exhibited DOL values of ∼ 2.7 and 0.75, respectively. The DOL of equistatin, estimated by MALDI‐TOF MS on a Bruker spectrometer (Germany), was ∼ 0.40. In six‐well culture plates cells were seeded and cultured for 3–5 days. Non‐confluent cells were then incubated in new medium containing 1 μm labelled protein. To the control cells was added an equal volume of NaCl/Pi. Cells were then incubated for 10 s, 5 min, 30 min, 2 h or 6 h at 4 or 37 °C. After incubation, cells were washed three times with NaCl/Pi and finally trypsinized at 37 °C for a minimum of 15 min. Cell fluorescence was measured by a FACS Calibur Flow Cytometer (Becton‐Dickinson, Franklin Lakes, NJ, USA). At least 3000 events were measured in each sample. Cells were gated to exclude cell debris and cell conglomerates. The median of the individual cell fluorescence was then calculated.
Immunocytochemistry
The internalization of cystatin C was also illustrated by immunocytochemistry. In six‐well culture plates 105 MCF‐7 or 6 × 104 A‐431 cells were seeded on cover slips (Knittel Glasbearbeitung GmbH, Braunschweig, Gemany) and then incubated for 2 days to reach 50–70% confluence. Cells were washed twice with NaCl/Pi and new culture medium containing 5 μm Alexa‐488 or Alexa‐568‐labelled recombinant cystatin C, or an equivalent volume of NaCl/Pi was added. After 6 h incubation the cells were washed with NaCl/Pi and fixed in methanol/acetone (1 : 1 v/v) or 4% paraformaldehyde in NaCl/Pi. Nuclei were stained with either propidium iodide or SytoxGreen.
Control experiments and specificity tests were performed for microscopical analyses, both of cells with internalized Alexa‐568 (Molecular Probes) conjugated cystatin C, and unlabelled cystatin C detected by immunocytochemistry. As control experiments of the cellular uptake of the Alexa‐568 conjugated cystatin C, cells were incubated with unlabelled cystatin C followed by immunocytochemical detection of cystatin C (both internalized and endogenous cystatin C were visualized). The primary antibody was polyclonal rabbit anti‐(human cystatin C) serum [20] and the secondary antibody used was an anti‐(rabbit‐IgG) made in goat and conjugated with Alexa‐568. Further control experiments included primary antibody omission and antigen absorption.
Microscope analyses
After incubation with cystatin C, conjugated with Alexa‐568 or Alexa‐488, cells were processed for microscope analyses, labelled with general nuclear markers or processed for immunocytochemistry. Labelled cells were transferred to glass slides, mounted and coverslipped in p‐phenylenediamine.
Cells were initially analysed with an epi‐fluorescence microscope (Olympus AX 60). Cystatin C (Alexa‐568 or Alexa‐488) and secondary antibodies (Alexa‐568), and fluorescent nuclear markers (Sytox Green, propidium iodide or DAPI, all Invitrogen) were employed. Images were grabbed digitally (Olympus DP70), separately for each individual spectral channel and then merged with the overlay function. CLSM analyses were performed with a Bio‐Rad MRC 1024, mounted on an inverted Nikon Diaphot 300 microscope. Another CLSM, a Zeiss LSM 510 Meta microscope, was used in some cases for excitation maxima at 405 nm in conjunction with DAPI as nuclear marker. During all CLSM analyses the settings were optimized for each fluorophore, and data acquisitions were obtained only by sequential scanning of individual fluorophores, which provided a total separation in the light collecting channels. The level of auto‐fluorescence recorded in all channels of non‐incubated cell populations was used as a background signal, adjusted for the settings of the individual channels. Optical slices (around 300 nm) were collected in Z‐steps through cells. The cellular localization (presence within the cytoplasm) of cystatin C conjugated with Alexa‐568 or Alexa‐488 was analysed as individual optical sections or merged images (image analyses in laser sharp or Zeiss lsm 510 software).
