

Abstract
This review focuses on the important contributions that macromolecular crystallography has made over the past 12 years to elucidating structures and mechanisms of the essential proteases of coronaviruses, the main protease (Mpro) and the papain‐like protease (PLpro). The role of X‐ray crystallography in structure‐assisted drug discovery against these targets is discussed. Aspects dealt with in this review include the emergence of the SARS coronavirus in 2002–2003 and of the MERS coronavirus 10 years later and the origins of these viruses. The crystal structure of the free SARS coronavirus Mpro and its dependence on pH is discussed, as are efforts to design inhibitors on the basis of these structures. The mechanism of maturation of the enzyme from the viral polyprotein is still a matter of debate. The crystal structure of the SARS coronavirus PLpro and its complex with ubiquitin is also discussed, as is its orthologue from MERS coronavirus. Efforts at predictive structure‐based inhibitor development for bat coronavirus Mpros to increase the preparedness against zoonotic transmission to man are described as well. The paper closes with a brief discussion of structure‐based discovery of antivirals in an academic setting.
Keywords: 3C‐like protease, autoprocessing, bat coronaviruses, high‐throughput screening, main protease, Middle East respiratory syndrome, papain‐like protease, protease maturation, severe acute respiratory syndrome, structure‐based inhibitor design
The coronavirus genome encodes two types of proteases, one chymotrypsin‐like main protease and one or two papain‐like protease(s), both of which are important targets for antiviral drug discovery. From just before the outbreak of the SARS coronavirus in 2002/2003 to the ongoing outbreak of MERS coronavirus, many crystal structures have been determined of these enzymes and their complexes with inhibitors, laying the basis for structure‐based lead optimization.
Abbreviations
- 3CLpro
3C‐like protease
- MERS
Middle East respiratory syndrome
- MERS‐CoV
MERS coronavirus
- Mpro
main protease
- Nsp
non‐structural protein
- PLpro
papain‐like protease
- SARS
severe acute respiratory syndrome
- SARS‐CoV
SARS coronavirus
- TGEV
transmissible gastroenteritis virus
SARS – a decade on
Eleven years ago, the world was shocked by the outbreak of the severe acute respiratory syndrome (SARS), which spread from its origin in the Southern Chinese province of Guangdong to Hong Kong and from there to about 30 countries in the world, of which Vietnam, Singapore, Taiwan and Canada (Toronto) were most affected. Also, the virus travelled from Hong Kong to Beijing, where alone more than 3000 SARS cases were recorded. Altogether, about 8000 cases have been registered worldwide, of whom about 10% did not survive. SARS was characterized by an atypical, severe pneumonia (for recent reviews commemorating the 2003 SARS outbreak and discussing the lessons learned, see 1, 2, 3, 4).
On 24 March 2003 a new coronavirus, appropriately named SARS coronavirus (SARS‐CoV), was described as the etiological agent causing the epidemic 5, 6, 7, 8. This virus was rapidly classified as an outlier of what were called group 2 coronaviruses at the time 9; according to the new nomenclature introduced a few years later (see for example 10), SARS‐CoV belongs to clade b of the genus Betacoronavirus.
Newly discovered and newly emerging human coronaviruses
Following the SARS epidemic, two new human coronaviruses have been discovered due to intensified research efforts targeting this previously neglected virus family. In 2004 human coronavirus NL63, a member of the genus Alphacoronavirus, was described 11, 12, followed by the discovery of HCoV HKU1, a clade‐a betacoronavirus, a year later 13. These viruses are widespread but do not cause severe disease in the majority of people infected by them 14. In September 2012 another novel human coronavirus, Middle East respiratory syndrome (MERS) coronavirus, was described 15. It had been detected in patients from Saudi Arabia and other countries on the Arab peninsula or in people who had a history of travel to the Middle East. The earliest cluster of MERS cases detected so far was in Jordan in April 2012, as shown retrospectively on the basis of patient samples. Symptoms of MERS include severe respiratory disease and often renal failure; as of 4 July 2014, 827 laboratory‐confirmed cases have been recorded, with 287 deaths (http://www.who.int). The case–fatality ratio of MERS is thus alarmingly high.
Where did the SARS and MERS coronaviruses come from?
In the case of SARS‐CoV, wild animals such as palm civets, sold as a delicacy on Chinese ‘wet markets’, were initially identified as the immediate source of the virus 16, but from 2005 insectivorous Rhinolophid bats came into focus as the original reservoir, from where the virus was possibly transmitted to civets and other market species and from them to humans 17, 18 (see 19 for a recent review on bat coronaviruses). However, it took until 2013 to discover a bat coronavirus that is more than 95% identical to SARS‐CoV and uses the same receptor on the surface of host cells, the angiotensin‐converting enzyme 2 (ACE2) 20. In the case of MERS coronavirus (MERS‐CoV), bats were again suspected to be the reservoir as a few coronaviruses with high sequence similarity to MERS‐CoV were discovered in African and European bats 21, 22, but in recent months the picture has changed somewhat and dromedary camels are now the main suspects of being the reservoir from where the zoonotic transmission into the human population originates 23, 24.
After the SARS epidemic was over, many scientists and policy‐makers, including even many virologists, believed that the event was unique and chances of repetition were extremely low. Thus, it must be said that more effort could (and should) have been made to develop small‐molecule compounds with anti‐coronavirus activity; this was hampered, however, by a sharp decline in funding of coronavirus research in many countries after 2005–2006, and lack of support from the scientific community. As a consequence, not all lessons that the SARS outbreak taught us were taken seriously (discussed in 1). But the recent – and still continuing – emergence of MERS‐CoV has illustrated that such an event can happen anywhere, at any time, given the large number of coronavirus species in Nature, of which we probably only know a fraction so far. Coronaviruses feature the largest RNA genome (about 30 kb; Fig. 1) known, and this genome is extremely flexible in terms of incorporation and deletion of gene products in response to evolutionary pressure such as the need to adapt to a new host. The coronavirus genome is also prone to recombination events, thereby adding further to its flexibility.
Figure 1.
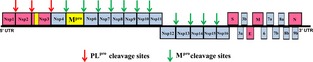
Schematic presentation of the genome of the SARS coronavirus. Occupying two‐thirds of the genome from the 5′ end, open‐reading frame 1 (ORF1) encodes two large polyproteins, pp1a and, through ribosomal frameshifting during translation, pp1ab. These polyproteins are processed into mature Nsps by the two proteases discussed here (indicated in yellow). The main protease (Mpro, also called 3C‐like protease, 3CL pro) is Nsp5, whereas the papain‐like protease (PL pro) is a part of Nsp3. The PL pro performs three cleavage reactions (red arrows) to release Nsp1, Nsp2 and Nsp3 (red), whereas the Mpro cleaves the polyprotein at 11 sites (cyan arrows) to release Nsp4–Nsp16 (cyan). The 3′‐terminal third of the genome codes for structural and accessory proteins.
