

Abstract
The 3C‐like protease of the severe acute respiratory syndrome (SARS) coronavirus has a C‐terminal extra domain in addition to the chymotrypsin‐fold adopted by piconavirus 3C proteases hosting the complete catalytic machinery. Previously we identified the extra domain to be involved in enzyme dimerization which has been considered essential for the catalytic activity. In an initial attempt to map out the extra‐domain residues critical for dimerization, we have systematically generated 15 point mutations, five deletions and one triple mutation and subsequently characterized them by enzymatic assay, dynamic light scattering, CD and NMR spectroscopy. The results led to identification of four regions critical for enzyme dimerization. Interestingly, Asn214Ala mutant with a significant tendency to form a monomer still retained ≈ 30% activity, indicating that the relationship between the activity and dimerization might be very complex. Very surprisingly, two regions (one over Ser284–Thr285–Ile286 and another around Phe291) were discovered on which Ala‐mutations significantly increased the enzymatic activities. Based on this, a super‐active triple‐mutant STI/A with a 3.7‐fold activity enhancement was thus engineered by mutating residues Ser284, Thr285 and Ile286 to Ala. The dynamic light scattering, CD and NMR characterizations indicate that the wild‐type (WT) and STI/A mutant share similar structural and dimerization properties, thus implying that in addition to dimerization, the extra domain might have other mechanisms to regulate the catalytic machinery. We rationalized these results based on the enzyme structure and consequently observed an interesting picture: the majority of the dimerization‐critical residues plus Ser284–Thr285–Ile286 and Phe291 are clustered together to form a nano‐scale channel passing through the central region of the enzyme. We therefore speculate that this channel might play a role in relaying regulatory effects from the extra domain to the catalytic machinery.
Abbreviations
- DLS
dynamic light scattering
- DTT
dithiothreitol
- FRET
fluorescence resonance energy transfer
- GST
glutathione S‐transferase
- IPTG
isopropyl‐1‐thio‐d‐galactopyranoside
- SARS
severe acute respiratory syndrome
- SARS 3CLp
SARS 3C‐like protease
A contagious human disease now called severe acute respiratory syndrome (SARS), characterized by high fever, malaise, rigor, headache, and nonproductive cough, suddenly appeared at the end of 2002 and then spread very rapidly to 29 countries [1, 2]. Until 2003, 8096 probable SARS cases with 774 deaths were documented (http://www.who.int/csr/sars/en/). The outbreak of this disease not only imposed a worldwide health hazard but also caused great damage to both the regional and global economies. To combat this unprecedented challenge, intense efforts from governmental agencies and academic scientists all over the world have been immediately directed to identifying its causative agent and to developing effective strategies to halt SARS. Indeed, shortly after a novel coronavirus was identified to be the pathogenic agent of SARS [3, 4]. Although now SARS has dramatically disappeared, the possibility still exists that SARS may come back. Moreover, besides SARS, other coronavirus members are also major causes of upper respiratory tract illness in humans [5, 6, 7, 8]. Also, the SARS outbreak was considered to carry essential elements of the bio‐terror attack [9, 10]. To this end, further study on the SARS and other coronaviruses is urgently needed; in particular so far no efficacious therapy or preventive treatment has been available.
Coronavirus belongs to the Coronaviridae, which are enveloped, positive‐stranded RNA viruses with the largest single‐stranded RNA genome (27–31 kb) among known RNA viruses [5, 6, 7, 8]. It is well known that in coronavirus the functional viral proteins required for genome replication and transcription are released from proteolytic cleavage of two very large replicative polyproteins encoded by the large replicase gene. The cleavage of the polyproteins is usually executed by two to three cysteine proteases, one with a chymotrypsin fold and the other two with a papain‐like topology [5, 6, 7, 8, 11, 12, 13]. In particular, the 33‐kDa ‘main proteinase’, or 3C‐like protease with a chymotrypsin‐fold is responsible for the cleavage of the majority of the sites and as a result served as a key target for drug design [11]. Recently two studies reported successful inhibitor design against coronavirus‐associated diseases [12, 13].
Immediately after the SARS outbreak in Singapore, we selected the 3CL protease as our SARS research target and identified its amino acid sequence out of the SARS Coronavirus genome [14, 15, 16]. Even at the very beginning, we were puzzled by the existence of a unique domain in the coronavirus 3CL protease in addition to chymotrypsin‐fold shared with the piconavirus 3C protease. In fact, the coronavirus 3CL protease with ≈ 310 residues was so named previously to highlight its similarity in the structure, enzymatic mechanism and specificity to the picornavirus 3C protease with ≈ 180 residues which only form a chymotrypsin‐fold hosting the complete catalytic machinery. However, some existing results at that time already implied that the extra domain in the coronavirus 3CL protease might play roles in the enzymatic activity [17, 18]. Therefore, we initiated a domain dissection approach to explore the function of the extra domain which eventually led to the first discovery of its critical involvement in dimerization [19] considered to be essential for the catalytic activity [20].
In an initial attempt to map the dimerization interface on the extra domain, we mutated all 15 single residues on the extra‐domain which have close contacts (= 7 Å) with any residues on another protomer to Ala, and constructed one triple‐mutation and five deletion mutants. The results obtained not only allowed the identification of the four regions critical for dimerization, but also led to the finding that the Asn214Ala mutant − which has a strong tendency to form monomer − still retained ≈ 30% activity. More surprisingly, we discovered two extra domain regions on which Ala‐mutations caused significant increases in proteolytic activity. The results strongly imply that the relationship between the catalytic activity and dimerization might be very complex and in addition to dimerization, the extra domain might have other mechanisms in the regulation of the catalytic machinery.
