

Abstract
For more than three decades, RNA recombination remained a puzzle and has only begun to be solved in the last few years. The available data provide evidence for a variety of RNA recombination mechanisms. Non-homologous recombination seems to be the most common for RNA. Recent experiments in both the in vitro and the in vivo systems indicate that this type of recombination may result from various transesterification reactions which are either performed by RNA molecules themselves or are promoted by some proteins. The high frequency of homologous recombination manifested by some RNA viruses can be easier explained by a replicative template switch.
Keywords: RNA recombination, RNA virus, Cell-free system, Transesterification reaction, Replicative template switch
1. Introduction
RNA recombination was discovered in the early 60s as an exchange of genetic material between closely related RNA viruses which co-infect the same cell [1], [2]. Since then, the formation of recombinant RNAs was observed in infected cells of every kingdom [3]. However, until recently, the molecular mechanism of this process remained an unsolved puzzle. In contrast to DNA recombination, no specific genes were associated with the recombinant formation that precluded dissection of the recombination mechanism by genetic tools; neither could the mechanism be studied biochemically because of the absence of cell-free experimental models. There was no answer even to such basic questions as to whether the recombination is accomplished by viral proteins (e.g. replicase), by host proteins or by RNA molecules themselves. Moreover, since RNA recombination was only observed in vivo, the possibility could not be excluded that the recombining species were not RNAs, but their cDNA copies.
Since RNA recombination is a very rare event, it can only be detected if there are means of selective amplification of recombinant molecules. Therefore, for a long time, RNA recombinations were exclusively observed in viral systems, but even in these systems, recombinants were occasionally detected consisting partially or entirely of cellular RNA segments [3], [4], [5]. It remained unclear whether any factors accompanying viral infection (e.g. presence of viral replicase) are required for recombinant formation or whether RNA recombination can occur in uninfected cells as well.
Apart from experimental difficulties, progress in this field was partially retarded by common belief that any type of RNA recombination must be due to a replicative template switch [3], [5], [6], [7], [8]. A breakthrough has been made during the last 2 years owing to the development of cell-free experimental models. It has appeared that RNAs can indeed recombine without DNA intermediates, that there are a variety of non-replicative mechanisms and that the existence of such mechanisms can be demonstrated in conventional in vivo systems.
2. Homologous versus non-homologous recombination
The easiest way to observe RNA recombination is to co-infect a cell with two related viruses which carry non-complementing genetic defects or selective markers, such that no virus multiplication is possible unless the viral genomes exchange their segments [3]. Recombination may be homologous, if both the parents and the progeny are homologous to each other around the cross-over site, or non-homologous, if it occurs otherwise. While homologous recombination preserves the genome structure, non-homologous recombination changes it. Since most of the viral genome comprises coding sequences or those essential for RNA replication and/or packaging into a virion, it is clear that most of the non-homologous recombinants will not survive. Therefore, experiments of this kind mostly reveal the homologous recombination producing an impression that homologous RNA recombination occurs more frequently than the non-homologous one [3]. As will be seen below, such a view is incorrect.
Non-homologous recombination can be observed by crossing deletion variants or fragments of viral RNAs which overlap at non-essential regions and whose recombination may restore the intact genome [9], [10], [11], [12]. It can also be observed by using an engineered virus carrying an insert in a non-essential region. Often, such an insert renders the virus viable but confers onto it a small-plaque phenotype. Occasionally, the insert is deleted by intrastrand recombination, resulting in the restoration of the wild-type phenotype, but the deletions are not necessarily precise [13], [14], [15]. Spontaneous deletions can also occur in wild-type viruses, resulting in the generation of defective interfering (DI) particles [16], [17]. DI particles were first described under the name of ‘inactive viruses’ as early as in 1943 [18], but only in the 70s was it recognized that these are generated by non-homologous RNA recombination. Since the deletions result in a defect or a lack of at least one of the viral genes, DI particles can only propagate if the same cell is co-infected with the intact authentic virus providing the missing proteins in trans and are detected as particles contaminating the virus preparation. DI particles have been named so due to their ability to interfere with replication of the helper virus [16], [17].