The coronavirus main protease (Mpro)
In this review, I will illuminate the question whether and how macromolecular crystallography contributed to the discovery of antivirals targeting proteins from the new viruses, SARS‐CoV and MERS‐CoV. In doing so, I will focus on the main antiviral drug targets, the coronavirus main protease (Mpro, also called the 3C‐like protease, 3CLpro) and the papain‐like protease (PLpro). Other enzymes of the coronaviruses, such as the helicase and the RNA‐dependent RNA polymerase, are also targets for antiviral drug discovery, but such efforts are limited so far because of the lack of crystal structures for these enzymes (see 25 for a recent review). The coronaviral proteases Mpro and PLpro are responsible for processing the huge polyproteins pp1a and pp1ab, which are encoded by open reading frame 1 (ORF1) of the coronavirus genome, into mature non‐structural proteins (Nsps), most of which form part of the coronaviral replication/transcription complex (Fig. 1; for information on other SARS‐CoV protein structures see 1, 25, 26, 27).
The Mpro is encoded by ORF1 as non‐structural protein 5 (Nsp5) and is responsible for no less than 11 cleavage sites in the polyproteins (Fig. 1). It is flanked by the proteins Nsp4 and Nsp6 which, along with parts of Nsp3, anchor the replication/transcription complex to double‐membrane vesicles that are derived from the endoplasmic reticulum membrane during the infection 28. Substrate cleavage by the Mpro follows the general pattern (small)‐X‐(L/F/M)‐Q↓(G/A/S)‐X (X ≡ any amino acid; ↓ cleavage site); in particular, the glutamine (Q) residue in the P1 position of the substrate is an absolute requirement. As no host‐cell proteases are known with this specificity, prospects for coming up with anti‐coronavirals without too many side‐effects are actually good.
Crystallographic studies on coronavirus Mpro prior to and during the SARS outbreak
My group had started working on the coronavirus Mpro around 1999. At that time, not a single crystal structure of a coronavirus protein had been determined. We first elucidated the crystal structure of the Mpro of transmissible gastroenteritis virus (TGEV), a porcine coronavirus that is fatal for young piglets. Published in 2002 29, the structure revealed that the Mpro is a dimer (cf. Fig. 2) in which the N‐terminus (the ‘N‐finger’) of one monomer helps shape the S1 substrate‐specificity pocket and the oxyanion hole of the other monomer; hence, dimerization is a prerequisite for catalytic activity. It also revealed the presence of an α‐helical domain (domain III) in addition to domains I and II, which together feature a chymotrypsin‐like fold and harbor the catalytic Cys…His dyad between them. Subsequently, we synthesized a chloromethylketone inhibitor and cocrystallized it with the TGEV Mpro in order to visualize the substrate‐binding site in detail 30. At the same time, we also determined the structure of the Mpro of human coronavirus 229E (HCoV 229E). When SARS‐CoV was identified and sequenced in the spring of 2003, we built the first homology model of the SARS‐CoV Mpro on the basis of the structure of the enzyme from HCoV 229E 30. We further suggested, on the basis of the binding mode of our chloromethylketone inhibitor, that the Michael acceptor compound AG7088 (rupintrivir), which was being developed by Pfizer as an inhibitor of the 3C protease of human rhinovirus 31, should be a good starting point for anti‐SARS drug design 30, 32. Later, this compound turned out not to have particularly high activity against SARS‐CoV in cell culture, but derivatives of this Michael acceptor lead turned out to exhibit good anti‐coronaviral activity in vitro and ex vivo 33, 34, 35. Towards the end of the SARS outbreak in Beijing (in June 2003), the crystal structure of the SARS‐CoV main protease itself was determined through a collaboration between the group of Zihe Rao in Beijing, who had recombinantly produced and crystallized the enzyme, and my group, both as the free protease (Fig. 2) and in complex with the chloromethylketone inhibitor that we had already used for the TGEV Mpro 36.
Figure 2.
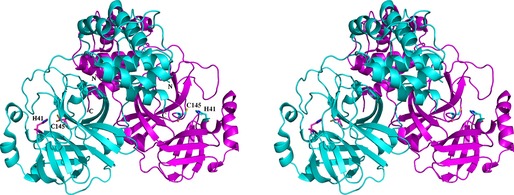
Stereo presentation of the structure of the SARS‐CoV Mpro dimer 36. The catalytic dyads of each subunit (Cys145…His41) are indicated, as are the N‐ and C‐termini. Note that the N‐terminus of the cyan polypeptide chain is located close to the substrate‐binding site of the purple subunit.
Influence of pH on the Mpro structure
The first structure of the free SARS‐CoV Mpro 36 was determined from crystals that had been grown at acidic pH (around 6.0); in this structure, one monomer of the Mpro dimer was in the active state and the other one in a catalytically incompetent conformation in which the S1 specificity pocket and the oxyanion hole were collapsed. When the same crystals were equilibrated in buffer of pH 7.4, both monomers were found in the active conformation, whereas at pH 8.0 the substrate‐binding site was less well defined due to increasing flexibility of the amino acid side‐chains involved. This phenomenon was explained by molecular dynamics simulations run with different protonation states for two key histidine residues (His163 and His172) involved in shaping the S1 substrate‐binding site 37. The pH–activity profile of the SARS‐CoV Mpro was found to be very probably determined by protonation of His163 (inactivation at acidic pH) and deprotonation of His172 (inactivation at basic pH) 37. The observation of the catalytically incompetent form (with the S1 site and the oxyanion hole collapsed) has occasionally been ascribed (e.g. 38) to the presence of five additional residues at the N‐terminus that remained from the cloning procedure; the phenomenon has not been observed with enzyme featuring authentic chain termini when crystalized in space group C2 39. We have determined structures of the SARS‐CoV Mpro with authentic chain termini from crystals grown with other symmetries and did observe the presence of both an active and an inactive monomer at low pH 40 (Verschueren et al., unpublished). The existence of a less active proform of the enzyme may allow control of the temporal order of processing the individual polyprotein cleavage sites to release intermediate and mature Nsps at the time in replication when they are needed. Unfortunately, the pH at the site of action of the Mpro, at the endoplasmic‐reticulum‐derived double‐membrane vesicles 28, is not known.
How does Mpro maturation work?
Before auto‐activation and liberation from the viral polyproteins pp1a and pp1ab, the Mpro (Nsp5) is an integral part of these polyproteins (Fig. 1). The mechanism of auto‐activation of the enzyme is not well understood (see 38 for a review). Several studies have used constructs carrying fluorescent proteins at both termini of the SARS‐CoV Mpro and connected to the enzyme by peptide sequences containing Mpro cleavage sites 41, 42. Such polyprotein models are usually monomeric, but dimer formation upon addition of substrates has been observed 41. We have found that upon mutation of three residues (Arg4, Glu290 and Arg298) involved in the monomer–monomer interface of the mature protease, the resulting monomeric enzyme can still perform N‐terminal autocleavage, while dimerization and trans‐cleavage activity are completely inhibited by the Glu290Arg and Arg298Glu mutations and partly so by the Arg4Glu mutation. Furthermore, the mature Glu290Arg mutant can resume N‐terminal autocleavage activity when mixed with an inactive Mpro species, whereas its trans‐cleavage activity remains absent. Therefore the N‐terminal autoprocessing of the Mpro appears to require only two ‘immature’ monomers approaching one another to form an ‘intermediate’ dimer structure and does not depend on the active dimer conformation existing in the mature protease 43. The octameric form of the immature Mpro, which features a three‐dimensional swap of the helical domain III of the enzyme 44, may play a role in the auto‐activation process.