Results
Expression and enzymatic activities of the wild‐type and mutated SARS 3C‐like proteases
We have succeeded in obtaining correct DNA constructs encoding all 15 single‐; one triple and five deletion mutants. These glutathione S‐transferase (GST)‐fusion constructs were transformed into Escherichia coli BL21 cell strain for overexpression. The results demonstrated that 15 single‐, one triple‐, ND(Δ1‐5) and LHD(Δ293‐306) were well expressed and soluble while the CD(Δ278‐306); LR(276–290/2G) and LR(276–290/4G) were not expressable. The cells carrying these three constructs grew much slower, indicating that the mutant proteins with the loop deleted might be toxic to cells, probably due to severe aggregation or amyloid formation. The expressed GST‐fusion proteins were purified by using the glutathione Sepharose affinity column and the pure mutated 3CL proteases were further obtained by in‐gel thrombin‐cleavage followed by FPLC gel‐filtration purification. The SDS/PAGE gel and MALDI‐TOF mass checking indicated that all recombinant proteins purified by this procedure were homogenous and intact (data not shown).
The enzymatic activities of the wild‐type and mutated proteases were measured by use of a FRET‐based assay at three different NaCl concentrations. Figure 1 presents the enzymatic activity profiles for the wild‐type and 18 mutated proteases in the assay buffer without NaCl. Based on this, the mutated proteases could be divided into four groups. The first group includes Glu288A, Asp289A, Glu290A, Arg298A, Gln299A, ND and LCD which showed dramatic losses of activities and retained < 10% of the wild‐type proteolytic activity. The second group contains Asn214Ala, Leu282Ala and Cys300Ala which had significant activity decreases but still preserve > 30% of the wild‐type activity while the third group consists of Thr280Ala, Gly283Ala and Ser301Ala which had no significant activity differences from the wild‐type protease. Very interestingly, the replacement of residues Ser284, Thr285, Ile286 or Phe291 by Ala gave rise to the mutated proteases with enzymatic activities higher than that of the wild‐type. Therefore, we constructed a triple‐mutant with three‐neighbouring residues Ser284, Thr285 and Ile286 all mutated to Ala and strikingly this led to a ‘super‐active’ SARS 3C‐like protease (SARS 3CLp) which possessed a 3.7‐fold enhanced activity. As such, we placed Ser284Ala, Thr285Ala, Ile286A, Phe291 and the super‐active mutants together as the fourth group.
Figure 1.
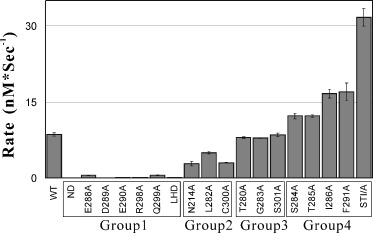
Enzymatic activities of the wild‐type (WT) and mutated SARS 3CLp. The FRET‐based activity assay was carried out by monitoring the increase of the emission fluorescence at 538 nm upon proteolytic cleavage of the substrate peptide Dabcyl‐KTSAVLQSGF RKME‐Edans. The reaction mixture contains 1 µm wild‐type or mutated protease and 3 µm fluorogenic substrate in a pH 7.0 buffer with 5.5 mm NaH2PO4 and 5 mm DTT. Each activity value was the average of four independent measurements. Based on the activities, all mutated proteases were categorized into four groups which are boxed and labelled.
It is also worthwhile to note that the enzymatic activities of the wild‐type and mutated proteases were all highly salt‐dependent. When 100 mm NaCl salt was introduced, only ≈ 50% of the enzymatic activities was preserved for almost all proteases. If the NaCl salt concentration was further increased up to 1050 mm, the residual activities accounted for < 10% for all proteases except the superactive mutant which still retained ≈ 16% activity (data not shown).
Dimerization properties characterized by DLS
Dynamic light scattering (DLS) was used to measure the apparent molecular mass resulting from monomer–dimer equilibrium of the wild‐type and mutated proteases at three different NaCl concentrations. As shown in Fig. 2, the super‐active mutant had an apparent molecular mass almost identical to that of the wild‐type protease. However, the apparent molecular masses of the mutated proteases in the first group were, on average, ≈ 22 kDa smaller than that of the wild‐type (56.3 kDa). This observation strongly suggested that these residues play an important role in the enzyme dimerization. Here it is particularly interesting to note that Asn214Ala which still retained ≈ 30% of the wild‐type proteolytic activity had a small apparent molecular mass (32.6 kDa), indicating that Asn214Ala had a dominant tendency to form a monomer. Therefore, at least four regions of the SARS 3CLp might be significantly associated with enzyme dimerization: (1) the N‐terminal residues 1–5 as previously identified [21, 22]; (2) the residue Asn214; (3) the region around residues Glu288‐Asp289‐Glu290 which had a close contact with the N‐terminus [21, 22]; and (4) the C‐terminal last helix region around residues Arg298–Gln299. Moreover, introduction of higher concentrations of NaCl statistically had no disrupting effect on enzyme dimerization (data not shown), indicating that the reduced activities in the presence of NaCl (Fig. 1) were not due to the disruption of dimerization.
Figure 2.