An important characteristic of recombination is its frequency. The frequency of homologous recombination greatly varies among different viruses. It is as high as 10−5/nucleotide (nt) in crosses between coronaviruses or picornaviruses belonging to the same serological type [3], but in RNA phages, the frequency is as low as 10−11/nt [19]. The frequency of non-homologous recombination was almost never reported, but it can be roughly estimated from the data presented in some papers. In each studied instance, the estimate gives the same value, about 10−5/nt, i.e. the same order as the frequency of the most efficient homologous recombination. Surprisingly, non-homologous recombination is equally efficient in picornaviruses [13], [20] manifesting the highest rate of homologous recombination, in RNA phages [15] producing homologous recombinants a million times less frequently and even in alphaviruses [12] in which homologous recombination was not detected at all [9].
The absence of any correlation in the frequencies of homologous and non-homologous recombination suggests that the mechanisms of these two processes may be entirely different. This conclusion conforms to the observation that some viral polymerase mutations differently affect homologous and non-homologous recombination [21].
3. Recombination in the purified Qβ system
Recently, several laboratories reported on the development of cell-free systems for studying RNA recombination [22], [23], [24], [25], [26], but only one paper describes a purified system in which the entire process, from mixing the parental strands and up to amplification of recombinant molecules, can be strictly controlled [23]. The reported system utilizes the pure replicase of RNA-containing bacteriophage Qβ, a unique RNA-directed RNA polymerase capable to exponentially amplify RNA in vitro [27]. This ability has been employed in the molecular colony technique where single RNA molecules are grown as colonies in an agarose gel containing Qβ replicase and rNTPs [28]. Like bacterial colonies, each RNA colony comprises the progeny of a single molecule, i.e. a clone [29].
Not any RNAs can be amplified by Qβ replicase, but only those carrying special 5′- and 3′-terminal structures which form the replicase recognition site and whose interaction presumably provides for the single strandedness of the template and the nascent RNA, thereby ensuring many rounds of replication [27]. The reported system [23] used, as recombination substrates, the 5′- and 3′-fragments of RQ135−1 RNA [30], a 134 nt long Qβ phage satellite RNA [28] which had arisen by recombination between cellular and viral sequences [30]. The fragments were obtained by breaking the RNA at a natural cross-over site and, to facilitate a replicase switch between the fragments, extending their truncated ends with homologous foreign sequences (Fig. 1). To initiate recombination between the fragments, they were mixed and applied to the replicase-containing agarose. The fragments themselves are not replicable, but recombination between them may produce replicable RNAs resulting in RNA colonies whose number reflects the number of recombinant molecules generated in the system.
Fig. 1.
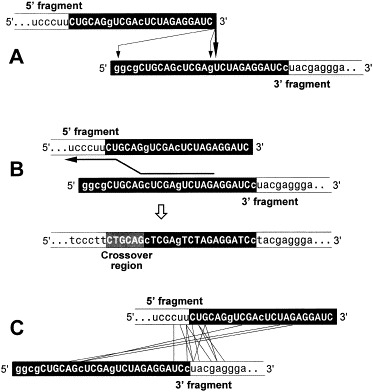
Joining the parental sequences in the recombinants generated in vitro in the presence of Qβ replicase and rNTPs (A), in the presence of AMV reverse transcriptase and dNTP (B) and in the absence of any enzyme and nt (C). Foreign extensions are shown in white letters, capital letters indicate the regions of homology.
RNA colonies did appear, directly demonstrating that RNA molecules can recombine without any DNA intermediates: those simply could not form because there were no deoxyribonucleotides. Recombination occurred under conditions (pH, temperature, salt and Mg2+ concentrations) similar to those in the cell. The recombination frequency was proportional to the product of the concentrations of the fragments. Extrapolation to the intracellular RNA concentration gives a value of 10−5–10−4/nt [23], i.e. close to the frequency of non-homologous recombination in RNA phages in vivo [15].