Discovery and design of Mpro inhibitors
A large number of crystal structures have been published of inhibitor complexes of the SARS‐CoV Mpro, of which only a few can be mentioned here. Many types of chemical warheads have been used to achieve covalent binding of peptidic or peptidomimetic inhibitors to the active‐site cysteine of the Mpro, including the halomethylketones 30, 36, 45 and Michael acceptor compounds (α,β‐unsaturated esters) 33, 34, 35 mentioned above, aldehydes 46, 47, 48, 49, α,β‐epoxyketones 50, 51, 52, nitriles 53 and phthalhydrazide ketones 54, 55. All of these compounds are peptidomimetics carrying electrophilic warheads, and several also efficiently inhibit SARS‐CoV replication in cell culture. Some of the inhibitors, such as for example halomethylketones, are certainly too reactive to be developed into drugs, as they are expected to exhibit considerable side‐effects. One might intuitively assume the same of aldehydes, but in fact peptide aldehyde inhibitors of thrombin (such as efegatran) did not show toxicity in clinical trials 56, 57. Also, it should be noted that two hepatitis C virus NS3/NS4A protease inhibitors introduced into the market in 2011, telaprivir and boceprivir, are peptidomimetics carrying the α‐ketoamide warhead 58. Finally, rupintrivir (AG7088) is an example of a Michael acceptor compound that was developed as an inhibitor of the 3C protease of human rhinovirus 31. There is a trend away from non‐covalent binders of target serine or cysteine proteases and towards covalent reversible or irreversible binders. Given the absolute requirement of the coronavirus Mpro for glutamine in the P1 position of the substrate, and the absence of human proteases with the same specificity, there is a good chance of developing coronavirus protease inhibitors carrying electrophilic warheads without having to expect too many side‐effects (see above). Figure 3 shows the binding of our broad‐spectrum Michael acceptor compound SG85, which we originally developed against the enterovirus 3C protease 59, in complex with the SARS‐CoV Mpro, as revealed by X‐ray crystallography (Zhu et al., unpublished; PDB code http://www.rcsb.org/pdb/search/structidSearch.do?structureId=3TNT). In agreement with the expectation outlined above, this compound shows no sign of toxicity in Huh‐T7 or Vero A cells (CC50 = 256 and 190 μm, respectively 59) or in mice (Leyssen, Neyts et al., unpublished), while it exhibits an IC50 of around 2 μm both against the isolated SARS‐CoV Mpro and in a SARS‐CoV replicon and of about 3.3 μm in SARS‐CoV‐infected Vero B4 cells (Zhu, Kusov, Muth et al., unpublished).
Figure 3.
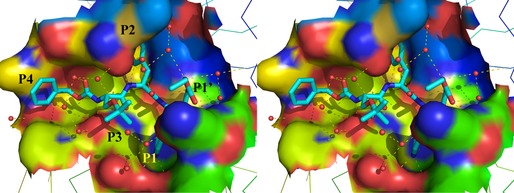
Stereo illustration of the Michael acceptor compound SG85 (Cbz–(tBu–O–)Ser–Phe–GlnLactam–CH=CH–CO–O–Et; 59) bound to the substrate‐binding site of the SARS‐CoV Mpro (Zhu et al., unpublished; PDB code http://www.rcsb.org/pdb/search/structidSearch.do?structureId=3TNT). The inhibitor is shown in cyan (for carbon), blue (for nitrogen) and red (for oxygen). Hydrogen bonds are indicated by dashed lines and water molecules by small red spheres. The P4–P1’ side‐chains of the inhibitor are labeled. The P1 side‐chain is buried in the S1 pocket and only the tip of its lactam moiety is visible in this illustration.
In addition, a number of non‐peptidic, reversible inhibitors of the main protease have been discovered by virtual screening and/or docking on the basis of the crystal structure; examples for such compounds are cinanserin 60, arylboronic acids 61, isatin derivatives 62, selected diarylsulfones 63 and a variety of others 63, 64. Other non‐peptidic inhibitors, such as benzotriazole esters 40, 65, 66 and non‐warheaded benzo 1, 2, 3triazoles 67, were discovered by screening of chemical libraries and subsequent optimization of the hits by medicinal chemistry. Chloropyridyl esters have been derived from the benzotriazole esters and found to have good antiviral activity in cell culture 68.
The SARS‐CoV papain‐like protease (PLpro): functions in the viral replication cycle and in antagonizing innate immunity
The other protease encoded by the SARS‐CoV genome, the papain‐like protease, is responsible for processing three cleavage sites in the N‐terminal part of the polyproteins, to produce mature Nsp1, Nsp2 and Nsp3 (Fig. 1). The cleavage specificity of the PLpro corresponds to the pattern (R/K)L(R/K)GG↓X. In addition, the enzyme is a deubiquitinase, i.e. it removes (poly)ubiquitin units from proteins tagged with them 69, 70. Ubiquitin carries the sequence LRLRGG at its C‐terminus, in perfect agreement with the coronavirus PLpro recognition motif. The deubiquitinase activity of the enzyme interferes, in an as‐yet unknown way, with the phosphorylation and nuclear import of interferon‐regulatory factor 3 (IRF3) and thereby prevents the production of type‐I interferons by the infected host cell 71, 72, 73. The SARS‐CoV PLpro has also been shown to have deISG15ylating activity 74, i.e. it removes ISG15 units from target proteins labeled this way (ISG, interferon‐stimulated gene product). Finally, the SARS‐CoV PLpro has been demonstrated to interfere with the nuclear factor κB pathway, i.e. it is an important weapon of the virus in its efforts to counteract the innate immune response of the infected host cell 73.
Crystallographic studies on the SARS‐CoV PLpro and inhibitor discovery
The crystal structure of the SARS‐CoV PLpro was reported by Ratia et al. 75. The enzyme consists of an N‐terminal ubiquitin‐like (Ubl) domain and a catalytic core domain that features an open‐right‐hand fold, with thumb, palm and fingers subdomains. At the tip of the fingers domain, a structural zinc ion is found within a zinc‐ribbon structure (Fig. 4). It took a number of years to obtain a crystalline complex between the SARS‐CoV PLpro and ubiquitin; only very recently, Chou et al. 76 published the structure of a complex between ubiquitin and a PLpro that had the catalytic cysteine residue replaced by serine, and Ratia et al. 77 reported the structure of the native SARS‐CoV PLpro in complex with ubiquitin aldehyde, where the C‐terminal aldehyde group forms a covalent bond with the catalytic Cys112 of the enzyme.
Figure 4.