In the live imaging experiments, 20 000 A‐431 cells were seeded in μ‐Slide ibiTreat wells (LRI Instrument AB, Lund, Sweden) and incubated overnight. The medium was changed and cystatin C–Alexa‐488 was added to a final concentration of 50 nm. After 15 min the cells were washed with NaCl/Pi and new medium containing 10 nm LysoTracker (Molecular Probes) was added. Live cells were analysed with an inverted fluorescence microscope (Olympus IX71) equipped with a large 37 °C incubator, thus heating the environment for stable conditions. A 60× apochromat oil immersion objective with a numerical aperture of 1.35 was used. Pictures were grabbed with a Hamamatsu Orca (Hamamatsu Photonics Norden AB, Solna, Sweden) monochromatic camera.
Quantification of endogenous and secreted cystatin C
In six‐well culture plates, 105 Capan‐1 cells were seeded and cultured to reach 50–70% confluence. The culture medium was changed and the cells were incubated for 6 or 24 h with new culture medium. Secreted cystatin C was determined in the culture medium and cells were lysed for quantification of the intracellular cystatin C level. The cystatin C concentration in the culture medium and lysate was measured using ELISA, as described previously [28]. Cystatin C levels were correlated to protein content in the cell lysates, determined for samples diluted 1 : 100 with Coomassie protein assay reagent (Pierce, Rockford, IL, USA). Time‐course experiments were carried out in a similar manner with cells incubated in medium containing 1 μm recombinant cystatin C.
Quantification of cystatin C uptake
The determination of basal cystatin C levels was followed by measuring the cellular content of active cysteine protease inhibitor after cystatin C incubation in a time‐scale manner. Capan‐1 cells were seeded in six‐well culture plates and cultured to reach 50–70% confluence. Cells were then washed twice with NaCl/Pi and new culture medium with 1 μm cystatin C or an equivalent volume of NaCl/Pi was added. Cells were incubated for 5, 30 min, 2 or 6 h, harvested and the cystatin C concentration in the lysate was measured by an ELISA, as described previously [28]. The level of cystatin C was correlated to protein content of the cell lysate determined by Coomassie protein assay reagent.
Western blotting
Capan‐1 cells were cultured as for the quantification of endogenous and secreted cystatin C. Cells were then washed twice with NaCl/Pi and new culture medium with 1 μm cystatin C or an equivalent volume of NaCl/Pi was added. Cells were incubated for 5, 30 min, 2 or 6 h before lysate preparation. Because of the rather low cystatin C content it had to be concentrated. Ten microlitres of CNBr‐Sepharose‐4B beads (Amersham Biosciences, Uppsala, Sweden) with coupled carboxymethylated papain (Sigma Aldrich, Steinway, Germany) was added to the lysate followed by 48 h incubation on a shaker at 4 °C [29]. After centrifugation, the supernatant was discarded and NuPage LDS sample buffer with NuPAGE sample reducing agent (Invitrogen) was added to the remaining gel pellet. The proteins were separated in a 4–12% SDS/PAGE gel (Novex, Invitrogen AB, Stockholm, Sweden) before electroblotting to a poly(vinylidene difluoride) membrane (Immobilon‐P, Millipore, Bedford, MA, USA). Blotted proteins were immunodetected using a polyclonal rabbit anti‐(human cystatin C) serum [20]. As secondary antibody a horseradish‐peroxidase conjugated goat anti‐(rabbit IgG) fraction (DAKO, Copenhagen, Denmark) was used. The blotted proteins were visualized by chemiluminescence (ECL Plus reagent; Amersham Biosciences, Piscataway, NJ, USA). As controls, two samples containing 100 and 10 ng cystatin C were added to the separating gel. Bands were quantified using nih image 1.63 software (NIH, Bethesda, MD, USA) and an Epson Expression 1600 scanner.
Cysteine protease inhibitor activity assays
Capan‐1 cells were cultured and incubated with or without the addition of 1 μm cystatin C in medium for 24 h. After lysate preparation in 1 mL lysis buffer, cells were centrifuged in order to remove cell debris. A portion of the supernatant was incubated at 95 °C for 5 min to denature the endogenous cysteine proteases. Under these conditions, the cystatins are stable [21, 30]. The denatured proteins were removed by centrifugation.