Sequencing of the recombinants has shown that all of them are indeed non-homologous despite the homology of the foreign extensions (Fig. 1A). In contrast, only homologous recombinants were formed in a control experiment, in which the same RNA fragments were incubated in the presence of dNTPs and a reverse transcriptase (Fig. 1B), an enzyme capable of switching between templates [31], implying that the mechanism of RNA recombination in the presence of Qβ replicase is different from template switch. The structure of recombinants suggested that they were generated by an attack of the 3′-terminus of the 5′-fragment at the phosphoester bonds within the 3′-fragment (Fig. 1A). Indeed, the recombination did not occur when the 3′-hydroxyl of the 5′-fragment (but not of the 3′-fragment) was eliminated and returned to the original level upon restoration of the hydroxyl (Fig. 2). In contrast to what is claimed in a recent review [8], these modifications did not introduce any bulky or otherwise unusual to RNA groups which could interfere with replicase binding (cf. Fig. 2). Moreover, the fact that similar modifications of a replicable RNA do not affect its replicability [23], [32] eliminates the possibility [8] that Qβ replicase requires the terminal 3′-hydroxyl for binding or RNA synthesis. Considered together, these results indicate that RNA recombination in the presence of Qβ replicase occurs via a transesterification reaction, rather than via a replicative template switch.
Fig. 2.
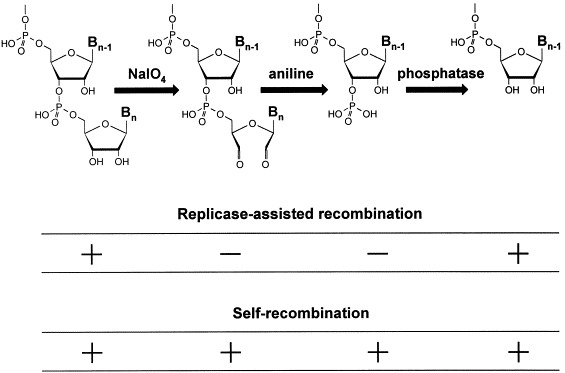
Terminal modifications of the 5′-fragment and their effects on RNA recombination in the cell-free Qβ system. Signs + and − indicate occurrence and absence of the recombination, respectively. Similar modifications of the 3′-fragment do not affect recombination.
The 3′-terminal modifications have recently been used as a tool for exploring the ability of RNAs to recombine in the absence of replicase [33]. In these experiments, a mixture of the RNA fragments was incubated in a salt buffer and, prior to applying to the Qβ replicase-containing agarose, it was melted (to destroy any non-covalent complexes) and oxidized with periodate (to prevent further recombination by the above mechanism). Thus, RNA colonies would only grow if recombination had occurred before the oxidation step. It has turned out that RNAs do recombine by themselves. The only requirement is the presence of Mg2+ ions at a millimolar concentration. However, such a self-recombination is entirely different from recombination in the presence of Qβ replicase. In this case, the fragments recombine by internal segments (Fig. 1C), the 3′-hydroxyls do not participate (Fig. 2) and the reaction is several orders of magnitude slower (Table 1). Moreover, because the chemical step is slow, it becomes rate-limiting and the reaction follows pseudo-first order kinetics. Similar reactions can also occur in cis, resulting in deletions of internal RNA segments [33].
Table 1.
Properties of the two recombination mechanisms in the in vitro Qβ system
| Self-recombination | Replicase-assisted recombination | |
| Reaction kinetics | Pseudo-first order | Second order |
| Rate-limiting step | Chemical reaction | Non-covalent interactions |
| Overall ratea, nt−1 h−1 | 10−9 | 10−4–10−5 |
| Nucleophilic attack | By 2′-OH | By 3′-OH |
| Reacting nt | Internal | 3′-Terminal+internal |
The rates have been extrapolated to the intracellular phage RNA concentration. Their values can be directly compared with the recombination frequency in RNA phages whose infection cycle takes about 1 h.
A conceivable mechanism of self-recombination includes an attack by 2′-hydroxyls, the attack being either intramolecular, leading to intermediate formation of the 2′,3′-cyclic phosphate and 5′-hydroxyl termini which are then cross-ligated, or intermolecular, resulting in a branched structure which is then copied by Qβ replicase to produce the final recombinant molecule. Distribution of cross-over sites revealed no sequence specificity (Fig. 1C) and the recombination rate did not change when the extension sequences were altered [33], indicating that the ability of self-recombination is not associated with some cryptic ribozyme structures. It follows that self-recombination is a general property of RNA and, inasmuch as it requires nothing but RNA itself and Mg2+, it must be ubiquitous in nature and involve both viral and cellular RNAs.