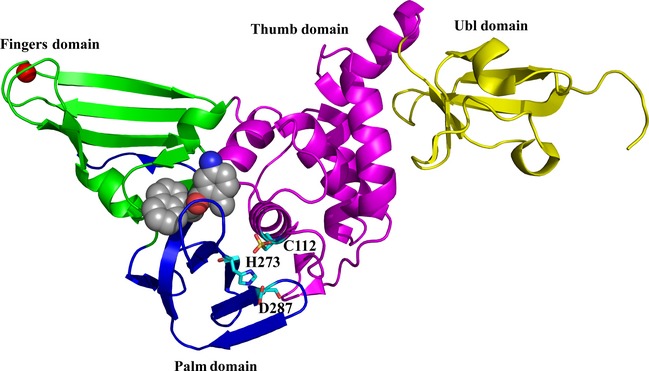
Structure of the SARS‐CoV PL pro in complex with the non‐peptidic inhibitor GRL0617 78. The domains of the enzyme are indicated and colored as follows: yellow, ubiquitin‐like (Ubl) domain; pink, thumb domain; blue, palm domain; cyan, fingers domain. The zinc ion bound at the tip of the fingers domain is colored red. The inhibitor (space‐filling presentation, with carbon in grey, nitrogen in blue and oxygen in red) binds to the S3 and S4 sites, far from the catalytic triad, C112–H273–D287 (cyan sticks).
The use of peptidomimetic inhibitors to block the SARS‐CoV PLpro is connected with the difficulty that such inhibitors would very likely also inhibit host‐cell deubiquitinases, so that severe side‐effects would have to be expected. Therefore, the search for inhibitors of the SARS‐CoV PLpro focused on screening chemical libraries for non‐peptidic, reversible inhibitors of the enzyme. This way, Ratia et al. 78 and Ghosh et al. 79, 80 identified hit compounds that were further optimized to yield inhibitors with submicromolar activities against the isolated enzyme and low‐micromolar activities in SARS‐CoV‐infected cell cultures (see also 81). The hit‐to‐lead optimization relied heavily on crystal structures of complexes between selected candidate inhibitors and the SARS‐CoV PLpro. Several of the inhibitors discovered this way, e.g. GRL0617 78, did not bind directly to the catalytic site of the protease but near the S3 and S4 sites (these are more spacious than the restricted S1 and S2 sites, which can accommodate exclusively glycine residues of the substrates, i.e. viral polyprotein or ubiquitin). Figure 4 shows the inhibitor GRL0617 (space‐filling presentation) bound to the S3 and S4 sites, far from the catalytic triad (cyan sticks).
Crystallographic and inhibitor discovery studies with bat coronavirus Mpro: increasing the preparedness against zoonotic transmission
As evidence was growing for a zoonotic transmission of SARS‐CoV from bats via intermediate hosts to humans 17, 18, we started to get interested in bat coronavirus main proteases as drug targets. Obviously, the goal is not to cure bats from their coronavirus infections (being the reservoir, most bats do not show any sign of disease when they carry coronaviruses), but we want to design inhibitors for these enzymes to have them ready in case of a zoonotic transmission of a bat coronavirus into the human population. The idea is to design and synthesize one or more lead compound(s) with broad‐spectrum anti‐coronaviral activity, which can immediately enter preclinical development in the case of a major epidemic. At the outset of this project, we selected three bat coronaviruses as representatives for coronavirus families: Bt‐CoV HKU8 as an alphacoronavirus 82, Bt‐CoV HKU4 as a betacoronavirus of clade c 83 and Bt‐HKU9 as a betacoronavirus of clade d 83, 84. (We excluded Betacoronavirus clades a and b as no bat coronaviruses of the former are known and clade b is already presented by the well‐studied SARS‐CoV.) So far, we have determined the crystal structures of the Mpros of Bt‐CoV HKU8 (Ma et al., unpublished) and HKU4 (Xiao et al., unpublished; PDB codes http://www.rcsb.org/pdb/search/structidSearch.do?structureId=2YNA, http://www.rcsb.org/pdb/search/structidSearch.do?structureId=2YNB) and have noticed that our above‐mentioned broad‐spectrum antiviral SG85, a Michael acceptor compound 59, inhibited the HKU4 (but not the HKU8) enzyme. Proof‐of‐principle for our ‘predictive’ approach came when MERS‐CoV emerged in 2012 and we found that SG85 was indeed a good inhibitor of this virus in cell culture (Xiao, de Wilde, Muth et al., to be published). BtCoV HKU4 turned out to be a close relative of MERS‐CoV, with 81% amino acid sequence identity (90% similarity) for the main protease.
The inactivity of SG85 against the HKU8 Mpro, however, also suggests that our inhibitors have to become more broad spectrum than they are at present. Ideally, one would like to have one broad‐spectrum antiviral at hand that would be efficacious against all coronavirus families. Modifications of SG85 with good activity against alphacoronaviruses are now under development in our laboratory.
Structure‐based inhibitor discovery against MERS coronavirus
Just as for SARS‐CoV, the main protease (Mpro or 3CLpro) and the papain‐like protease (PLpro) are prime targets for the development of antivirals against the newly emerging MERS‐CoV. A three‐dimensional structure was described for the Mpro shortly after the discovery of the new virus 85, but unfortunately atomic coordinates have not been deposited in the Protein Data Bank. The same publication describes the SARS‐CoV Mpro inhibitor N3, a Michael acceptor compound 33, as a good inhibitor of the MERS‐CoV Mpro 85. The structure of the papain‐like protease of the new virus has also been determined 86. The enzyme features significant differences from the SARS‐CoV PLpro. Thus, the stabilization of the oxyanion intermediate of the proteolytic reaction catalyzed by the MERS‐CoV PLpro appears to be different from the mechanism proposed for the SARS‐CoV PLpro 75. In papain‐like proteases, the oxyanion is commonly stabilized by two hydrogen bonds from the enzyme, one donated by the main‐chain amide of the catalytic residue, here Cys111, and the other from a glutamine or asparagine side‐chain five or six residues N‐terminal to the catalytic cysteine. In SARS‐CoV PLpro, the corresponding side‐chain is that of Trp107, which is proposed to donate a hydrogen bond to the oxyanion from the indole nitrogen 75. But in the MERS‐CoV PLpro this tryptophan is replaced by Leu106, which lacks hydrogen‐bonding capability. Interestingly, the Leu106Trp mutation of the MERS‐CoV PLpro increases the peptidolytic and deubiquitinating activities of the enzyme by factors of 60 and 3.4, respectively 86, indicating that the protease has not been optimized for maximum activity during evolution of the virus. Other differences between the SARS‐CoV PLpro and the MERS‐CoV PLpro include the S3 and S5 specificity subsites. These subsites accommodate arginine residues of ubiquitin in the SARS‐CoV PLpro‐ubiquitin complex 76, 77 and arginine or lysine at the PLpro cleavage sites in the viral polyprotein. Accordingly, the subsites are dominated by negatively charged amino acid side‐chains in the SARS‐CoV enzyme, i.e. Asp165 in the S3 site and Glu168 in the S5 site. However, in the MERS‐CoV PLpro, the latter residue is replaced by the positively charged Arg168. Hence, direct extrapolation from the structure of the SARS‐CoV PLpro‐ubiquitin complex 76, 77 to ubiquitin recognition by the MERS‐CoV enzyme is not possible; rather, the crystal structure of the complex has to be awaited.