E‐64 titration
The activity of the papain used (Sigma Aldrich) was determined by titration with the irreversible cysteine protease inhibitor E‐64 (trans‐epoxysuccinyl‐l‐leucylamido‐(4‐guanidino)‐butane, Sigma Aldrich). Z‐Phe‐Arg‐AMC (Bachem, Bubendorf, Switzerland) was used as substrate. To a buffer mix (final concentrations, 0.1 m phosphate, pH 6.5, 1 mm dithiothreitol, 1 mm EDTA) containing 5 μL papain (1 μg·mL−1), was added 0–10 μL of E‐64. The final volume was adjusted to 93 μL with 0.01% Brij‐35 (Sigma Aldrich). The mixture was allowed to incubate for 10 min before the addition of 7 μL substrate (200 μm). The fluorescence was measured in a plate reader (Fluoroskan Ascent, Thermo Electron, Vantaa, Finland) at 355 nm excitation and 460 nm emission. The difference between fluorescence at the start and following 30 min incubation for each sample, reflecting the remaining enzyme activity for each concentration of E‐64 when compared with a sample without E‐64 added, was used to construct the titration curve.
Papain and cathepsin B inhibition
The cysteine protease inhibitory capacity in cell lysates was determined by measuring the inhibition of papain as well as cathepsin B. To measure the capacity of the cell lysate to inhibit papain, 88 μL buffer mix as above, containing 5 μL papain (1 μg·mL−1), was used, to which 5 μL of the cell lysate was added. The mixture was allowed to incubate for 10 min before the addition of 7 μL substrate (Z‐Phe‐Arg‐AMC; 200 μm). As positive and negative controls, instead of cell lysate, 5 μL of 0.10 μm E‐64 or 5 μL of 0.01% Brij‐35 was used, respectively. Fluorescence was measured in a plate reader at the start and end of a 30‐min incubation, as above. To measure the inhibition of cathepsin B (kindly provided by J. Mort, Shriners Hospital, Montreal, Quebec, Canada), 83 μL buffer mix as above, containing 5 μL cathepsin B (20 nm) and 10 μL cell lysate was used. As positive and negative controls, instead of cell lysate, 10 μL of 0.10 μm E‐64 or 10 μL of 0.01% Brij‐35 was used. The mixture was allowed to incubate for 10 min before the addition of 7 μL of substrate (Z‐Phe‐Arg‐AMC; 200 μm).
Uptake competition
Capan‐1 cells were seeded, grown and treated as above except that unlabelled cystatin C was added (10, 20 or 50 μm) just before the addition of Alexa‐488‐labelled cystatin C (1 μm). Cells were then incubated for 4 h at 37 °C. Cell fluorescence was measured by a FACS Calibur Flow Cytometer. The median of the individual cell fluorescence was then calculated and the result was related to the fluorescence of the cells which were incubated without a competing, unlabelled inhibitor.
Supporting information
Fig. S1. Confocal microscopy of cancer cells incubated with cystatin C
Fig. S2. Immunolabelling of cystatin C in A–431 cells by a specific polyclonal rabbit antiserum.
Please note: Blackwell Publishing are not responsible for the content or functionality of any supplementary material supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Acknowledgements
This study was supported by the Swedish Foundation for International Cooperation in Research and Higher Education (PD2001‐72), the Swedish Research Council (project 09915), the Swedish Cancer Society, A Österlund’s foundation, Magnus Bergvall’s foundation and the Crafoord foundation, Spanish Ministry of Education and Science (BIO2007‐6846‐C02). We thank the Institute of Cell and Organism Biology, Lund University, for kindly letting us use their Zeiss LSM 510 Meta microscope.