Despite its low rate, RNA self-recombination might play an important evolutionary role both in the prebiotic RNA world and in the contemporary DNA world. At a rate of 10−9/nt/h, a new recombinant RNA could arise in a human cell every minute, yielding up to 1020 recombinant molecules during the life span of the human body. Reverse transcription and integration [34] of even a minute fraction of the rearranged sequences would change the genome significantly. Moreover, the reaction rate in the cell milieu might be much higher: cellular proteins might non-specifically enhance self-recombination as they promote the action of ribozymes [35], [36].
Thus, the experiments in the cell-free Qβ system have demonstrated the existence of at least two different non-replicative mechanisms of RNA recombination, one being performed by RNA itself and the other being somehow promoted by Qβ replicase. The mechanism of the replicase action and whether other viral or cellular RNA polymerases can similarly promote reactions between RNA molecules remain to be determined.
4. Template switch versus the breaking and joining mechanism
The generation of a recombinant sequence can be conceived in two different ways: via breaking the parental sequences and joining the resulting fragments or via de novo synthesis by replicase which switches to another template after it has copied a portion of the first template.
The replicative template switch mechanism has been commonly accepted as the only possibility for generating recombinant RNAs since the work of Kirkegaard and Baltimore on homologous recombination in poliovirus [37], which is widely cited as the most strong evidence in favor of this mechanism. They crossed the wild-type virus (guanidine sensitive and temperature resistant) with a double mutant (guanidine resistant and temperature sensitive) and looked for a recombinant progeny that was both guanidine and temperature resistant. A peculiarity of their experiment was that viruses infected the cells in two consecutive steps, so that superinfection was performed under conditions inhibiting the replication of the first virus, the duration of the steps being adjusted to ensure equal yields of the parental viruses by the end of the experiment. An unexpected observation was that the yield of recombinants differed more than 100-fold depending on which restrictive factor was applied at superinfection. Specifically, the number of recombinants was high when the cells were superinfected with the wild-type virus at an elevated temperature and low when the cells were superinfected with the mutant virus in the presence of guanidine. According to the authors, such an asymmetric result could not be produced by a breaking and joining mechanism inasmuch as the parental genomes were equally abundant in each case, but was a consequence of asymmetric action of the restrictive factors on RNA synthesis. By referring to unpublished data, they claimed that while an elevated temperature inhibited the synthesis of only the negative strands of the mutant virus, the synthesis of both the negative and positive strands of the wild-type virus was suppressed in the presence of guanidine. Hence, as required by the template switch mechanism, recombination appeared to depend on the ongoing RNA synthesis.
However, these arguments do not seem to be strong enough. Since the negative strands comprise only 2% of the total viral RNA [38] and are presumably involved in double-stranded replicative intermediates [5], recombination by the breaking and joining mechanism may entirely depend on the availability of the positive strands. Yet, the positive strands synthesized at the first step may not be available during superinfection because of their packaging or involvement into polyribosomes. Hence, recombination by this mechanism may depend on whether the positive strands of each parental virus are synthesized during superinfection and will not occur in the presence of guanidine when synthesis of only the mutant positive strands is allowed. In addition, the possibility cannot be ruled out that the observed asymmetry resulted from a direct suppression of recombination by guanidine.
Thus, the template switch mechanism has never been unequivocally demonstrated for RNA recombination. Nevertheless, it may operate in some viruses, such as picorna- and coronaviruses that manifest an unusually high rate of homologous recombination. Switching between templates was directly demonstrated for retroviral reverse transcriptases [31], [39] and for DNA polymerases during PCR [40]. In each of these cases, template switch is possible due to a particular mechanism for dissociating the nascent strand base-paired to its template: the RNA template degradation by the RNAse H activity of reverse transcriptase and the heat-induced melting of DNA duplexes during PCR. Obviously, neither of these means is available for RNA viruses. However, some RNA viruses might have evolved special mechanisms to overcome the duplex problem, e.g. the use of ATP-fueled helicases.