Concluding remarks
The response of the crystallographic community to the SARS outbreak has occasionally been described as ‘swift’; however, to be realistic, it should be noted that had we not determined the structures of the TGEV and HCoV‐229E Mpro, including that of an inhibitor complex, prior to the SARS outbreak, the response would probably have been significantly slower. Nevertheless, I hope that I was able in this review to illustrate the important role played by X‐ray crystallography in elucidating the three‐dimensional structures of two important targets for the discovery and development of anti‐coronavirus drugs, the main protease (Mpro) and the papain‐like protease (PLpro). In fact, most of the peptidomimetic inhibitors of the Mpro were designed on the basis of the structural knowledge of the enzyme, whereas several non‐peptidic inhibitors were identified by using the crystal structure of the target for virtual screening of chemical libraries. The known inhibitors of the PLpro, on the other hand, are mostly based on original hits identified in high‐throughput screening or virtual screening campaigns against the recombinant enzyme, which were subsequently optimized according to their docking to the SARS‐CoV PLpro or to the crystal structure of their complex with the target. However, none of the compounds directed against the coronavirus proteases has gone through a complete preclinical development program, mainly because of a sharp decline in funding in most countries in 2005–2006. Nonetheless, some of the inhibitors described so far are good starting points for development in the case of future zoonotic transmissions of coronaviruses into the human population, or in the case of a continuation of the MERS outbreak.
It is occasionally argued that drug discovery should remain a domain of the pharmaceutical industry and not a priority in academia, as the former is undoubtedly better at it. However, it should be realized that big pharma generally has little interest in emerging RNA viruses, because these typically cause self‐limiting rather than chronic disease. Yet, these viruses potentially pose a big threat to man, as we were impressively taught by the SARS coronavirus 1, and we are well advised to increase our preparedness in view of the increasing frequency of outbreaks caused by these viruses 87, 88. Academic institutions have important tasks in these efforts at increasing preparedness, as far as the preclinical discovery phase of the drug development process is concerned 89. Macromolecular crystallography will undoubtedly continue to play a major role in these efforts.
Acknowledgements
I am grateful to Linlin Zhang and Jian Lei for help with the figures. I am indebted to the past and present coworkers in my coronaviral protease research group, in particular Kanchan Anand, Stefan Anemüller, Shuai Chen, Shyla George, Yuri Kusov, Jian Lei, Daizong Lin, Qingjun Ma, Jeroen R. Mesters, Ksenia Pumpor, Jinzhi Tan, Koen Verschueren, Linlin Zhang and Lili Zhu. I also acknowledge my past and present collaborators, in particular Christian Drosten (Bonn), Hualiang Jiang (Shanghai), Hong Liu (Shanghai), Johan Neyts (Leuven), Leo L. M. Poon (Hong Kong), Jörg Rademann (Berlin), Zihe Rao (Beijing), Xu Shen (Shanghai), Eric J. Snijder (Leiden) and John Ziebuhr (then at Würzburg). Work from my laboratory described in this article was supported by the European Commission (projects SEPSDA, VIZIER and SILVER), the German Center for Infection Research (DZIF) and the Deutsche Forschungsgemeinschaft.
This article is dedicated to my academic teacher, Professor Wolfram Saenger, on the occasion of his 75th birthday.
References
- 1. Hilgenfeld R & Peiris JSM (2013) From SARS to MERS: 10 years of research on highly pathogenic human coronaviruses. Antiviral Res 100, 286–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cheng VC, Chan JF, To KK & Yuen KY (2013) Clinical management and infection control of SARS: lessons learned. Antiviral Res 100, 407–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Graham RL, Donaldson EF & Baric RS (2013) A decade after SARS: strategies for controlling emerging coronaviruses. Nat Rev Microbiol 11, 836–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Koplan JP, Butler‐Jones D, Tsang T & Yu W (2013) Public health lessons from severe acute respiratory syndrome a decade later. Emerg Infect Dis 19, 861–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Peiris JS, Lai ST, Poon LL, Guan Y, Yam LY, Lim W, Nicholls J, Yee WK, Yan WW, Cheung MT et al, SARS Study Group (2003) Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet 361, 1319–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ksiazek TG, Erdman D, Goldsmith CS, Zaki SR, Peret T, Emery S, Tong S, Urbani C, Comer JA, Lim W et al, SARS Working Group (2003) A novel coronavirus associated with severe acute respiratory syndrome. N Engl J Med 348, 1953–1966. [DOI] [PubMed] [Google Scholar]
- 7. Drosten C, Günther S, Preiser W, van der Werf S, Brodt HR, Becker S, Rabenau H, Panning M, Kolesnikova L, Fouchier RA et al (2003) Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N Engl J Med 348, 1967–1976. [DOI] [PubMed] [Google Scholar]
- 8. Kuiken T, Fouchier RA, Schutten M, Rimmelzwaan GF, van Amerongen G, van Riel D, Laman JD, de Jong T, van Doornum G, Lim W et al (2003) Newly discovered coronavirus as the primary cause of severe acute respiratory syndrome. Lancet 362, 263–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Snijder EJ, Bredenbeek PJ, Dobbe JC, Thiel V, Ziebuhr J, Poon LL, Guan Y, Rozanov M, Spaan WJ & Gorbalenya AE (2003) Unique and conserved features of genome and proteome of SARS‐coronavirus, an early split‐off from the coronavirus group 2 lineage. J Mol Biol 331, 991–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Woo PC, Huang Y, Lau SK & Yuen KY (2010) Coronavirus genomics and bioinformatics analysis. Viruses 2, 1804–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. van der Hoek L, Pyrc K, Jebbink MF, Vermeulen‐Oost W, Berkhout RJ, Wolthers KC, Wertheim‐van Dillen PM, Kaandorp J, Spaargaren J & Berkhout B (2004) Identification of a new human coronavirus. Nat Med 10, 368–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fouchier RA, Hartwig NG, Bestebroer TM, Niemeyer B, de Jong JC, Simon JH & Osterhaus AD (2004) A previously undescribed coronavirus associated with respiratory disease in humans. Proc Natl Acad Sci USA 101, 6212–6216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Woo PC, Lau SK, Chu CM, Chan KH, Tsoi HW, Huang Y, Wong BH, Poon RW, Cai JJ, Luk WK et al (2005) Characterization and complete genome sequence of a novel coronavirus, coronavirus HKU1, from patients with pneumonia. J Virol 79, 884–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pyrc K, Berkhout B & van der Hoek L (2007) The novel human coronaviruses NL63 and HKU1. J Virol 81, 3051–3057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus AD & Fouchier RA (2012) Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med 367, 1814–1820. [DOI] [PubMed] [Google Scholar]