References
- 1. Barrett AJ (1999) Handbook of Proteolytic Enzymes. Academic Press, London. [Google Scholar]
- 2. Mohamed MM & Sloane BF (2006) Cysteine cathepsins: multifunctional enzymes in cancer. Nat Rev Cancer 6, 764–775. [DOI] [PubMed] [Google Scholar]
- 3. Sloane BF, Dunn JR & Honn KV (1981) Lysosomal cathepsin B: correlation with metastatic potential. Science 212, 1151–1153. [DOI] [PubMed] [Google Scholar]
- 4. Abrahamson M, Alvarez‐Fernandez M & Nathanson CM (2003) Cystatins. Biochem Soc Symp 70, 179–199. [DOI] [PubMed] [Google Scholar]
- 5. Levicar N, Strojnik T, Kos J, Dewey RA, Pilkington GJ & Lah TT (2002) Lysosomal enzymes, cathepsins in brain tumour invasion. J Neur Oncol 58, 21–32. [DOI] [PubMed] [Google Scholar]
- 6. Heidtmann HH, Salge U, Abrahamson M, Bencina M, Kastelic L, Kopitar‐Jerala N, Turk V & Lah TT (1997) Cathepsin B and cysteine proteinase inhibitors in human lung cancer cell lines. Clin Exp Metastasis 15, 368–381. [DOI] [PubMed] [Google Scholar]
- 7. Keppler D, Sameni M, Moin K, Mikkelsen T, Diglio CA & Sloane BF (1996) Tumor progression and angiogenesis: cathepsin B and C. Biochem Cell Biol 74, 799–810. [DOI] [PubMed] [Google Scholar]
- 8. Corticchiato O, Cajot JF, Abrahamson M, Chan SJ, Keppler D & Sordat B (1992) Cystatin C and cathepsin B in human colon carcinoma: expression by cell lines and matrix degradation. Int J Cancer 52, 645–652. [DOI] [PubMed] [Google Scholar]
- 9. Cox JL, Sexton PS, Green TJ & Darmani NA (1999) Inhibition of B16 melanoma metastasis by overexpression of the cysteine proteinase inhibitor cystatin C. Melanoma Res 9, 369–374. [DOI] [PubMed] [Google Scholar]
- 10. Sexton PS & Cox JL (1997) Inhibition of motility and invasion of B16 melanoma by the overexpression of cystatin C. Melanoma Res 7, 97–101. [DOI] [PubMed] [Google Scholar]
- 11. Coulibaly S, Schwihla H, Abrahamson M, Albini A, Cerni C, Clark JL, Ng KM, Katunuma N, Schlappack O, Glossl J et al. (1999) Modulation of invasive properties of murine squamous carcinoma cells by heterologous expression of cathepsin B and cystatin C. Int J Cancer 83, 526–531. [DOI] [PubMed] [Google Scholar]
- 12. Shridhar R, Zhang J, Song J, Booth BA, Kevil CG, Sotiropoulou G, Sloane BF & Keppler D (2004) Cystatin M suppresses the malignant phenotype of human MDA‐MB‐435S cells. Oncogene 23, 2206–2215. [DOI] [PubMed] [Google Scholar]
- 13. Zhang J, Shridhar R, Dai Q, Song J, Barlow SC, Yin L, Sloane BF, Miller FR, Meschonat C, Li BD et al. (2004) Cystatin M: a novel candidate tumor suppressor gene for breast cancer. Cancer Res 64, 6957–6964. [DOI] [PubMed] [Google Scholar]
- 14. Xing R, Wu F & Mason RW (1998) Control of breast tumor cell growth using a targeted cysteine protease inhibitor. Cancer Res 58, 904–909. [PubMed] [Google Scholar]
- 15. Merz GS, Benedikz E, Schwenk V, Johansen TE, Vogel LK, Rushbrook JI & Wisniewski HM (1997) Human cystatin C forms an inactive dimer during intracellular trafficking in transfected CHO cells. J Cell Physiol 173, 423–432. [DOI] [PubMed] [Google Scholar]
- 16. Abrahamson M (1994) Cystatins. Methods Enzymol 244, 685–700. [DOI] [PubMed] [Google Scholar]
- 17. Korant BD, Brzin J & Turk V (1985) Cystatin, a protein inhibitor of cysteine proteases alters viral protein cleavages in infected human cells. Biochem Biophys Res Commun 127, 1072–1076. [DOI] [PubMed] [Google Scholar]