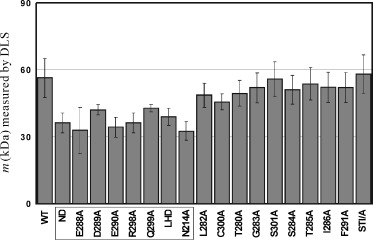
The apparent molecular mass (m) of the wild‐type and mutated SARS 3C‐like protease (SARS 3CLp). The apparent molecular mass of the wild‐type and mutated 3C‐like proteases in a rapid equilibrium between a monomer and dimer were measured by use of dynamic light scattering at 20 °C on a DynaPro‐MS/X instrument (Protein Solutions Inc.). The protein samples with a concentration of 100 µm were prepared in a pH 7.0 buffer containing 5.5 mm NaH2PO4, 5 mm DTT. The values of apparent molecular mass were calculated and averaged from 10 measurements using the built‐in analysis software. The mutants with significantly decreased apparent molecular mass are boxed.
Structural properties characterized by CD and NMR
Far‐UV CD spectra were collected for the wild‐type and mutated proteases to evaluate their secondary structures. As seen in Fig. 3, although the Ala‐mutations and fragment deletions induced some changes, the changes were not very significant. Therefore, it is very unlikely that a significant change in the secondary structure occurred upon mutation and deletion.
Figure 3.
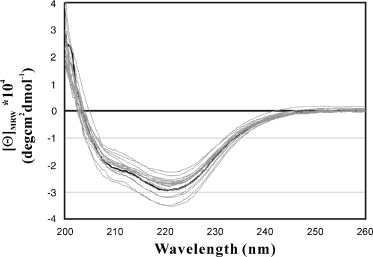
Far‐UV CD spectra of the wild‐type and mutated SARS 3CLp. The far‐UV CD spectra were collected at 20 °C in 10 mm phosphate buffer at pH 7.0 containing ≈ 5 µm proteins and 5 mm DTT. The CD spectrum of the wild‐type protease are shown in black and the spectra of the mutated protease are shown in grey.
The tertiary packing of the wild‐type and mutated proteases was first assessed by 1D 1H NMR spectra. As indicated in Fig. 4A–D, similar to the wild‐type protease, all mutants gave rise to several NMR resonance peaks at a very upfield region (−1.1 to 0.5 p.p.m), indicating that they may own well‐packed tertiary structures. A detailed examination of the NMR spectra revealed that the mutants in the first group plus Asn214Ala had spectra with sharper resonance peaks than the wild‐type protease, consistent with the observation that these mutants had significantly smaller average relative molecular mass than the wild‐type because the NMR resonance peak width is size‐dependent and a larger protein will give rise to broader NMR peaks due to the short T2 (transverse relaxation time) [23, 24]. Interestingly, regardless of the DLS relative molecular mass, all mutant proteases gave rise to one but not two sets of upfield resonance peaks similar to the wild‐type, indicating that the monomer–dimer equilibrium is a fast exchange process on the NMR time scale [23, 24].
Figure 4.
The structural properties of the wild‐type and mutated 3CLp assessed by 1D 1H NMR. One‐dimensional 1H NMR spectra were acquired at 20 °C in a pH 7.0 buffer containing 5.5 mm NaH2PO4, 5 mm DTT. The aliphatic side‐chain regions were shown for the wild‐type and group 1 mutated proteases (A); wild‐type and group 2 (B); wild‐type and group 3 (C); wild‐type and group 4 (D). The upfield NMR resonance peaks resulting from a tight tertiary packing are indicated by arrows.
To gain more detailed insights, wild‐type, ND, Glu288Ala, Glu290Ala, Arg298Ala, Gln299Ala, LHD and STI/A mutants were further 15N‐labelled and subjected to HSQC assessment. As seen in Fig. 5A, the HSQC spectrum of the wild‐type protease are very broad and only a small portion of resonance peaks are visible, consistent with our previous report [19]. This is mostly due to the very large relative molecular mass for the wild‐type protease in a dimmer‐dominant state. Interestingly, the STI/A mutant had a HSQC spectrum (Fig. 5B) very similar to that of the wild‐type, indicating that both STI/A and wild‐type proteases shared similar structural and dimerization properties. On the other hand, Arg298Ala and ND mutants which were shown by DLS to have a monomer‐like relative molecular mass (Fig. 2) had very‐dispersed HSQC spectra with many resonance peaks detectable (Figs 5C and E). This observation indicates that the two mutants were in monomer‐dominant states which were also well‐structured [25, 26, 27]. When our manuscript was under a previous review, a paper was published in which both ND and LHD mutants were demonstrated to have a great tendency to form a monomer by analytic ultracentrifuge analysis [28]. Interestingly, although here the LHD mutant also had a well‐dispersed HSQC spectrum (Fig. 5D), the visible HSQC resonance peaks were less than those of Arg298Ala and ND mutants. Therefore, in the current case, it appears that the HSQC line‐width was modulated both by the average relative molecular mass resulting from fast intermolecular monomer–dimer equilibrium as well as by intramolecular conformational exchanges. It is very likely that although the LHD mutant had a dominant tendency to form a monomer [28], the deletion of the last helix may provoke µs–ms conformational exchanges to some degree which caused some HSQC peaks too broad to be clearly distinguished from the noise signals. However, if the HSQC spectral level was lowered, it could be found that most HSQC peaks of the ND, Glu288Ala, Glu290Ala, Arg298Ala, Gln299Ala, LHD and STI/A are almost superimposable to those of the wild‐type protease, as exemplified by Fig. 5F in which the HSQC spectra of the Arg298Ala and wild‐type proteases were superimposed at a lower spectral level. These results, together with CD spectra, indicated that very likely − over the well‐packed regions such as chymotrypsin‐fold and the C‐terminal extra domain − no dramatic change occurred for the secondary structures and tertiary structures upon mutation or deletion. However, certainly the mutation and deletion resulted in minor structural changes as well as conformational exchanges on the µs–ms time scale to different degrees which consequently made some HSQC too broad to be detected, as we recently discovered on the CHABII molecule [25, 26].