- 16. Guan Y, Zheng BJ, He YQ, Liu XL, Zhuang ZX, Cheung CL, Luo SW, Li PH, Zhang LJ, Guan YJ et al (2003) Isolation and characterization of viruses related to the SARS coronavirus from animals in southern China. Science 302, 276–278. [DOI] [PubMed] [Google Scholar]
- 17. Li W, Shi Z, Yu M, Ren W, Smith C, Epstein JH, Wang H, Crameri G, Hu Z, Zhang H et al (2005) Bats are natural reservoirs of SARS‐like coronaviruses. Science 310, 676–678. [DOI] [PubMed] [Google Scholar]
- 18. Lau SK, Woo PC, Li KS, Huang Y, Tsoi HW, Wong BH, Wong SS, Leung SY, Chan KH & Yuen KY (2005) Severe acute respiratory syndrome coronavirus‐like virus in Chinese horseshoe bats. Proc Natl Acad Sci USA 102, 14040–14045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Drexler JF, Corman VM & Drosten C (2013) Ecology, evolution and classification of bat coronaviruses in the aftermath of SARS. Antiviral Res 101, 45–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ge XY, Li JL, Yang XL, Chmura AA, Zhu G, Epstein JH, Mazet JK, Hu B, Zhang W, Peng C et al (2013) Isolation and characterization of a bat SARS‐like coronavirus that uses the ACE2 receptor. Nature 503, 535–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Annan A, Baldwin HJ, Corman VM, Klose SM, Owusu M, Nkrumah EE, Badu EK, Anti P, Agbenyega O, Meyer B et al (2013) Human betacoronavirus 2c EMC/2012‐related viruses in bats, Ghana and Europe. Emerg Infect Dis 19, 456–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ithete NL, Stoffberg S, Corman VM, Cottontail VM, Richards LR, Schoeman MC, Drosten C, Drexler JF & Preiser W (2013) Close relative of human Middle East respiratory syndrome coronavirus in bat, South Africa. Emerg Infect Dis 19, 1697–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Reusken CB, Haagmans BL, Müller MA, Gutierrez C, Godeke GJ, Meyer B, Muth D, Raj VS, Smits‐de Vries L, Corman VM et al (2013) Middle East respiratory syndrome coronavirus neutralising serum antibodies in dromedary camels: a comparative serological study. Lancet Infect Dis 13, 859–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Haagmans BL, Al Dhahiry SH, Reusken CB, Raj VS, Galiano M, Myers R, Godeke GJ, Jonges M, Farag E, Diab A et al (2014) Middle East respiratory syndrome coronavirus in dromedary camels: an outbreak investigation. Lancet Infect Dis 14, 140–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Subissi L, Imbert I, Ferron F, Collet A, Coutard B, Decroly E & Canard B (2014) SARS‐CoV ORF1b‐encoded nonstructural proteins 12−16: replicative enzymes as antiviral targets. Antiviral Res 101, 122–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hilgenfeld R, Tan J, Chen S, Shen X & Jiang H (2008) Structural proteomics of emerging viruses: the examples of SARS‐CoV and other coronaviruses In Structural Proteomics and Its Impact on the Life Sciences (Sussman J. & Silman I, eds), pp. 361–433. World Scientific, Singapore. [Google Scholar]
- 27. Li F (2013) Receptor recognition and cross‐species infections of SARS coronavirus. Antiviral Res 100, 246–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Knoops K, Kikkert M, van den Worm SH, Zevenhoven‐Dobbe JC, van der Meer Y, Koster AJ, Mommaas AM & Snijder EJ (2008) SARS‐coronavirus replication is supported by a reticulovesicular network of modified endoplasmic reticulum. PLoS Biol 6, e226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Anand K, Palm GJ, Mesters JR, Siddell SG, Ziebuhr J & Hilgenfeld R (2002) Structure of coronavirus main proteinase reveals combination of a chymotrypsin fold with an extra alpha‐helical domain. EMBO J 21, 3213–3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Anand K, Ziebuhr J, Wadhwani P, Mesters JR & Hilgenfeld R (2003) Coronavirus main proteinase (3CLpro) structure: basis for design of anti‐SARS drugs. Science 300, 1763–1767. [DOI] [PubMed] [Google Scholar]
- 31. Matthews DA, Dragovich PS, Webber SE, Fuhrman SA, Patick AK, Zalman LS, Hendrickson TF, Love RA, Prins TJ, Marakovits JT et al (1999) Structure‐assisted design of mechanism‐based irreversible inhibitors of human rhinovirus 3C protease with potent antiviral activity against multiple rhinovirus serotypes. Proc Natl Acad Sci USA 96, 11000–11007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Anand K, Yang H, Bartlam M, Rao Z & Hilgenfeld R (2005) Coronavirus main proteinase: target for antiviral drug therapy In Coronaviruses with Special Emphasis on First Insights Concerning SARS (Schmidt A, Wolf MH. & Weber O, eds), pp. 173–199. Birkhäuser, Basel. [Google Scholar]
- 33. Yang H, Xie W, Xue X, Yang K, Ma J, Liang W, Zhao Q, Zhou Z, Pei D, Ziebuhr J et al (2005) Design of wide‐spectrum inhibitors targeting coronavirus main proteases. PLoS Biol 3, 1742–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ghosh AK, Xi K, Grum‐Tokars V, Xu X, Ratia K, Fu W, Houser KV, Baker SC, Johnson ME & Mesecar AD (2007) Structure‐based design, synthesis, and biological evaluation of peptidomimetic SARS‐CoV 3CLpro inhibitors. Bioorg Med Chem Lett 17, 5876–5880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lee CC, Kuo CJ, Ko TP, Hsu MF, Tsui YC, Chang SC, Yang S, Chen SJ, Chen HC, Hsu MC et al (2009) Structural basis of inhibition specificities of 3C and 3C‐like proteases by zinc‐coordinating and peptidomimetic compounds. J Biol Chem 284, 7646–7655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yang H, Yang M, Ding Y, Liu Y, Lou Z, Zhou Z, Sun L, Mo L, Ye S, Pang H et al (2003) The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc Natl Acad Sci USA 100, 13190–13195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tan J, Verschueren KHG, Anand K, Shen J, Yang M, Xu Y, Rao Z, Bigalke J, Heisen B, Mesters JR et al (2005) pH‐dependent conformational flexibility of the SARS‐CoV main proteinase (Mpro) dimer: molecular dynamics simulations and multiple X‐ray structure analyses. J Mol Biol 354, 25–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Xia B & Kang X (2011) Activation and maturation of SARS‐CoV main protease. Protein Cell 2, 282–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Xue X, Yang H, Shen W, Zhao Q, Li J, Yang K, Chen C, Jin Y, Bartlam M & Rao Z (2007) Production of authentic SARS‐CoV Mpro with enhanced activity: application as a novel tag‐cleavage endopeptidase for protein overproduction. J Mol Biol 366, 965–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Verschueren KHG, Pumpor K, Anemüller S, Chen S, Mesters JR & Hilgenfeld R (2008) A structural view of the inactivation of the SARS‐coronavirus main proteinase by benzotriazole esters. Chem Biol 15, 597–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Li