While the involvement of the replicative template switch in RNA recombination has yet to be demonstrated, evidence for non-replicative mechanisms of the breaking and joining type is now available from the in vitro experiments in the Qβ system. The question inevitably arises: to what extent are the conclusions made from in vitro data valid for the natural systems [8]? In this regard, recent experiments on recombination between non-replicable and non-translatable fragments of the poliovirus RNA [41] deserve special attention. Upon transfecting appropriate cells, the fragments did produce viable viral genomes, demonstrating that non-replicative recombination can occur in vivo. The results show that, similar to self-recombination, the 3′-hydroxyls do not participate in the reaction and suggest an involvement of the 2′,3′-cyclic phosphate intermediates. The recombination is non-homologous and, importantly, its frequency is similar to the standard frequency of non-homologous RNA recombination in vivo. The results have also revealed recombinational ‘hot spots’ associated with ribozyme-like RNA structures. Apart from self-recombination, replicase- and ribozyme-assisted recombination, there may exist other non-replicative mechanisms for rearranging the RNA sequences. For example, a mechanism including RNAse-catalyzed cleavage and ligation has been proposed [42].
Acknowledgements
I thank Helena Chetverina for helpful discussions. This work was supported by INTAS Grant 97-10348, Grant 99-04-48672 from the Russian Foundation for Basic Research and by an International Research Scholar’s award from the Howard Hughes Medical Institute.
References
- 1.Hirst G.K. Cold Spring Harbor Symp. Quant. Biol. 1962;27:303–309. doi: 10.1101/sqb.1962.027.001.028. [DOI] [PubMed] [Google Scholar]
- 2.Ledinko N. Virology. 1963;180:107–119. doi: 10.1016/0042-6822(63)90145-4. [DOI] [PubMed] [Google Scholar]
- 3.Lai M.M.C. Microbiol. Rev. 1992;56:61–79. doi: 10.1128/mr.56.1.61-79.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chetverin A.B. Semin. Virol. 1997;8:121–129. [Google Scholar]
- 5.Agol V.I. Semin. Virol. 1997;8:77–84. [Google Scholar]
- 6.Jarvis T.C, Kirkegaard K. Trends Genet. 1991;7:186–191. doi: 10.1016/0168-9525(91)90434-R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wimmer E, Hellen C.U.T, Cao X. Annu. Rev. Genet. 1993;27:353–436. doi: 10.1146/annurev.ge.27.120193.002033. [DOI] [PubMed] [Google Scholar]
- 8.Nagy P.D, Simon A.E. Virology. 1997;235:1–9. doi: 10.1006/viro.1997.8681. [DOI] [PubMed] [Google Scholar]
- 9.Weiss B.G, Schlesinger S. J. Virol. 1991;65:4017–4025. doi: 10.1128/jvi.65.8.4017-4025.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Onodera S, Qiao X, Gottlieb P, Strassman J, Frilander M, Mindich L. J. Virol. 1993;67:4914–4922. doi: 10.1128/jvi.67.8.4914-4922.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nagy P.D, Bujarski J.J. Proc. Natl. Acad. Sci. USA. 1993;90:6390–6394. doi: 10.1073/pnas.90.14.6390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hajjou M, Hill K, Subramaniam S.V, Hu J.Y, Raju R. J. Virol. 1996;70:5153–5164. doi: 10.1128/jvi.70.8.5153-5164.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kuge S, Kawamura N, Nomoto A. J. Virol. 1989;63:1069–1075. doi: 10.1128/jvi.63.3.1069-1075.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pilipenko E.V, Gmyl A.P, Agol V.I. Nucleic Acids Res. 1995;23:1870–1875. doi: 10.1093/nar/23.11.1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Olsthoorn R.C.L, van Duin J. J. Virol. 1996;70:729–736. doi: 10.1128/jvi.70.2.729-736.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Holland, J.J. (1985) in: Virology (Fields, B.N., Ed.), pp. 77–99, Raven Press, New York.
- 17.Schlesinger, S. (1988) in: RNA Genetics (Domingo, E., Holland, J.J. and Ahlquist, P., Eds.), Vol. II, pp. 167–185, CRC Press, Boca Raton, FL.