Figure 5.
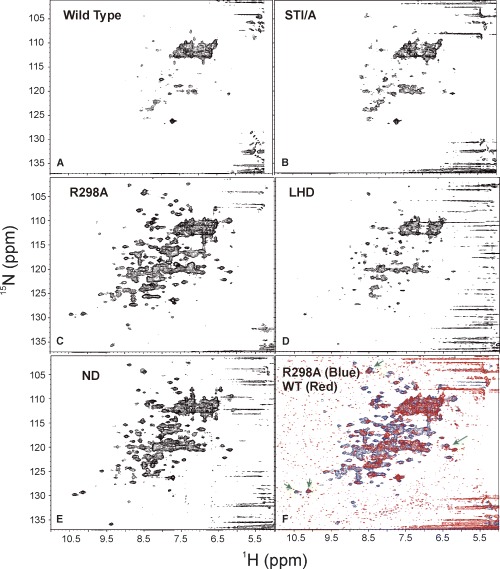
The structural properties of the wild‐type and mutated 3CLp assessed by two‐dimensional 1H‐15N HSQC NMR. Two‐dimensional 1H‐15N HSQC NMR spectra were acquired for 15N isotope‐labelled wild‐type and mutated proteases at 20 °C in a pH 7.0 buffer containing 5.5 mm NaH2PO4, 5 mm DTT. (A) HSQC spectrum for wild‐type 3CL protease; (B) for super‐active STI/A mutant; (C) for R298A mutant; (D) for last‐helix deleted mutant LHD; (E) for the N‐terminal five‐residue deleted mutant ND; and (F) superimposition of the HSQC spectra of R298A mutant (blue) and wild‐type (red).
Discussion
Due to the central role in the virus replication, the SARS 3CLp has now been established as a critical target for design of anti‐SARS drugs and consequently extensive studies were reported on its structural and enzymatic properties [11‐13,19, 20, 21, 22, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37]. By a protein dissection approach, we first demonstrated the critical role of the extra domain in both dimerization and activity of the SARS 3CLp [19]. Because dimerization was extensively considered to be essential for enzymatic activity [20], more investigations have been directed towards understanding the dimerization–activity relationship of the SARS 3CLp [22, 28, 37]. Recently the role of the N‐ and C‐terminal residues in dimerization has been assessed by analytic ultracentrifugation analysis [28]. Strikingly, in this report [28], two mutated proteases exactly corresponding to our ND and LHD mutants were both demonstrated by analytic ultracentrifuge analysis to have a great tendency to form a monomer.
Our current results suggest that in addition to well‐recognized N terminus and its contact residues Glu290 [21, 22, 28], other extra‐domain regions are also critical for enzyme dimerization: namely the region around Asn214, region over Glu288–Asp289–Glu290, and the region over Arg298–Gln299 on the C‐terminal last helix. More strikingly, our study has revealed that the mutant Asn214Ala still retained ≈ 30% activity although it had a tendency similar to ND and LHD to form a monomer (according to our results to the monomer–dimer distribution study of ND and LHD mutants) [28]. This result implies that the relationship between dimerization and activity might be very complex and needs to be carefully addressed in the future. If the residues critical for dimerization were mapped back to the dimeric structure of the SARS 3CLp, a very interesting picture is observed. As seen in Fig. 6A, the dimerization regions are clustered together to form a tertiary packing core in the middle of the dimeric enzyme. In fact in the core region, only two direct intermonomer contacts exist, namely between Glu290 of one protomer and Arg4 of another. Indeed, the importance of the Arg4–Glu290 interaction in dimerization has been reported previously [22]. The role of the identified residues other than Arg4 and Glu290 in dimerization might be in maintaining the correct side‐chain conformations of the residues Glu290 and Arg4 by interacting with them. Indeed, within the same protomer, Asn214 has close contacts with Phe3 and Gln299 while Glu288 closely contacts to Lys5. Alternatively, some newly identified residues might contribute to dimerization by interacting with other nonextra‐domain residues which are not explored in the present study. It is also of note that although the dimerization interface appears relatively large, a single‐mutation in particular on Asn214, Glu288, Asp289, Arg298 or Gln299 is sufficient to significantly disrupt enzyme dimerization.
Figure 6.