C, Qi Y, Teng X, Yang Z, Wei P, Zhang C, Tan L, Zhou L, Liu Y & Lai L (2010) Maturation mechanism of severe acute respiratory syndrome (SARS) coronavirus 3C‐like proteinase. J Biol Chem 285, 28134–28140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Muramatsu T, Kim YT, Nishii W, Terada T, Shirouzu M & Yokoyama S (2013) Autoprocessing mechanism of severe acute respiratory syndrome coronavirus 3C‐like protease (SARS‐CoV 3CLpro) from its polyproteins. FEBS J 280, 2002–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chen S, Jonas F, Shen C & Hilgenfeld R (2010) Liberation of SARS‐CoV main protease from the viral polyprotein: N‐terminal autocleavage does not depend on the mature dimerization mode. Protein Cell 1, 59–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhang S, Zhong N, Xue F, Kang X, Ren X, Chen J, Jin C, Lou Z & Xia B (2010) Three‐dimensional domain swapping as a mechanism to lock the active conformation in a super‐active octamer of SARS‐CoV main protease. Protein Cell 1, 371–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bacha U, Barrila J, Gabelli SB, Kiso Y, Mario Amzel L & Freire E (2008) Development of broad‐spectrum halomethyl ketone inhibitors against coronavirus main protease 3CLpro . Chem Biol Drug Des 72, 34–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Al‐Gharabli SI, Shah ST, Weik S, Schmidt MF, Mesters JR, Kuhn D, Klebe G, Hilgenfeld R & Rademann J (2006) An efficient method for the synthesis of peptide aldehyde libraries employed in the discovery of reversible SARS coronavirus main protease (SARS‐CoV Mpro) inhibitors. ChemBioChem 7, 1048–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Schmidt MF, Isidro‐Llobet A, Lisurek M, El‐Dahshan A, Tan J, Hilgenfeld R & Rademann J (2008) Sensitized detection of inhibitory fragments and iterative development of non‐peptidic protease inhibitors by dynamic ligation screening. Angew Chem Int Ed Engl 47, 3275–3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhu L, George S, Schmidt MF, Al‐Gharabli SI, Rademann J & Hilgenfeld R (2011) Peptide aldehyde inhibitors challenge the substrate specificity of the SARS‐coronavirus main protease. Antiviral Res 92, 204–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Akaji K, Konno H, Mitsui H, Teruya K, Shimamoto Y, Hattori Y, Ozaki T, Kusunoki M & Sanjoh A (2011) Structure‐based design, synthesis, and evaluation of peptide‐mimetic SARS 3CL protease inhibitors. J Med Chem 54, 7962–7973. [DOI] [PubMed] [Google Scholar]
- 50. Lee TW, Cherney MM, Huitema C, Liu J, James KE, Powers JC, Eltis LD & James MN (2005) Crystal structures of the main peptidase from the SARS coronavirus inhibited by a substrate‐like aza‐peptide epoxide. J Mol Biol 353, 1137–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lee TW, Cherney MM, Liu J, James KE, Powers JC, Eltis LD & James MN (2007) Crystal structures reveal an induced‐fit binding of a substrate‐like aza‐peptide epoxide to SARS coronavirus main peptidase. J Mol Biol 366, 916–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Goetz DH, Choe Y, Hansell E, Chen YT, McDowell M, Jonsson CB, Roush WR, McKerrow J & Craik CS (2007) Substrate specificity profiling and identification of a new class of inhibitor for the major protease of the SARS coronavirus. Biochemistry 46, 8744–8752. [DOI] [PubMed] [Google Scholar]
- 53. Chuck CP, Chen C, Ke Z, Wan DC, Chow HF & Wong KB (2013) Design, synthesis and crystallographic analysis of nitrile‐based broad‐spectrum peptidomimetic inhibitors for coronavirus 3C‐like proteases. Eur J Med Chem 59, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhang J, Pettersson HI, Huitema C, Niu C, Yin J, James MN, Eltis LD & Vederas JC (2007) Design, synthesis, and evaluation of inhibitors for severe acute respiratory syndrome 3C‐like protease based on phthalhydrazide ketones or heteroaromatic esters. J Med Chem 50, 1850–1864. [DOI] [PubMed] [Google Scholar]
- 55. Yin J, Niu C, Cherney MM, Zhang J, Huitema C, Eltis LD, Vederas JC & James MN (2007) A mechanistic view of enzyme inhibition and peptide hydrolysis in the active site of the SARS‐CoV 3C‐like peptidase. J Mol Biol 371, 1060–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Steinmetzer T, Hauptmann J & Stürzebecher J (2001) Advances in the development of thrombin inhibitors. Expert Opin Investig Drugs 10, 845–864. [DOI] [PubMed] [Google Scholar]
- 57. Klootwijk P, Lenderink T, Meij S, Boersma H, Melkert R, Umans VA, Stibbe J, Müller EJ, Poortermans KJ, Deckers JW et al (1999) Anticoagulant properties, clinical efficacy and safety of efegatran, a direct thrombin inhibitor, in patients with unstable angina. Eur Heart J 20, 1101–1111. [DOI] [PubMed] [Google Scholar]
- 58. Pawlotsky JM (2013) Treatment of chronic hepatitis C: current and future. Curr Top Microbiol Immunol 369, 321–342. [DOI] [PubMed] [Google Scholar]
- 59. Tan J, George S, Kusov Y, Perbandt M, Anemüller S, Mesters JR, Norder H, Coutard B, Lacroix C, Leyssen P et al (2013) 3C protease of enterovirus 68: structure‐based design of Michael acceptor inhibitors and their broad‐spectrum antiviral effects against picornaviruses. J Virol 87, 4339–4351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Chen L, Gui C, Luo X, Yang Q, Gunther S, Scandella E, Drosten C, Bai D, He X, Ludewig B et al (2005) Cinanserin is an inhibitor of the 3C‐like proteinase of severe acute respiratory syndrome coronavirus and strongly reduces virus replication in vitro . J Virol 79, 7095–7103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Bacha U, Barrila J, Velazquez‐Campoy A, Leavitt SA & Freire E (2004) Identification of novel inhibitors of the SARS coronavirus main protease 3CLpro . Biochemistry 43, 4906–4912. [DOI] [PubMed] [Google Scholar]
- 62. Chen LR, Wang YC, Lin YW, Chou SY, Chen SF, Liu LT, Wu YT, Kuo CJ, Chen TS & Juang SH (2005) Synthesis and evaluation of isatin derivatives as effective SARS coronavirus 3CL protease inhibitors. Bioorg Med Chem Lett 15, 3058–3062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lu IL, Mahindroo N, Liang PH, Peng YH, Kuo CJ, Tsai KC, Hsieh HP, Chao YS & Wu SY (2006) Structure‐based drug design and structural biology study of novel nonpeptide inhibitors of severe acute respiratory syndrome coronavirus main protease. J Med Chem 49, 5154–5161. [DOI] [PubMed] [Google Scholar]
- 64. Tsai KC, Chen SY, Liang PH, Lu IL, Mahindroo N, Hsieh HP, Chao YS, Liu L, Liu D, Lien W et al (2006) Discovery of a novel family of SARS‐CoV protease inhibitors by virtual screening and 3D‐QSAR studies. J Med Chem 49, 3485–3495. [DOI] [PubMed] [Google Scholar]