- 18. Collins AR & Grubb A (1991) Inhibitory effects of recombinant human cystatin C on human coronaviruses. Antimicrob Agents Chemother 35, 2444–2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bjorck L, Grubb A & Kjellen L (1990) Cystatin C, a human proteinase inhibitor, blocks replication of herpes simplex virus. J Virol 64, 941–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Abrahamson M, Barrett AJ, Salvesen G & Grubb A (1986) Isolation of six cysteine proteinase inhibitors from human urine. Their physicochemical and enzyme kinetic properties and concentrations in biological fluids. J Biol Chem 261, 11282–11289. [PubMed] [Google Scholar]
- 21. Hall A, Hakansson K, Mason RW, Grubb A & Abrahamson M (1995) Structural basis for the biological specificity of cystatin C. Identification of leucine 9 in the N‐terminal binding region as a selectivity‐conferring residue in the inhibition of mammalian cysteine peptidases. J Biol Chem 270, 5115–5121. [DOI] [PubMed] [Google Scholar]
- 22. Alvarez‐Fernandez M, Barrett AJ, Gerhartz B, Dando PM, Ni J & Abrahamson M (1999) Inhibition of mammalian legumain by some cystatins is due to a novel second reactive site. J Biol Chem 274, 19195–19203. [DOI] [PubMed] [Google Scholar]
- 23. Hasegawa A, Naruse M, Hitoshi S, Iwasaki Y, Takebayashi H & Ikenaka K (2007) Regulation of glial development by cystatin C. J Neurochem 100, 12–22. [DOI] [PubMed] [Google Scholar]
- 24. Anderson RG, Brown MS & Goldstein JL (1977) Role of the coated endocytic vesicle in the uptake of receptor‐bound low density lipoprotein in human fibroblasts. Cell 10, 351–364. [DOI] [PubMed] [Google Scholar]
- 25. Abrahamson M, Dalboge H, Olafsson I, Carlsen S & Grubb A (1988) Efficient production of native, biologically active human cystatin C by Escherichia coli . FEBS Lett 236, 14–18. [DOI] [PubMed] [Google Scholar]
- 26. Hass GM, Nau H, Biemann K, Grahn DT, Ericsson LH & Neurath H (1975) The amino acid sequence of a carboxypeptidase inhibitor from potatoes. Biochemistry 14, 1334–1342. [DOI] [PubMed] [Google Scholar]
- 27. Strukelj B, Lenarcic B, Gruden K, Pungercar J, Rogelj B, Turk V, Bosch D & Jongsma MA (2000) Equistatin, a protease inhibitor from the sea anemone Actinia equina, is composed of three structural and functional domains. Biochem Biophys Res Commun 269, 732–736. [DOI] [PubMed] [Google Scholar]
- 28. Olafsson I, Lofberg H, Abrahamson M & Grubb A (1988) Production, characterization and use of monoclonal antibodies against the major extracellular human cysteine proteinase inhibitors cystatin C and kininogen. Scand J Clin Lab Invest 48, 573–582. [DOI] [PubMed] [Google Scholar]
- 29. Anastasi A, Brown MA, Kembhavi AA, Nicklin MJ, Sayers CA, Sunter DC & Barrett AJ (1983) Cystatin, a protein inhibitor of cysteine proteinases. Improved purification from egg white, characterization, and detection in chicken serum. Biochem J 211, 129–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lenney JF, Tolan JR, Sugai WJ & Lee AG (1979) Thermostable endogenous inhibitors of cathepsins B and H. Eur J Biochem 101, 153–161. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Confocal microscopy of cancer cells incubated with cystatin C
Fig. S2. Immunolabelling of cystatin C in A–431 cells by a specific polyclonal rabbit antiserum.
Please note: Blackwell Publishing are not responsible for the content or functionality of any supplementary material supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item