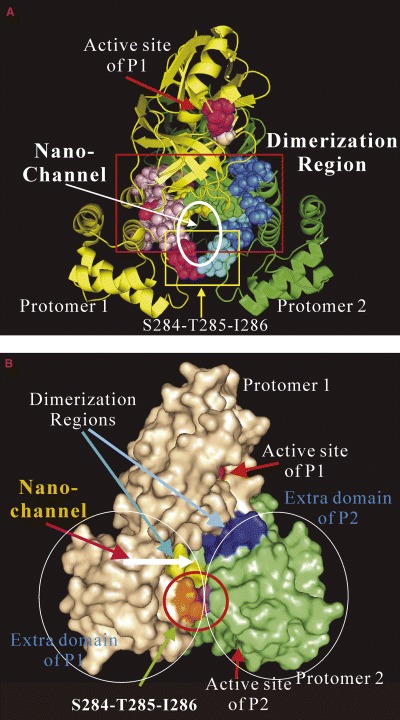
Visualization of residues critical for dimerization and regulation. (A) Ribbon representation of the dimeric SARS 3CLp with the residues identified critical for dimerization and regulation drawn in the sphere mode. The N‐terminal five residues of protomer 1 are shown in yellow while those of protomer 2 are shown in green. Dimerization‐critical residues on the extra domain are in pink for protomer 1 and blue for protomer 2. The hotpink spheres are used to indicate residues Ser284–Thr285–Ile286 and Phe291 for protomer 1 and cyan for protomer 2. The proposed nano‐channel is indicated. The active site residues His41 and Cys145 are shown in red and light‐brown, respectively. (B) Surface representation with residues shown in the same way. The two Ser284–Thr285–Ile286 loops from each of the protomers contact closely with each other to constitute the bottom wall of the channel.
Another striking and surprising result in the present study is the discovery of two regions (Ser284–Thr285–Ile286 and Phe291) on which reducing the side‐chain volumes significantly boosted enzymatic activity. Based on this observation, a super‐active mutant with a 3.7‐fold enhancement of proteolytic activity was engineered by mutating three residues Ser284–Thr285–Ile286 to Ala. As judged from the DLS results and NMR HSQC spectra, the triple‐mutant has structural and dimerization properties similar to those of the wild‐type protease. This observation strongly implies that in addition to dimerization, the extra domain might have other mechanisms for regulating the catalytic machinery. It would be of significant interest in the future to introduce the mutations into viral infectious clones to test whether these mutations will increase the viral fitness or viral replication capacity.
As seen in Fig. 6A, the two extra‐domain enhancing regions are far away from the active sites of both protomers. This raises a very interesting question as to how these regions can achieve regulation of the catalytic machinery which is far away. Possibly, the slight but long‐range alterations of structure and dynamics of the enzyme, as well as enzyme–solvent interactions upon mutation could account for the observed regulatory effects. As seen in Fig. 6A and B the majority of the dimerization residues plus Phe291, Ser284–Thr285–Ile286 are clustered together to form a molecular channel passing through the middle region of the enzyme. Owing to its molecular‐scale size, we thus call it a nano‐channel which has a diameter of ≈12 Å and a surface area of ≈ 360 Å2, with the up‐wall length of ≈ 18 Å and bottom‐wall length of ≈ 7 Å. The upper wall of the channel appears to have connectivity to the inner cavities while the two ends of the channel are also further connected to the surface cavities of the enzyme molecule which eventually lead to the active‐site pockets. Interestingly Ala mutations of the residues Ser284–Thr285–Ile286 which significantly enhanced catalytic activity would remove the bottom wall of the channel. Based on this, we thus speculate that this channel might serve as a regulator sitting in the central region of the enzyme, and play a role in relaying the regulatory effect from the extra domain to the catalytic machinery. Certainly, the future determination of the high‐resolution structures of the mutant proteases such as ND, Arg298Ala, Asn214 and super‐active mutant STI/A may shed light on the molecular mechanism as to why dimerization is critical for the activity of the wild‐type protease as well as to why Ala mutations of Ser284, Thr285 and Ile286 can dramatically enhance the catalytic activity.
In summary, our study revealed several previously unknown phenomena associated with the coronavirus 3CL protease which highlight the regulatory roles of the extra domain on the catalytic machinery. Interestingly, although the piconavirus 3C protease shares a basic catalytic machinery with the coronavirus 3CL protease [38, 39, 40, 41], it appears that the latter places its catalytic machinery under extensive regulations by the extra domain. Our results thus illustrate an interesting example showing how nature can nicely engineer a regulatable 3CL protease by introducing an additional domain to the catalytic machinery.
Experimental procedures
Selection of residues for site‐directed mutagenesis
In an attempt to identify potential extra‐domain residues involved in dimerization, we decided to conduct alanine site‐directed mutagenesis on all extra‐domain residues which have distances equal to or less than 7 Å with any other residues on another protomer of the SARS 3CLp dimeric structure. Thus, we selected one deposited crystallographic structure of the SARS 3CLp (PDB code: 1Q2W) as a template, and subsequently added hydrogen atoms to the structure using graphic software yasara (http://www.yasara.org). A TCL/TK script was prepared to calculate the distances between the residues of the extra‐domain and those on another protomer. Consequently a total of 15 residues were listed out including Asn214, Thr280, Leu282, Gly283, S284, Thr285, Ile286, Glu288, Asp289, Glu290, Phe291, Arg298, Gln299, Cys300 and Ser301. Since a closer examination of the 3D structures including 1Q2W and other [21] revealed that both N‐ and C‐termini had a large number of contacts with the 15 selected residues, we generated three N‐ and C‐terminal deletion constructs, namely ND(Δ1‐5) with N‐terminal residues 1–5 deleted; LHD(Δ293‐306) with the last helix (residues 293–306) deleted; CD(Δ278‐306) with the C‐terminal residues 278–306 deleted. Moreover, the examination also indicated extensive contacts between the loop regions (residues 276–290) on the two protomers, we also made two loop‐replacement constructs, with the loop residues 276–290 replaced by two Gly residues for LR(276–290/2G) and by four Gly residues for LR(276–290/4G).