- 65. Wu CY, King KY, Kuo CJ, Fang JM, Wu YT, Ho MY, Liao CL, Shie JJ, Liang PH & Wong CH (2006) Stable benzotriazole esters as mechanism‐based inactivators of the severe acute respiratory syndrome 3CL protease. Chem Biol 13, 261–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Hilgenfeld R & Pumpor K (2006) Sometimes intermediates do the job!. Chem Biol 13, 235–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Turlington M, Chun A, Tomar S, Eggler A, Grum‐Tokars V, Jacobs J, Daniels JS, Dawson E, Saldanha A, Chase P et al (2013) Discovery of N‐(benzo[1,2,3]triazol‐1‐yl)‐N‐(benzyl)acetamidophenyl) carboxamides as severe acute respiratory syndrome coronavirus (SARS‐CoV) 3CLpro inhibitors: identification of ML300 and noncovalent nanomolar inhibitors with an induced‐fit binding. Bioorg Med Chem Lett 23, 6172–6177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Ghosh AK, Gong G, Grum‐Tokars V, Mulhearn DC, Baker SC, Coughlin M, Prabhakar BS, Sleeman K, Johnson ME & Mesecar AD (2008) Design, synthesis and antiviral efficacy of a series of potent chloropyridyl ester‐derived SARS‐CoV 3CLpro inhibitors. Bioorg Med Chem Lett 18, 5684–5688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Barretto N, Jukneliene D, Ratia K, Chen Z, Mesecar AD & Baker SC (2005) The papain‐like protease of severe acute respiratory syndrome coronavirus has deubiquitinating activity. J Virol 79, 15189–15198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Lindner HA, Fotouhi‐Ardakani N, Lytvyn V, Lachance P, Sulea T & Ménard R (2005) The papain‐like protease from the severe acute respiratory syndrome coronavirus is a deubiquitinating enzyme. J Virol 79, 15199–15208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Devaraj SG, Wang N, Chen Z, Chen Z, Tseng M, Barretto N, Lin R, Peters CJ, Tseng CT, Baker SC et al (2007) Regulation of IRF‐3‐dependent innate immunity by the papain‐like protease domain of the severe acute respiratory syndrome coronavirus. J Biol Chem 282, 32208–32221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Frieman M, Ratia K, Johnston RE, Mesecar AD & Baric RS (2009) Severe acute respiratory syndrome coronavirus papain‐like protease ubiquitin‐like domain and catalytic domain regulate antagonism of IRF3 and NF‐κB signaling. J Virol 83, 6689–6705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Clementz MA, Chen Z, Banach BS, Wang Y, Sun L, Ratia K, Baez‐Santos YM, Wang J, Takayama J, Ghosh AK et al (2010) Deubiquitinating and interferon antagonism activities of coronavirus papain‐like proteases. J Virol 84, 4619–4629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Lindner HA, Lytvyn V, Qi H, Lachance P, Ziomek E & Ménard R (2007) Selectivity in ISG15 and ubiquitin recognition by the SARS coronavirus papain‐like protease. Arch Biochem Biophys 466, 8–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Ratia K, Saikatendu KS, Santarsiero BD, Barretto N, Baker SC, Stevens RC & Mesecar AD (2006) Severe acute respiratory syndrome coronavirus papain‐like protease: structure of a viral deubiquitinating enzyme. Proc Natl Acad Sci USA 103, 5717–5722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Chou CY, Lai HY, Chen HY, Cheng SC, Cheng KW & Chou YW (2014) Structural basis for catalysis and ubiquitin recognition by the severe acute respiratory syndrome coronavirus papain‐like protease. Acta Crystallogr D Biol Crystallogr 70, 572–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Ratia K, Kilianski A, Baez‐Santos YM, Baker SC & Mesecar A (2014) Structural basis for the ubiquitin‐linkage specificity and deISGylating activity of SARS‐CoV papain‐like protease. PLoS Pathog 10, e1004113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Ratia K, Pegan S, Takayama J, Sleeman K, Coughlin M, Baliji S, Chaudhuri R, Fu W, Prabhakar BS, Johnson ME et al (2008) A noncovalent class of papain‐like protease/deubiquitinase inhibitors blocks SARS virus replication. Proc Natl Acad Sci USA 105, 16119–16124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Ghosh AK, Takayama J, Aubin Y, Ratia K, Chaudhuri R, Baez Y, Sleeman K, Coughlin M, Nichols DB, Mulhearn DC et al (2009) Structure‐based design, synthesis, and biological evaluation of a series of novel and reversible inhibitors for the severe acute respiratory syndrome‐coronavirus papain‐like protease. J Med Chem 52, 5228–5240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Ghosh AK, Takayama J, Rao KV, Ratia K, Chaudhuri R, Mulhearn DC, Lee H, Nichols DB, Baliji S, Baker SC et al (2010) Severe acute respiratory syndrome coronavirus papain‐like novel protease inhibitors: design, synthesis, protein‐ligand X‐ray structure and biological evaluation. J Med Chem 53, 4968–4979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Baez‐Santos YM, Barraza SJ, Wilson MW, Agius M, Mielech AM, Davis NM, Baker SC, Larsen SD & Mesecar AD (2014) X‐ray structural and biological evaluation of a series of potent and highly selective inhibitors of human coronavirus papain‐like proteases. J Med Chem 57, 2392–2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Chu DK, Peiris JS, Chen H, Guan Y & Poon LL (2008) Genomic characterizations of bat coronaviruses (1A, 1B and HKU8) and evidence for co‐infections in Miniopterus bats. J Gen Virol 89, 1282–1287. [DOI] [PubMed] [Google Scholar]
- 83. Woo PC, Wang M, Lau SK, Xu H, Poon RW, Guo R, Wong BH, Gao K, Tsoi HW, Huang Y et al (2007) Comparative analysis of twelve genomes of three novel group 2c and group 2d coronaviruses reveals unique group and subgroup features. J Virol 81, 1574–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Lau SK, Poon RW, Wong BH, Wang M, Huang Y, Xu H, Guo R, Li KS, Gao K, Chan KH et al (2010) Coexistence of different genotypes in the same bat and serological characterization of Rousettus bat coronavirus HKU9 belonging to a novel Betacoronavirus subgroup. J Virol 84, 11385–11394. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 85. Ren Z, Yan L, Zhang N, Guo Y, Yang C, Lou Z & Rao Z (2013) The newly emerged SARS‐like coronavirus HCoV‐EMC also has an ‘Achilles’ heel’: current effective inhibitor targeting a 3C‐like protease. Protein Cell 4, 248–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Lei J, Mesters JR, Drosten C, Anemüller S, Ma Q & Hilgenfeld R (2014) Crystal structure of the MERS‐coronavirus papain‐like protease reveals unusual, potentially druggable active‐site features. Antiviral Res 109, 72–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Coutard B, Gorbalenya AE, Snijder EJ, Leontovich AM, Poupon A, De Lamballerie X, Charrel R, Gould EA, Gunther S, Norder H et al (2008) The VIZIER project: preparedness against pathogenic RNA viruses. Antiviral Res 78, 37–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Coutard B & Canard B (2010) The VIZIER project: overview; expectations; and achievements. Antiviral Res 87, 85–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Hilgenfeld R (2010) Structure‐based antivirals for emerging and neglected RNA viruses: an emerging field for medicinal chemistry in academia. Future Med Chem 2, 1061–1067. [DOI] [PubMed] [Google Scholar]