- 18.Henle W, Henle G. Science. 1943;98:87–89. doi: 10.1126/science.98.2534.87. [DOI] [PubMed] [Google Scholar]
- 19.Palasingam K, Shaklee P.N. J. Virol. 1992;66:2435–2442. doi: 10.1128/jvi.66.4.2435-2442.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cole C.N, Smoler D, Wimmer E, Baltimore D. J. Virol. 1971;7:478–485. doi: 10.1128/jvi.7.4.478-485.1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Figlerowicz M, Nagy P.D, Bujarski J.J. Proc. Natl. Acad. Sci. USA. 1997;94:2073–2078. doi: 10.1073/pnas.94.5.2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Qiao X, Qiao J, Mindich L. Virology. 1997;227:103–110. doi: 10.1006/viro.1996.8311. [DOI] [PubMed] [Google Scholar]
- 23.Chetverin A.B, Chetverina H.V, Demidenko A.A, Ugarov V.I. Cell. 1997;88:503–513. doi: 10.1016/S0092-8674(00)81890-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tang R.S, Barton D.J, Flanegan J.B, Kirkegaard K. RNA. 1997;3:624–633. [PMC free article] [PubMed] [Google Scholar]
- 25.Duggal R, Cuconati A, Gromeier M, Wimmer E. Proc. Natl. Acad. Sci. USA. 1997;94:13786–13791. doi: 10.1073/pnas.94.25.13786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nagy P.D, Zhang C, Simon A.E. EMBO J. 1998;17:2392–2403. doi: 10.1093/emboj/17.8.2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chetverin A.B, Spirin A.S. Prog. Nucleic Acid Res. Mol. Biol. 1995;51:225–270. doi: 10.1016/s0079-6603(08)60880-6. [DOI] [PubMed] [Google Scholar]
- 28.Chetverin A.B, Chetverina H.V, Munishkin A.V. J. Mol. Biol. 1991;222:3–9. doi: 10.1016/0022-2836(91)90729-p. [DOI] [PubMed] [Google Scholar]
- 29.Chetverina H.V, Chetverin A.B. Nucleic Acids Res. 1993;21:2349–2353. doi: 10.1093/nar/21.10.2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Munishkin A.V, Voronin L.A, Ugarov V.I, Bondareva L.A, Chetverina H.V, Chetverin A.B. J. Mol. Biol. 1991;221:463–472. doi: 10.1016/0022-2836(91)80067-5. [DOI] [PubMed] [Google Scholar]
- 31.Negroni M, Riccheti M, Nouvel P, Buc H. Proc. Natl. Acad. Sci. USA. 1995;92:6971–6975. doi: 10.1073/pnas.92.15.6971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rensing U, August J.T. Nature. 1969;224:853–856. doi: 10.1038/224853a0. [DOI] [PubMed] [Google Scholar]
- 33.Chetverina H.V, Demidenko A.A, Ugarov V.I, Chetverin A.B. FEBS Lett. 1999;450:89–94. doi: 10.1016/s0014-5793(99)00469-x. [DOI] [PubMed] [Google Scholar]
- 34.Brosius J, Tiedge H. Virus Genes. 1995;11:163–179. doi: 10.1007/BF01728656. [DOI] [PubMed] [Google Scholar]
- 35.Herschlag D, Khosla M, Tsuchihashi Z, Karpel R.L. EMBO J. 1994;13:2913–2924. doi: 10.1002/j.1460-2075.1994.tb06586.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Coetzee T, Herschlag D, Belfort M. Genes Dev. 1994;8:1575–1588. doi: 10.1101/gad.8.13.1575. [DOI] [PubMed] [Google Scholar]
- 37.Kirkegaard K, Baltimore D. Cell. 1986;47:433–443. doi: 10.1016/0092-8674(86)90600-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Novak J.E, Kirkegaard K. J. Virol. 1991;65:3384–3387. doi: 10.1128/jvi.65.6.3384-3387.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Diaz L, DeStefano J.J. Nucleic Acids Res. 1996;24:3086–3092. doi: 10.1093/nar/24.15.3086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Frohman, M.A. and Martin, G.R. (1990) in: PCR Protocols (Innis, M.A., Ed.), pp. 228–236, Academic Press, New York.
- 41.Gmyl, A.P., Belousov, E.V., Maslova, S.V., Khitrina, E.V., Chetverin, A.B. and Agol, V.I. (1999) J. Virol. 73(11), in press. [DOI] [PMC free article] [PubMed]
- 42.Tsagris M, Tabler M, Singer H.L. Nucleic Acids Res. 1991;19:1605–1612. doi: 10.1093/nar/19.7.1605. [DOI] [PMC free article] [PubMed] [Google Scholar]