All Ala‐mutation and deletion constructs were obtained by manipulation of the wild‐type SARS 3CLp construct obtained previously [19]. By using DNA oligonucleotides listed in the Table S1, the single‐, triple‐ and loop‐replacement mutations were also successfully made by QuikChange® Site‐Directed Mutagenesis Kit (Stratagene, La Jolla, CA, USA), and the N‐, C‐termini deletions were achieved by PCR. The DNA fragments obtained were then inserted into the pGEX‐4T‐1 GST‐fusion expression vector (Amersham Biosciences, GE Healthcare, Little Chalfont, UK) using the BamHI/XhoI restriction sites. The sequences of all constructs were confirmed by DNA automated sequencing.
Expression and purification of the wild‐type and mutated SARS 3CLp
All of the above DNA constructs were transformed into E. coli BL21 to overexpress the GST fusion proteins as previously described [19]. Briefly, the cells were cultured at 37 °C until the absorbance at 600 nm reached ≈ 0.7. Then 0.5 mm isopropyl‐1‐thio‐d‐galactopyranoside (IPTG) was added into Luria–Bertani cell culture medium to induce the foreign protein expression at 22 °C overnight. The harvested cells were sonicated in the lysis buffer to release soluble GST proteins, which were subsequently purified using glutathione Sepharose (Amersham Biosciences). The in‐gel cleavage of the fusion proteins was performed at room temperature by incubating the fusion proteins attached to the Sepharose beads with bovine thrombin. The released recombinant proteins were further purified by a AKTA FPLC machine (Amersham Biosciences) on a gel filtration column (HiLoad 16/60 Superdex 200) equilibrated and eluted with a buffer at pH 7.4 containing 50 mm NaH2PO4, 150 mm NaCl and 28.8 mmβ‐mercaptoethanol. The eluted peak corresponding to 3CLp or its mutants was collected and buffer‐exchanged to a pH 7.0 buffer containing 10 mm NaH2PO4, 0.01% NaN3, and 5 mm dithiothreitol (DTT) for storage using Amicon Ultra‐15 centrifugal filter devices (5 kDa cutoff, Millipore, Billerica, MA, USA).
For detailed assessment of the structural properties of wild‐type, Glu288A, Asp289A, Glu290A, Arg298A, Gln299A, NDL, LHD and STI/A mutant proteases by heteronuclear 1H‐15N HSQC experiment, the proteins were prepared in 15N‐labelled forms using a similar expression protocol except for growing E. coli cells in minimal M9 media instead of the 2YT media, with an addition of (15NH4)2SO4 for 15N‐labelling. The intactness of the native and mutant proteases was confirmed by SDS/PAGE and MALDI‐TOF MS. The protein concentration was determined by the denaturant method as previously described [42, 43].
Chemical synthesis of the fluorogenic substrate peptides
The substrate peptide with a pair of internally quenched fluorescent groups Dabacyl and Edans in a sequence of Dabcyl‐KTSAVLQSGF RKME‐Edans was chemically synthesized and purified by HPLC on a RP C18 column (Vydac).
Enzymatic activity assays
The enzymatic activities of the wild‐type and mutated SARS 3CL proteases were measured by a fluorescence resonance energy transfer (FRET)‐based assay using fluorogenic substrate peptide previously described [29]. Briefly, the reaction mixture contained 1 µm protease and 3 µm fluorogenic substrate in a pH 7.0 buffer with 5.5 mm NaH2PO4 and 5 mm DTT. The enzyme activity was measured by monitoring the increase of the emission fluorescence at a wavelength of 538 nm with excitation at 355 nm using a Perkin‐Elmer LS‐50B luminescence spectrometer. For each mutant protease, the activity measurements were carried out at three different NaCl concentrations: 0 mm, 100 mm and 1050 mm while under each condition four independent measurements were performed.
DLS, CD and NMR spectroscopy
The dimerization properties of the wild‐type and all mutated 3CL proteases were assessed by use of dynamic light scattering at 20 °C on a DynaPro‐MS/X instrument (Protein Solutions Inc., Lakewood, NJ, USA). Briefly, the protein samples were dissolved in a pH 7.0 buffer containing 5.5 mm NaH2PO4, 5 mm DTT to reach a final concentration of 100 µm. For each protein, its apparent relative molecular mass was measured at three different NaCl concentrations: 0, 100 and 1050 mm. The values of apparent relative molecular mass were calculated and averaged from at least 10 measurements using the built‐in analysis software.
CD experiments were performed on a Jasco J‐810 spectropolarimeter equipped with thermal controller. The proteins were dissolved in 5.5 mm phosphate buffer at pH 7.0 with a concentration of 5 µm. Far‐UV CD spectra from 190 to 260 nm were collected using 1‐mm path length cuvette with a 0.1‐nm spectral resolution at 20 °C. Five independent scans were averaged for each sample.
One‐dimensional 1H NMR spectra for the unlabelled proteases and heteronuclear 2D 1H–15N HSQC spectra for isotope labelled proteases were acquired with a concentration of ≈ 100 µm as previously described [19] at 20 °C in a pH 7.0 buffer containing 5.5 mm NaH2PO4, 5 mm DTT on a Bruker 500 MHz NMR spectrometer equipped with both an actively shielded cryoprobe and pulse field gradient units. The structure display and manipulation were achieved by use of the graphic software pymol (http://www.pymol.org).
Acknowledgements
We thank Dr DX Liu at IMCB, Singapore for helpful comments. This research is supported by Faculty of Science, National University of Singapore Grants R154000208112 and R154000192101 to J. Song.
Supporting information
Supporting info item
References
- 1. Lee ND, Hui A, Wu P, Chan P, Cameron GM, Joynt A, Ahuja MY, Yung CB, Leung KF, To SF, et al. (2003) A major outbreak of severe acute respiratory syndrome in Hong Kong. N Engl J Med 348. [DOI] [PubMed] [Google Scholar]
- 2. Poutanen SMDE, Low B, Henry S, Finkelstein D, Rose K, Green R, Tellier R, Draker D, Adachi M, Ayers AK, et al. (2003) Identification of severe acute respiratory syndrome in Canada. N Engl J Med 348, 1995–2005. [DOI] [PubMed] [Google Scholar]
- 3. Drosten C, Gunther S, Preiser W, van der Werf S, Brodt HR, Becker S, Rabenau H, Panning M, Kolesnikova L, Fouchier RA et al. (2003) Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N Engl J Med 348, 1967–1976. [DOI] [PubMed] [Google Scholar]
- 4. Ksiazek TG, Erdman D, Goldsmith CS, Zaki SR, Peret T, Emery S, Tong S, Urbani C, Comer JA, Lim W et al. (2003) A novel coronavirus associated with severe acute respiratory syndrome. N Engl J Med 348, 1953–1966. [DOI] [PubMed] [Google Scholar]
- 5. Lai MMC & Cavanagh D (1997) The molecular biology of coronaviruses. Adv Virus Res 48, 1–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ziebuhr J, Snijder EJ & Gorbalenya AE (2000) Virus‐encoded proteinases and proteolytic processing in the Nidovirales. J Gen Virol 81, 853–879. [DOI] [PubMed] [Google Scholar]
- 7. Ziebuhr J (2003) Basically SARS is not a surprise. Coronaviruses lie in wait almost everywhere. Krankenpfl J 41, 114–115. [PubMed] [Google Scholar]
- 8. Ziebuhr J (2004) Molecular biology of severe acute respiratory syndrome coronavirus. Curr Opin Microbiol 7, 412–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Weber SG, Bottei E, Cook R & O'Connor M (2004) SARS, emerging infections, and bioterrorism preparedness. Lancet Infect Dis 4, 483–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tham KY (2004) An emergency department response to severe acute respiratory syndrome: a prototype response to bioterrorism. Ann Emerg Med 43, 6–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Anand K, Ziebuhr J, Wadhwani P, Mesters JR & Hilgenfeld R (2003) Coronavirus main proteinase (3CLpro) structure: basis for design of anti‐SARS drugs. Science 300, 1763–1767. [DOI] [PubMed] [Google Scholar]
- 12. Yang H, Xie W, Xue X, Yang K, Ma J, Liang W, Zhao Q, Zhou Z, Pei D, Ziebuhr J, et al. (2005) Design of wide‐spectrum inhibitors targeting coronavirus main proteases PLoS Biol 3, e324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen L, Gui C, Luo X, Yang Q, Gunther S, Scandella E, Drosten C, Bai D, He X, Ludewig B et al. (2005) Cinanserin is an inhibitor of the 3C‐like proteinase of severe acute respiratory syndrome coronavirus and strongly reduces virus replication in vitro. J Virol 79, 7095–7103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Marra MA, Jones SJ, Astell CR, Holt RA, Brooks‐Wilson A, Butterfield YS, Khattra J, Asano JK, Barber SA, Chan SY et al. (2003) The Genome sequence of the SARS‐associated coronavirus. Science 300, 1399–1404. [DOI] [PubMed] [Google Scholar]
- 15. Rota PA, Oberste MS, Monroe SS, Nix WA, Campagnoli R, Icenogle JP, Penaranda S, Bankamp B, Maher K, Chen MH et al. (2003) Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science 300, 1394–1399. [DOI] [PubMed] [Google Scholar]
- 16. Ruan YJ, Wei CL, Ee AL, Vega VB, Thoreau H, Su ST, Chia JM, Ng P, Chiu KP, Lim L, et al. (2003) Comparative full‐length genome sequence analysis of 14 SARS coronavirus isolates and common mutations associated with putative origins of infection. Lancet 361, 1779–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Anand K, Palm GJ & Mesters J (2002) R, Siddell, S. G., Ziebuhr, J. & Hilgenfeld, R. Structure of coronavirus main proteinase reveals combination of a chymotrypsin fold with an extra alpha‐helical domain. EMBO J 21, 3213–3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ziebuhr J, Heusipp G & Siddell SG (1997) Biosynthesis, purification, and characterization of the human coronavirus 229E, 3C‐like proteinase. J Virol 71, 3992–3997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shi J, Wei Z & Song J (2004) Dissection study on the severe acute respiratory syndrome 3C‐like protease reveals the critical role of the extra domain in dimerization of the enzyme: defining the extra domain as a new target for design of highly specific protease inhibitors. J Biol Chem 279, 24765–24773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fan K, Wei P, Feng Q, Chen S, Huang C, Ma L, Lai B, Pei J, Liu Y, Chen J & Lai L (2004) Biosynthesis, purification, and substrate specificity of severe acute respiratory syndrome coronavirus 3C‐like proteinase. J Biol Chem 279, 1637–1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yang H, Yang M, Ding Y, Liu Y, Lou Z, Zhou Z, Sun L, Mo LYeS, Pang H, Gao GF, Anand K, Bartlam M, Hilgenfeld R & Rao Z (2003) The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc Natl Acad Sci USA 100, 13190–13195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chou CY, Chang HC, Hsu WC, Lin TZ, Lin CH & Chang GG (2004) Quaternary structure of the severe acute respiratory syndrome (SARS) coronavirus main protease. Biochemistry 43, 14958–14970. [DOI] [PubMed] [Google Scholar]
- 23. Wagner G (1993) Prospects for NMR of large proteins. J Biomol NMR 3, 375–385. [DOI] [PubMed] [Google Scholar]
- 24. Tugarinov V, Hwang PM & Kay LE (2004) Nuclear magnetic resonance spectroscopy of high‐molecular‐weight proteins. Annu Rev Biochem 73, 107–146. [DOI] [PubMed] [Google Scholar]
- 25. Song J, Jamin N, Gilquin B, Vita C & Menez A (1999 February) (1999) A gradual disruption of tight side‐chain packing: 2D, 1H‐NMR characterization of acid‐induced unfolding of CHABII. Nat Struct Biol 6(2), 129–134. [DOI] [PubMed] [Google Scholar]
- 26. Wei Z & Song J (2005) Molecular mechanism underlying the thermal stability and pH‐induced unfolding of CHABII. J Mol Biol 348, 205–218. [DOI] [PubMed] [Google Scholar]
- 27. Ran X & Song J (2005) Structural insight into the binding diversity between the Tyr‐phosphorylated human ephrinBs and Nck2 SH2 domain. J Biol Chem 280, 19205–19212. [DOI] [PubMed] [Google Scholar]
- 28. Hsu WC, Chang HC, Chou CY, Tsai PJ, Lin PI & Chang GG (2005) Critical assessment of important regions in the subunit association and catalytic action of the severe acute respiratory syndrome coronavirus main protease. J Biol Chem 280, 22741–22748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kuo CJ, Chi YH, Hsu JT & Liang PH (2004) Characterization of SARS main protease and inhibitor assay using a fluorogenic substrate. Biochem Biophys Res Commun 318, 862–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bacha U, Barrila J, Velazquez‐Campoy A, Leavitt SA & Freire E (2004) Identification of novel inhibitors of the SARS coronavirus main protease 3CLpro. Biochemistry 43, 4906–4912. [DOI] [PubMed] [Google Scholar]
- 31. Blanchard JE, Elowe NH, Huitema C, Fortin PD, Cechetto JD, Eltis LD & Brown ED (2004) High‐throughput screening identifies inhibitors of the SARS coronavirus main proteinase. Chem Biol 11, 1445–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kao RY, To AP, Ng LW, Tsui WH, Lee TS, Tsoi HW & Yuen KY (2004) Characterization of SARS‐CoV main protease and identification of biologically active small molecule inhibitors using a continuous fluorescence‐based assay. FEBS Lett 576, 325–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Parera M, Clotet B & Martinez MA (2004) Genetic screen for monitoring severe acute respiratory syndrome coronavirus 3C‐like protease. J Virol 78, 14057–14061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hsu MF, Kuo CJ, Fang JM, Shie JJ, Chang KT, Chang HC, Chou CC, Ko TP, Shr HL, Chang GG, Wu YT, Wang AH & Liang PH (2005) Mechanism of the maturation process of SARS‐CoV 3CL protease. J Biol Chem 280, 31257–31266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kuang WF, Chow LP, Wu MH & Hwang LH (2005) Mutational and inhibitive analysis of SARS coronavirus 3C‐like protease by fluorescence resonance energy transfer‐based assays. Biochem Biophys Res Commun 331, 1554–1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fan K, Ma L, Han X, Liang H, Wei P, Liu Y & Lai L (2005) The substrate specificity of SARS coronavirus 3C‐like proteinase. Biochem Biophys Res Commun 329, 934–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chen S, Chen L, Tan J, Du Chen JL, Sun T, Shen J, Chen K, Jiang H & Shen X (2005) Severe acute respiratory syndrome coronavirus 3C‐like proteinase N terminus is indispensable for proteolytic activity but not for enzyme dimerization. Biochemical and thermodynamic investigation in conjunction with molecular dynamics simulations. J Biol Chem 280, 164–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Allaire M, Chernaia MM, Malcolm BA & James MN (1994) Picornaviral 3C cysteine proteinases have a fold similar to chymotrypsin‐like serine proteinases. Nature 369, 72–76. [DOI] [PubMed] [Google Scholar]
- 39. Malcolm BA (1995) The picornaviral 3C proteinases: cysteine nucleophiles in serine proteinase folds. Protein Sci 4, 1439–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Khan AR, Khazanovich‐Bernstein N, Bergmann EM & James MN (1999) Structural aspects of activation pathways of aspartic protease zymogens and viral 3C protease precursors. Proc Natl Acad Sci USA 96, 10968–10975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Seipelt J, Guarne A, Bergmann E, James M, Sommergruber W, Fita I & Skern T (1999) The structures of picornaviral proteinases. Virus Res 62, 159–168. [DOI] [PubMed] [Google Scholar]
- 42. Edelhoch H (1967) Spectroscopic determination of tryptophan and tyrosine in proteins. Biochemistry 6, 1948–1954. [DOI] [PubMed] [Google Scholar]
- 43. Pace CN, Vajdos F, Fee L, Grimsley G & Gray T (1995) How to measure and predict the molar absorption coefficient of a protein. Protein Sci 4, 2411–2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting info item